
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fully bio-based aromatic–aliphatic copolyesters: poly(butylene furandicarboxylate-co-succinate)s obtained by ring opening polymerization†
Juan Carlos
Morales-Huerta
,
Claudio Batista
Ciulik
,
Antxon Martínez
de Ilarduya
and
Sebastián
Muñoz-Guerra
 *
*
Department d'Enginyeria Química, Universitat Politècnica de Catalunya, ETSEIB, Diagonal 647, 8028 Barcelona, Spain. E-mail: sebastian.munoz@upc.edu
First published on 30th November 2016
Abstract
Poly(butylene 2,5-furandicarboxylate-co-succinate) copolyesters (coPBFxSy) have been synthesized by ring opening polymerization (ROP). Cyclic butylene 2,5-furandicarboxylate and butylene succinate oligomer mixtures, to be used as monomers for ROP, were prepared by high dilution condensation and enzymatic cyclization reactions, respectively. Two different catalytic systems, tin dioctanoate and supported Candida antarctica lipase B (CALB), were used for polymerization. Thus two series of copolyesters covering the whole range of compositions were prepared and their properties comparatively examined. In both cases, random copolyesters with compositions close to those used in their respective feeds were obtained. The influence of composition on reaction kinetics with respect to time and temperature was evaluated for the two series. Chemically catalyzed ROP rendered copolyesters with Mw in the ∼50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–65000 g mol−1 range, whereas values between 15000 and 45000 g mol−1 were attained when the ROP reaction was assisted by CALB. The thermal behavior of coPBFxSy obtained by ROP was similar to that reported for such copolymers prepared by melt polycondensation. They all start to decompose above 300 °C and display melting enthalpy and temperatures that decrease with copolymerization, attaining minimum values when the comonomer contents are approximate to balance. On the contrary, the glass-transition temperature increased almost linearly with the content of butylene furandicarboxylate units, covering the whole range of values between those of the two parent homopolyesters. Small deviations in thermal properties observed between the two series could be attributed to their differences in molecular weights. Hydrolytic and enzymatic degradation studies revealed that coPBFxSy became more degradable with increasing content of succinic units, whereas the homopolyester PBF remained practically unaffected when incubated under similar conditions.
000–65000 g mol−1 range, whereas values between 15000 and 45000 g mol−1 were attained when the ROP reaction was assisted by CALB. The thermal behavior of coPBFxSy obtained by ROP was similar to that reported for such copolymers prepared by melt polycondensation. They all start to decompose above 300 °C and display melting enthalpy and temperatures that decrease with copolymerization, attaining minimum values when the comonomer contents are approximate to balance. On the contrary, the glass-transition temperature increased almost linearly with the content of butylene furandicarboxylate units, covering the whole range of values between those of the two parent homopolyesters. Small deviations in thermal properties observed between the two series could be attributed to their differences in molecular weights. Hydrolytic and enzymatic degradation studies revealed that coPBFxSy became more degradable with increasing content of succinic units, whereas the homopolyester PBF remained practically unaffected when incubated under similar conditions.
Introduction
2,5-Furandicarboxylic acid (FDCA) is an aromatic diacid that is industrially prepared by catalytic oxidation of 5-methylfurfural, an aldehyde easily obtained from naturally-occurring hexoses. FDCA stands out as a highly promising bio-based monomer able to replace traditional aromatic diacids of fossil-fuel origin in the production of polyesters and other polycondensates.1–3 It is the aromatic nature of the furanic ring that provides enough stiffness to the molecule as to be able to substitute the phthalate units without a deleterious effect on chain mobility. The excellence of FDCA for the preparation of bio-based polyesters and other condensation polymers has been evidenced in a worthy review recently published by Sousa et al.4 To date, several poly(alkylene 2,5-furandicarboxylate) homopolyesters have been synthesized and it has been shown that their properties compare well with those displayed by their poly(alkylene terephthalate) analogs. In addition, FDCA, either as the diacid itself or as its dimethyl ester, has been copolymerized with a number of either aromatic or aliphatic diacids to render furanoate-containing copolyesters with satisfactory properties.5–10 The most recent progress attained in the production of bio-based 2,5-furandicarboxylate polyesters have been reviewed by Bikiaris et al.11Poly(butylene succinate) (PBS) is a fully bio-based aliphatic polyester that is achieving amazing popularity because it displays good thermal and mechanical behavior in addition to being biodegradable.12–14 Copolymerization has been the approach frequently used to render PBS-based materials with higher performance, in particular with improved thermal properties.15–18 This is really challenging since renewable compounds with the appropriate constitution are not easily accessible. Recently, several copolyesters of PBS containing monocyclic or bicyclic carbohydrate units with improved thermal properties and displaying biodegradability have been reported.19–23 Nevertheless, the use of FDCA as a comonomer of succinic acid (SA) to produce fully bio-based aromatic–aliphatic copolyesters has been scarcely investigated. To our knowledge the contribution by Wu et al.6 describing a series of poly(butylene furandicarboxylate-co-succinate) copolyesters, the work by Jacquel et al.9 evidencing the biodegradability of some of these copolymers, and the recent paper by Lomelí-Rodríguez et al.24 on poly(propylene furandicarboxylate-co-succinate) copolyesters are the only references dealing with these systems that are available in the accessible literature.
Until very recently the polymerization method invariably applied for the synthesis of both PBF and PBS and their copolyesters was the conventional melt polycondensation using either the diacid or the dimethyl ester of FDCA and SA.25–28 Nevertheless Loos et al.29 have reported a series of furanic aliphatic polyesters successfully produced via enzymatic polycondensation of the dimethyl ester of FDCA with various potentially renewable aliphatic diols using Candida antarctica Lipase B (CALB).
The application of the ROP method to the synthesis of PBS was reported for the first time by Japanese researchers using as the feed a mixture of cyclic butylene succinate oligomers that were either synthesized by enzymatic cyclization30–32 or recovered as a subproduct in the preparation of PBS by melt polycondensation.33 On the other hand, the first papers reporting the synthesis of PBF homopolyesters by ROP were published at the end of last year,34,35 and the first work using ROP for the preparation of PBF copolyesters has just been published; in that work a series of poly(butylene furandicarboxylate-co-terephthalate) copolyesters was synthesized and their properties evaluated as a function of the comonomer composition.36
Entropically driven ring opening polymerization (ED-ROP) is a well-established technique that has been successfully used for the polymerization of cyclic oligoesters.37–39 The specific advantages of ROP are a high polymerization rate that allows the application of relatively mild reaction conditions and the low generation of reaction subproducts with subsequent minimization of undesirable discoloration. In this paper we applied for the first time the ED-ROP methodology to the synthesis of fully bio-based aromatic–aliphatic poly(butylene furandicarboxylate-co-succinate) copolyesters (coPBFxSy). The synthesis of the cyclic oligomers required for feeding has been accomplished by cyclization condensation, and their copolymerization was performed in the bulk using either tin dioctanoate (chemical process) or supported Candida antarctica (enzymatic process). The two ROP processes are critically compared and the two respectively produced copolyester series are extensively characterized; in particular their thermal properties are evaluated and collated to each other as well as to those reported for the same polymers obtained by conventional melt polycondensation.
2. Experimental
2.1. Materials
2,5-Furandicarboxylic acid (FDCA, >98% purity) was purchased from Satachem (China). 1,4-Butanediol (BD), thionyl chloride (SOCl2, 99%), lipase porcine pancreas, di-azabicyclo[2.2.2] octane (DABCO, 99%) and tin dioctanoate (Sn(Oct)2, 99%) were purchased from Sigma-Aldrich Co. Dimethyl succinate (DMS, 99%) and triethylamine (Et3N, 98%) were purchased from Panreac. Novozym 435 (lipase B Candida antarctica CALB) was a gift of Novozymes. Solvents used for reaction, isolation and purification were of high-purity grade and used as received except dichloromethane (DCM), tetrahydrofuran (THF), diphenyl ether and dichlorobenzene (DCB), which were dried on 3 Å molecular sieves. DABCO catalyst was purified by sublimation.2.2. Measurements
Elemental analyses were carried out at the Servei de Microanàlisi at IQAC (Barcelona). Tests were made in a Flash 1112 elemental microanalyser (A5), which was calibrated with appropriate standards of known composition. C and H contents were determined by the dynamic flash combustion method using He as the carrier gas. Results were given in (w/w) percentages and in duplicates. 1H and 13C NMR spectra were recorded on a Bruker AMX-300 spectrometer at 25 °C, operating at 300.1 and 75.5 MHz, respectively. For NMR analysis, monomers and intermediate compounds were dissolved in deuterated chloroform (CDCl3) and polymers in a 1:8 (v/v) mixture of trifluoroacetic acid (TFA) and CDCl3, and spectra were internally referenced to tetramethylsilane (TMS). About 10 and 50 mg of the sample in 1 mL of solvent were used for 1H and 13C NMR, respectively. Sixty-four scans were recorded for 1H and between 1000 and 10000 scans were recorded for 13C NMR. High-performance liquid chromatography (HPLC) analysis was performed at 25 °C in a Waters apparatus equipped with a UV detector of Applied Biosystems, operating at a wavelength of 254 nm, and a Scharlau Science column (Si60, 5 μm; 250 × 4.6 mm). Cyclic oligomers (1 mg) were dissolved in chloroform (1 mL) and eluted with hexane:1,4-dioxane 70:30 (v/v) at a flow rate of 1.0 mL min−1. Molecular weight analysis was performed by GPC on a Waters equipment provided with RI and UV detectors. 100 μL of 0.1% (w/v) sample solution was injected and chromatographed with a flow of 0.5 mL min−1 of 1,1,1,3,3,3-hexafluoroisopropanol (HFIP). HR5E and HR2 Waters linear Styragel columns (7.8 mm × 300 mm, pore size 103–104 Å) packed with crosslinked polystyrene and protected with a precolumn were used. Molar mass average and distributions were calculated against PMMA standards. Intrinsic viscosities were measured from polymer solutions in DCA using an Ubbelohde viscometer thermostated at 25 ± 0.1 °C. Matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectra were recorded in a 4700 Proteomics Analyzer instrument (Applied Biosystems) of the Proteomics Platform of Barcelona Science Park, University of Barcelona. Spectrum acquisition was performed in the MS reflector positive-ion mode. About 0.1 mg of the sample was dissolved in 50 μL of DCM, and 2 μL of this solution was mixed with an equal volume of an anthracene solution in DCM (10 mg mL−1), and the mixture was left to evaporate to dryness on the stainless steel plate of the analyser. The residue was then covered with 2 μL of a solution of 2,5-dihydroxibenzoic acid in acetonitrile:H2O (1:1) containing 0.1% TFA, and the mixture was left to dry prior to exposure to the laser beam.
