
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the causes of thermal hysteresis using tunable Nagg micelles with linear and brush-like thermoresponsive coronas†
L. D.
Blackman
a,
M. I.
Gibson
 *ab and
R. K.
O'Reilly
*a
*ab and
R. K.
O'Reilly
*a
aDept. of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: r.k.o-reilly@warwick.ac.uk
bWarwick Medical School, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: m.i.gibson@warwick.ac.uk
First published on 9th August 2016
Abstract
Self-assembled thermoresponsive polymers in aqueous solution have great potential as smart, switchable materials for use in biomedical applications. In recent years, attention has turned to the reversibility of these polymers’ thermal transitions, which has led to debate over what factors influence discrepancies in the transition temperature when heating the system compared to the temperature obtained when cooling the system, known as the thermal hysteresis. Herein, we synthesize micelles with tunable aggregation numbers (Nagg) whose cores contain poly(n-butyl acrylate-co-N,N-dimethylacrylamide) (p(nBA-co-DMA)) and four different thermoresponsive corona blocks, namely poly(N-isopropylacrylamide) (pNIPAM), poly(N,N-diethylacrylamide) (pDEAm), poly(diethylene glycol monomethyl ether methacrylate) (pDEGMA) and poly(oligo(ethylene glycol) monomethyl ether methacrylate) (pOEGMA). By studying their thermoresponsive behavior, we elucidate the effects of changing numerous important characteristics both in the thermoresponsive chain chemistry and architecture, and in the structure of their self-assemblies. Our findings demonstrate large deviations in the reversibility between the self-assemblies and the corresponding thermoresponsive homopolymers; specifically we find that micelles whose corona consist of polymers with a brush-like architecture (pDEGMA and pOEGMA) exhibit irreversible phase transitions at a critical chain density. These results lead to a deeper understanding of stimuli-responsive self-assemblies and demonstrate the potential of tunable Nagg micelles for uncovering structure–property relationships in responsive polymer systems.
Introduction
The understanding and utilization of stimuli-responsive polymer materials has opened up new avenues in potential applications such as their use as drug delivery vehicles,1–5 smart surface coatings,6,7 for detection of specific molecules or ions,6,8–11 and for enhanced oil recovery12 to name just a few. These materials respond dramatically to subtle environmental changes such as light, pH, CO2, glucose or redox changes.7,13–19 One of the most studied and utilized stimuli in the literature is temperature as it offers a simple means of non-invasively altering the polymers’ environment.14,19–21 These thermoresponsive polymers exhibit a change in their solubility over a temperature range and reports of thermoresponsive behavior in both aqueous and organic solutions exist. The critical temperature at which this change in solubility occurs is known as an upper or lower critical solution temperature (UCST or LCST respectively).19,21 Above the UCST or below the LCST, the polymer and solvent exist as a single phase. Phase separation occurs if a polymer solution is heated above its LCST or cooled below its UCST and is normally observable by variable temperature turbidimetry, microcalorimetry, or NMR spectroscopy. This class of polymer materials has gathered interest in the medical field by virtue of the fact that they can exploit subtle temperature differences, for example between healthy or cancerous cells, for diagnostic, therapeutic or theranostic purposes,1,22,23 or for the design of smart injectable hydrogels for tissue engineering.24,25 Of these thermoresponsive polymer systems, poly(N-isopropylacrylamide) (pNIPAM) in aqueous solution is one of the most widely studied.20,26 This is because the polymer exhibits an LCST close to body temperature and this transition temperature is relatively insensitive to other environmental conditions such as pH or salt concentration making it an attractive candidate for biological applications.26 Although an important polymer, of which new potential applications are still being realized, several limitations currently exist including its lack of biodegradability, and its slow reversibility in certain systems, known as a thermal hysteresis;26 the latter of which may limit its applicability as a fully reversible, switchable smart material. As such, new thermoresponsive homo- and copolymers have been sought with lower or negligible hysteresis such that the transition occurs at the same temperature regardless of whether the system is being heated or cooled.27–33 In studying new thermoresponsive polymers, a greater understanding of the factors surrounding thermal hysteresis has been developed. For instance, structurally it has been shown that the hydrogen bonding character of the polymer side chain