
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA PHEA-g-PEO well-defined graft copolymer exhibiting the synchronous encapsulation of both hydrophobic pyrene and hydrophilic Rhodamine 6G†
Fangxu
Sun
,
Guolin
Lu
,
Chun
Feng
,
Yongjun
Li
and
Xiaoyu
Huang
*
Key Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, People's Republic of China. E-mail: xyhuang@sioc.ac.cn; Fax: +86-21-64166128; Tel: +86-21-54925310
First published on 17th November 2016
Abstract
A well-defined graft copolymer consisting of a poly(2-hydroxyethyl acrylate) (PHEA) backbone and poly(ethylene oxide) (PEO) side chains was synthesized by successive reversible addition–fragmentation chain transfer (RAFT) polymerization and atom transfer nitroxide radical coupling (ATNRC) reaction. RAFT homopolymerization of a Cl-containing acrylate monomer, 2-hydroxyethyl 2-((2-chloropropanoyloxy)methyl)acrylate (HECPMA), was first performed in a controlled way to afford a well-defined PHEA-based backbone with a low polydispersity (Mw/Mn = 1.08). The target poly(2-hydroxyethyl acrylate)-g-poly(ethylene oxide) (PHEA-g-PEO) graft copolymer with a narrow molecular weight distribution (Mw/Mn = 1.16) was then obtained by ATNRC reaction between the pendant –OCOCH(CH3)Cl group in every repeated unit of PHEA-based backbone and PEO with a TEMPO end group via the grafting-onto strategy, using CuCl/PMDETA as a catalytic system. The critical micelle concentrations (cmcs) of the obtained graft copolymer in pure water, brine, and aqueous Na2SO4 solution were determined by the fluorescence probe technique using N-phenyl-1-naphthylamine as probe and micellar morphologies in aqueous media were visualized by TEM. It was found that PHEA-g-PEO graft copolymer self-assembled into large compound micelles in aqueous media, which can encapsulate hydrophilic Rhodamine 6G and hydrophobic pyrene separately or simultaneously.
Introduction
Recently, reversible-deactivation radical polymerizations (RDRP), including nitroxide mediated radical polymerization (NMRP),1,2 atom transfer radical polymerization (ATRP),3–6 reversible addition–fragmentation chain transfer (RAFT) polymerization,7,8 and single-electron-transfer living radical polymerization (SET-LRP),9,10 have been widely investigated and applied to synthesize diverse polymers with different architectures. Combining certain RDRP strategies is a good way to construct polymers with complicated structures and compositions, such as graft,11 hyperbranched,12 cyclic,13 dendritic,14 and star-shaped copolymers,15,16 which can be used in biomedical materials,17 nanotechnology,18 catalysis,19etc. The synthesis of graft copolymers via three common strategies including grafting-through,20–22 grafting-onto,11,23,24 and grafting-from25,26 is a typical example of the combination of certain controlled polymerization approaches. The grafting-onto strategy is a promising way to construct graft copolymers because their structures and compositions can be tuned precisely. Thanks to the development of coupling reactions with high efficiency, this strategy has become a convenient and efficient way to synthesize graft copolymers.24,27,28Click chemistry,29 in which Cu-catalyzed azide/alkyne cycloaddition (CuAAC) reaction has been studied most deeply, has been widely employed in polymer chemistry nowadays.29–32 However, the photosensitive and unstable azide compounds used in CuAAC would pose a security threat to people and need to be operated carefully.33
Combining NMRP and ATRP, Huang et al. put forward a new coupling strategy, atom transfer nitroxide radical coupling (ATNRC),33,34 in which graft copolymers can be obtained by the coupling reaction between the polymeric backbone containing halide atoms and the side chains containing 2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPO) radicals in the presence of CuBr and ligands. ATNRC is quite different from ATRP because CuBr in ATNRC is not a catalyst, but a reactant.4,34 The carbon radicals on the backbone would be captured by TEMPO-containing polymeric chains immediately to form an alkoxyamine between two polymers. The coupling efficiency of ATNRC can be as high as 90%, even comparable to CuAAC reaction, therefore ATNRC has been often employed as an efficient way to construct well-defined polymers, such as graft, star, and block copolymers.35–39
Poly(ethylene oxide) (PEO) is often chosen as the hydrophilic part of amphiphilic copolymers40–43 since PEO is a kind of nontoxic, nonionic, semi-crystalline polymer with excellent solubility in aqueous media.44 Besides, it also displays excellent chemical and biological properties including non-covalent complexation ability, good biocompatibility, lack of immunogenicity, and resistance to both protein adsorption and cellular adhesion.45–48 Particularly, the incorporation of a PEO segment into a macromolecule can obviously modulate the properties of polymeric solution.49 ATNRC is a convenient way to graft PEO side chains to the polymeric backbone because well-defined PEO homopolymers with defined terminal functionalities are commercially available.33,50 Graft copolymers bearing PEO side chains have been obtained via ATNRC effectively with a tunable density of PEO side chains.33,51–54
As for graft copolymers based on poly(2-hydroxyethyl acrylate) (PHEA)/poly(2-hydroxyethyl methylacrylate) (PHEMA) as the polymeric backbone, their synthesis is a tough task because the pendant hydroxyls are usually utilized to connect the side chains via ring opening polymerization (ROP),55 ATRP56–58 or “click chemistry”,24etc. In addition, PHEMA has always been regarded as one kind of hydrophilic copolymers. In 2004, however, Armes et al. discovered that the solubility of PHEMA-based copolymers was closely related with polymerization degree, temperature, and composition.59 Thus, an interesting question arises from those studies, what would be the self-assembly behavior in aqueous phase of the graft copolymers bearing PHEA/PHEMA as the backbone and PEO as the side chains?
On the basis of a newly developed trifunctional monomer, 2-hydroxyethyl 2-((2-chloropropanoyloxy)methyl)acrylate (HECPMA) bearing a –OCOCH(CH3)Cl moiety and a hydroxyl simultaneously,60 the pendant hydroxyls can remain intact while grafting the side chains. Herein, we report the synthesis of a poly(2-hydroxyethyl acrylate)-g-poly(ethylene oxide) (PHEA-g-PEO) graft copolymer, bearing a PHEA backbone via ATNRC (Scheme 1). PHECPMA homopolymer with a pendant halogen-containing –OCOCH(CH3)Cl group in every repeated unit was first obtained through RAFT homopolymerization of HECPMA monomer. PEO homopolymer with TEMPO terminal functionality was then grafted onto PHECPMA backbone by ATNRC to give the well-defined PHEA-g-PEO graft copolymer. Interestingly, PHEA-g-PEO graft copolymer could self-assemble in aqueous media to form large compound micelles, able to encapsulate hydrophobic and hydrophilic agents.