The thermal behaviour of cyclic compounds and polymers were examined by differential scanning calorimetry (DSC), using a Perkin-Elmer Pyris apparatus. The thermograms were obtained from 4–6 mg samples at heating and cooling rates of 10 °C min−1 under a nitrogen flow of 20 mL min−1. Indium and zinc were used as standards for temperature and enthalpy calibration. The glass transition temperature (Tg) was taken as the inflection point of the heating DSC traces recorded at 20 °C min−1 from melt-quenched samples, and the melting temperature (Tm) was taken as the maximum of the endothermic peak appearing on heating traces. Thermogravimetric analysis was performed on a Mettler-Toledo TGA/DSC 1 Star System under a nitrogen flow of 20 mL min−1 at a heating rate of 10 °C min−1 and within a temperature range of 30 to 600 °C.
X-Ray diffraction patterns from powdered samples coming directly from synthesis were recorded on a PANalytical X'Pert PRO MPD θ/θ diffractometer using CuKα radiation of wavelength 0.1542 nm. Micrographs were recorded using an Olimpus BX 51 polarizing optical microscope provided with a Linkam THMS 600 stage. For observation, 10 mg of each sample were dissolved in 1 mL of chloroform or HFIP, and aliquots of 0.2 mL were slowly evaporated on a glass slide.
2.3. Synthesis of cyclic oligomers
:10 (v/v) as the eluent. Yield 75%. 1H NMR (δ ppm, CDCl3, 300 MHz): 7.25 (s, 2H), 4.40 (t, 4H), 1.92 (q, 4H). 13C NMR (δ ppm, CDCl3, 75.5 MHz): 157.9, 146.5, 118.7, 64.8, 25.4.
2.4. Synthesis of polymers
Blank assays were carried out for comparative purposes. For this, a mixture of 0.24 mmol (49.3 mg) of c(BF)n and 0.94 mmol (161.7 mg) of c(BS)n without any catalyst added was made to react at 150 and 220 °C for 12 and 24 h, respectively. The evolution of the reaction was followed as above and the resulting products were analysed by NMR.
3. Results and discussion
The strategy followed in this work to produce the poly(butylene 2,5-furandicarboxylate-co-succinate) copolyesters is depicted in Scheme 1. Firstly cyclic oligomers of both butylene 2,5-furandicarboxylate, c(BF)n, and butylene succinate, c(BF)n, were synthesized, and secondly, mixtures of the two types of cycles were polymerized through ring opening reactions catalysed either chemically or enzymatically.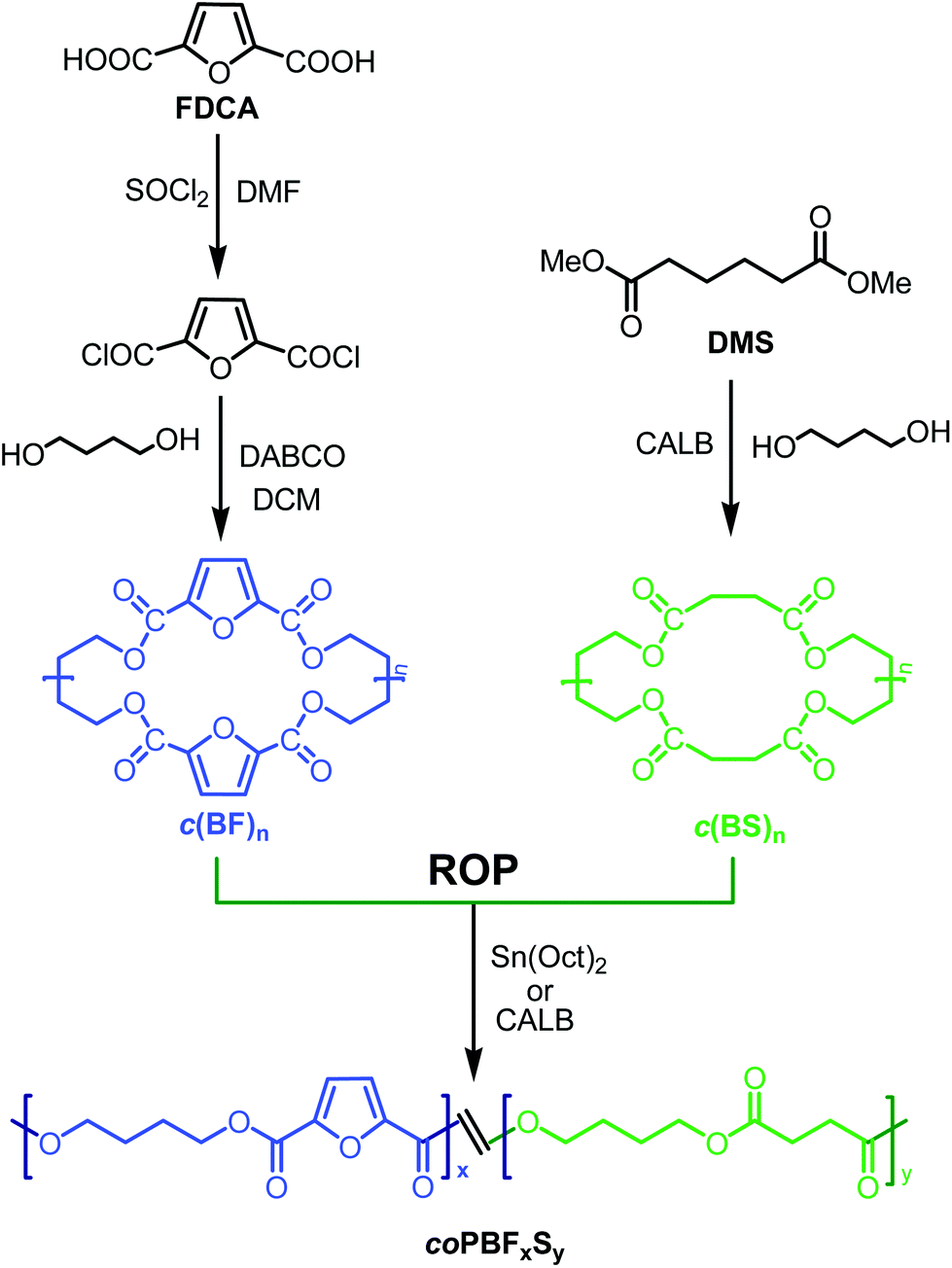 | ||
| Scheme 1 Synthesis route to poly(butylene furandicarboxylate-co-succinate) copolyesters by ROP. | ||
3.1. Synthesis of cyclic oligomers
In a recent study we reported for the first time the synthesis of cyclic (butylene 2,5-furandicarboxylate) oligomers by both high dilution condensation (HDC) and cyclodepolymerization (CD) methods. Both methods were suitable for c(BF)n synthesis but the former afforded the oligomeric mixture in slightly higher yield and more enriched with the cyclic dimer.35 In the present work we have made use of the HDC method to produce c(BF)n, with the results shown in Table 1. After purification by column chromatography, the 1H NMR analysis (Fig. 1a) was in full agreement with the expected structure and showed no signal arising from linear species. The HPLC chromatogram (Fig. 1b) displayed three peaks corresponding to cyclic dimers, trimers and tetramers in an approximate 8:4:1 ratio, although the MALDI-TOF analysis (Fig. 1c) revealed the presence of cyclic species of higher sizes in minor amounts. Nevertheless, this mixture without further fractionation was used for ROP since our previous kinetics study on the polymerization of c(BF)n showed no relevant differences when cycles of different sizes or their mixtures were used for feeding.35
