
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of the zwitterion structure on the thermo-responsive behaviour of poly(sulfobetaine methacrylates)†
Viet
Hildebrand
a,
André
Laschewsky
*ab,
Michael
Päch
c,
Peter
Müller-Buschbaum
d and
Christine M.
Papadakis
d
aInstitut für Chemie, Universität Potsdam, Karl-Liebknechtstr. 24-25, 14476 Potsdam-Golm, Germany. E-mail: laschews@uni-potsdam.de
bFraunhofer Institute for Applied Polymer Research IAP, Geiselbergstr. 69, 14476 Potsdam-Golm, Germany
cUP Transfer GmbH, Am Neuen Palais 10, 14469 Potsdam, Germany
dPhysik-Department, Lehrstuhl für Funktionelle Materialien/Fachgebiet Physik Weicher Materie, Technische Universität München, James-Franck-Str. 1, 85748 Garching, Germany
First published on 16th November 2016
Abstract
A series of new sulfobetaine methacrylates, including nitrogen-containing saturated heterocycles, was synthesised by systematically varying the substituents of the zwitterionic group. Radical polymerisation via the RAFT (reversible addition–fragmentation chain transfer) method in trifluoroethanol proceeded smoothly and was well controlled, yielding polymers with predictable molar masses. Molar mass analysis and control of the end-group fidelity were facilitated by end-group labeling with a fluorescent dye. The polymers showed distinct thermo-responsive behaviour of the UCST (upper critical solution temperature) type in an aqueous solution, which could not be simply correlated to their molecular structure via an incremental analysis of the hydrophilic and hydrophobic elements incorporated within them. Increasing the spacer length separating the ammonium and the sulfonate groups of the zwitterion moiety from three to four carbons increased the phase transition temperatures markedly, whereas increasing the length of the spacer separating the ammonium group and the carboxylate ester group on the backbone from two to three carbons provoked the opposite effect. Moreover, the phase transition temperatures of the analogous polyzwitterions decreased in the order dimethylammonio > morpholinio > piperidinio alkanesulfonates. In addition to the basic effect of the polymers’ precise molecular structure, the concentration and the molar mass dependence of the phase transition temperatures were studied. Furthermore, we investigated the influence of added low molar mass salts on the aqueous-phase behaviour for sodium chloride and sodium bromide as well as sodium and ammonium sulfate. The strong effects evolved in a complex way with the salt concentration. The strength of these effects depended on the nature of the anion added, increasing in the order sulfate < chloride < bromide, thus following the empirical Hofmeister series. In contrast, no significant differences were observed when changing the cation, i.e. when adding sodium or ammonium sulfate.
Introduction
Our knowledge and understanding of thermo-responsive, water-soluble polymers showing a lower critical solution (LCST) behaviour1–6 is, notwithstanding a number of remaining questions,6 rather advanced compared to those exhibiting upper critical solution temperature (UCST) behaviour.7–9 In fact, LCST behaviour is a common phenomenon of non-ionic polymers in aqueous solution.2 Moreover, their transition temperatures can be easily fine-tuned via their molecular structure, as they can be synthesised not only by various polymerisation methods directly from the respective monomers,1,2 but also by copolymerisation3,10–12 or by post-polymerisation modification.4,13–15 In contrast, the number of polymers showing UCST behaviour in water is quite limited.7,8 These polymers mostly comprise polyzwitterions, in particular of the class of polysulfobetaines.16–18 Although their thermo-responsiveness has been explored occasionally, actually quite some time ago,19–23 this feature was considered rather a nuisance to their use (e.g. for producing biocompatible or low-fouling materials). Only recently has interest in the UCST behaviour of polysulfobetaines gained more impetus, mostly in order to enlarge the scope of the “traditional” thermo-responsive polymers with LCST behaviour15,24–30 or to obtain stimuli-responsive materials of increasing complexity.31–40Chemically well-defined polysulfobetaines are most conveniently prepared via free radical polymerisation of the underlying monomers.16,18 Yet, only a few such monomers are commercially available at present. The most popular among these are 3-((3-methacrylamidopropyl)dimethylammonio)propane-1-sulfonate (“SPP”), and in particular 3-((2-methacryloyloxyethyl)dimethylammonio)propane-1-sulfonate (“SPE”, 1). The molecular blueprint of the latter offers a number of variables for systematic structural variations (Scheme 1), and subsequently for investigation of the resulting thermo-responsive behaviour.18 Still, little work on such variations has been reported to date,22,29,41–51 and studies to elucidate their effects on the phase behaviour of these poly(methacrylate)s are scarcer still.22,29,52–54 Even the aqueous-phase behaviour of the basic polymer of 1, denoted herein as P-1, has not been fully established, as the reported data are apparently conflicting.21,24,27,29,54–57 Characteristically, the phase transition temperature of P-1 in aqueous solution depends not only on its molar mass and concentration, but also on the presence of small amounts of additives (in particular of low molar mass electrolytes) as well as on the chemical defects in the polymer chains. In fact, the latter is an important obstacle for establishing reliable transition temperatures when producing polysulfobetaines under harsh reaction conditions, or indeed by post-polymerisation modification.
 | ||
| Scheme 1 Generalised structure of simple sulfobetaine methacrylates with their structural variables x, y, R1 and R2. | ||
In this context, we recently prepared a set of poly(sulfobetaine methacrylamides) related to the commercial monomer SPP by reversible addition–fragmentation chain transfer (RAFT) polymerisation.28,30 By varying the spacer groups between the cationic ammonium and the anionic sulfonate groups, we were able to study their aqueous solution behaviour and dependence on temperature and on the concentration of selected inorganic salts. While strong effects on the phase transition temperature were induced by apparently small structural modifications, the observed increase – or decrease – in the temperature did not follow a simple obvious logic. In the present work, we extend these studies to an analogous series of zwitterionic poly(methacrylates) structurally related to the arguably most employed sulfobetaine monomer 1, using the homopolymer (P-1) as a reference (Fig. 1, cf. Scheme 1). In detail, we varied, within narrow limits, the spacer length between the polymerisable and the ammonium moieties (variable x), the nature of the two variable substituents on the ammonium nitrogen (variables R1 and R2), and the spacer group length between the ammonium and the sulfonate groups (variable y). For instance, we incorporated the cationic ammonium moiety of the zwitterionic groups into saturated heterocycles, which has been rarely done up to now.44,58 When applying the simple logic of adding incrementally hydrophilic and hydrophobic elements to the basic structure of 1, one would expect that the overall hydrophilicity of the monomers would decrease with the increasing x, y and sizes of R1 and R2. Accordingly, for a given molar mass, the UCSTs of the corresponding polymers should increase concomitantly. While polymers derived from monomers 5 have been studied occasionally,29,45,46,48,50,59–61 and polymers from monomer 4 have been described only briefly,25,49,51,62 data on the UCST behaviour of these polymers are more scarce, or are even missing completely. Monomers 2, 3 and 6–8, and their polymers are new compounds. By studying polymers P-1 to P-8 in water and in aqueous salt solutions, we aimed to reveal the structure–property relationships concerning the occurrence and relative positions of their UCSTs in water, specifically in order to facilitate the tailoring of polyzwitterions with specific transition temperatures in the future.
 | ||
| Fig. 1 Chemical structures of the zwitterionic sulfobetaine methacrylate monomers studied and of the RAFT agent used. | ||
Experimental section
Materials
Information on the sources, quality and purification of the standard chemicals used are provided in the ESI.† The fluorophore-labelled chain transfer agent 2-(6-(dimethylamino)-1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)ethyl-4-cyano-4-(((phenethylthio) carbonothioyl) thio) pentanoate (CTA) was synthesised as described previously.28 The tertiary amine-functionalised methacrylate intermediates: 2-(piperidin-1-yl)ethyl methacrylate, 2-morpholinoethyl methacrylate and 3-(dimethylamino)propyl methacrylate were synthesised by transesterification of the methylmethacrylate by adopting a literature procedure (cf. ESI†).63 Deionised water was further purified by a Millipore Milli-Q Plus water purification system (resistivity 18 MΩ cm−1).Monomer synthesis
1H NMR (300 MHz, D2O, 298 K): δ (ppm) = 1.60–2.30 (m, 11H, –CH2–CH2–CH2– piperidine (C4, C3, C5)), ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CH3, –CH2–C–SO3−), 3.00 (t, J = 7.2 Hz, 2H, –CH2–SO3−), 3.40–3.54 (t, J = 5.6 Hz, 4H, –CH2–N+–CH2– piperidine (C2, C6)), 3.54–3.68 (m, 2H, –N+–CH2–), 3.80–3.90 (m, 2H, –COO–C–CH2–N+–), 4.64 (t, 2H, J = 4.6 Hz, –COO–CH2–), 5.80 (s, 1H, CHC–COO– (trans)), 6.17 (s, 1H, CHC–COO– (cis)).