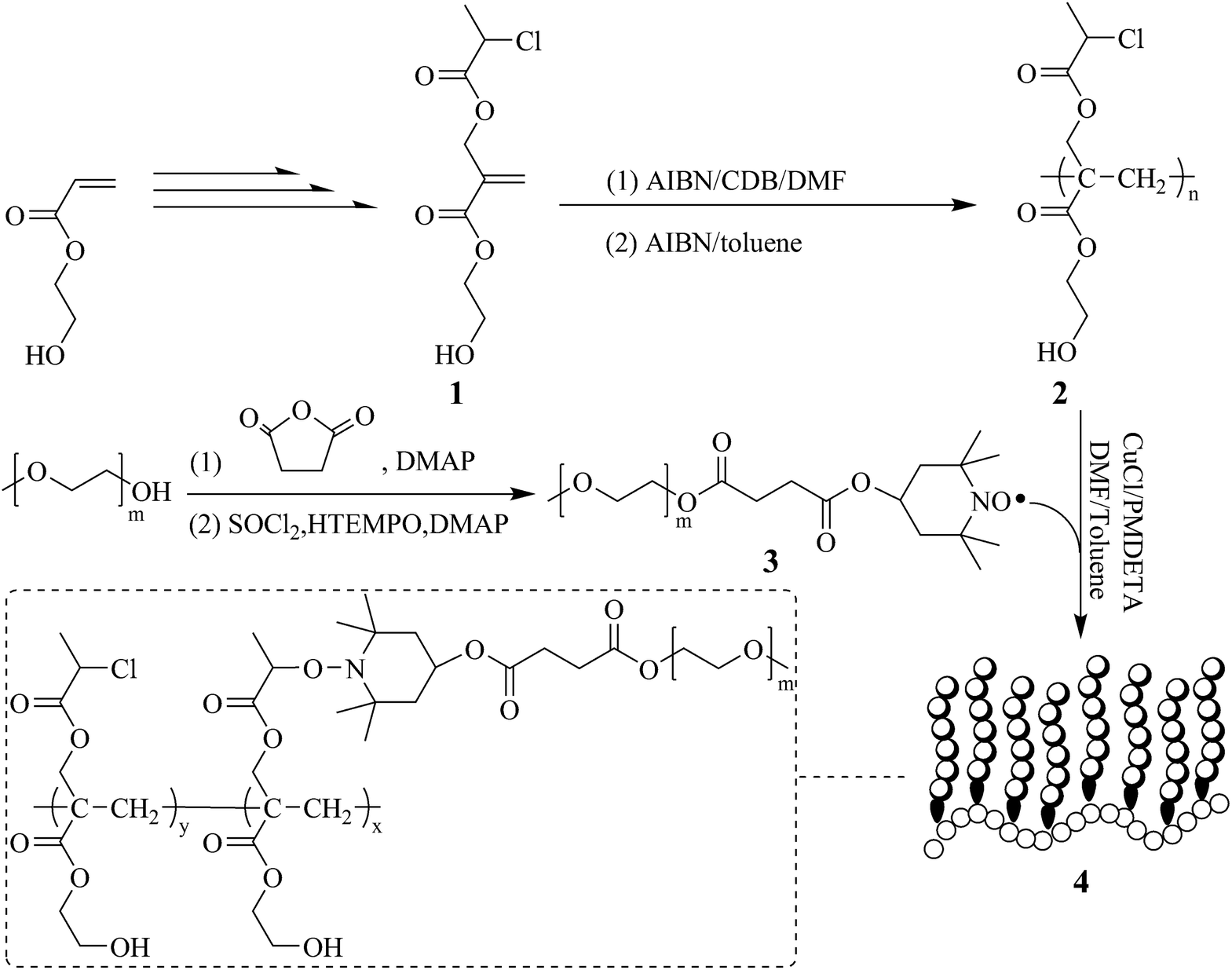 | ||
| Scheme 1 Synthetic route of PHEA-g-PEO well-defined graft copolymer. | ||
Results and discussion
Preparation of PHEA-based well-defined backbone
It has been extensively reported that ethylene oxide, a cyclic monomer, cannot be polymerized via RDRP, which means that graft copolymers bearing PEO side chains cannot be constructed through the most popular grafting-from approach using RDRP. On the other hand, well-defined PEO homopolymers with precisely determined molecular weights, narrow molecular weight distributions, and controllable end functionalities are commercially available so that the grafting-onto strategy using the coupling reactions with high efficiency such as click chemistry and ATNRC is generally used for constructing graft copolymers with PEO side chains.24,33,52–54In the current case, the desired graft copolymer is PHEA-g-PEO in which every repeated unit of the backbone possesses a pendant hydroxyl, that is to say PEO side chains cannot be attached onto the backbone through the usual ester linkage. To solve this puzzle, HECPMA monomer60 developed by us in 2014 which comprised a polymerizable double bond, a halogen-containing –OCOCH(CH3)Cl group, and a hydroxyl was herein employed. The first amphiphilic graft copolymer containing a hydrophilic PHEA backbone, PHEA-g-PS, was obtained through the grafting-from strategy60via ATRP of styrene initiated by –OCOCH(CH3)Cl group in every repeated unit of PHECPMA homopolymer; and this graft copolymer is quite different from PHEMA/PHEA-based graft copolymers24,55–58 whose backbone was usually hydrophobic because the pendant hydroxyls of the backbone were utilized for connecting the side chains. Moreover, halide (Cl) atom of –OCOCH(CH3)Cl group in every repeated unit of PHECPMA homopolymer can also be an effective component in ATNRC reaction so as to be treated with terminal TEMPO radical of the polymeric chain for affording the corresponding graft copolymer through the grafting-onto strategy. Herein, we assume that PHEA-g-PEO graft copolymer can be formed via ATNRC reaction between Cl atom of –OCOCH(CH3)Cl group in every repeated unit of PHECPMA backbone and TEMPO end functionality of PEO side chains.
Firstly, a well-defined PHEA-based backbone possessing a pendant hydroxyl in every repeated unit was constructed via RAFT homopolymerization of HECMPA 1 monomer using CDB61 as chain transfer agent (CTA). ATRP is not suitable for the homopolymerization of HECPMA because ATRP of functional monomers comprising a halogen-containing initiating group would result in an undesirable hyperbranched polymer, not a linear polymer.62 RAFT polymerization with excellent tolerance of various functionalities63,64 was employed for the homopolymerization of HECPMA monomer since it would not interfere with –OCOCH(CH3)Cl group during the polymerization process. As HECPMA is an acrylate monomer, CDB might be the appropriate CTA for its RAFT homopolymerization.65 A pink powder with dithiobenzoate end group (inset a in Fig. 1) was first obtained, which would pose a negative influence on the following ATNRC reaction. Then, excessive AIBN (32.9 eq.) was used to eliminate the terminal dithiobenzoate group,66 affording a white PHECPMA 2 homopolymer (inset b in Fig. 1) and the disappearance of the signal of dithiobenzoate end group at 510 nm in UV/vis absorbance spectrum showed the complete removal of dithiobenzoate end group.66
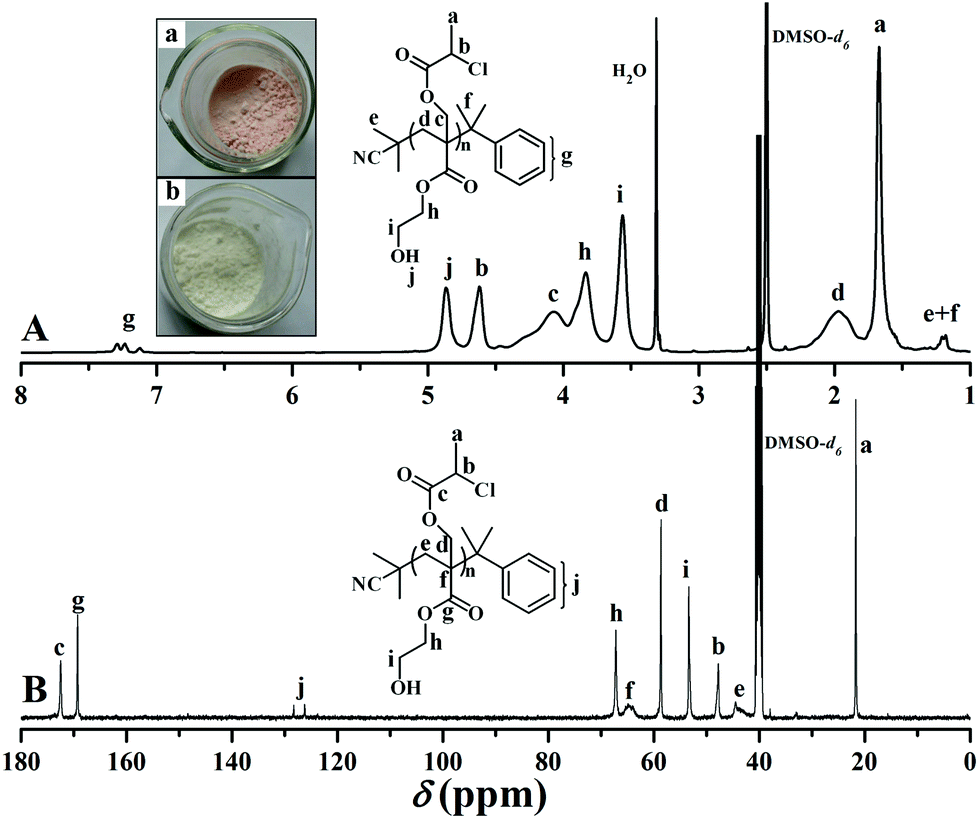 | ||
| Fig. 1 1H (A) and 13C (B) NMR spectra of PHECPMA 2 in DMSO-d6. | ||
PHECPMA 2 homopolymer was characterized by GPC, FT-IR, 1H NMR, and 13C NMR and the details can be found in our previous report.60 In particular, 1H NMR spectrum of the homopolymer (Fig. 1A) exhibited the absence of the signals of the double bond of HECPMA monomer at 5.95 and 6.46 ppm, which closely matched the disappearance of the peaks at 129.0 and 134.4 ppm in 13C NMR spectrum (Fig. 1B), verifying the successful polymerization of HECPMA monomer. RAFT mechanism of the polymerization was illustrated by the multiplet between 7.11 ppm and 7.30 ppm in 1H NMR spectrum plus the peaks at 126.0 and 128.1 ppm in 13C NMR spectrum belonged to the phenyl end group. The proton resonance signals of the –OCOCH(CH3)Cl group were located at 4.63 and 1.66 ppm and the corresponding carbon resonance signals were located at 21.8 (CHClCH3) and 47.8 (CHClCH3) ppm, respectively, which confirmed the retention of –OCOCH(CH3)Cl group during RAFT polymerization and the elimination of terminal dithiobenzoate group. The proton resonance signal of hydroxyl also appeared at 4.88 ppm in Fig. 1A. Thus, all these evidences clearly witnessed the formation of a well-defined PHECPMA 2 homopolymer bearing a pendant –OCOCH(CH3)Cl group and hydroxyl in every repeated unit. Moreover, because every HECPMA repeated unit consists of both a hydrophilic hydroxyl and hydrophobic –OCOCH(CH3)Cl moiety, PHECPMA 2 homopolymer may behave like a kind of amphiphilic homopolymer.67 Indeed, this homopolymer is water-soluble with low solubility and it self-assembled into aggregates with a hydrodynamic diameter (Dh) of about 80 nm with the rising of the concentration of the homopolymer in aqueous media.