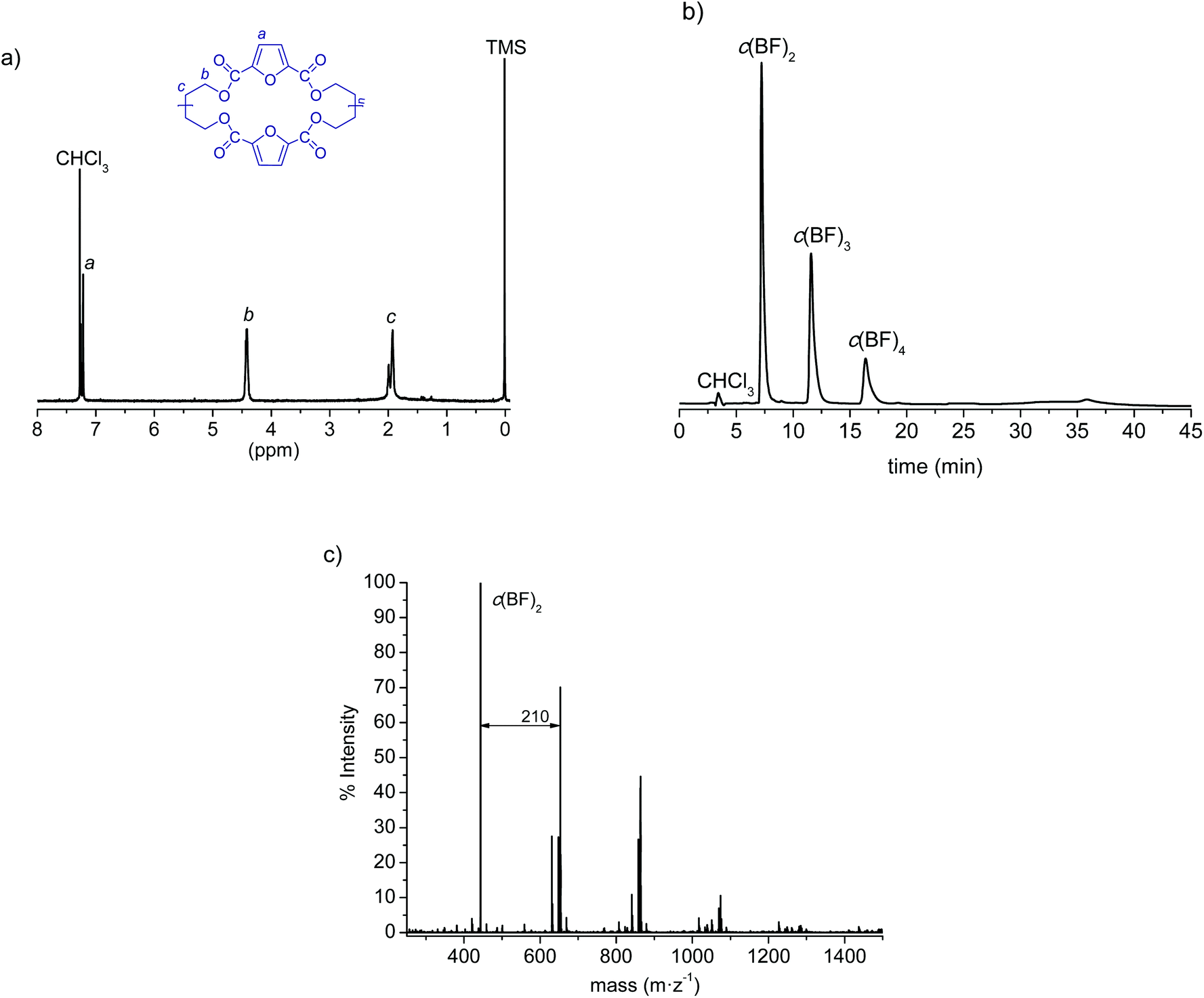 | ||
| Fig. 1 (a) 1H NMR, (b) HPLC and (c) MALDI-TOF spectra for c(BF)n. | ||
| Methoda | Yield (%) | Compositionb (2/3/4) |
T
mc (°C) |
o
T
5%c (°C) |
T
dc (°C) |
|
|---|---|---|---|---|---|---|
| a HDC: high dilution cyclization using DABCO/Et3N; EC: enzymatic cyclization using CALB. b Relative content (w/w) of the reaction product in cyclic dimers, trimers and tetramers as measured by HPLC. c Melting and decomposition temperatures measured by DSC and TGA. | ||||||
| c(BF)n | HDC | 67 | 61/31/8 | 147 | 276 | 387 |
| c(BS)n | EC | 70 | 50/40/10 | 105 | 266 | 340 |
The synthesis of c(BS)n was enzymatically performed as well, and the obtained results are given in Table 1. We applied the method reported by Sugihara et al.30 which consists of cyclizing a mixture of BD and DMS dissolved in toluene by the action of CALB. The 1H NMR and MALDI-TOF spectra as well as the HPLC chromatogram of the cyclization reaction product without being subjected to purification are shown in Fig. 2. The mixture is composed almost exclusively of cyclic species with sizes ranging from dimers to nonamers; cyclic dimers, trimers and tetramers are in an approximate 5:4:1 ratio, and their sum amounts to more than 90% of the total oligomers present in the mixture. Here again the oligomer mixture without fractionation was used for ROP since kinetics studies carried out by Kondo et al.40 have shown that polymerization results are not substantially affected by the cycle size.
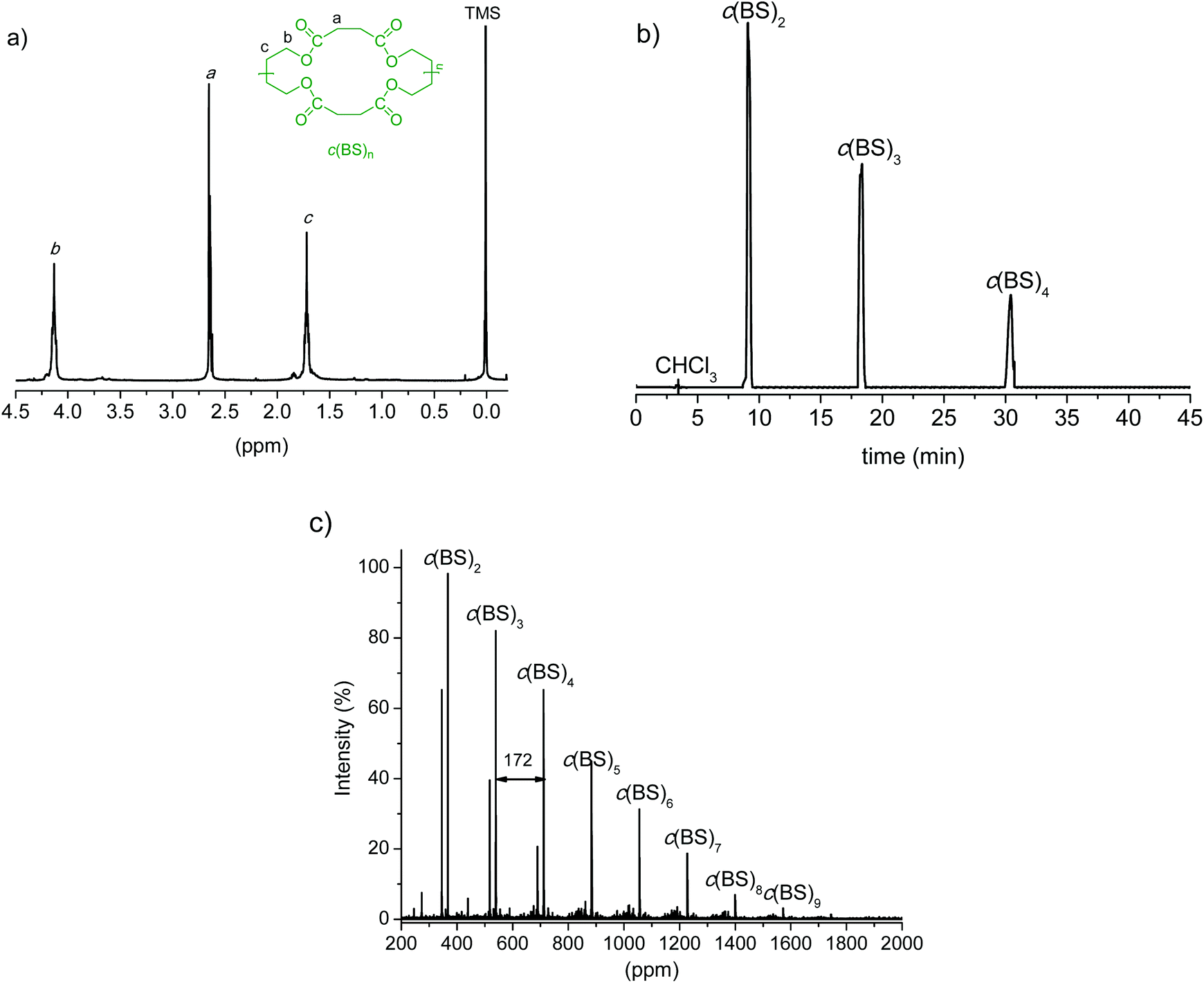 | ||
| Fig. 2 (a) 1H NMR, (b) HPLC and (c) MALDI-TOF spectra for c(BS)n. | ||
Both c(BF)n and c(BS)n mixtures are white semicrystalline powders with melting temperatures of 147 °C and 105 °C, respectively. They start to decompose by the effect of heating at temperatures above 250 °C, following a single-step degradation process that displays its maximum rate at temperatures between 300 and 400 °C. The TGA traces recorded from these mixtures along with their derivative curves are available in the ESI (Fig. SI-3†).
3.2. Synthesis of coPBFxSy copolyesters
:20, 50:50 and 20:80) in addition to the two pristine cyclic oligomer fractions were examined. The results are shown in Fig. 3a, where Mw is plotted against six temperatures increasing 20 or 10 °C from 180 up to 230 °C. In general a steady increase in the molecular weight of the resulting polymer was observed for all compositions as the temperature was raised. It was noticeable however that the Mw of both the copolyester coPBF80S20 and the homopolyester PBF levelled off at temperatures above ∼200 °C, probably due to the higher thermal sensitivity to degradation of the furan-based compounds. Nevertheless, the relevant outcome of this study is that the molecular weight of the resulting polyester increases with the content of the feed in c(BF)n at whichever reaction temperature. This result brings into evidence the higher reactivity of the furan-based cyclic oligomers in chemically catalysed ROP.