C–CH3, –CH2–C–SO3−), 3.00 (t, J = 7.2 Hz, 2H, –CH2–SO3−), 3.40–3.54 (t, J = 5.6 Hz, 4H, –CH2–N+–CH2– piperidine (C2, C6)), 3.54–3.68 (m, 2H, –N+–CH2–), 3.80–3.90 (m, 2H, –COO–C–CH2–N+–), 4.64 (t, 2H, J = 4.6 Hz, –COO–CH2–), 5.80 (s, 1H, CHC–COO– (trans)), 6.17 (s, 1H, CHC–COO– (cis)).
13C NMR (75 MHz, D2O, 298 K): δ (ppm) = 18.2 (–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2–C–SO3−), 18.3 (–C–H3), 20.3 (H2–C–H2– piperidine (C3, C5)), 21.5 (–C–H2–C– piperidine (C4)), 48.2 (–H2–SO3−), 57.8 (–COO–C–H2–), 58.3 (–N+–H2–), 58.9 (–COO–H2–), 61.3 (–H2–N+–H2– piperidine (C2, C6)), 128.7 (H2), 136.2 (–COO–), 169.5 (–COO–).
H2–C–SO3−), 18.3 (–C–H3), 20.3 (H2–C–H2– piperidine (C3, C5)), 21.5 (–C–H2–C– piperidine (C4)), 48.2 (–H2–SO3−), 57.8 (–COO–C–H2–), 58.3 (–N+–H2–), 58.9 (–COO–H2–), 61.3 (–H2–N+–H2– piperidine (C2, C6)), 128.7 (H2), 136.2 (–COO–), 169.5 (–COO–).
HR-MS (ESI): calculated: 319.1500 [M]+; found: 320.1524 [M + H]+.
Elemental analysis (C14H25NO5S, Mr = 319.42): calculated: C = 52.64%, H = 7.89%, N = 4.39%, S = 10.04%; found: C = 52.77%, H = 7.97%, N = 4.39%, S = 10.04%.
FT-IR (selected bands, cm−1): 3022 ν(N+–CH2), 2961 ν(CH3), 1722 ν(CO), 1637 ν(CC), 1155 νas(SO3−), 1034 νs(SO3−).
1H NMR (300 MHz, D2O, 298 K): δ (ppm) = 1.95 (s, 3H, C–CH3), 2.26 (m, 2H, –CH2–C–SO3−), 3.01 (t, J = 7.2 Hz, 2H, –CH2–SO3−), 3.60–3.80 (m, 6H, –CH2–N+–CH2– morpholine (C2, C6), –N+–CH2–), 3.96–4.20 (m, 6H, –COO–C–CH2–N+–, –CH2–O–CH2– morpholine (C3, C5)), 4.68 (t, 2H, J = 4.0 Hz, –COO–CH2–), 5.80 (s, 1H, CHC–COO– (trans)), 6.15 (s, 1H, CHC–COO– (cis)).
13C NMR (75 MHz, D2O, 298 K): δ (ppm) = 18.2 (–H2–C–SO3−), 18.3 (–C–H3), 48.0 (H2–SO3−), 53.2 (–N+–H2–), 58.8 (–COO–H2–), 59.9 (–H2–N+–H2– morpholine (C2, C6)), 61.1 (–H2–O–H2– morpholine (C3, C5)), 64.8 (–COO–C–H2–), 128.7 (H2), 136.0 (–COO–), 169.4 (–COO–).
HR-MS (ESI): calculated: 321.1200 [M]+; found: 322.1319 [M + H]+.
Elemental analysis (C13H23NO6S, Mr = 321.39): calculated: C = 48.58%, H = 7.21%, N = 4.36%, S = 9.98%; found: C = 48.60%, H = 7.21%, N = 4.34%, S = 9.94%.
FT-IR (selected bands, cm−1): 3020 ν(N+–CH2), 2962 ν(CH3), 1723 ν(CO), 1636 ν(CC), 1156 νas(SO3−), 1033 νs(SO3−).
1H NMR (300 MHz, D2O, 298 K): δ (ppm) = 1.95 (s, 3H, C–CH3), 2.15–2.35 (m, 4H, COO–C–CH2–, –CH2–C–SO3−), 3.01 (t, J = 7.1 Hz, 2H, –CH2–SO3−), 3.17 (s, 6H, –N+–(CH3)2), 3.45–3.60 (m, 4H, –CH2–N+–CH2–), 4.31 (t, 2H, J = 5.8 Hz, –COO–CH2–), 5.76 (s, 1H, CHC–COO– (trans)), 6.17 (s, 1H, CHC–COO– (cis)).
13C NMR (75 MHz, D2O, 298 K): δ (ppm) = 18.3 (–C–H3), 19.1 (–H2–C–SO3−), 22.7 (COO–C–H2–), 48.2 (–H2–SO3−), 51.7 (–N+–(H3)2), 62.3 (–N+–H2–), 62.8 (–COO–H2–), 63.2 (–H2–N+–), 128.0 (H2), 136.7 (–COO–), 170.5 (–COO–).
HR-MS (ESI): calculated: 293.1300 [M]+; found: 316.1181 [M + Na]+.
Elemental analysis (C12H23NO5S, Mr = 293.38): calculated: C = 49.13%, H = 7.90%, N = 4.77%, S = 10.93%; found: C = 49.00%, H = 7.81%, N = 4.78%, S = 10.90%.
FT-IR (selected bands, cm−1): 3041 ν(N+–CH3), 2972 ν(CH3), 1708 ν(CO), 1627 ν(CC), 1189 νas(SO3−), 1037 νs(SO3−).
1H NMR (300 MHz, D2O, 298 K): δ (ppm) = 1.72–2.10 (m, 7H, –CH2–CH2–C–SO3−, C–CH3), 2.99 (t, J = 7.5 Hz, 2H, –CH2–SO3−), 3.21 (s, 6H, –N+–(CH3)2), 3.45 (m, 2H, –N+–CH2–), 3.81 (m, 2H, –COO–C–CH2–N+–), 4.66 (t, 2H, J = 4.6 Hz, –COO–CH2–), 5.81 (s, 1H, CHC–COO– (trans)), 6.18 (s, 1H, CHC–COO– (cis)).
13C NMR (75 MHz, D2O, 298 K): δ (ppm) = 18.3 (–C–H3), 22.0 and 22.1 (–H2–H2–C–SO3−), 51.0 (–C–H2–SO3−), 52.3 (–N+–(H3)2), 59.4 (–COO–H2–), 63.4 (–COO–C–H2–), 65.7 (–N+–H2–), 128.7 (H2), 136.2 (–COO–), 169.4 (–COO–).
HR-MS (ESI): calculated: 293.1300 [M]+; found: 316.1164 [M + Na]+.
Elemental analysis (C12H23NO5S, Mr = 293.38): calculated: C = 49.13%, H = 7.90%, N = 4.77%, S = 10.93%; found: C = 49.03%, H = 7.95%, N = 4.80%, S = 10.92%.
FT-IR (selected bands, cm−1): 3033 ν(N+–CH3), 2960 ν(CH3), 1713 ν(CO), 1636 ν(CC), 1169 νas(SO3−), 1035 νs(SO3−).
1H NMR (400 MHz, D2O, 298 K): δ (ppm) = 1.60–2.00 (m, 13H, –CH2–CH2–CH2– piperidine (C4, C3, C5)), –CH2–CH2–C–SO3−, C–CH3), 2.95 (t, J = 7.2 Hz, 2H, –CH2–SO3−), 3.38–3.70 (m, 6H, –CH2–N+–CH2– piperidine (C2, C6), –N+–CH2–), 3.80 (t, 2H, J = 4.6 Hz, COO–C–CH2–N+–), 4.60 (t, 2H, J = 4.6 Hz, –COO–CH2–), 5.78 (s, 1H, CHC–COO– (trans)), 6.14 (s, 1H, CHC–COO– (cis)).
13C NMR (125 MHz, D2O, 298 K): δ (ppm) = 18.3 (–C–H3), 20.3 (–H2–C–H2– piperidine (C3, C5)), 21.0 (–C–H2–C– piperidine (C4)), 21.6 (–H2–C–SO3−), 22.3 (–H2–C–C–SO3−), 51.1 (–H2–SO3−), 57.7 (–COO–C–H2–), 58.9 (–COO–H2–), 59.8 (–N+–H2–), 61.3 (–H2–N+–H2– piperidine (C2, C6)), 128.7 (H2), 136.2 (–COO–), 169.6 (–COO–).
HR-MS (ESI): calculated: 333.1600 [M]+; found: 334.1673 [M + H]+.
Elemental analysis (C15H27NO5S, Mr = 333.44): calculated: C = 54.03%, H = 8.16%, N = 4.20%, S = 9.61%; found: C = 54.40%, H = 8.10%, N = 4.20%, S = 9.92%.
FT-IR (selected bands, cm−1): 3010 ν(N+–CH2), 2967 ν(CH3), 1712 ν(CO), 1627 ν(CC), 1160 νas(SO3−), 1035 νs(SO3−).