The absolute molecular weight of the homopolymer (Mn,GPC/MALS) was determined by GPC/MALS with a value of 8223 g mol−1 so that the number of HECPMA repeated unit was calculated according to eqn (1) (187 and 236.5 refer to the molecular weights of terminal CTA moiety and HECPMA monomer, respectively). The result showed that PHECPMA 2 homopolymer possesses 34.0 –OCOCH(CH3)Cl groups.
| nHECPMA = (Mn,GPC/MALS − 187)/236.5 | (1) |
Characterization of PEO-TEMPO
Commercially available PEO-OH (Mn = 2000 g mol−1) was firstly converted to PEO-COOH through the esterification of terminal hydroxyl using succinic anhydride. The broad peak at 3507 cm−1 and the sharp band at 1734 cm−1 in FT-IR spectrum after the esterification (Fig. 2A) were attributed to the newly incorporated carboxyl, and the peaks located at 2.60, 3.55, and 4.21 ppm in 1H NMR spectrum belonged to the protons of the linkage between PEO and carboxyl, which evidenced that the terminal hydroxyl of PEO had been transformed into carboxyl. Furthermore, the signal at 173.9 ppm in 13C NMR spectrum also distinctly affirmed the introduction of carboxyl.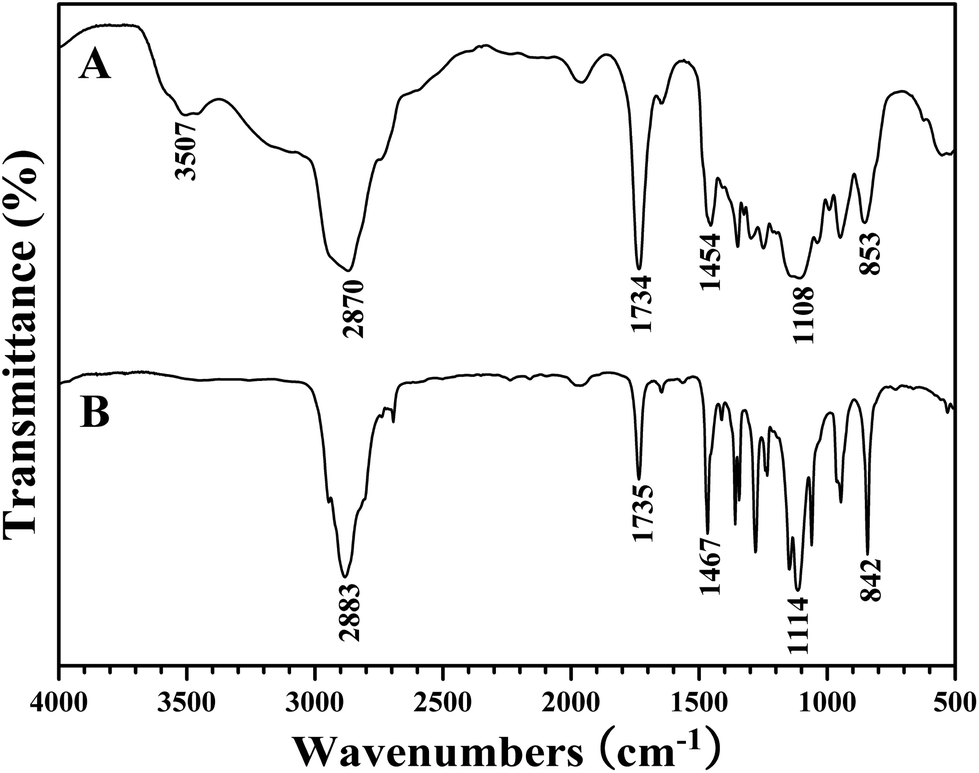 | ||
| Fig. 2 FT-IR spectra of PEO-COOH (A) and PEO-TEMPO (B). | ||
Next, treated with SOCl2, PEO-COOH was converted to PEO-COCl almost quantitatively followed by its in situ reaction with HTEMPO to afford the desired PEO-TEMPO. The color change from colorless to orange was the first piece of evidence on the demonstration of successful preparation of PEO-TEMPO. In FT-IR spectrum (Fig. 2B), the disappearance of the broad peak at 3507 cm−1 pointed out the complete conversion of carboxyl. Meanwhile, the resonance signals at 1.15, 1.53, and 1.85 ppm in 1H NMR spectrum (Fig. 3A) were attributed to the protons of CH3 and CH2 of TEMPO moiety, and the peaks at 19.9 (CH3) and 60.9 (C(CH3)2) ppm in 13C NMR spectrum (Fig. 3B) also verified the incorporation of TEMPO. The covalent connection between TEMPO and PEO was demonstrated by the peak at 2.60 ppm originating from 4 protons of CO2CH2CH2CO2. Thus, it is clear that PEO was successfully functionalized with TEMPO end group, giving PEO-TEMPO 3.
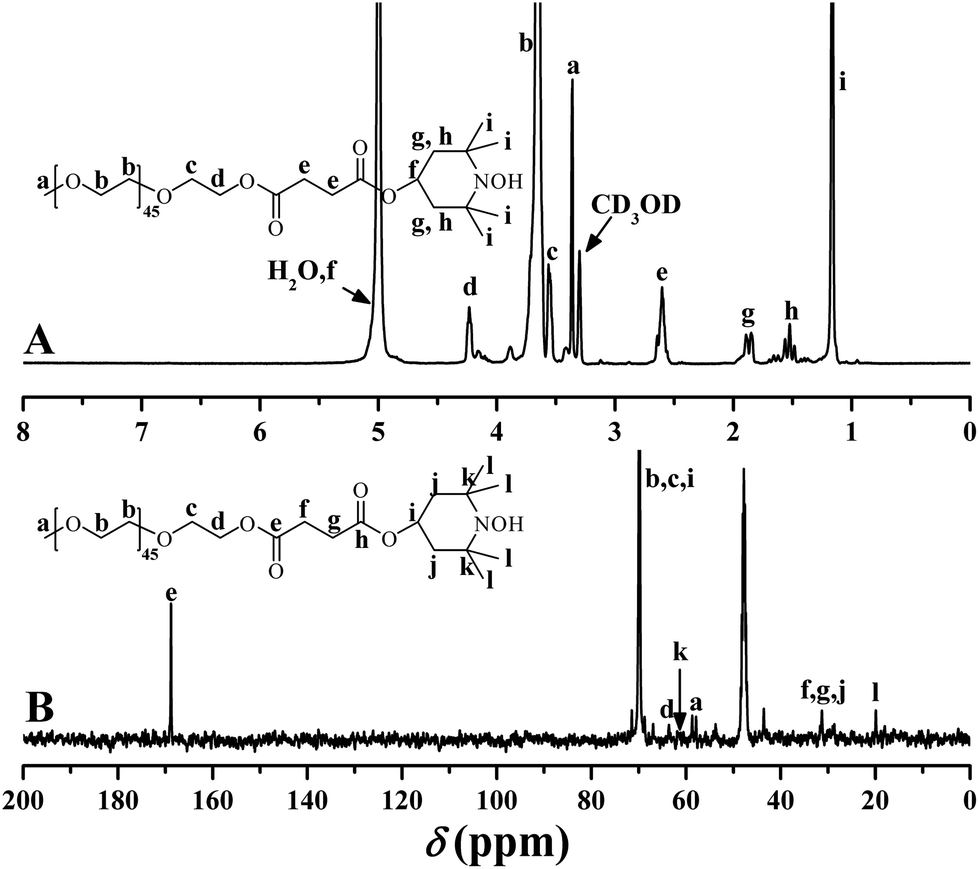 | ||
| Fig. 3 1H (A) and 13C (B) NMR spectra of PEO-TEMPO 3 in CD3OD. | ||
Synthesis of PHEA-g-PEO well-defined graft copolymer
The desired PHEA-g-PEO graft copolymer was constructed via the grafting-onto strategy by a Cu(I)-mediated ATNRC reaction since the pendant –OCOCH(CH3)Cl groups in PHECPMA 2 were designed to directly couple with the terminal TEMPO functionality of PEO-TEMPO 3. Herein, ATNRC reaction23,33–39 was performed in DMF/toluene (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v = 1:1) at 90 °C using CuCl/PDMAEA as catalytic system and the results are summarized in Table 1. Dialysis was utilized to exclude the unreacted PEO-TEMPO 3 as affirmed by GPC curve after purification (Fig. 4) where the trace of PEO-TEMPO 3 was absent. The molecular weight of the purified product was much higher than that of PHECPMA, which demonstrated the occurrence of a coupling reaction. Moreover, it should be pointed out that GPC curve showed a unimodal and symmetric eluent peak without tailing or shoulder, this indicating the absence of intra- or inter-termination during the coupling process;68 and the polydispersity was as low as 1.16, which illustrated that the structure of the product was well-defined.