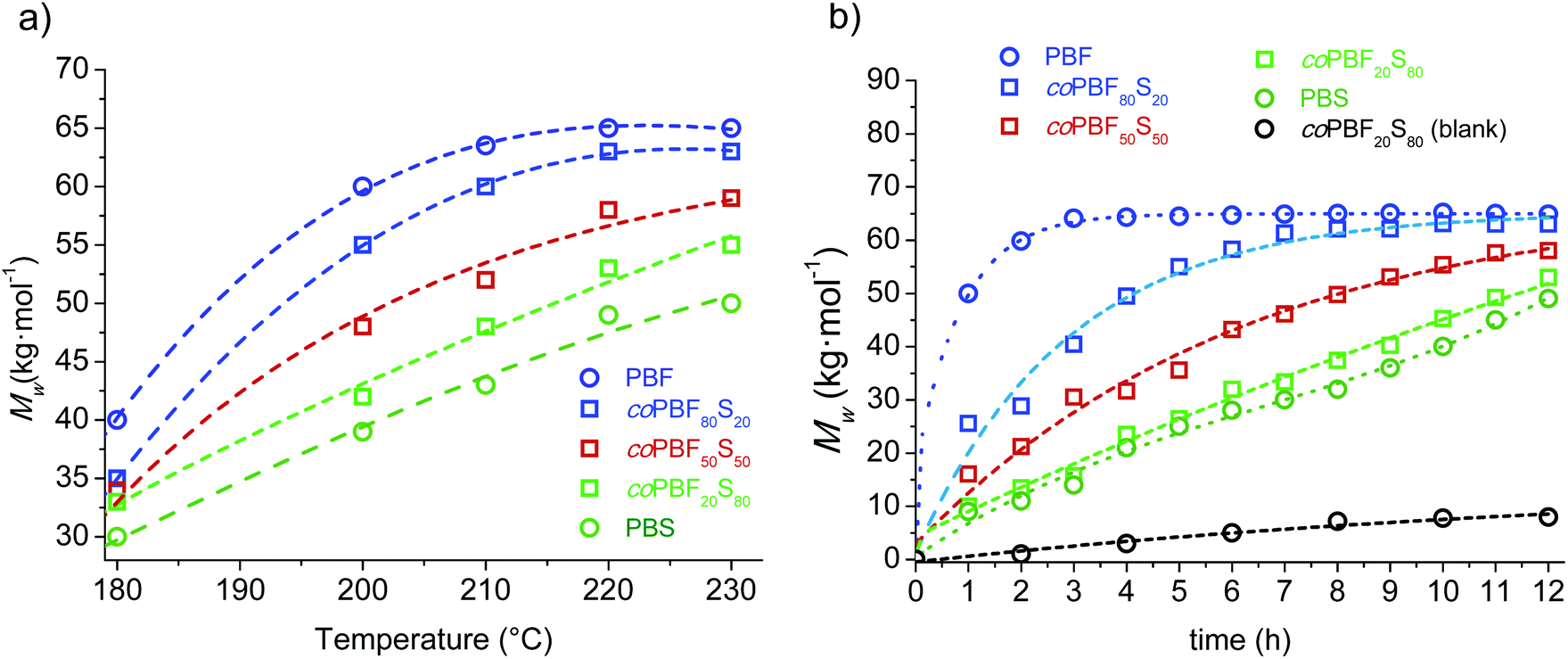 | ||
| Fig. 3 Effect of temperature and time on Sn(Oct)2 catalyzed ROP of mixtures of c(BF)n and c(BS)n carried out in the melt. (a) Mw of coPBFxSy against reaction temperature after 12 h of reaction time. (b) Evolution of Mw with reaction time at 220 °C. | ||
The evolution of the molecular weight along the reaction time was then examined for the polymerization of the same oligomer cyclic mixtures carried out at 220 °C, and the obtained results are shown in Fig. 3b. The trends shown by PBF and PBS are remarkably different; the c(BF)n fraction polymerized initially very quickly to reach an almost constant molecular weight of about 65000 g mol−1 after 2–3 h of reaction, whereas c(BS)n polymerized following a nearly linear trend to render a PBS with a Mw near 45000 g mol−1 after 12 h of reaction. The behaviour observed for the copolyesters were intermediate between those displayed by the two homopolymers, with the profile being closer to one or the other according to the composition.
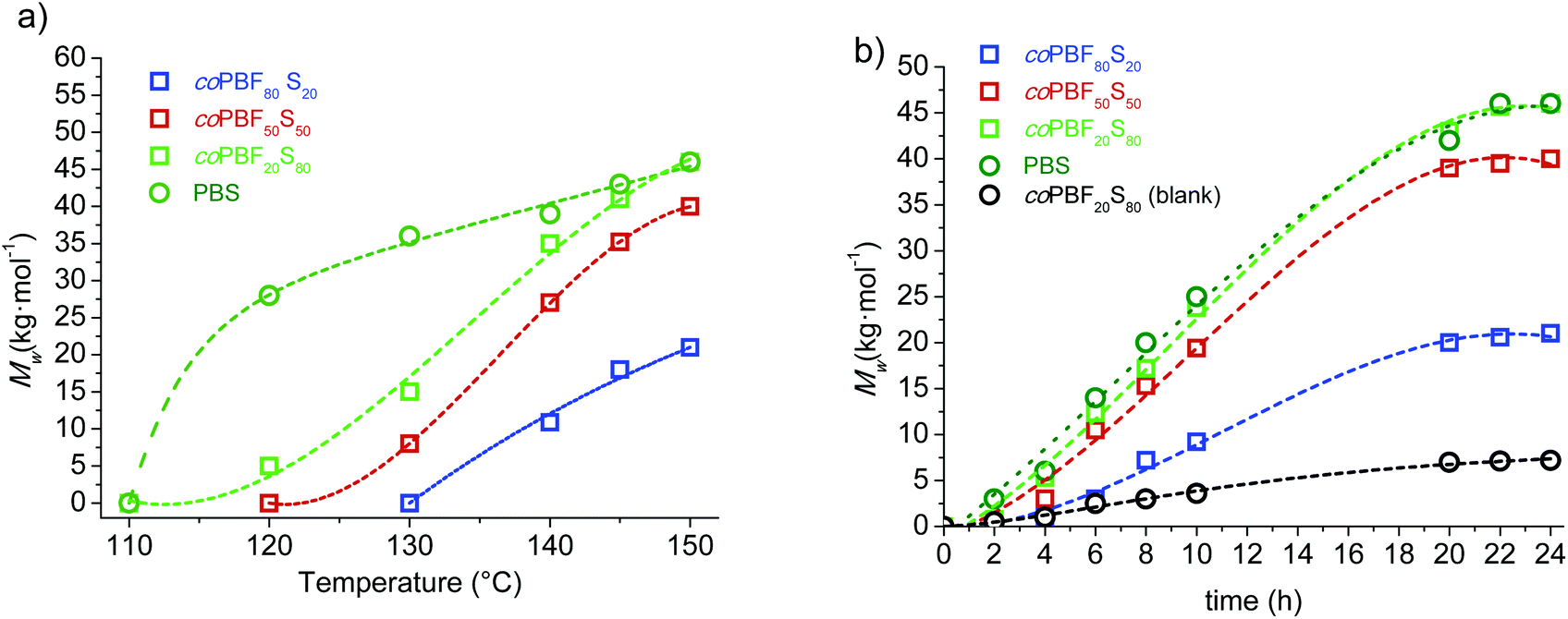 | ||
| Fig. 4 Effect of temperature and time on CALB assisted ROP of mixtures of c(BF)n and c(BS)n in the melt. (a) Mw of PBFxSy against reaction temperature for a reaction time of 24 h. (b) Evolution of Mw with reaction time at 150 °C. | ||
The trend followed by the molecular weight with increasing reaction temperature was not so predictable as in the case of chemocatalyzed ROP. The profile shape changed significantly with the composition, and differences among the Mw values largely vary with temperature. Notwithstanding, it was interestingly noticed that polymer growing catalyzed by C. antarctica was more favoured as the content of butylene succinate increased, which is exactly opposite to the behaviour observed in tin catalysed reactions. In fact, minimum Mw values were reached at all assayed temperatures for the PBF80S20, which is the copolyester with the highest content of butylene furandicarboxylate units attainable by this method.
The evolution of the molecular weight of the polyesters with time in the CALB assisted ROP of c(BF)n/c(BS)n mixtures at 150 °C along a period of 24 h is depicted in Fig. 4b. The Mwvs. time profiles are of similar shape for the four assayed polyesters with the slope decreasing with the content of BF units. Differences in the polymerization rates become more apparent as the reaction progresses, and the molecular weights reached at the end of the process were lower or higher than those obtained by chemocatalysis depending on the composition.
In order to appraise the possible occurrence of self-polymerization of the c(BF)n/c(BS)n by ring opening reaction induced by heating, a series of blank assays, i.e., assays conducted under the same reaction conditions but in the absence of any catalyst, was carried out with the mixture c(BF)n/c(BS)n (20:80). The profiles Mwvs. either temperature or time that were obtained from these assays under chemically or enzymatically catalyzed conditions are included in Fig. 3b and 4b for comparison. The highest molecular weight reached in these assays was about 5000 g mol−1, corresponding to oligomers of about 10 monomeric units. The 1H NMR spectra of the feeding mixture and the reaction product of the ROP carried out either without catalyst or in the presence of CALB are compared in Fig. 5. This comparison reveals that c(BF)n remained unreacted in the blank whereas an appreciable amount of c(BS)n cycles disappeared. Conversely, both c(BF)n and c(BS)n reacted almost completely in the presence of CALB.