1H NMR (400 MHz, D2O, 298 K): δ (ppm) = 1.75–2.00 (m, 7H, –CH2–CH2–C–SO3−, C–CH3), 2.97 (t, J = 7.4 Hz, 2H, –CH2–SO3−), 3.55–3.72 (m, 6H, –CH2–N+–CH2– morpholine (C2, C6), –N+–CH2–), 3.99 (t, 2H, J = 3.9 Hz, –COO–C–CH2–N+–), 4.10 (m, 4H, –CH2–O–CH2– morpholine (C3, C5)), 4.65 (t, 2H, J = 3.5 Hz, –COO–CH2–), 5.79 (s, 1H, CHC–COO– (trans)), 6.14 (s, 1H, CHC–COO– (cis)).
13C NMR (125 MHz, D2O, 298 K): δ (ppm) = 18.3 (–C–H3), 20.9 (–H2–C–C–SO3−), 22.1 (H2–C–SO3−), 51.0 (–H2–SO3−), 58.2 (–COO–C–H2–), 58.8 (–COO–H2–), 59.8 (–H2–N+–H2– morpholine (C2, C6)), 60.2 (–N+–H2–), 61.1 (H2–O–H2– morpholine (C3, C5)), 128.8 (H2), 136.3 (–COO–), 169.5 (–COO–).
HR-MS (ESI): calculated: 335.1400 [M]+; found: 336.1465 [M + H]+.
Elemental analysis (C14H25NO6S, Mr = 335.42): calculated: C = 50.13%, H = 7.51%, N = 4.18%, S = 9.56%; found: C = 50.08%, H = 7.49%, N = 4.19%, S = 9.53%.
FT-IR (selected bands, cm−1): 3008 ν(N+–CH2), 2968 ν(CH3), 1715 ν(CO), 1629 ν(CC), 1171 νas(SO3−), 1030 νs(SO3−).
1H NMR (300 MHz, D2O, 298 K): δ (ppm) = 1.70–2.40 (m, 9H, –CH2–CH2–SO3−), C–CH3, COO–C–CH2–), 2.99 (t, J = 7.4 Hz, 2H, –CH2–SO3−), 3.13 (s, 6H, –N+–(CH3)2), 3.30–3.60 (m, 4H, –CH2–N+–CH2–), 4.31 (t, 2H, J = 5.8 Hz, –COO–CH2–), 5.77 (s, 1H, CHC–COO– (trans)), 6.17 (s, 1H, CHC–COO– (cis)).
13C NMR (75 MHz, D2O, 298 K): δ (ppm) = 18.3 (C–H3), 21.9 (H2–C–C–SO3−), 22.1 (H2–C–SO3−), 22.7 (COO–C–H2–), 51.0 (–H2–SO3−), 51.7 (–N+–(H3)2), 62.2 (–H2–N+–), 62.8 (–COO–H2–), 64.4 (–N+–H2–), 128.0 (H2), 136.0 (–COO–), 169.4 (–COO–).
HR-MS (ESI): calculated: 307.1500 [M]+; found: 330.1338 [M + Na]+.
Elemental analysis (C13H25NO5S, Mr = 307.41): calculated: C = 50.79%, H = 8.20%, N = 4.56%, S = 10.43%; found: C = 50.80%, H = 8.22%, N = 4.55%, S = 10.44%.
FT-IR (selected bands, cm−1): 3031 ν(N+–CH3), 2956 ν(CH3), 1715 ν(CO), 1636 ν(CC), 1171 νas(SO3−), 1034 νs(SO3−).
Polymer synthesis
All the RAFT polymerisations of the sulfobetaine monomers were performed following a general procedure in which the sulfobetaine monomer, RAFT agent and initiator V-501 in TFE, after purging with N2 for 30 min, were polymerised at 75 °C for a given time span. For purification, the mixture was poured into 10-fold its volume of methanol. The precipitate was collected, dissolved in TFE and precipitated into methanol (repeated twice). After drying in vacuo, the polymers were obtained as hygroscopic amorphous yellow solids (colour intensity decreasing with the increasing molar mass).The purified homopolymers were characterised by 1H NMR, UV-vis and IR spectroscopies as well as by TGA, DSC and turbidimetry measurements. The individual samples were named P-monomern, with n being the number average degree of polymerisation that was theoretically calculated by using eqn (1).
Examples for the RAFT polymerisation of all the monomers are provided in the ESI.†
Methods
The standard analytical equipment used is described in the ESI.†The approximate monomer conversions were determined from the 1H NMR spectra of the crude polymerisation mixtures. The theoretically expected number average molar masses Mtheon were calculated according to eqn (1):
 | (1) |
The number average molar masses of the polymers were determined by UV/Vis spectroscopic end-group analysis in trifluoroethanol solution using the maximum of the absorbance band of the naphthalimide chromophore at about 444 nm with the extinction coefficient ε of 1.98 × 104 L mol−1 cm−1, and assuming that every polymer carries one naphthalimide moiety.28 The molar concentration of the naphthalimide chromophore, and thus of the polymer, was calculated using the Lambert–Beer law (eqn (2)), therefrom calculating the molar masses viaeqn (3):
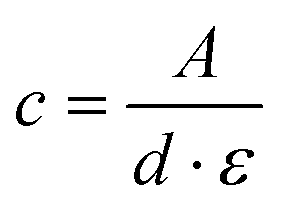 | (2) |
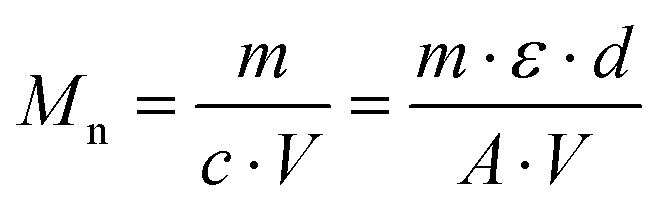 | (3) |
Cloud points were determined by turbidimetry using a Varian Cary 50 UV-vis spectrophotometer, equipped with a single cell Peltier thermostated cell holder, using 1 cm × 1 cm quartz cuvettes. Measurements were performed at a wavelength of 800 nm, together with heating and cooling rates of 0.5 K min−1. Aqueous polymer solutions of various concentrations were prepared in D2O, in Millipore water or aqueous salt solutions. The cloud point was taken as the temperature where the normalised transmittance of the solution in the cooling run started to decrease suddenly, i.e. where the transmittance was reduced by 5% relative to the difference between the maximum and minimum transmittance in the run.
Results and discussion
Monomers 2–8 (Fig. 1) were synthesised following standard procedures, the key step being the final ring-opening quaternisation of the methacrylate functionalised tertiary amine precursors by the sultones16,18 in dry acetonitrile. This strategy has the advantage of yielding the final zwitterionic monomers in high purity by a simple precipitation or crystallisation, and in particular, free of inorganic salts. While such salt contaminations may affect the water-solubility of polymers dramatically, once introduced, they may be difficult to remove due to their high affinity to sulfobetaines. Here, no particular problems were encountered, though even when alkylating the amines with the less reactive butane sultone.29,41 High yields were obtained not only for monomers 4, 5 and 8 with the established substitution patterns of N-alkyl-N,N-dimethylammoniopropanesulfonates and -butanesulfonates, but also for the sulfobetaines derived from the sterically more demanding heterocyclic tertiary amines, 2–3 and 6–7. The features of the monomers’ 1H and 13C NMR spectra (cf. Fig. S3–S16†) correspond to the reported spectra of the related monomers.29,44,46,64,65Polymers P-1n–P-8n (cf.Fig. 1, with n indicating the theoretically calculated number average degree of polymerisation) were synthesised by RAFT polymerisation at 75 °C in trifluoroethanol, in order to establish homogeneous reaction conditions for both the zwitterionic monomers and the much less polar RAFT agent CTA. The polymerisations in trifluoroethanol proceeded smoothly, in agreement with the literature on other sulfobetaine monomers.28–30,41,46,66 The ratio of monomer to the fluorophore-functionalised CTA in the reaction mixtures was varied between 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 up to 600:1, in order to modulate the molar masses, while the ratio between CTA and the azoinitiator was always kept constant as 5:1 (Table 1). Under such conditions, the vast majority of the polymer chains were initiated by the “R”-residue of the RAFT agent and consequently labelled by the fluorophore.28,30 This enabled not only a facile tracking of the polymers during the various experiments by virtue of their strong fluorescence, but also the ability to apply end-group analysis to support the molecular characterisation.67 As shown previously,28,30 the signals of both the aromatic moieties of the “R”- and the “Z” end groups could be resolved and quantified in the 1H NMR spectra for moderate molar masses (<105), while the UV/vis absorbance band of the naphthalimide chromophore enabled quantification of the “R” end groups, even for high molar masses. This enabled the facile and quite reliable determination of the absolute number average molar mass (Mn) of the polyzwitterions as well as of the extent of the preservation of the RAFT-active end groups.