:v = 1:1) at 90 °C using CuCl/PDMAEA as catalytic system and the results are summarized in Table 1. Dialysis was utilized to exclude the unreacted PEO-TEMPO 3 as affirmed by GPC curve after purification (Fig. 4) where the trace of PEO-TEMPO 3 was absent. The molecular weight of the purified product was much higher than that of PHECPMA, which demonstrated the occurrence of a coupling reaction. Moreover, it should be pointed out that GPC curve showed a unimodal and symmetric eluent peak without tailing or shoulder, this indicating the absence of intra- or inter-termination during the coupling process;68 and the polydispersity was as low as 1.16, which illustrated that the structure of the product was well-defined.
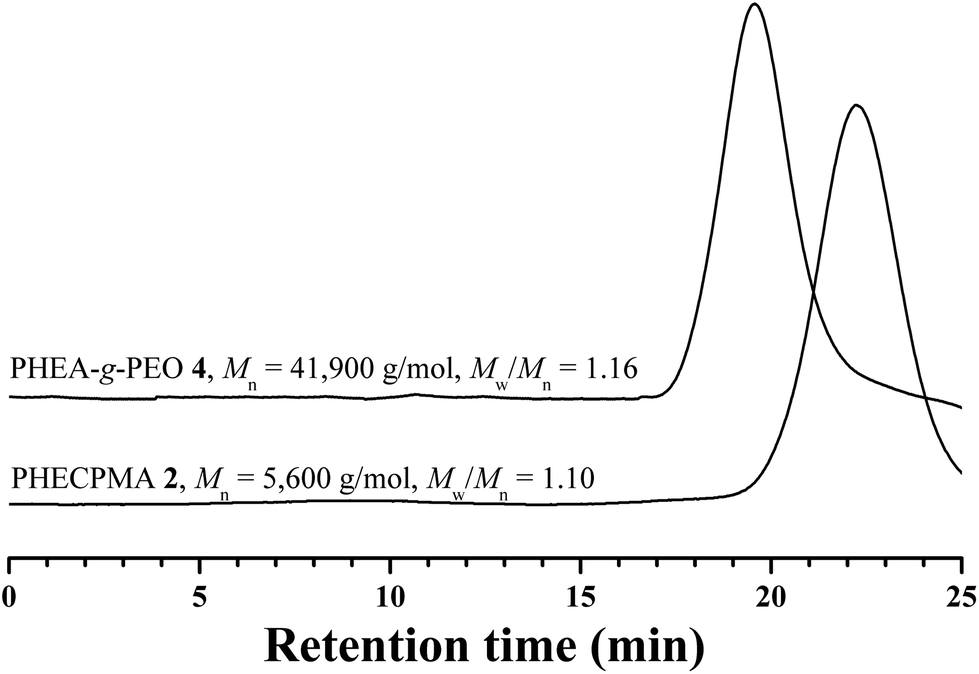 | ||
| Fig. 4 GPC curves of PHECPMA 2 and PHEA-g-PEO 4 in DMF. | ||
| [TEMPO]:[CuCl] |
M
n,GPCb (g mol−1) |
M
w/Mnb |
n
PEOc |
D
graftd (%) |
|---|---|---|---|---|
|
a Synthesized from PHECPMA 2 (Mn,GPC/MALS = 8223 g mol−1, Mw/Mn = 1.08, NHECPMA = 34.0) and PEO-TEMPO 3 in DMF/toluene (v:v = 1:1) using CuCl/PMDETA, [PMDETA]:[CuCl] = 1:1, temperature: 90 °C, time: 3 h.
b Measured by GPC in DMF at 35 °C.
c The number of grafted PEO side chains obtained from 1H NMR.
d The grafting density of PEO side chain obtained from 1H NMR.
|
||||
| 1.2:1 |
41900 |
1.16 | 11.0 | 32.4 |
1H NMR spectrum of the purified product in CDCl3 is shown in Fig. 5A. The peaks appearing at 2.62 (4H, O2CCH2CH2CO2), 3.38 (3H, OCH3), 3.56 (2H, CO2CH2CH2O), 3.64 (4H, OCH2CH2), and 4.25 (2H, CO2CH2CH2O) ppm were typical proton resonance signals of PEO side chains while the proton resonance signal originating from the polyacrylate-based backbone was located at 2.06 (2H, CH2CCO2) ppm. The presence of a piperidine linkage between PHEA backbone and PEO side chains was clearly evidenced by the proton resonance signals at 0.89, 1.21 (12H, CH3 of TEMPO), 1.25, 1.38 (4H, CH2 of TEMPO), and 5.00 (1H, CH of TEMPO) ppm. The minor peak at 3.88 (2H, CO2CH2CH2OH) ppm plus the strong peak at 3430 cm−1 in FT-IR spectrum verified the retention of pendant hydroxyls in the polyacrylate-based backbone after ATNRC reaction, i.e. hydroxyl is inert during the Cu(I)-mediated coupling reaction.
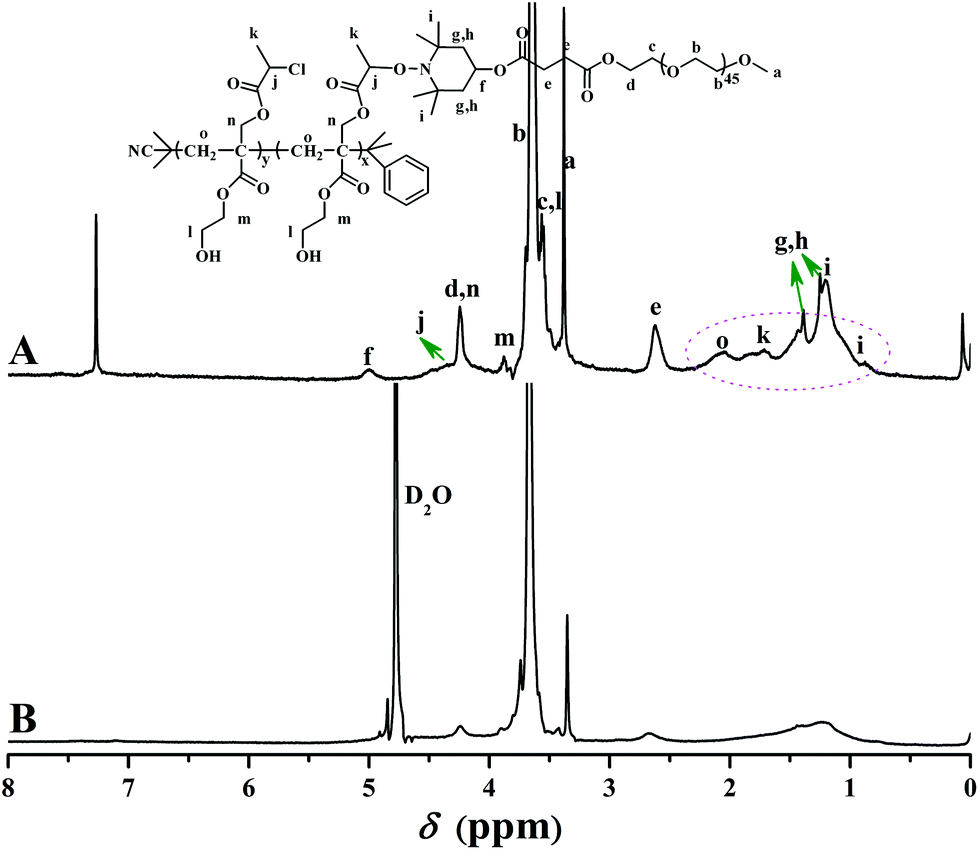 | ||
| Fig. 5 1H NMR spectra of PHEA-g-PEO 4 in CDCl3 (A) and D2O (B). | ||
For graft copolymers with a non-linear structure, molecular weight, which is the basis for the calculation of the number of side chains, measured by GPC using linear polystyrene as standard, is certainly much different from the ‘actual’ value according to previous reports.21,69,70 Therefore, the number of grafted PEO side chains of PHEA-g-PEO 4 graft copolymer (nPEO), which was built through the grafting-onto approach, was estimated by 1H NMR instead of conventional GPC according to eqn (2) (Se and Sm are the integrated areas of peaks “e” at 2.62 ppm and “m” at 3.88 ppm in Fig. 5A, 34 is the number of HECPMA repeated units in the backbone). The result is 11.0 as listed in Table 1 so that the grafting density of PEO side chain was calculated to be 32.4% according to eqn (3). This grafting efficiency was similar to the moderate coupling efficiencies (36–75%) of CuAAC “grafting-onto” reaction.71 The relatively low coupling efficiency in the current work could be attributed to the steric hindrance of the dense structure of the graft copolymer.34,71 Thus, it is clear that PHEA-g-PEO 4 graft copolymer possesses a well-defined structure: a poly(2-hydroxyethyl acrylate) backbone (34.0 repeated units) and 11.0 grafted PEO side chains of a certain length (44.8 repeated units per side chain).