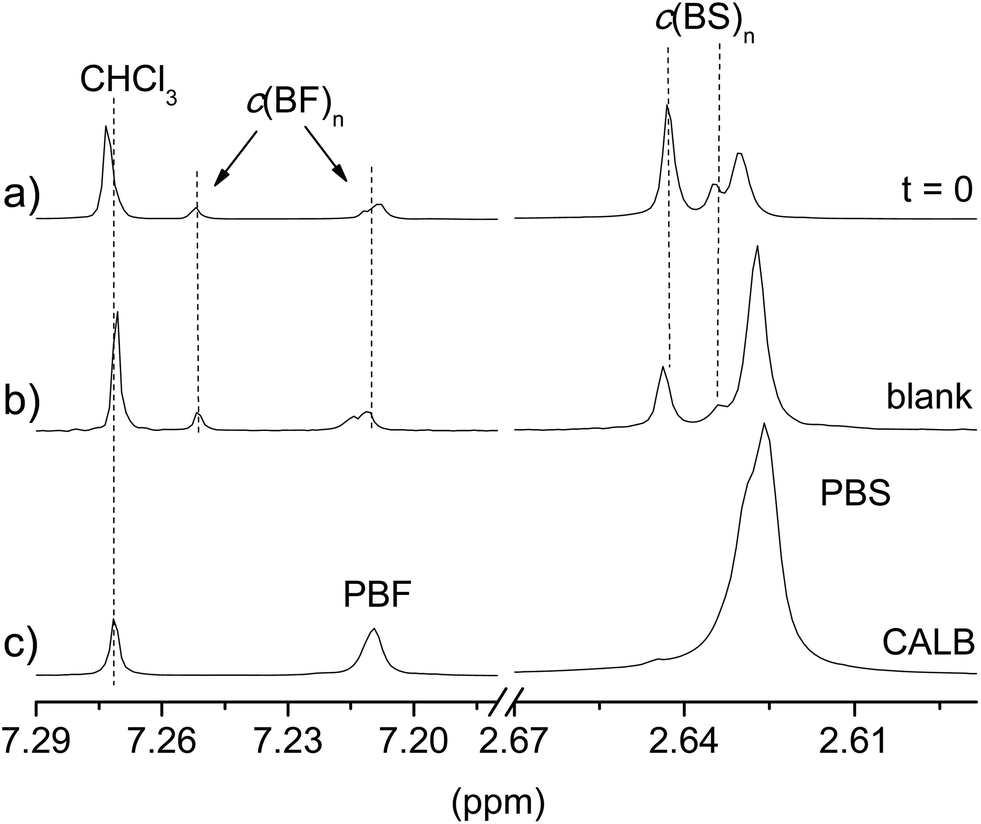 | ||
| Fig. 5 Enlarged 7.3–7.18 and 2.68–2.60 ppm regions of the 1H NMR spectra of the initial c(BF)n/c(BS)n (20:80) mixture (a), after reaction at 150 °C for 24 h in the absence of catalyst (b), and with CALB added (c). | ||
3.3. Chemical structure and microstructure of copolyesters
The chemical structure, including both composition and microstructure as well as the molecular weight, of the homopolyesters and copolyesters obtained by the two ROP methods applied in this work were systematically determined, and results are gathered in Table 2. These data refer to polyesters synthesized with Sn(Oct)2 as the catalyst at 230 °C for 12 h, and to those synthesized with the assistance of CALB at 150 °C for 24 h. The chemical constitution of all the polyesters was ascertained by NMR spectroscopy. The 1H NMR spectra of the copolyester PBF50S50 prepared by either chemo-ROP or enzym-ROP are shown in Fig. 6 for illustrative purposes. Both spectra are almost undistinguishable with all peaks duly assigned and displaying no signals other than those expected for the predicted chemical constitution. Both the 1H and 13C NMR spectra of the two whole sets of synthesized polyesters are given in the ESI (Fig. SI-1 and SI-2†).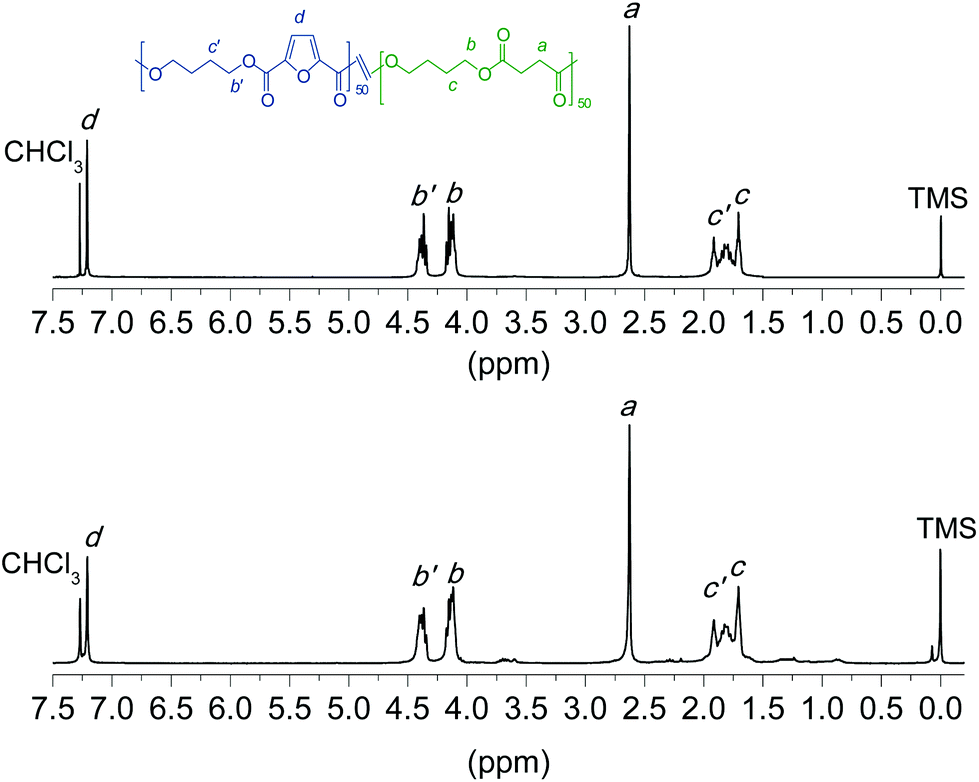 | ||
| Fig. 6 1H NMR spectra of coPBF50S50 obtained by chemically (top) and enzymatically (bottom) catalyzed ROP. | ||
| Copolyester | Yield (%) | CompositionbXBF/YBS (mol mol−1) | Molecular weightc | Dyad contentd (mol%) | Average sequence lengthe | R | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| M w (g mol−1) | Đ | [η] (dL g−1) | FBF | FBS + SBF | SBS | n BF | n BS | ||||
| a Copolyesters synthesized using Sn(Oct)2 (top row) and CALB (bottom row). b Copolyester compositions determined from signals observed in the 1H NMR spectra. c M w, Đ; and η determined by GPC and Ubbelohde viscosity, respectively. d Determined by integration of the α-methylene signals in the butylene segments in the 13C NMR spectra. e Average lengths of homogeneous BF and BS sequences and randomness (R) calculated by applying a statistical approach.41 f Calculated for coPBF65S35: C, 56.73%; H, 5.47%; Found: C, 53.37%; H: 5.54%. Calculated for coPBF58S42: C, 56.40%; H, 5.67%; Found: C, 56.06%; H, 5.96%. | |||||||||||
| PBF | 95 | 100/0 | 65000 |
1.98 | 0.76 | — | — | — | — | — | — |
| — | 100/0 | — | — | — | — | — | — | — | — | — | |
| coPBF90S10 | 92 | 94/6 | 64000 |
1.79 | 0.74 | 93.2 | 2.6 | 4.2 | 6.58 | 1.30 | 0.92 |
| — | — | — | — | — | — | — | — | — | — | — | |
| coPBF80S20 | 93 | 84/16 | 63000 |
1.85 | 0.74 | 66.7 | 28.2 | 5.1 | 5.73 | 1.36 | 0.91 |
| 65 | 81/19 | 21000 |
1.30 | 0.25 | 67.8 | 25.2 | 7.0 | 6.38 | 1.56 | 0.80 | |
| coPBF60S40 | 90 | 65/35f | 62000 |
1.90 | 0.71 | 39.5 | 48.8 | 11.7 | 2.62 | 1.48 | 1.06 |
| 70 | 58/42f | 23000 |
1.30 | 0.30 | 35.3 | 45.1 | 19.6 | 2.57 | 1.87 | 0.92 | |
| coPBF50S50 | 90 | 51/49 | 58000 |
1.79 | 0.62 | 27.0 | 48.6 | 48.7 | 2.11 | 2.00 | 0.97 |
| 91 | 49/51 | 40000 |
1.60 | 0.55 | 15.7 | 47.6 | 36.6 | 1.66 | 2.53 | 0.99 | |
| coPBF40S60 | 90 | 43/57 | 56000 |
1.95 | 0.65 | 16.7 | 52.2 | 31.1 | 1.64 | 2.19 | 1.07 |
| 92 | 41/59 | 43000 |
1.70 | 0.55 | 13.4 | 41.3 | 65.9 | 1.65 | 3.19 | 0.91 | |
| coPBF20S80 | 89 | 22/78 | 53000 |
1.90 | 0.54 | 5.2 | 31.3 | 63.5 | 1.33 | 5.06 | 0.95 |
| 91 | 22/78 | 46000 |
1.50 | 0.53 | 3.2 | 33.7 | 63.0 | 1.19 | 4.73 | 1.05 | |
| coPBF10S90 | 88 | 12/88 | 53000 |
1.89 | 0.60 | 0 | 22.6 | 77.4 | 1.00 | 7.8 | 1.13 |
| 91 | 11/89 | 47000 |
1.50 | 0.53 | 1.3 | 15.6 | 83.1 | 1.17 | 11.6 | 0.94 | |
| PBS | 95 | 0/100 | 49000 |
1.97 | 0.55 | — | — | — | — | — | — |
| 92 | 0/100 | 46000 |
1.40 | 0.51 | — | — | — | — | — | — | |
The molar composition in BF and BS units of the two series of copolyesters was determined by quantification of the signals b and b′ arising from the α-methylene of the butylene furandicarboxylate and butylene succinate units, respectively, and the obtained values are listed in Table 2. In support of the NMR determination, the elemental analysis of the copolyester coPBF60S40 obtained by the two methods was carried out as an example. The results were fully consistent with the predicted chemical constitution and composition determined by NMR for both cases (Table 2).
A comparative inspection of the feed and copolyester compositions reveals that deviations between them are higher when the ROP is chemically catalysed, and that in this case, they correspond to BS unit losses. Furthermore, the differences become more noticeable as the content of BF increases, with the loss in BS becoming around 40% for the coPBF90S10 copolyester. Conversely, the observed deviations were less than 5% for the copolyesters synthesized by enzymatically assisted ROP. These results are consistent with the different relative reactivity of the c(BF)n and c(BS)n species in the two polymerization methods, although reasons due to differences in volatility should not be fully neglected provided that much higher temperatures are used in the chemical procedure.
The molecular weights measured by GPC against polystyrene standards were in the 65000–50000 and 47000–20000 g mol−1 ranges for polyesters obtained by chemically and enzymatically catalysed ROP, with dispersities significantly lower in the latter case. Intrinsic viscosities were in agreement with the Mw values determined by GPC. The trends observed for the molecular weights in the two series are opposite to each other according again to the different relative reactivities displayed by c(BF)n and c(BS)n in each of the two polymerization methods.
The microstructure of the copolyesters was elucidated by 13C NMR analysis, taking benefit of the sensitivity of the α-carbon of the butylene segment to the distribution sequence at the dyad level. The four possible dyads, FBF, SBS, and FBS/SBF, appear within the 60.5–58.5 ppm region (Fig. SI-4†) as four well-resolved peaks whose areas were integrated for each copolyester composition, and the resulting values used in the corresponding equations to calculate the number average sequence length. The results are listed in Table 2 for the two copolyester series, where it can be seen that the nBF and nBS values vary steadily with the composition in opposite directions, and both approximate to 2 for the 50:50 composition. The degree of randomness is between 0.9 and 1.1, indicating that an approximate random distribution of BF and BS units is present in all the copolyesters. The conclusion that a coPBFxSy random copolyester is genuinely generated in the ROP of mixtures of c(BF)n and c(BS)n whether it is chemically or enzymatically catalyzed cannot be straightforwardly drawn from such results since randomization by transesterification is very probable to occur at the high temperatures used in these processes. In order to clarify this point, ROP of the c(BF)n/c(BS)n (50:50) mixture assisted by CALB was accomplished in solution at 50 °C, a temperature at which the occurrence of transesterification reactions may be neglected. The coPBF50S50 copolyester resulting in these conditions was obtained in a 80% yield and had a Mw of 14000 g mol−1 and a Đ of 1.8. Its 13C NMR spectrum was similar to that recorded from the copolyester obtained a high temperature; specifically, no differences can be appreciated when they are compared in the 60.5–58.5 ppm region (Fig. 7, inset). It can be therefore concluded that the ROP of c(BF)n/c(BS) renders random copolyesters whatever the applied reaction conditions.