:1 up to 600:1, in order to modulate the molar masses, while the ratio between CTA and the azoinitiator was always kept constant as 5:1 (Table 1). Under such conditions, the vast majority of the polymer chains were initiated by the “R”-residue of the RAFT agent and consequently labelled by the fluorophore.28,30 This enabled not only a facile tracking of the polymers during the various experiments by virtue of their strong fluorescence, but also the ability to apply end-group analysis to support the molecular characterisation.67 As shown previously,28,30 the signals of both the aromatic moieties of the “R”- and the “Z” end groups could be resolved and quantified in the 1H NMR spectra for moderate molar masses (<105), while the UV/vis absorbance band of the naphthalimide chromophore enabled quantification of the “R” end groups, even for high molar masses. This enabled the facile and quite reliable determination of the absolute number average molar mass (Mn) of the polyzwitterions as well as of the extent of the preservation of the RAFT-active end groups.
| Sample | Monomer | Molar ratio monomer:CTA:V-501 |
m (monomer) [g] | m CTA [g] | m V-501 [g] | t [h] |
|---|---|---|---|---|---|---|
| P- 1 85 | 1 | 100:1:0.2 |
5.0 | 0.109 | 0.010 | 2.5 |
| P- 1 270 | 1 | 300:1:0.2 |
5.0 | 0.036 | 0.003 | 7.5 |
| P- 1 575 | 1 | 600:1:0.2 |
5.0 | 0.018 | 0.002 | 15 |
| P- 1 585 | 1 | 600:1:0.2 |
5.0 | 0.018 | 0.002 | 15 |
| P- 2 95 | 2 | 100:1:0.2 |
2.5 | 0.049 | 0.005 | 19 |
| P- 2 250 | 2 | 600:1:0.2 |
1.0 | 0.003 | 0.001 | 18 |
| P- 2 330 | 2 | 400:1:0.2 |
1.0 | 0.005 | 0.001 | 12 |
| P- 2 485 | 2 | 600:1:0.2 |
1.0 | 0.003 | 0.001 | 18 |
| P- 3 65 | 3 | 100:1:0.2 |
2.0 | 0.038 | 0.004 | 19 |
| P- 3 95 | 3 | 100:1:0.2 |
1.0 | 0.019 | 0.002 | 2.7 |
| P- 3 230 | 3 | 300:1:0.2 |
1.0 | 0.006 | 0.001 | 9 |
| P- 3 585 | 3 | 600:1:0.2 |
1.0 | 0.003 | 0.001 | 18 |
| P- 4 75 | 4 | 100:1:0.2 |
1.0 | 0.027 | 0.002 | 2 |
| P- 4 295 | 4 | 300:1:0.2 |
1.0 | 0.007 | 0.001 | 6 |
| P- 4 480 | 4 | 500:1:0.2 |
1.0 | 0.004 | 0.001 | 10 |
| P- 4 585 | 4 | 600:1:0.2 |
2.0 | 0.007 | 0.001 | 12 |
| P- 5 50 | 5 | 050:1:0.2 |
1.0 | 0.041 | 0.004 | 2 |
| P- 5 80 | 5 | 080:1:0.2 |
1.0 | 0.026 | 0.002 | 2 |
| P- 5 95 | 5 | 100:1:0.2 |
2.0 | 0.041 | 0.004 | 2 |
| P- 5 282 | 5 | 300:1:0.2 |
1.0 | 0.007 | 0.001 | 6 |
| P- 6 80 | 6 | 100:1:0.2 |
1.0 | 0.018 | 0.002 | 2.5 |
| P- 6 250 | 6 | 300:1:0.2 |
1.0 | 0.006 | 0.001 | 7.5 |
| P- 6 420 | 6 | 500:1:0.2 |
1.0 | 0.004 | 0.001 | 12.5 |
| P- 6 500 | 6 | 600:1:0.2 |
1.9 | 0.006 | 0.001 | 15 |
| P- 7 85 | 7 | 100:1:0.2 |
1.0 | 0.018 | 0.002 | 3 |
| P- 7 260 | 7 | 300:1:0.2 |
1.0 | 0.006 | 0.001 | 9 |
| P- 7 430 | 7 | 500:1:0.2 |
1.0 | 0.004 | 0.001 | 15 |
| P- 7 520 | 7 | 600:1:0.2 |
1.4 | 0.004 | 0.001 | 18 |
| P- 8 100 | 8 | 100:1:0.2 |
5.0 | 0.099 | 0.009 | 19 |
| P- 8 290 | 8 | 300:1:0.2 |
2.0 | 0.013 | 0.002 | 3 |
| P- 8 480 | 8 | 500:1:0.2 |
1.0 | 0.004 | 0.001 | 5 |
| P- 8 540 | 8 | 600:1:0.2 |
3.2 | 0.012 | 0.001 | 6 |
The results of the various polymerisations are compiled in Table 2. The data show that, notwithstanding that high conversions were achieved, the determined molar mass values Mn agree well with the theoretically expected ones Mtheon, and that the end-group fidelity is high, thus suggesting a well-controlled polymerisation. Accordingly, a set of well-defined, fluorescence-labelled samples of poly(sulfobetaine methacrylates) up to molar masses of 200 kDa was prepared.
| Sample | Conversiona | M n [kg mol−1] | Ratio Z/R (NMR) | |||
|---|---|---|---|---|---|---|
| Theoretical | By 1H NMR (via Z-group) | By 1H NMR (via R-group) | By UV-vis (via R-group)b | |||
| a Determined by 1H NMR analysis of the reaction mixture. b Determined in TFE. c Signal intensity too weak to allow reliable integration. | ||||||
| P- 1 85 | 0.86 | 25 | 29 | 26 | 31 | 0.9 |
| P- 1 270 | 0.90 | 76 | n.d.c | n.d.c | 88 | — |
| P- 1 575 | 0.95 | 161 | n.d.c | n.d.c | 160 | — |
| P- 1 585 | 0.97 | 164 | n.d.c | n.d.c | 179 | — |
| P- 2 95 | 0.94 | 30 | 104 | 26 | 27 | 0.3 |
| P- 2 250 | 0.42 | 78 | 111 | 125 | 136 | 0.9 |
| P- 2 330 | 0.83 | 103 | n.d.c | n.d.c | 100 | — |
| P- 2 485 | 0.81 | 151 | n.d.c | n.d.c | 145 | — |
| P- 3 65 | 0.66 | 22 | 41 | 28 | 27 | 0.7 |
| P- 3 95 | 0.96 | 32 | 35 | 35 | 35 | 1.0 |
| P- 3 230 | 0.77 | 75 | n.d.c | n.d.c | 109 | — |
| P- 3 585 | 0.97 | 188 | n.d.c | n.d.c | 197 | — |
| P- 4 75 | 0.75 | 23 | 24 | 24 | 23 | 1.0 |
| P- 4 295 | 0.98 | 87 | n.d.c | n.d.c | 74 | — |
| P- 4 480 | 0.96 | 141 | n.d.c | n.d.c | 145 | — |
| P- 4 585 | 0.97 | 172 | n.d.c | n.d.c | 185 | — |
| P- 5 50 | 0.94 | 14 | 22 | 16 | 14 | 0.7 |
| P- 5 80 | 0.96 | 23 | 35 | 26 | 24 | 0.7 |
| P- 5 95 | 0.94 | 28 | 43 | 32 | 29 | 0.7 |
| P- 5 280 | 0.94 | 83 | n.d.c | n.d.c | 83 | — |
| P- 6 80 | 0.82 | 28 | 32 | 29 | 24 | 0.9 |
| P- 6 250 | 0.83 | 84 | 93 | 85 | 73 | 0.9 |
| P- 6 420 | 0.84 | 141 | n.d.c | n.d.c | 120 | — |
| P- 6 500 | 0.83 | 167 | n.d.c | n.d.c | 141 | — |
| P- 7 85 | 0.86 | 30 | 30 | 30 | 27 | 1.0 |
| P- 7 260 | 0.86 | 87 | 96 | 86 | 87 | 0.9 |
| P- 7 430 | 0.86 | 145 | n.d.c | n.d.c | 147 | — |
| P- 7 520 | 0.86 | 175 | n.d.c | n.d.c | 181 | — |
| P- 8 100 | 0.99 | 31 | 37 | 31 | 33 | 0.9 |
| P- 8 290 | 0.97 | 90 | n.d.c | n.d.c | 88 | — |
| P- 8 480 | 0.96 | 148 | n.d.c | n.d.c | 155 | — |
| P- 8 540 | 0.89 | 166 | n.d.c | n.d.c | 160 | — |
The 1H NMR spectra of the various polymers showed characteristic broadening of the signals and the consumption of the methacrylate double bonds, but corresponded otherwise well to the ones of the underlying monomers (cf.Fig. 2 and 3). R- and Z-end groups were visible as long as the molar masses did not exceed 100 kDa (cf. Fig. S17†). From the shape and relative intensities of the signals between 0.5 and 1.2 ppm of the methyl group attached to the polymer backbone, the tacticities of the polymers could be estimated.68 They were very similar for all samples, suggesting roughly a ratio of 3/2 for the syndiotactic (signal centred at about 0.9 ppm) and atactic (signal centred at about 1.1 ppm) triades present, with only a few (<10%) isotactic (signal centred at about 1.25 ppm) triades formed, as encountered typically for the free radical polymerisation of methacrylates at the applied reaction temperature.68 Notwithstanding that the polymerisations were conducted in a fluorinated alcohol, the spectra gave no indication of an excessive formation of syndiotactic triades.68,69
 | ||
| Fig. 2 1H NMR spectra of selected sulfobetaine methacrylate monomers D2O (solvent signal at 4.78 ppm): ammoniopropanesulfonates (a) 3 and (b) 4, and ammoniobutanesulfonates: (c) 5, and (d) 6. | ||
 | ||
| Fig. 3 1H NMR spectra of poly(sulfobetaine methacrylate)s P-1–P-8 in D2O with added NaCl (solvent signal at 4.78 ppm); from top to bottom: P-1270, P-2250, P-3230, P-4295, P-5280, P-6250, P-7260 and P-8290. | ||
Thermogravimetric analysis (TGA) of the polysulfobetaines showed that decomposition accompanied by mass loss starts at about 300 °C. Differential scanning calorimetry (DSC) measurements revealed no thermal transition for any of the polymers before degradation starts. This is in agreement with reports on most sulfobetaine homopolymers.43,44,70
The solubility of the polymers P-1 to P-8 in water was investigated by turbidimetry. With the remarkable exception of polymer P-2, which is water-soluble over the full range of 0–100 °C, the aqueous solutions of all polysulfobetaines studied exhibited a miscibility gap, with an UCST-type phase transition in water as well as in deuterated water (Table 3). In both H2O and D2O, the clouding transitions were all sharp and the curves were reproducible. As the hysteresis of the transitions for heating and cooling runs was marginal (≤1 °C), we conclude that the binodal and spinodal lines of the polysulfobetaine/water phase diagram coincide virtually. Table 3 summarises the cloud points, which should correspond very closely to the true phase transition temperatures.