| nPEO = 34Se/2Sm | (2) |
| Dgraft = nPEO/34 | (3) |
Self-assembly of PHEA-g-PEO graft copolymer
In 2004, Armes et al. systematically investigated the solubility of copolymers based on PHEMA and they found that it was associated with molecular weight, composition, and temperature,59 which meant that the backbone of PHEA-g-PEO 4 graft copolymer, PHEA, was not absolutely water-soluble. As it was mentioned before that PHECPMA 2 homopolymer behaves like a kind of amphiphilic homopolymer, and only one third of hydrophobic –OCOCH(CH3)Cl moiety was consumed for coupling, we can deduce that PHEA backbone with 22.4 hydrophobic –OCOCH(CH3)Cl groups may act as a hydrophobic core in aqueous media, which offers an opportunity for studying the self-assembly behavior in aqueous media.First of all, critical micelle concentrations (cmcs) of PHEA-g-PEO 4 graft copolymer in aqueous media and different salt solutions ([NaCl] = 0.2 and 0.8 mol L−1, [Na2SO4] = 0.3 mol L−1) were measured by fluorescence spectroscopy using PNA as probe (Fig. 6). The fluorescence spectrum of PNA is sensitively influenced by the environment and the polarity of its surrounding.72,73 In the presence of micelles, PNA can be solubilized within the interior of the hydrophobic part so that the fluorescence intensity will increase with the ascending of the concentration of polymer. Fig. 6A shows the relationship of the fluorescence intensity of PNA (I/I0) with the concentration of PHEA-g-PEO 4 graft copolymer in aqueous media. Apparently, I/I0 rose sharply while the concentration of PHEA-g-PEO 4 graft copolymer exceeded a certain value, this indicating the incorporation of PNA probe into the hydrophobic core of micelles.72 Thus, cmc of PHEA-g-PEO 4 graft copolymer in aqueous media was determined to be the intersection of two straight lines with a value of 2.51 × 10−6 g mL−1. As shown in Fig. 6B–D, cmcs of PHEA-g-PEO 4 graft copolymer in different salt solutions are all higher than that in pure water, especially for two brine solutions (4.57 × 10−6 g mL−1 for [NaCl] = 0.2 M and 6.02 × 10−6 g mL−1 for [NaCl] = 0.8 M), which shows that cmc of PHEA-g-PEO 4 graft copolymer could be affected by adding an inorganic salt to increase the ion strength of solution. This effect might impact the hydrophobic segment to make the cmc rise or drop. In addition, all four cmcs are much lower than those of low molecular weight surfactants; however, they are comparable to those of polymeric amphiphiles.74,75
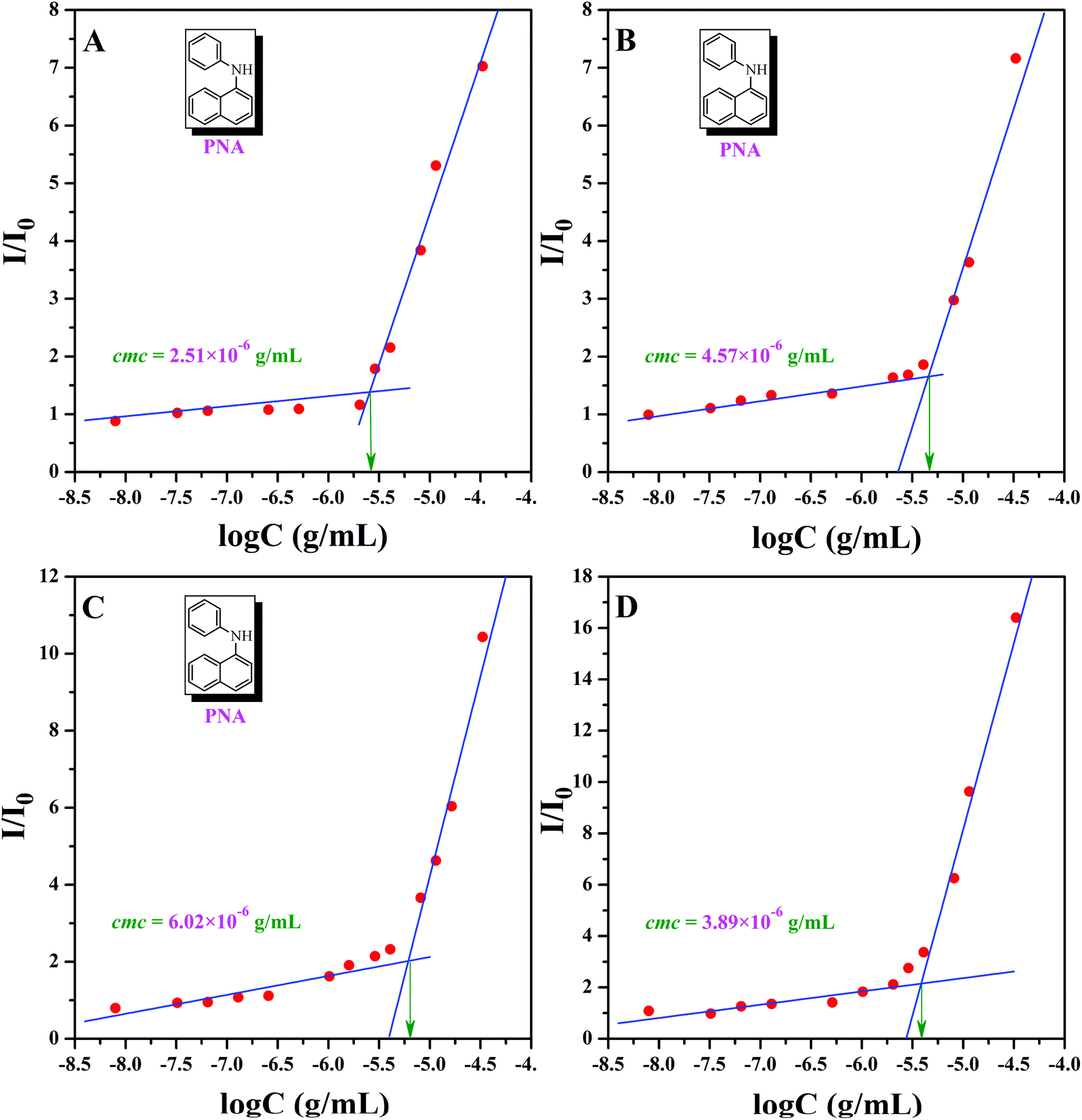 | ||
| Fig. 6 Dependence of fluorescence intensity ratio of PNA emission band at 418 nm ([PNA] = 2 × 10−6 g mL−1) on the concentration of PHEA-g-PEO 4 graft copolymer in pure water (A), 0.2 M NaCl (B), 0.8 M NaCl (C), and 0.3 M Na2SO4 (D). | ||
Aqueous and salt solutions of PHEA-g-PEO 4 graft copolymer with a concentration of 0.1 mg mL−1 (above cmc) were then prepared for TEM and DLS measurements. Micellar morphologies were visualized under TEM and it can be seen from Fig. 7 that PHEA-g-PEO 4 graft copolymer aggregated into spherical micelles in both pure water and salt solutions, whose sizes were distributed between 145.0 nm and 187.0 nm as determined by DLS in various aqueous media as shown in Fig. 8. Particularly, the micellar size in pure water was larger than those in salt solutions, which also evidenced that salt had an influence on the self-assembly behavior of PHEA-g-PEO 4 graft copolymer in aqueous media. The chemical composition of the micelles formed by PHEA-g-PEO 4 graft copolymer in aqueous media, i.e. the building segment of the core and corona, was determined by 1H NMR in D2O. 1H NMR is a common and convenient way to observe the solubility of a copolymer since the proton resonance signals of the insoluble segment would decrease significantly, even disappear, while the proton resonance signals of the soluble segment would remain unchanged. It can be seen from Fig. 5B that the characteristic proton resonance signals corresponding to PHEA backbone including peaks “j”, “k”, “m”, and “o” became attenuated apparently compared to those in Fig. 5A, however the typical proton resonance signals originating from PEO side chains such as peaks “a”, “b”, and “c” did not change at all. This result clearly supported our deduction that PHEA backbone with 22.4 hydrophobic –OCOCH(CH3)Cl groups acted as hydrophobic core and PEO side chains acted as hydrophilic corona in the current case; in contrast, PHEA backbone possessing pendent hydroxyls (all –OCOCH(CH3)Cl groups were consumed for initiation) acted as hydrophilic corona and PS side chains acted as hydrophobic core for PHEA-g-PS amphiphilic graft copolymer reported in 2014.60