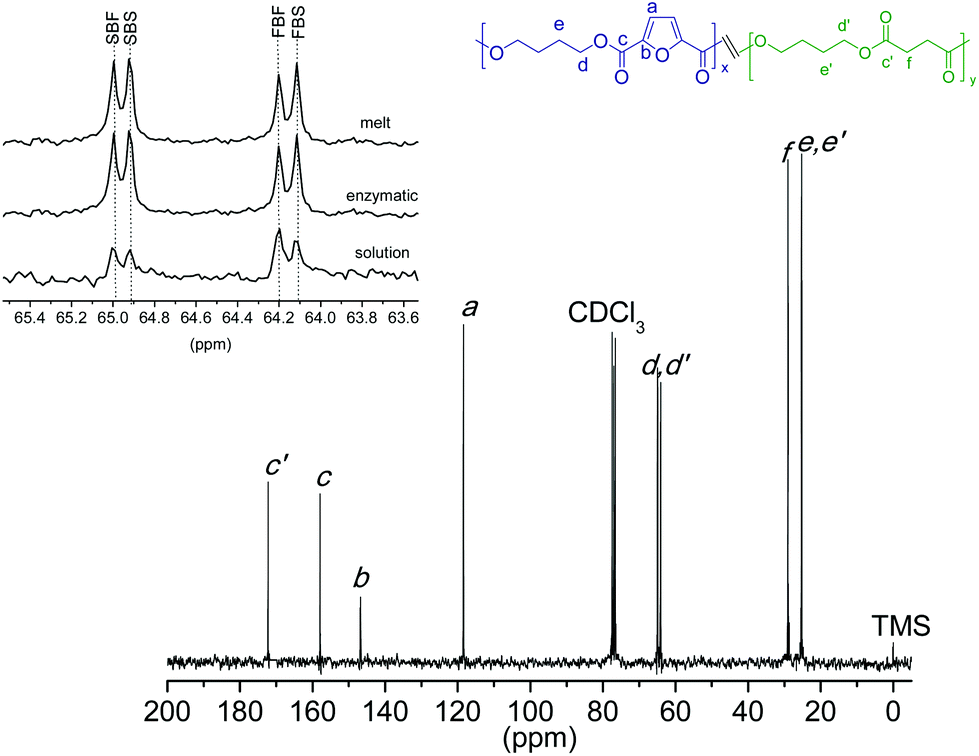 | ||
| Fig. 7 13C NMR spectra of coPBF50S50 obtained by ROP at 50 °C in solution catalyzed by Sn(Oct)2. Inset: enlarged 60.5–58.5 ppm region compared for different methods of polymerization. | ||
3.4. Thermal properties
The thermal properties of the polyesters and copolyesters composing the two series prepared by ROP were comparatively examined by TGA and DSC, and the most relevant parameters resulting from the study are collected in Table 3. Moreover, data are also compared with those reported by Wu et al., which were obtained from polyesters with similar compositions but prepared by two-step melt polycondensation.6 The TGA weight loss–T traces of all the polyesters synthesized in this work as well as their derivative curves are available in the ESI (Fig. SI-5†). In all cases thermal decomposition proceeded through a single step taking place within the 390–400 °C range and without significant differences between the two series. The onset decomposition temperatures of the copolyesters were between 350 and 340 °C, and polymers prepared by enzymatic ROP invariably started to decompose at a few degrees below those obtained by ROP catalysed by Sn(Oct)2, which is reasonable due to their lower molecular weight. The residue left after heating at 600 °C under a nitrogen flow was significantly smaller in the enzymatically synthesized polyesters. This is a much-expected result because of the absence of metallic components in these polymers. The thermal decomposition data obtained for the ROP-synthesized coPBFxSy are essentially coincident with those reported for this family of copolyesters obtained by polycondensation, with slight differences in magnitudes being explainable by differences in molecular weight or experimental inaccuracy.a
| Copolyster | TGAb | DSCc | ||||||
|---|---|---|---|---|---|---|---|---|
| o T d (°C) | max T d (°C) | W (%) | T g (°C) | T m 1 (°C) | ΔHm1 (J g−1) | T m 2 (°C) | ΔHm2 (J g−1) | |
| a Values in the upper and lower rows correspond to polyesters obtained by tin and lipase catalyzed ROP, respectively. b o T d is the onset degradation temperature for 5% of weight lost and maxTd is temperature at which degradation happens at the maximum rate. c 1 and 2 refer to data registered at the first heating and second heating, respectively. | ||||||||
| PBF | 370 | 400 | 7 | 42 | 170 | 40 | 170 | 35 |
| — | — | — | — | — | — | — | — | |
| coPBF90S10 | 350 | 395 | 8 | 28 | 166 | 38 | 164 | 30 |
| — | — | — | — | — | — | — | ||
| coPBF80S20 | 350 | 395 | 7 | 23 | 146 | 33 | 146 | 27 |
| 348 | 396 | 5 | 20 | 140 | 15 | 140 | 17 | |
| coPBF60S40 | 343 | 397 | 6 | 7 | 114 | 13 | — | — |
| 342 | 397 | 5 | 5 | 126 | 7 | — | — | |
| coPBF50S50 | 349 | 400 | 5 | −1 | 95 | 11 | — | — |
| 341 | 399 | 1 | −6 | 86 | 4 | — | — | |
| coPBF40S60 | 347 | 399 | 6 | −13 | 60 | 6 | — | — |
| 344 | 399 | 2 | −20 | 72 | 10 | — | — | |
| coPBF20S80 | 343 | 399 | 5 | −23 | 90 | 55 | 93 | 32 |
| 338 | 400 | 3 | −23 | 93 | 30 | 93 | 51 | |
| coPBF10S90 | 340 | 401 | 3 | −23 | 100 | 71 | 105 | 49 |
| 337 | 400 | 2 | −25 | 102 | 55 | 103 | 63 | |
| PBS | 350 | 399 | 3 | −30 | 112 | 106 | 114 | 82 |
| 348 | 398 | 3 | −40 | 112 | 60 | 111 | 87 | |
The thermal transitions, i.e. glass and melting/crystallization, were examined by DSC. The DSC traces of the two series registered at heating and cooling from the melt are included in the ESI (Fig. SI-6†), and the measured thermal parameters are listed in Table 3. All the pristine copolyesters displayed crystallinity with enthalpy values decreasing as the content of BF and BS units becomes more equilibrated, so that only those copolyesters containing more than 40% of one or another unit are able to crystallize from the melt. A representative selection of polarizing optical micrographs showing the crystalline textures displayed by these copolyesters upon crystallization is available in the ESI (Fig. SI-7†). The XRD analysis of the copolyesters gave results in agreement with the DSC data. The scattering profiles registered from crystalline coPBFxSy and from the two homopolyesters PBF and PBS prepared by chemically catalyzed ROP are compared in Fig. 8. It is clearly seen that the characteristic scattering produced by one or another homopolyester is present in the copolyester profiles, with more or less intensity depending on the composition. For BF/BS ratios close to 1, the crystallinity becomes so low that no discrete scattering is perceivable on the XRD profiles of the copolyesters. As it is commonly found, the crystal structure of the homopolyesters is fully retained by the copolyesters containing limited amounts of the second comonomer, beyond which they become essentially amorphous.