| Sample | Cloud point [°C] | Sample | Cloud point [°C] | ||
|---|---|---|---|---|---|
| In H2O | In D2O | In H2O | In D2O | ||
| P- 1 85 | 41 | 50 | P- 5 50 | 82 | 94 |
| P- 1 270 | 55 | 68 | P- 5 80 | >100 | >100 |
| P- 1 575 | 67 | 80 | P- 5 95 | >100 | >100 |
| P- 1 585 | 71 | 86 | P- 5 280 | >100 | >100 |
| P- 2 95 | < 0 | < 0 | P- 6 80 | < 0 | 17 |
| P- 2 250 | < 0 | < 0 | P- 6 250 | 4 | 28 |
| P- 2 330 | < 0 | < 0 | P- 6 420 | 11 | 36 |
| P- 2 485 | < 0 | < 0 | P- 6 500 | 15 | 40 |
| P- 3 65 | < 0 | 17 | P- 7 85 | 70 | 75 |
| P- 3 95 | 24 | 34 | P- 7 260 | 88 | 94 |
| P- 3 230 | 38 | 48 | P- 7 430 | >100 | >100 |
| P- 3 585 | 47 | 56 | P- 7 520 | >100 | >100 |
| P- 4 75 | < 0 | 7 | P- 8 100 | 41 | 47 |
| P- 4 295 | 5 | 14 | P- 8 290 | >100 | >100 |
| P- 4 480 | 8 | 18 | P- 8 480 | >100 | >100 |
| P- 4 585 | 10 | 20 | P- 8540 | >100 | >100 |
The observed cloud points varied markedly with the precise chemical structure of the polysulfobetaines, as expected. For a given structure, the values increased with the molar mass over the full range studied (DPn ≤ 600) (Table 3, see also Fig. S18†). Also, they increased notably with the polymer concentration, at least up to concentrations of 50 g L−1 (Fig. 4, see also Fig. S18†). Though this finding seems rather unsurprising, it contrasts with a minimum of the cloud point reported for P-1 within this concentration range.21 Whereas the concentration dependence seems to level off already at concentrations of about 25 g L−1 for some polymers (P-1, P-6, P-7), no levelling was seen for P-5 and for P-8 even at 50 g L−1 (Fig. 4, see also Fig. S18†). With respect to the chemical structure, we found that polymer P-2 was fully soluble in water even for the sample with the highest molar mass (Table 3). For P-3, P-4 and P-6, water-solubility over the full temperature range of 0–100 °C was only observed for the samples with the lowest molar mass; otherwise, cloud points were found. In contrast, the polymer samples of P-5, P-7 and P-8 with a low molar mass showed cloud points, whereas samples with high molar masses were insoluble in water over the full temperature range.
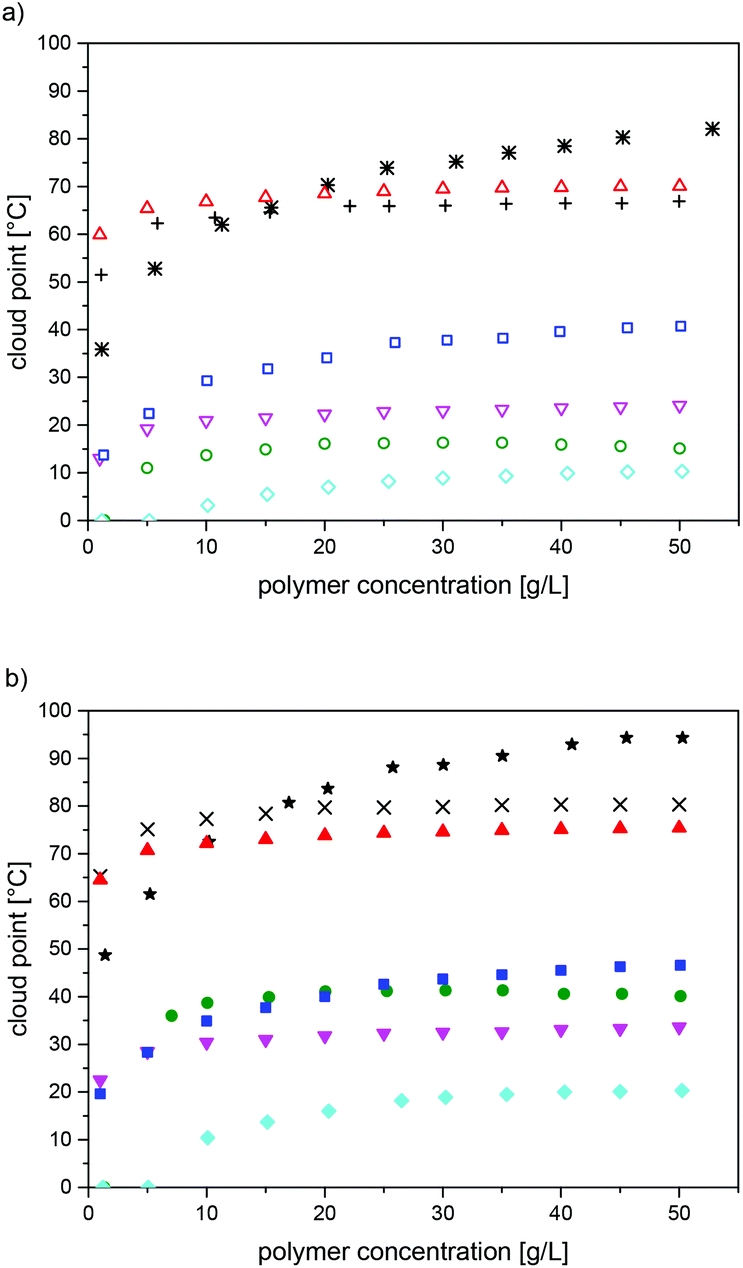 | ||
| Fig. 4 Concentration dependence of the UCST-type cloud points of the polymers P-1575 (+, ×), P-395 (∇, ▼), P-4585 (◊, ◆), P-550 (✲, ★), P-6500 (○, ●), P-785 (△, ▲), and P-8100 (□, ■): (a) in H2O (open symbols), (b) in D2O (full symbols). | ||
For all the thermo-responsive polymers, pronounced H-D isotope effects were found (Fig. 4), in analogy to the closely related poly(sulfobetaine methacrylamides).28,30 The differences observed between the cloud points in H2O and in D2O were about 6 °C in the cases of P-7 and P-8, and about 10 °C in the cases of P-1, P-3, P-4 and P-5. The highest difference of about 25 °C was found for P-6. The reasons for the marked, but in its extent notably varying H-D-effect, are not clear at present.
A synopsis of the relative water-solubilities of the various polysulfobetaines synthesised reveals that as a general feature, cloud points are considerably higher for the poly(ammonio-butanesulfonate)s than for their poly(ammoniopropanesulfonate) analogues (Table 3), i.e. we observed for a given molar mass that P-1 < P-5, P-2 < P-6, P-3 < P-7, and P-4 < P-8. This finding agrees with previous studies on the pair P-1/P-5,20,29,53 and matches well with reports on analogous pairs of acrylamide- and methacrylamide-based polysulfobetaines.15,30,71 One might be tempted to explain this observation by an increased overall hydrophobicity of the poly(ammoniobutanesulfonates) due to the additional methylene group in the spacer separating the cationic and the anionic groups of the betaine moiety. However, such a simplistic explanation seems to fall too short, because the few data available for poly(ammonioethanesulfonate) analogues indicate that their solubility in water is inferior to that of their poly(ammoniopropanesulfonate) analogues,72 in agreement with studies on analogous ammonioalkanesulfonate surfactant series.73 Steric reasons facilitating, or hampering, respectively, intra-chain ion pairing were suggested as a possible explanation. Interestingly, in the case of the structurally closely related ammonioalkanoates, theoretical studies predict the maximum hydrophilicity of the zwitterionic group being for separation of the anionic and the cationic groups by 6–8 methylene groups, due to balancing the hydration and dehydration energies of the ionic and the spacer groups, respectively.74 A rich series of poly(ammonioalkanesulfonate methacrylates) with dimethylene up to dodecamethylene (C12) spacer groups was reported recently50 and could possibly shed more light on this aspect; however, this has not been studied yet concerning their water-solubility.