 | ||
| Fig. 7 TEM images of micelles formed by PHEA-g-PEO 4 graft copolymer in pure water (A), 0.2 M NaCl (B), 0.8 M NaCl (C), and 0.3 M Na2SO4 (D) with a concentration of 0.1 mg mL−1. | ||
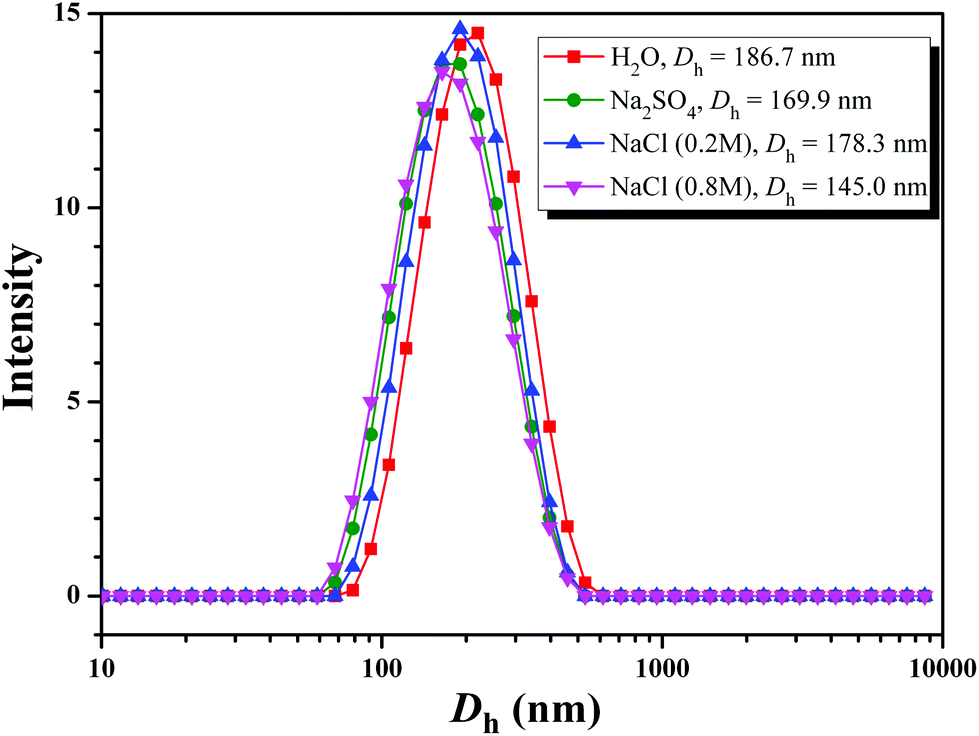 | ||
| Fig. 8 Hydrodynamic diameter distributions of PHEA-g-PEO 4 graft copolymer in various solutions with a concentration of 0.1 mg mL−1. | ||
Given the fact that the total lengths of PEG side chain and PHEA backbone were much smaller than half of Dh, these observations indicated that large compound micelles, not usual spherical micelles, were supposed to be formed in aqueous media, where PEO side chains formed the corona of micelles and the core consisted of numerous reverse micelles with islands of PEO segments in the continuous phase of PHEA segments.76,77
For common spherical micelles with a hydrophobic core and hydrophilic corona, only hydrophobic compounds can be encapsulated within the core of micelles. Nevertheless, according to the aforementioned results, PHEA-g-PEO 4 graft copolymer aggregated into large compound micelles in aqueous media so that it can be supposed that the hydrophilic and hydrophobic domains within the core of large compound micelles might be able to encapsulate both hydrophobic and hydrophilic agents. Therefore, pyrene soluble in the hydrophobic domain and water-soluble Rhodamine 6G (R6G) were employed as model guest molecules for examining the encapsulating ability of large compound micelles formed by PHEA-g-PEO 4 graft copolymer. We firstly checked the encapsulation capacity of aqueous micellar solution of copolymer 4 for hydrophobic compound in aqueous media using pyrene as a model hydrophobic agent78 and the loading content of pyrene, 9.7 μg pyrene per mg micelle, was determined by UV/vis absorption spectroscopy using a standard curve at 337 nm. The aqueous solution of pyrene (after filtration to remove the insoluble pyrene) showed almost no UV absorption of pyrene (Fig. 9A) while the typical UV absorption of pyrene appeared at 337 nm in UV/vis absorption spectrum of aqueous micellar solution of copolymer 4 (Fig. 9A), which illustrated that micelles could sequester pyrene from water so as to solubilize pyrene within the hydrophobic domains of the core.79,80
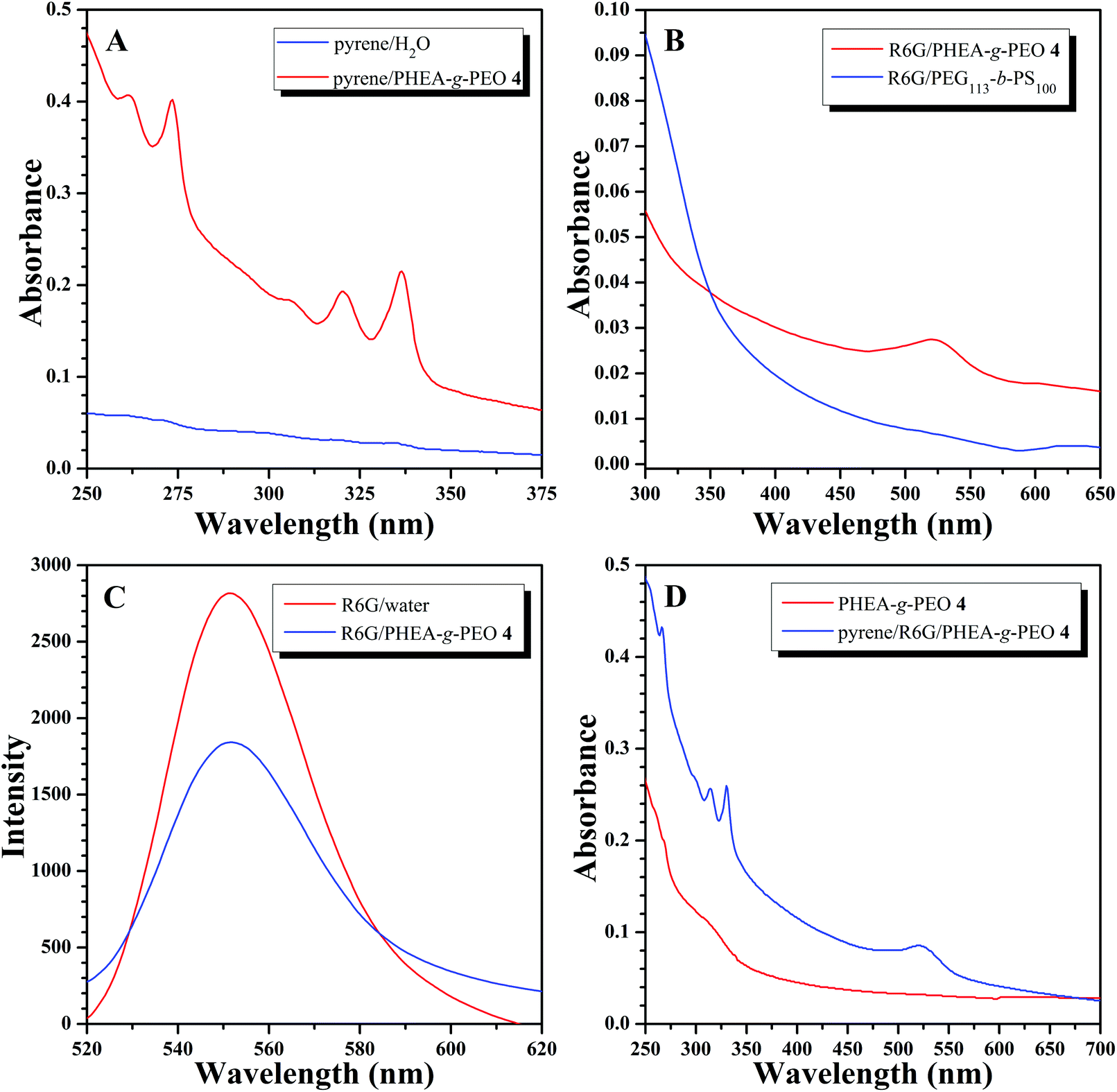 | ||
| Fig. 9 (A) UV/vis absorption spectra of pyrene in water and aqueous micellar solution of PHEA-g-PEO 4. (B) UV/vis absorption spectra of R6G in aqueous micellar solutions of PHEA-g-PEO 4 and PEG113-b-PS100. (C) Fluorescence emission spectra of R6G in water and an aqueous micellar solution of PHEA-g-PEO 4. (D) UV/vis absorption spectra of pyrene and R6G in an aqueous micellar solution of PHEA-g-PEO 4, and the micellar solution of graft copolymer 4 for the control experiment. | ||
Next, R6G was used as a model hydrophilic agent to check the encapsulation ability of aqueous micellar solution of copolymer 4 for hydrophilic compound. The loading content of R6G, 3.68 μg R6G per mg micelle, was also determined by UV/vis absorption spectroscopy using a standard curve at 522 nm. To check whether the amine group of R6G reacted with the pendant Cl in the graft copolymer, we recovered the graft copolymer from the micellar solution after loading with R6G by lyophilization and performed GPC measurement of the obtained graft copolymer using a UV detector (522 nm). Indeed, there is no detectable signal at 522 nm, the maximum absorbance for R6G, which meant that no R6G was covalently attached to the graft copolymer via a reaction between amine group of R6G and Cl of graft copolymer. The characteristic absorption peak of R6G was located at 522 nm in UV/vis absorption spectrum of R6G-containing aqueous micellar solution of copolymer 4 (after dialysis to remove free R6G) as shown in Fig. 9B, however, this peak could not be found in UV/vis absorption spectrum of R6G-containing aqueous micellar solution of PEG113-b-PS100 diblock copolymer (Fig. 9B), which formed common spherical micelles with an average hydrodynamic diameter of 73 nm in aqueous media.79 This fact obviously verified that the large compound micelles formed by copolymer 4 can capture R6G within the hydrophilic domains of the core of large compound micelles,81,82 while the common spherical micelles formed by PEG113-b-PS100 diblock copolymer cannot encapsulate R6G.78 It is well-recognized that the fluorescence intensity of R6G in aqueous micellar solution will be much lower than that of R6G in pure water solution.78,79Fig. 9C shows that the