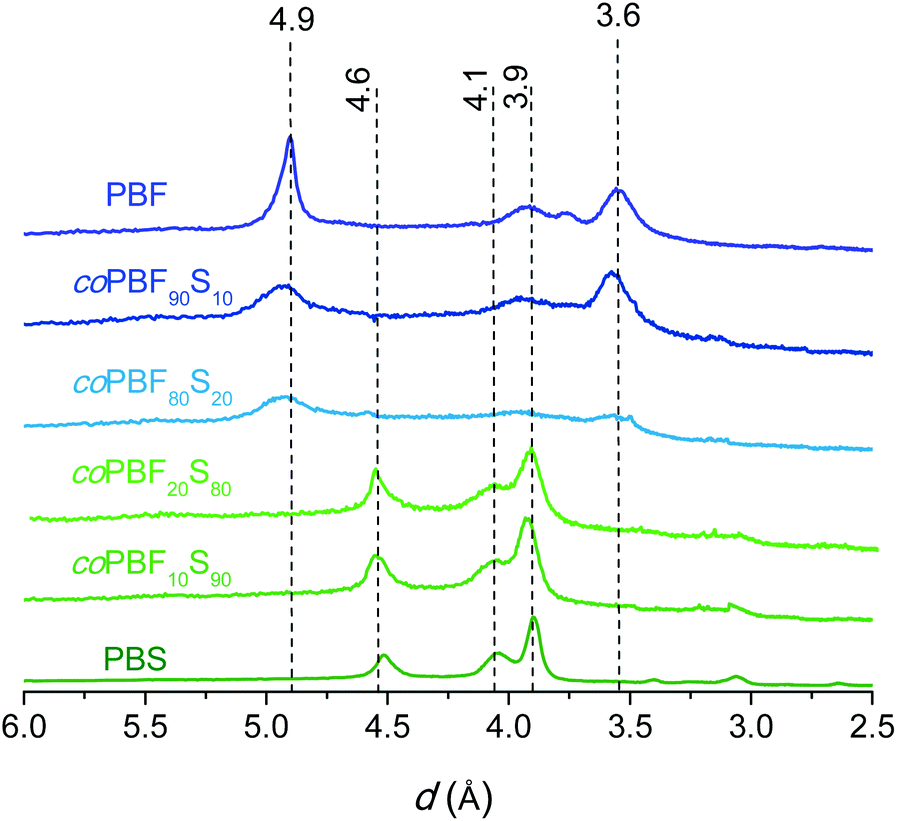 | ||
| Fig. 8 Powder X-ray diffraction profiles of coPBFxSy synthesized by ROP with Sn(Oct)2. | ||
The melting and glass-transition temperatures of the coPBFxSy copolyesters are properties of high relevance regarding their possible applications as thermoplastics. The consequence of incorporating the furanoate units in the PBS is to modify both the Tm and Tg. The variation of Tm with composition for the two series of coPBFxSy copolyesters respectively prepared by the two ROP methods here described is plotted in Fig. 9a. In this plot, it is clearly shown how Tm decreases along the BS-high composition range to invert the trend when the copolyester starts to become enriched with BF units. This behavior is very similar for the two series, and is also similar to that described for the same copolyesters prepared by melt polycondensation, with the slight deviations observed among them being attributable to differences in molecular weights or of experimental origin. A similar trend is observed for the melting enthalpy, which is readily explained by the incompatibility of BF and BS to cocrystallize. Nevertheless it is worthy to notice the presence of some crystallinity in the copolyesters containing approximately similar amounts of the two units, which brings into evidence the ability of BF and BS short sequences for crystallizing. Similar behavior has been recently reported for random copolyesters made of butylene furanoate and butylene terephthalate units.36
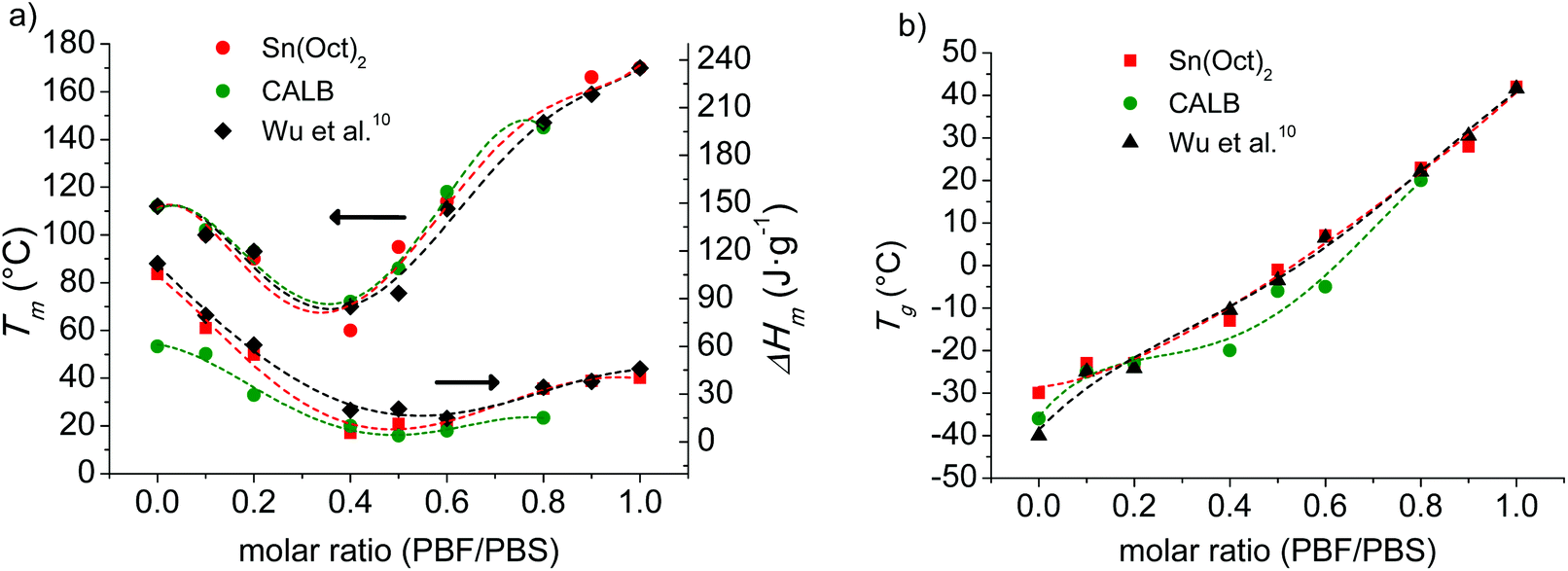 | ||
| Fig. 9 T m (a) and Tg (b) of coPBFxSy as a function of composition. Data reported by Wu et al.6 are also plotted for comparison. | ||
All the copolyesters display a single Tg in agreement with the random monomer distribution determined by NMR. The variation of Tg with the composition is plotted in Fig. 9b for the two series as well as for the series made by melt polycondensation reported by Wu et al.6 In all cases the Tg of PBS goes up continuously with the content of BF units, which should be expected for the increase in chain stiffness. These results demonstrate the possibility of tuning the Tg of the copolyesters by adjusting the composition. Again small discrepancies are observed among copolyesters produced by different procedures. Such deviations become more apparent for the copolyesters with intermediate compositions produced by enzyme catalyzed ROP, which are those presenting higher differences in molecular weight.
3.5. Hydrolytic and enzymatic degradation
It has been reported that PBS is hydrolysable and also biodegradable,20,42,43 whereas PBF is known to be highly resistant to hydrolysis and apparently insensitive to biodegradation,44 in agreement with the behaviour commonly displayed by aromatic polyesters. It is also known that the degradability of aromatic polyesters may be enhanced by inserting at random aliphatic ester units in the polymer chain. In fact the enzymatic degradation of random poly(butylene furanoate-co-adipate) copolyesters has been recently demonstrated, although a minimum content of 50% of adipate units was required to perceive the degradation effects.44In order to evaluate the degradability of PBF as a function of the incorporation of succinate units, the homopolyesters PBF and PBS and the copolyesters coPBF60S40 and coPBF40S60 were subjected in parallel to degradation assays. Samples were incubated at 37 °C and either pH 2.0 or pH 7.4, in the latter case both in the presence and in the absence of porcine pancreas lipase. The changes taking place in both the sample weight and molecular weight along an incubation period of 30 days were recorded and plotted in Fig. 10. According to what has been reported, PBF remained practically unchanged under all assayed conditions whereas PBS was more or less degraded depending on the pH and whether the enzyme is present or not, showing a maximum of losses at around 25%. As expected, the results obtained for the copolyesters depended on the composition, with degradation being faster as the content of succinate units increased. The degradation rate of the copolyesters was similar at the two assayed pH values, whereas a significant increase was observed in the presence of lipase where both the sample weight and molecular weight losses were around 20%. The conclusion that may be drawn from this preliminary study is that the degradability of PBF can be increased and controlled by the proper adjustment of the amount of succinate units that are incorporated by copolymerization.
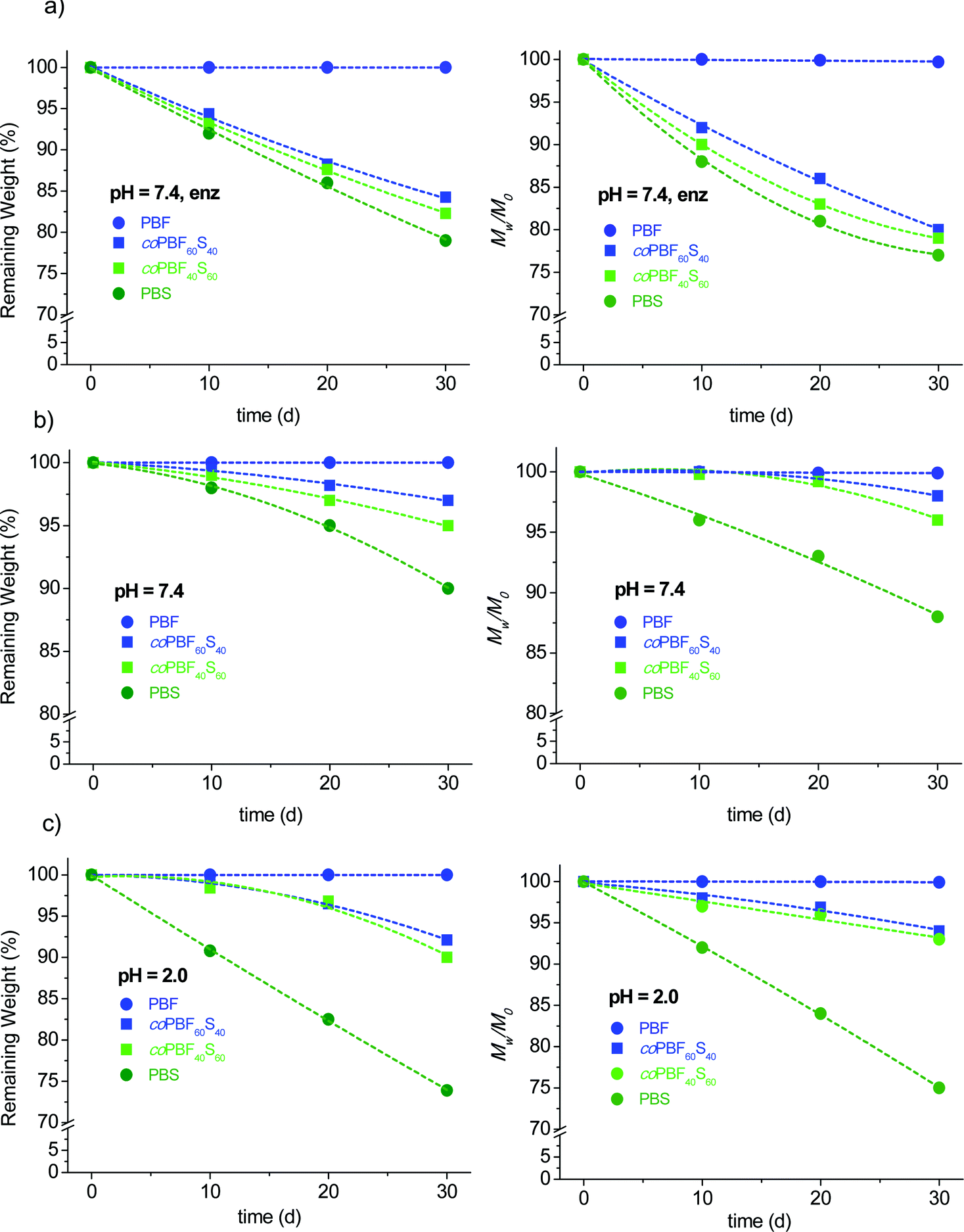 | ||
| Fig. 10 Enzymatic (a) and hydrolytic degradation (pH 7.4 (b) and pH 2.0 (c)) of coPBFxSy. Remaining weight (left) and molecular weight (right) vs. incubation time. | ||
4. Conclusions
The results attained in this work demonstrate for the first time that fully bio-based poly(butylene 2,5-furandicarboxylate-co-butylene succinate) copolyesters can be successfully synthesized by ROP. Furthermore, the reaction can be either chemically or enzymatically activated by using Sn(Oct)2 or CALB as the catalyst, respectively. A series of copolyesters covering the whole range of compositions including the two parent homopolyesters could be prepared by chemically catalyzed ROP, whereas only copolyesters containing up to 80 mol% of butylene furanoate units could be prepared by the enzymatic procedure. The ROP method has been proven to be suitable for providing high molecular weight poly(butylene 2,5-furandicarboxylate-co-succinate) copolyesters under reaction conditions milder than those required for melt polycondensation. However, the ROP reactions catalyzed by lipase proceed at significantly higher rates, generating copolyesters of lower molecular weights. The composition of the copolyesters produced by ROP can be readily controlled by adjusting the composition of the feed, and their microstructure is random in all cases. The differences in thermal properties between the two series (obtained by chemically and enzymatically catalyzed ROP) are small and reasonable according to their differences in molecular weights. The preliminary results obtained in this work on biodegradability will contribute to encourage the interest in these copolyesters as fully bio-based materials suitable for short-term applications.Comparison of our results with those reported by Wu et al.6 for the same copolyesters produced by melt polycondensation leads us to conclude that the ROP method is a promising option for the synthesis of coPBFxSy copolyesters. However, the true potential of the method in an industrial context will be determined by the accessibility to the cyclic oligomers required for polymerization. The c(BS)n oligomers are generated as a minor subproduct in the industrial production of PBS by melt polycondensation so that they are readily accessible at large scale. Conversely, the only source today of c(BF)n is their synthesis in the laboratory, meaning their short-term availability in large amounts is more than unlikely. Nevertheless, the development of PBF chemical recycling processes based on cyclodepolymerization in the not too distant future could be envisaged as a source of these cyclic oligomers. In fact, this method is one of the routes that can be used in the laboratory for their preparation.