When comparing the effect of the substituents on the ammonium group on the phase transition, we noticed that cloud points decrease as P-1 > P-3 > P-2, and P-5 > P-7 > P-6, i.e. in the order of dimethylammonio → morpholinio → piperidinioalkanesulfonate. Apparently, increasing the steric hindrance on the ammonium group decreases the phase transition temperature, which also seems to match some early observations on other polysulfobetaines.20 Nevertheless, the polymers P-3 and P-7 containing the morpholine building block show higher cloud points than their analogues P-2 and P-6 containing the piperidine building block, which has a similar steric demand but is a priori more hydrophobic. Hence, a mere steric effect should result in lower cloud points for the poly(morpholinioalkanesulfonates), which, however, is opposite to our findings. The strong effect of the sulfobetaine moiety's precise structure on thermo-responsiveness is evident, but the result cannot be rationalised by a few plain rules.
The lack of a facile structure–property relationship is corroborated when analysing the relative water-solubilities of the pair P-4 and P-8 compared to the pair P-1 and P-5. These two polymer pairs are only distinguished by the number of methylene groups, namely three and two, respectively, that separate the nitrogen atom of their zwitterionic moiety from their polymethacrylate backbone. Yet we find again that such apparently small modifications induce big effects, and that the formally more hydrophobic polymers P-4 and P-8, as judged from their chemical structures, exhibit much lower cloud points. In fact, this finding is consistent with previous studies that reported a smaller miscibility gap for the polysulfobetaine analogue with 11 methylene units between the backbone and ammonium group in comparison to P-1, despite seeming a priori to be much more hydrophobic.22,52 The observations possibly indicate that the increased mobility and steric hindrance of the zwitterionic side chain weaken the Coulomb interaction between the ammonium and the sulfonate groups, thereby lowering the UCST of the polymers.
Comparing the phase transition temperatures for the reference polymer P-1 measured by us with the various values reported in the literature, which differ strongly from one another,21,24,27,29,35,54–57 we note that our values for a given molar mass and concentration are throughout the highest. Model studies suggest that the rather bulky and hydrophobic dye-labelled end groups may be responsible for an increase in the cloud point by a few degrees,28,29 but cannot justify the much larger discrepancies encountered. As addressed already in the introduction, the phase transition temperature of P-1 is known to be quite sensitive to chemical defects as well as to the presence of certain inorganic salts. Indeed, in one case, the partial hydrolysis of P-1 was declared.24 Chemical defects, such as an incomplete, though close to quantitative conversion of tertiary amine groups into sulfobetaine moieties, can account for the much lower values reported, when P-1 was made by a post-polymerisation modification.29,56 The remaining salt contaminants when conducting the polymerisation in the presence of inorganic salts,27,35,57 or when using persulfate initiators,21,24,54,55 may explain the other deviating data. In one case, however, we cannot offer a reasonably plausible explanation for the mismatch between our's and the reported cloud point values,29 as the chosen polymerisation approach and conditions were very similar (trifluoroethanol solution, no salt added, use of azo initiators and of hydrophobic RAFT agents). Similarly to P-1, the cloud points found for the ammoniobutanesulfonate analogue P-5 were also considerably higher in our study than the previously reported values.29 Here again, we cannot offer presently a convincing explanation for this discrepancy, as none of the plausible possible reasons, as discussed above, can be invoked. Nevertheless, not taking the differences in the absolute values of the cloud points for P-1 and P-5 into account, the results of the two studies are consistent in so far as the cloud points are systematically higher for the ammoniobutanesulfonate P-5 compared to the standard P-1.
In any case, the comparison of the various polymers with their systematically varied structure of the sulfobetaine moiety reveals that at present it is not possible to predict reasonably the phase transition characteristics of newly made polysulfobetaine variants, even for apparently small chemical changes made. The steric demands and flexibility of the side chains appear to affect the phase behaviour in aqueous solution more than the additional hydrophilic or hydrophobic molecular fragments. Note also, that the UCST-type cloud points of the poly(sulfobetaine methacrylate) pair P-4 and P-8 were significantly lower than the reported cloud points of the analogous pair of poly(sulfobetaine methacrylamides) made under identical polymerisation conditions and bearing the identical substituents and spacer groups, except for the amide moiety instead of the ester.30 This finding, which is opposite to the a priori more hydrophobic character of the ester moiety compared to amides, underlines the failure of a simple hydrophilic–hydrophobic group contribution analysis for rationalising the water-solubility of polysulfobetaines.
As the strong effect of the added low molar mass electrolytes on the water-solubility of poly(zwitterions) is well known, we investigated also the effect of selected inorganic salts on the thermo-responsivity of the poly(sulfobetaine methacrylates). The evolutions of the cloud points in H2O containing NaCl, NaBr, Na2SO4 and (NH4)2SO4 are shown in Fig. 5 for one example of each of the studied polymers. For a given polymer, samples of different molar masses showed always the same patterns (cf. Fig. S19–S23†). As a common feature, small amounts of added salt make a notable impact. The effect depends notably on the nature of the anion in the order SO42− < Cl− < Br−, in agreement with the empirical Hofmeister series.75,76 In contrast, the chemical nature of the added cations seems to be of minor importance, as for a given anion, namely SO42−, we do not observe significant differences between the sodium and ammonium salts. Polymers P-1, P-4, P-5, P-6 and P-8 were subject to a straightforward salting-in behaviour. Their transition temperatures decreased continuously with increasing the amount of added salt following the same pattern, whereby salting-in effectivity increases in the order (NH4)2SO4 ≈ Na2SO4 < NaCl < NaBr. In the case of the latter two salts, the cloud points fell eventually below the freezing point, reaching this point for salt concentrations always lower than 0.1 M (Fig. 5a, c–e and g), i.e. below the molarity of the physiological salt solution. In contrast, high concentrations (>0.3 M) of sulfates induced a small increase in the cloud point, i.e. they make the polymer solubility pass through a maximum.
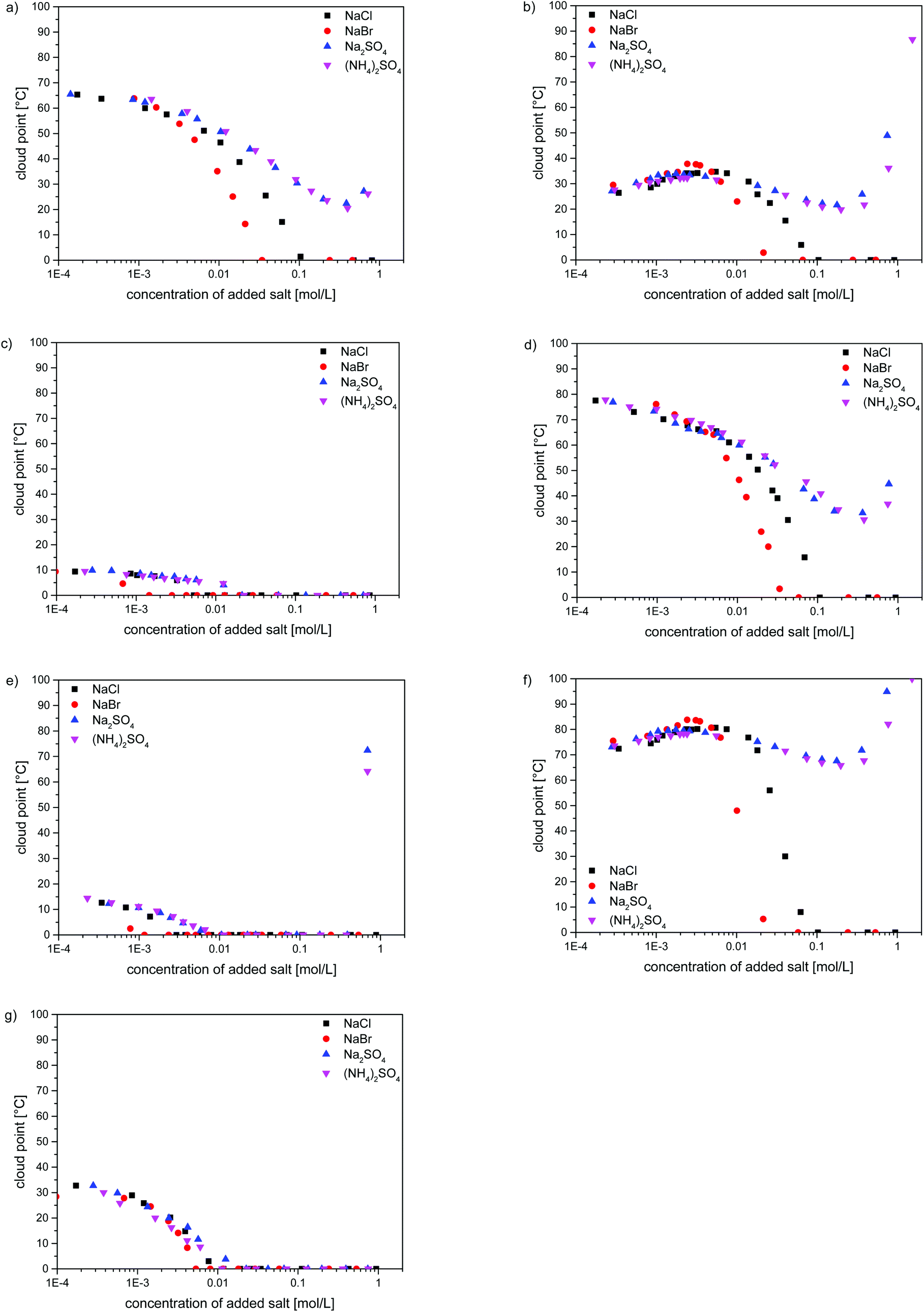 | ||
| Fig. 5 Evolution of UCST-type cloud points of the polymer solutions in H2O (50 g L−1) containing inorganic salts for (a) P-1575, (b) P-395, (c) P-4585, (d) P-550, (e) P-6500, (f) P-785, and (g) P-8100. (■) = NaCl, (●) = NaBr, (▲) = Na2SO4, (▼) = (NH4)2SO4. | ||
The salt effects on the water-solubility of polymers P-3 and P-7, containing the morpholinium group, were somewhat more complex (Fig. 5b and f). First, we observed a slight increase of the cloud point with increasing salt concentration, up to the lower mM range. Then, the cloud points passed through a maximum, i.e. water-solubility passes through a minimum. Such maxima of the cloud points upon the addition of small amounts of salt have been noticed for some other polysulfobetaines,22,24,28,30 but the reasons remain a matter of discussion.24 Beyond the maximum, the cloud points of P-3 and P-7 followed the same pattern as for the other polysulfobetaines when the amounts of salt were further increased: in the case of NaBr and NaCl, the cloud points decreased eventually below freezing point, whereas high concentrations (>0.3 M) of sulfates provoked again a small increase of the cloud point, i.e. they make the polymer solubility pass through a maximum (Fig. 5).