fluorescence intensity of R6G-containing aqueous micellar solution of copolymer 4 is just 65% that of pure aqueous solution of R6G with the same apparent concentration of R6G, which clearly indicated the self-quenching effect of R6G in the micellar solution, that is, R6G was indeed located inside the micelles formed by copolymer 4, and not free in water. Moreover, both pyrene and R6G can be encapsulated by aqueous micellar solution of copolymer 4 simultaneously. Both typical UV absorption peaks appeared at 332 (pyrene) and 522 (R6G) nm in UV/vis absorption spectrum of R6G/pyrene-containing aqueous micellar solution of copolymer 4 (Fig. 9D), which were absent in UV/vis absorption spectrum of aqueous micellar solution of copolymer 4 (Fig. 9D), indicating the co-solubilization of R6G and pyrene model loading agents within the core of micelles formed by copolymer 4. All these results demonstrated that large compound micelles formed by copolymer 4 can separately or simultaneously uptake hydrophobic and hydrophilic compounds, different from common spherical micelles, which was similar to the previous literature.78
Conclusions
We have presented the detailed synthesis of PHEA-g-PEO well-defined graft copolymer with a narrow molecular weight distribution (Mw/Mn = 1.16) through sequential RAFT polymerization and ATNRC reaction via the grafting-onto strategy, using a Cl-containing HECPMA trifunctional monomer as starting material. The whole synthesis process avoided post-polymerization functionality transformation since the pendant –OCOCH(CH3)Cl and hydroxyl groups of HECPMA acrylate monomer could remain inert during RAFT homopolymerization. With the successful preparation of PHECPMA homopolymer, the versatility of ATNRC can enable the substitution of PEO segments by other polymeric chains bearing TEMPO end functionality, which obviously paves a convenient way for developing new well-defined graft copolymers bearing PHEA backbone. The self-assembly behavior of PHEA-g-PEO graft copolymer in aqueous media was investigated by TEM, and large compound micelles with PEO as corona were formed in aqueous solution. In particular, the multi-component structure of large compound micelles formed by PHEA-g-PEO graft copolymer could be used as a multi-compartment delivery vehicle for the separate or simultaneous uptake of hydrophobic and hydrophilic compounds, which was verified to be able to separately or simultaneously package pyrene and R6G compounds in its different nanodomains.Acknowledgements
The authors acknowledge the financial support from National Natural Science Foundation of China (21274162), Strategic Priority Research Program of Chinese Academy of Sciences (XDB20020000), and Shanghai Scientific and Technological Innovation Project (16JC1402500 and 16520710300).Notes and references
- J. Li, X. Chen and Y. C. Chang, Langmuir, 2005, 21, 9562–9567 CrossRef CAS PubMed.
- L. Hong, J. He, Y. Chen and T. Kakuchi, Polym. Chem., 2016, 7, 3599–3607 RSC.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- S. R. Samanta, R. Cai and V. Percec, Polym. Chem., 2015, 6, 3259–3270 RSC.
- X. Pang, L. Zhao, C. Feng, R. Wu, H. Ma and Z. Lin, Polym. Chem., 2013, 4, 2025–2032 RSC.
- B. Mo, H. Liu, X. Zhou and Y. Zhao, Polym. Chem., 2015, 6, 3489–3501 RSC.
- Q. Tang, J. Chen, Y. Zhao and K. Zhang, Polym. Chem., 2015, 6, 6659–6663 RSC.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS PubMed.
- J. Burdynska, Y. Li, A. V. Aggarwal, S. Höger, S. S. Sheiko and K. Matyjaszewski, J. Am. Chem. Soc., 2014, 136, 12762–12770 CrossRef CAS PubMed.
- J. Teng and E. R. Zubarev, J. Am. Chem. Soc., 2003, 125, 11840–11841 CrossRef CAS PubMed.
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18–37 CrossRef CAS PubMed.
- H. R. Culver, S. D. Steichen, M. Herrera-Alonso and N. A. Peppas, Langmuir, 2016, 32, 5629–5636 CrossRef CAS PubMed.
- L. Lempke, A. Ernst, F. Kahl, R. Weberskirch and N. Krause, Adv. Synth. Catal., 2016, 358, 1491–1499 CrossRef CAS.
- Y. Xia, B. D. Olsen, J. A. Kornfield and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 18525–18532 CrossRef CAS PubMed.
- L. N. Gu, Z. Shen, S. Zhang, G. L. Lu, X. H. Zhang and X. Y. Huang, Macromolecules, 2007, 40, 4486–4493 CrossRef CAS.
- A. Cappelli, M. Paolino, G. Grisci, G. Giuliani, A. Donati, R. Mendichi, A. C. Boccia, F. Samperi, S. Battiato, E. Paccagnini, E. Giacomello, V. Sorrentino, M. Licciardi, G. Giammona and S. Vomero, Polym. Chem., 2011, 2, 2518–2527 RSC.
- Y. G. Li, Y. Q. Zhang, D. Yang, Y. J. Li, J. H. Hu, C. Feng, S. J. Zhai, G. L. Lu and X. Y. Huang, Macromolecules, 2010, 43, 262–270 CrossRef CAS.