Acknowledgements
Financial support for this research was given by MINECO with grant MAT-2012-38044-CO3-03. J. C. Morales-Huerta thanks CONACYT (Mexico) for the Ph.D. grant awarded. C. B. Ciulik wishes to acknowledge CNPq (National Research Council)-Brazil for the financial support. The authors are indebted to Abdelilah Alla for her assistance with the XRD experiments and data processing. The authors are also thankful to Novozymes for providing the Novozym 435 that has been used in this work.References
- A. Gandini, Polym. Chem., 2010, 1, 245–251 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2006, 107, 2411–2502 CrossRef PubMed.
- C. Vilela, A. F. Sousa, A. Fonseca, A. Serra, J. Cohello, C. Freire and A. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. Freire, G. J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- J. Ma, Y. Pang, M. Wang, J. Xu, H. Ma and X. Nie, J. Mater. Chem., 2012, 22, 3457–3461 RSC.
- L. Wu, R. Mincheva, Y. Xu, J. M. Raquez and P. Dubois, Biomacromolecules, 2012, 13, 2973–2981 CrossRef CAS PubMed.
- C. H. Wilsens, B. A. Noordover and S. Rastogi, Polymer, 2014, 55, 2432–2439 CrossRef CAS.
- M. Matos, A. F. Sousa, A. Fonseca, C. Freire, J. Coehlo and A. Silvestre, Macromol. Chem. Phys., 2014, 215, 2175–2184 CrossRef CAS.
- N. Jacquel, R. Saint-Loup, J. P. Pascault, A. Rousseau and F. Fenouillot, Polymer, 2015, 59, 234–242 CrossRef CAS.
- M. Soccio, M. Costa, N. Lotti, M. Gazzano, V. Siracusa, E. Salatelli, P. Manaresi and A. Munari, Eur. Polym. J., 2016, 81, 397–412 CrossRef CAS.
- G. Z. Papageorgiou, D. G. Papageorgiou, Z. Terzopoulou and D. N. Bikiaris, Eur. Polym. J., 2016, 83, 202–229 CrossRef CAS.
- J. Xu and G. Bao-Hua, Biotechnol. J., 2010, 5, 1149–1163 CrossRef CAS PubMed.
- I. Bechthold, K. Bretz, S. Kabasci, R. Kopitzky and A. Springer, Chem. Eng. Technol., 2008, 31, 647–654 CrossRef CAS.
- M. Gigli, M. Fabbri, N. Lotti, R. Gamberini, B. Rimini and A. Munari, Eur. Polym. J., 2016, 75, 431–460 CrossRef CAS.
- M. Bautista, A. Martínez de Ilarduya, A. Alla and S. Muñoz-Guerra, Polymers, 2015, 7, 1232–1247 CrossRef CAS.
- Q. Liu and X. M. Zhou, J. Macromol. Sci., Part A: Pure Appl. Chem., 2015, 52, 625–629 CrossRef CAS.
- L. W. Ren, Y. S. Wang, J. Ge, D. N. Lu and Z. Liu, Macromol. Chem. Phys., 2015, 216, 636–640 CrossRef CAS.
- Y. J. Sun, L. B. Wu, Z. Y. Bu, B. G. Li, N. X. Li and J. M. Dai, Ind. Eng. Chem. Res., 2014, 53, 10380–10386 CrossRef CAS.
- E. Zakharova, A. Alla, A. Martínez de Ilarduya and S. Muñoz-Guerra, RSC Adv., 2015, 5, 46395–46404 RSC.
- E. Zakharova, C. Lavilla, A. Alla, A. Martínez de Ilarduya and S. Muñoz-Guerra, Eur. Polym. J., 2014, 61, 263–273 CrossRef CAS.
- R. T. Duan, Q. X. He, X. Dong, D. F. Li, X. L. Wang and Y. Z. Wang, ACS Sustainable Chem. Eng., 2016, 4, 350–362 CrossRef CAS.
- S. L. Li, F. Wu, Y. Yang, Y. Z. Wang and J. B. Zeng, Polym. Adv. Technol., 2015, 26, 1003–1013 CrossRef CAS.
- L. C. Tan, Y. W. Chen, W. Zhou, J. C. Wei and S. W. Ye, J. Appl. Polym. Sci., 2011, 121, 2291–2300 CrossRef CAS.
- M. Lomelí-Rodríguez, M. Martín-Molina, M. Jiménez-Pardo, Z. Nasim-Afzal, S. I. Cauët, T. E. Davies, M. Rivera-Toledo and J. A. Lopez-Sanchez, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2876–2887 CrossRef.
- J. Zhu, J. Cai, W. Xie, P. Chen, M. Gazzano, M. Scandola and R. Gross, Macromolecules, 2013, 46, 796–804 CrossRef CAS.
- A. Gandini and T. M. Lacerda, Prog. Polym. Sci., 2015, 48, 1–39 CrossRef CAS.
- R. J. I. Knoop, W. Vogelzang, J. Van Haveren and D. S. Van Es, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4191–4199 CrossRef CAS.
- (a) M. Ishii, M. Okazaki, Y. Shibasaki, M. Ueda and T. Teranishi, Biomacromolecules, 2001, 2, 1267–1270 CrossRef CAS PubMed; (b) R. Mani, M. Bhattacharya, C. Leriche, L. Nie and S. Bassi, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3232–3239 CrossRef CAS.
- Y. Jiang, A. J. Woortman, G. O. A. van Ekenstein and K. Loos, Polym. Chem., 2015, 6, 5198–5211 RSC.
- (a) S. Sugihara, K. Toshima and S. Matsumura, Macromol. Rapid Commun., 2006, 27, 203 CrossRef CAS; (b) S. Matsumura, Macromol. Biosci., 2002, 2, 105–126 CrossRef CAS.
- T. Yagihara and S. Matsumura, Polymers, 2012, 4, 1259–1277 CrossRef.
- H. Ebata, K. Toshima and S. Matsumura, J. Synth. Org. Chem. Jpn., 2008, 66, 673–382 CrossRef CAS.
- C. W. Lee, K. Masutani and Y. Kimura, Polymer, 2014, 55, 5673–5679 CrossRef CAS.
- D. Pfister, G. Storti, F. Tancini, L. I. Costa and M. Morbidelli, Macromol. Chem. Phys., 2015, 216, 2141–2146 CrossRef CAS.
- J. C. Morales-Huerta, A. Martínez de Ilarduya and S. Muñoz-Guerra, Polymer, 2016, 87, 148–158 CrossRef.
- J. C. Morales-Huerta, A. Martínez de Ilarduya and S. Muñoz-Guerra, ACS Sustainable Chem. Eng., 2016, 4, 4965–4973 CrossRef CAS.
- P. Hodge, Chem. Rev., 2014, 114, 2278–2312 CrossRef CAS PubMed.
- D. J. Brunelle, Cyclic Oligomer Chemistry, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1151–1164 CrossRef CAS.
- S. Strandman, J. E. Gautrot and X. X. Zhu, Polym. Chem., 2011, 2, 791–799 RSC.
- A. Kondo, S. Sugihara, M. Kuwahara, K. Toshima and K. Matsumura, Macromol. Biosci., 2008, 8, 533–539 CrossRef CAS PubMed.
- J. C. Randall, in Polymer Sequence Determination, New York, 1977, pp. 42–62 Search PubMed.
- M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS.
- J. Xu and B.-H. Guo, Biotechnol. J., 2010, 5, 1149–1163 CrossRef CAS PubMed.
- W. Zhou, X. Wang, B. Yang, Y. Xu, W. Zhang, Y. Zhang and J. Ji, Polym. Degrad. Stab., 2013, 98, 2177–2183 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py01879c |
| This journal is © The Royal Society of Chemistry 2017 |