The strong dependence of the cloud points of P-1 on the concentration and on the type of added inorganic salts, in particular on the anion type in close correlation to the Hofmeister series, is well established.20–23,43,57,77–82 Therefore, the strong effects of the added salts and their relative impacts, observed for structurally closely related polymers P-2–P-8 are not surprising. Still, the widespread perception that the interaction of salts with polysulfobetaines can be treated as a general salting-in effect is not adequate, as evident from the evolution of the cloud points as a function of salt concentration. Maxima and minima can occur in dependence of the precise zwitterion structure, as also reported for certain other polysulfobetaines.22,28,30 Such a complex behaviour may have severe implications when employing polysulfobetaines, e.g. in systems that are not closed and where material exchange can take place, as typically encountered in the biomedical field. The understanding of the underlying reasons remains, despite initial attempts,24,57 a challenge.
Conclusions
Beyond being modulated by molar mass and concentration, the UCST-type thermo-responsive behaviour of poly(sulfobetaine methacrylate) is extremely sensitive to apparently small changes of the chemical structure of the polyzwitterions. The effects observed can, however, not be described or predicted by a simple rule. While water-solubility is generally enhanced for the poly(ammoniopropanesulfonates) compared to their poly(ammoniobutanesulfonate) analogues, it can be also enhanced when increasing the – formally hydrophobic – alkyl spacer between the polymer backbone and the zwitterionic moiety. Moreover, the incorporation of heterocyclic ammonium groups improved the solubility in water. However, the extent of the effect did not correlate with the formal relative hydrophilicities of the heterocycles introduced. Accordingly, a simple addition of hydrophilic and hydrophobic group contributions clearly fails to describe the structural effects on the phase behaviour encountered. In fact, the best water-solubility is obtained for the polyzwitterions derived from the – formally – least hydrophilic amine building block within the series, namely from piperidine. These observations possibly indicate that the increased mobility and steric requirements of the zwitterionic side chain weaken the Coulomb interaction between the ammonium and the sulfonate groups, thereby lowering the UCST of the polymers. In any case, our results show that structural variations, even small ones, of the polyzwitterion structure are an appropriate strategy, if particularly hydrophilic (or vice versa, less hydrophilic) polyzwitterions are searched for, as, e.g. is the case for low-fouling applications. Furthermore, the aqueous thermo-responsive behaviour is strongly modulated by adding inorganic salts. The modulation, however, cannot be understood as a straightforward salting-in effect, as the impact of the salts depends in a complex way on both the amount and the precise chemical nature of the ions added, in particular of the anions.Altogether, our study demonstrates the rich thermo- and salt-responsive behaviour that polysulfobetaines derived from monomer units even with a rather simple structure may offer in aqueous media. Nevertheless, we are still far from a thorough understanding of the phenomena, or of being able to establish rules that would enable a reliable prediction of the behaviour of new variants for such stimuli-responsive polymers.
Acknowledgements
We thank D. Schanzenbach (Universität Potsdam) and E. Wischerhoff (Fraunhofer IAP) for support during polymer analysis and for stimulating discussions. The work was supported by Deutsche Forschungsgemeinschaft (DFG) under grants La611/11-1, Mu1487/17-1 and Pa771/14-1.References
- S. Aoshima and S. Kanaoka, Adv. Polym. Sci., 2007, 210, 169–208 CrossRef.
- V. Aseyev, H. Tenhu and F. Winnik, Adv. Polym. Sci., 2011, 242, 29–89 CrossRef CAS.
- G. Vancoillie, D. Frank and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1074–1095 CrossRef CAS.
- P. Schattling, F. D. Jochum and P. Theato, Polym. Chem., 2014, 5, 25–36 RSC.
- S. Strandman and X. X. Zhu, Prog. Polym. Sci., 2015, 42, 154–176 CrossRef CAS.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- J. Seuring and S. Agarwal, Macromol. Rapid Commun., 2012, 33, 1898–1920 CrossRef CAS PubMed.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- Q. Zhang and R. Hoogenboom, Prog. Polym. Sci., 2015, 48, 122–142 CrossRef CAS.
- J. H. Priest, S. L. Murray, R. J. Nelson and A. S. Hoffman, ACS Symp. Ser., 1987, 350, 255–264 CrossRef CAS.
- Ö. Akdemir, N. Badi, S. Pfeifer, Z. Zarafshani, A. Laschewsky, E. Wischerhoff and J.-F. Lutz, ACS Symp. Ser., 2009, 1023, 189–202 CrossRef.
- J. Weiss, A. Li, E. Wischerhoff and A. Laschewsky, Polym. Chem., 2012, 3, 352–361 RSC.
- A. Laschewsky, E. D. Rekai and E. Wischerhoff, Macromol. Chem. Phys., 2001, 202, 276–286 CrossRef CAS.
- N. ten Brummelhuis and H. Schlaad, Polym. Chem., 2011, 2, 1180–1184 RSC.
- Y. Zhu, R. Batchelor, A. B. Lowe and P. J. Roth, Macromolecules, 2016, 49, 672–680 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Chem. Rev., 2002, 102, 4177–4189 CrossRef CAS PubMed.
- S. Kudaibergenov, W. Jaeger and A. Laschewsky, Adv. Polym. Sci., 2006, 201, 157–224 CrossRef CAS.
- A. Laschewsky, Polymers, 2014, 6, 1544–1601 CrossRef CAS.
- J. C. Salamone, W. Volksen, A. P. Olson and S. C. Israel, Polymer, 1978, 19, 1157–1162 CrossRef CAS.
- V. M. Monroy Soto and J. C. Galin, Polymer, 1984, 25, 254–262 CrossRef.
- D. N. Schulz, D. G. Peiffer, P. K. Agarwal, J. Larabee, J. J. Kaladas, L. Soni, B. Handwerker and R. T. Garner, Polymer, 1986, 27, 1734–1742 CrossRef CAS.
- P. Köberle, A. Laschewsky and T. D. Lomax, Makromol. Chem., Rapid Commun., 1991, 12, 427–433 CrossRef.
- M. B. Huglin and M. A. Radwan, Makromol. Chem., 1991, 192, 2433–2445 CrossRef CAS.
- P. Mary, D.D. Bendejacq, M.-P. Labeau and P. Dupuis, J. Phys. Chem. B, 2007, 111, 7767–7777 CrossRef CAS PubMed.
- F. O. Obiweluozor, A. GhavamiNejad, S. Hashmi, M. Vatankhah-Varnoosfaderani and F. J. Stadler, Macromol. Chem. Phys., 2014, 215, 1077–1091 CrossRef CAS.
- P. A. Woodfield, Y. Zhu, Y. Pei and P. J. Roth, Macromolecules, 2014, 47, 750–762 CrossRef CAS.
- H. Willcock, A. Lu, C. F. Hansell, E. Chapman, I. R. Collins and R. K. O'Reilly, Polym. Chem., 2014, 5, 1023–1030 RSC.