- H. F. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 6633–6639 CrossRef CAS PubMed.
- G. Cavallaro, M. Licciardi, M. D. Stefano, G. Pitarresi and G. Giammona, Macromolecules, 2009, 42, 3247–3257 CrossRef CAS.
- B. S. Sumerlin, D. Neugebauer and K. Matyjaszewski, Macromolecules, 2005, 38, 702–708 CrossRef CAS.
- C. Feng, Y. J. Li, D. Yang, J. H. Hu, X. H. Zhang and X. Y. Huang, Chem. Soc. Rev., 2011, 40, 1282–1295 RSC.
- A. C. Engler, H. I. Lee and P. T. Hammond, Angew. Chem., Int. Ed., 2009, 48, 9334–9338 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- J. F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- T. P. Lodge, Macromolecules, 2009, 42, 3827–3829 CrossRef CAS.
- W. C. Lin, Q. Fu, Y. Zhang and J. L. Huang, Macromolecules, 2008, 41, 4127–4135 CrossRef CAS.
- Q. Fu, W. C. Lin and J. Huang, Macromolecules, 2008, 41, 2381–2387 CrossRef CAS.
- Y. Deng, S. Zhang, G. L. Lu and X. Y. Huang, Polym. Chem., 2013, 4, 1289–1299 RSC.
- G. W. Wang and J. L. Huang, Polym. Chem., 2014, 5, 277–308 RSC.
- D. Yang, C. Feng and J. H. Hu, Polym. Chem., 2013, 4, 2384–2394 RSC.
- Q. Fu, G. W. Wang, W. C. Lin and J. L. Huang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 986–990 CrossRef CAS.
- R. M. Sun, G. W. Wang, C. Liu and J. L. Huang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1930–1938 CrossRef CAS.
- S. Sugihara, S. P. Armes and A. L. Lewis, Angew. Chem., Int. Ed., 2010, 49, 3500–3503 CrossRef CAS PubMed.
- J. Z. Du and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 12800–12801 CrossRef CAS PubMed.
- S. Pioge, L. Fontaine, C. Gaillard, E. Nicol and S. Pascual, Macromolecules, 2009, 42, 4262–4272 CrossRef CAS.
- J. Zhu and R. C. Hayward, J. Am. Chem. Soc., 2008, 130, 7496–7502 CrossRef CAS PubMed.
- A. C. French, A. L. Thompson and B. G. Davis, Angew. Chem., Int. Ed., 2009, 48, 1248–1252 CrossRef CAS PubMed.
- Y. Akiyama, H. Otsuka, Y. Nagasaki, M. Kato and K. Kataoka, Bioconjugate Chem., 2000, 11, 947–950 CrossRef CAS PubMed.
- S. D. Xiong, L. Li, J. Jiang, L.-P. Tong, S. Wu, Z.-S. Xu and P. K. Chu, Biomaterials, 2010, 31, 2673–2685 CrossRef CAS PubMed.
- C. V. Bonduelle and E. R. Gillies, Macromolecules, 2010, 43, 9230–9233 CrossRef CAS.
- H. Yin, S. W. Kang and Y. H. Bae, Macromolecules, 2009, 42, 7456–7464 CrossRef CAS.
- G. Wanka, H. Hoffmann and W. Ulbricht, Macromolecules, 1994, 27, 4145–4159 CrossRef CAS.
- S. Boisse, J. Rieger, A. Di-Cicco, P. A. Albouy, C. Bui, M. H. Li and B. Charleux, Macromolecules, 2009, 42, 8688–8696 CrossRef CAS.
- Y. G. Li, M. Du, Y. J. Li, L. Sui, G. L. Lu and X. Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4783–4789 CrossRef CAS.
- Y. G. Li, M. Du, Y. Q. Zhang, Y. J. Li, L. Sui, G. L. Lu and X. Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1890–1899 CrossRef CAS.
- Y. G. Li, Y. Q. Zhang, S. J. Zhai, Y. Deng, H. M. Xiong, G. L. Lu and X. Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 23–34 CrossRef CAS.
- X. H. Zhang, Z. Shen, C. Feng, D. Yang, Y. G. Li, J. H. Hu, G. L. Lu and X. Y. Huang, Macromolecules, 2009, 42, 4249–4256 CrossRef CAS.
- S. Villarroya, J. Zhou, K. J. Thurecht and S. M. Howdle, Macromolecules, 2006, 39, 9080–9086 CrossRef CAS.
- Y. Cai, Y. Tang and S. P. Armes, Macromolecules, 2004, 37, 9728–9737 CrossRef CAS.
- J. N. Kizhakkedathu and D. E. Brooks, Macromolecules, 2003, 36, 591–598 CrossRef CAS.
- H. I. Lee, K. Matyjaszewski, S. Yu and S. S. Sheiko, Macromolecules, 2005, 38, 8264–8271 CrossRef CAS.
- J. V. M. Weaver, I. Bannister, K. L. Robinson, X. Bories-Azeau, S. P. Armes, M. Smallridge and P. McKenna, Macromolecules, 2004, 37, 2395–2403 CrossRef CAS.
- X. Y. Jiang, G. L. Lu, C. Feng and X. Y. Huang, Polym. Chem., 2014, 5, 4915–4925 RSC.
- G. Moad, J. Chiefari, Y. K. Chong, J. Krstina, R. T. A. Mayadunne, A. Postma, E. Rizzardo and S. H. Thang, Polym. Int., 2000, 49, 993–1001 CrossRef CAS.
- K. Matyjaszewski, S. G. Gaynor and A. H. E. Muller, Macromolecules, 1997, 30, 7034–7041 CrossRef CAS.
- S. Pal, S. G. Roy and P. De, Polym. Chem., 2014, 5, 1275–1284 RSC.
- M. Lejars, A. Margaillan and C. Bressy, Polym. Chem., 2013, 4, 3282–3292 RSC.
- S. H. Wang, Q. X. Shen, M. H. Nawaz and W. A. Zhang, Polym. Chem., 2013, 4, 2151–2157 RSC.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- S. Basu, D. R. Vutukuri, S. Shyamroy, B. S. Sandanraj and S. Thayumanavan, J. Am. Chem. Soc., 2004, 126, 9890–9891 CrossRef CAS PubMed.
- Q. Fu, Z. N. Zhang, W. C. Lin and J. L. Huang, Macromolecules, 2009, 42, 4381–4383 CrossRef CAS.
- Z. F. Jia, Q. Fu and J. L. Huang, Macromolecules, 2006, 39, 5190–5193 CrossRef CAS.
- D. Peng, X. H. Zhang and X. Y. Huang, Macromolecules, 2006, 39, 4945–4947 CrossRef CAS.
- N. V. Tsarevsky, S. A. Bencherif and K. Matyjaszewski, Macromolecules, 2007, 40, 4439–4445 CrossRef CAS.
- L. C. You, F. Z. Lu, Z. C. Li, W. Zhang and F. M. Li, Macromolecules, 2003, 36, 1–4 CrossRef CAS.
- P. S. Xu, H. D. Tang, S. Y. Li, J. Ren, E. V. Kirk, W. J. Murdoch, M. Radosz and Y. Q. Shen, Biomacromolecules, 2004, 5, 1736–1744 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- S. B. Clendenning, S. Fournier-Bidoz, A. Pietrangelo, G. C. Yang, S. J. Han, P. M. Brodersen, C. M. Yip, Z. H. Lu, G. A. Ozin and I. Manners, J. Mater. Chem., 2004, 14, 1686–1690 RSC.
- L. F. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168–3181 CrossRef CAS.
- C. Feng, G. L. Lu, Y. J. Li and X. Y. Huang, Langmuir, 2013, 29, 10922–10931 CrossRef CAS PubMed.
- B. B. Xu, G. X. Gu, C. Feng, X. Jiang, J. H. Hu, G. L. Lu, S. Zhang and X. Y. Huang, Polym. Chem., 2016, 7, 613–624 RSC.
- T. P. Lodge, A. Rasdal, Z. B. Li and M. A. Hillmyer, J. Am. Chem. Soc., 2005, 127, 17608–17609 CrossRef CAS PubMed.
- E. N. Savariar, S. V. Aathimanikandan and S. Thayumanavan, J. Am. Chem. Soc., 2006, 128, 16224–16230 CrossRef CAS PubMed.
- Y. R. Que, C. Feng, S. Zhang and X. Y. Huang, J. Phys. Chem. C, 2015, 119, 1960–1970 CAS.
- H. M. Jung, K. E. Price and D. T. McQuade, J. Am. Chem. Soc., 2003, 125, 5351–5355 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py01595f |
| This journal is © The Royal Society of Chemistry 2017 |