- V. Hildebrand, A. Laschewsky and D. Zehm, J. Biomater. Sci., Polym. Ed., 2014, 25, 1602–1618 CrossRef CAS PubMed.
- Y. Zhu, J.-M. Noy, A. B. Lowe and P. J. Roth, Polym. Chem., 2015, 6, 5705–5718 RSC.
- V. Hildebrand, A. Laschewsky and E. Wischerhoff, Polym. Chem., 2016, 7, 731–740 RSC.
- M. Arotçaréna, B. Heise, S. Ishaya and A. Laschewsky, J. Am. Chem. Soc., 2002, 124, 3787–3793 CrossRef.
- Y. Maeda, H. Mochiduki and I. Ikeda, Macromol. Rapid Commun., 2004, 25, 1330–1334 CrossRef CAS.
- M. Mertoglu, S. Garnier, A. Laschewsky, K. Skrabania and J. Storsberg, Polymer, 2005, 46, 7726–7740 CrossRef CAS.
- J. D. Flores, X. Xu, N. J. Treat and C. L. McCormick, Macromolecules, 2009, 42, 4941–4945 CrossRef CAS.
- Y.-J. Shih, Y. Chang, A. Deratani and D. Quemener, Biomacromolecules, 2012, 13, 2849–2858 CrossRef CAS PubMed.
- Md. R. Rodríguez-Hidalgo, C. Soto-Figueroa and L. Vicente, Soft Matter, 2013, 9, 5762–5770 RSC.
- Q. Zhang, X. Tang, T. Wang, F. Yu, W. Guo and M. Pei, RSC Adv., 2014, 4, 24240–24247 RSC.
- V. A. Vasantha, S. Jana, S. S.-C. Lee, C.-S. Lim, S. L.-M. Teo, A. Parthiban and J. G. Vancso, Polym. Chem., 2015, 6, 599–606 RSC.
- T. Matsumoto, T. Ichikawa and H. Ohno, Polym. Chem., 2016, 7, 1230–1233 RSC.
- J. Virtanen, M. Arotçaréna, B. Heise, S. Ishaya, A. Laschewsky and H. Tenhu, Langmuir, 2002, 18, 5360–5365 CrossRef CAS.
- V. M. Monroy-Soto and J. C. Galin, Polymer, 1984, 25, 121–128 CrossRef CAS.
- A. Laschewsky and I. Zerbe, Polymer, 1991, 32, 2070–2080 CrossRef CAS.
- P. Köberle, A. Laschewsky and D. van den Boogaard, Polymer, 1992, 33, 4029–4039 CrossRef.
- P. Köberle and A. Laschewsky, Macromolecules, 1994, 27, 2165–2179 CrossRef.
- J. Cardoso, R. Manrique, M. Albores-Velasco and A. Huanosta, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 479–488 CrossRef CAS.
- M. Gauthier, T. Carrozzella and A. Penlidis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 511–523 CrossRef CAS.
- J. Cardoso, O. Soria-Arteche, G. Vazquez, O. Solorza and I. Gonzalez, J. Phys. Chem. C, 2010, 114, 14261–14268 CAS.
- Y. Terayama, M. Kikuchi, M. Kobayashi and A. Takahara, Macromolecules, 2011, 44, 104–111 CrossRef CAS.
- S. Hashmi, A. GhavamiNejad, F. O. Obiweluozor, M. Vatankhah-Varnoosfaderani and F. J. Stadler, Macromolecules, 2012, 45, 9804–9815 CrossRef CAS.
- D. Kratzer, L. Barner, C. Friedmann, S. Bräse and J. Lahann, Eur. J. Org. Chem., 2014, 8064–8071 CrossRef CAS.
- A. GhavamiNejad, C. H. Park and C. S. Kim, Biomacromolecules, 2016, 17, 1213–1223 CrossRef CAS PubMed.
- A. Laschewsky and I. Zerbe, Polymer, 1991, 32, 2081–2086 CrossRef CAS.
- J. Gao, G. Zhai, Y. Song and B. Jiang, J. Appl. Polym. Sci., 2008, 107, 3548–3556 CrossRef CAS.
- Y.-J. Shih and Y. Chang, Langmuir, 2010, 26, 17286–17294 CrossRef CAS PubMed.
- L. Chen, Y. Honma, T. Mizutani, D.-J. Liaw, J. P. Gong and Y. Osada, Polymer, 2000, 41, 141–147 CrossRef CAS.
- J. V. M. Weaver, S. P. Armes and V. Bütün, Chem. Commun., 2002, 38, 2122–2123 RSC.
- F. Wang, J. Yang and J. Zhao, Polym. Int., 2015, 64, 999–1005 CrossRef CAS.
- P. Anton and A. Laschewsky, Macromol. Rapid Commun., 1991, 12, 189–196 CrossRef CAS.
- J. Cardoso, R. Montiel and A. Huanosta-Tera, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 1152–1160 CrossRef CAS.
- W. Ding, C. Lv, Y. Sun, X. Liu, T. Yu, G. Qu and H. Luan, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 432–440 CrossRef CAS.
- Y. Higaki, J. Nishida, A. Takenaka, R. Yoshimatsu, M. Kobayashi and A. Takahara, Polym. J. Jpn., 2015, 47, 811–818 CrossRef CAS.
- G. S. Georgiev, A. A. Tzoneva, L. G. Lyutov and I. K. Petkov, Nonlinear Opt., Quantum Opt., 2004, 31, 347–361 CAS.
- V. Bette, H. Bergmann, J. Petzoldt and F. Höfer, US Pat, 20120077977A1, 2012, 6 pages Search PubMed.
- A. Laschewsky, R. Touillaux, P. Hendlinger and A. Vierengel, Polymer, 1995, 36, 3045–3049 CrossRef CAS.
- P. Kasák, Z. Kroneková, I. Krupa and I. Lacík, Polymer, 2011, 52, 3011–3020 CrossRef.
- B. Yu, A. B. Lowe and K. Ishihara, Biomacromolecules, 2009, 10, 950–958 CrossRef CAS PubMed.
- K. Skrabania, A. Miasnikova, A. M. Bivigou-Koumba, D. Zehm and A. Laschewsky, Polym. Chem., 2011, 2, 2074–2083 RSC.
- Y. Isobe, K. Yamada, T. Nakano and Y. Okamoto, Macromolecules, 1999, 32, 5979–5981 CrossRef CAS.
- İ. Değirmenci, Ş. Eren, V. Aviyente, B. De Sterck, K. Hemelsoet, V. Van Speybroeck and M. Waroquier, Macromolecules, 2010, 43, 5602–5610 CrossRef.
- M. Galin, E. Marchal, A. Mathis, B. Meurer, Y. M. Monroy Soto and J. C. Galin, Polymer, 1987, 28, 1937–1944 CrossRef CAS.
- K. Haraguchi, J. Ning and G. Li, Eur. Polym. J., 2015, 68, 630–640 CrossRef CAS.
- T. A. Wielema and J. B. F. N. Engberts, Eur. Polym. J., 1987, 23, 947–950 CrossRef CAS.
- J. G. Weers, J. F. Rathman, F. U. Axe, C. A. Crichlow, L. D. Foland, D. R. Scheuing, R. J. Wiersema and A. G. Zielske, Langmuir, 1991, 7, 854–867 CrossRef CAS.
- H. Du and X. Qian, J. Comput. Chem., 2016, 37, 877–885 CrossRef CAS PubMed.
- P. S. C. Yanjie-Zhang, Curr. Opin. Chem. Biol., 2006, 10, 658–663 CrossRef PubMed.
- A. Salis and B. W. Ninham, Chem. Soc. Rev., 2014, 43, 7358–7377 RSC.
- D.-J. Liaw, W.-F. Lee, Y.-C. Whung and M.-C. Lin, J. Appl. Polym. Sci., 1987, 34, 999–1011 CrossRef CAS.
- R. Knoesel, M. Ehrmann and J. C. Galin, Polymer, 1993, 34, 1925–1932 CrossRef CAS.
- H. Wang, T. Hirano, M. Seno and T. Sato, Eur. Polym. J., 2003, 39, 2107–2114 CrossRef CAS.
- G. Georgiev, A. Tzoneva, L. Lyutov, S. Iliev, I. Kamenova, V. Georgieva, E. Kamenska and A. Bund, Macromol. Symp., 2004, 210, 393–401 CrossRef CAS.
- M. Kobayashi, Y. Terayama, M. Kikuchi and A. Takahara, Soft Matter, 2013, 9, 5138–5148 RSC.
- T. Ye, Y. Song and Q. Zheng, Colloid Polym. Sci., 2016, 294, 389–397 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of tertiary amine bearing methacrylates; 1H NMR, 13C NMR, 1H–1H COSY and 1H–13C HMQC NMR spectra of chain transfer agent and monomers; polymer 1H NMR spectrum illustrating the limits of end group detection; molar mass dependent evolutions of the polymer cloud points with concentration in H2O, and as function of added inorganic salts. See DOI: 10.1039/c6py01220e |
| This journal is © The Royal Society of Chemistry 2017 |