has a marked effect on the reversibility.34,35 In the case of pNIPAM, it has been shown using multiple angle light scattering, ultrasensitive differential scanning calorimetry and FT-IR analysis that the polymer undergoes several distinct conformations in the globule-to-coil transition owing to intramolecular hydrogen bonding between the amide repeat units, which leads to irreversibility in this system.34–36 Similar analysis has been performed on a polymer analogue that cannot form polymer–polymer hydrogen bonding interactions, namely poly(N,N-diethylacrylamide) (pDEAm), which did not exhibit the same behavior.37 It has also been postulated that the glass transition temperature (Tg) may play a role in the reversibility of thermoresponsive polymer systems.21,38 Evidence suggests that polymers that exhibit a Tg below their LCST show transitions with lower thermal hysteresis than those whose LCST is above the Tg because of the increased mobility of the chains, which can facilitate easier redissolution.21,38 Polymers whose transition temperature is below their Tg are partially vitrified in the globular state so exhibit much slower rehydration kinetics.39 A loose trend in increasing hysteresis with Tg was also observed by Seuring and Agarwal in UCST type polymers.40The advent of “living” polymerization and reversible-deactivation radical polymerization (RDRP) techniques has given access to amphiphilic block copolymers.41–43 These form higher order self-assembled polymer structures, such as micelles, worms and vesicles, and are only synthetically possible because of the ability to covalently attach incompatible domains (blocks) that micro-phase separate both in the bulk and in a selective solvent for one of the domains.44–46 There has been great interest in the synthesis and utilization of self-assembled amphiphilic block copolymers that include stimuli-responsive polymers as one or more of the blocks, for instance the incorporation of thermoresponsive blocks.47–54 These can undergo reversible morphological transitions such as micelles to vesicles, vesicles to unimers and micelles to unimers.49–54 However, when using pNIPAM as the responsive block, it has been shown that these transitions occur very slowly,53,55 with micelle to vesicle transitions occurring in the order of days to several weeks. Further study revealed that using a core-forming block containing methyl acrylate instead of tert-butyl acrylate resulted in a faster micelle to vesicle transition by lowering the Tg of the core-forming block.56 Additionally, Jiang and Chen et al. have shown that the reversibility in this system could be further improved by using a core-forming alkyl end group57 as opposed to the hydrophobic polymer blocks used in the studies by our group. These micelles were only lightly associated, which facilitated the rearrangement of the pNIPAM block by increasing the overall mobility of the chains.
In recent years, attention has turned to the effect of subtle changes in the structure of self-assemblies on the thermoresponsive behavior. Wang and Li synthesized a series of n-dodecyl terminated pNIPAM polymers with various molecular weights.58 These were self-assembled into micelles in aqueous solution, whose cores comprised solely of the terminal alkyl chain. The aggregation number (Nagg), defined as the number of polymer chains per micelle, was then altered by adding the anionic surfactant sodium n-dodecyl sulfate (SDS), which could associate to the end groups of the polymer chains, thereby lowering the average Nagg. It was found that the addition of SDS to the micelles increased the thermal transition temperature by reducing the overall hydrophobicity of the polymer end groups. The effect of SDS increasing the cloud point of the pNIPAM chains was more pronounced in lower molecular weight pNIPAM chains because their transition temperatures were initially most affected by the presence of the hydrophobic end group.58 Zhang and coworkers looked at the effect of the corona conformation within ABA and AB block copolymers with hydrophobic poly(styrene) A blocks and thermoresponsive pNIPAM B blocks, which formed either flower-like micelles with looped pNIPAM coronas, or classical crew-cut micelles respectively.59 It was found that micelles with a looped corona topology had a lower transition temperature, which occurred over a smaller temperature range than those formed from the AB diblock copolymer, as assessed by turbidimetry and 1H NMR spectroscopy of the dilute solutions. This was a consequence of the differences in the loss of entropy associated with the collapse of the corona chains between each set of micelles.59 Both these examples show that simply the arrangement of the chains in the micellar structure plays a dominant role in determining the overall thermoresponsive behavior.
Recently, we reported the synthesis of micelles formed from AB diblock copolymers with a thermoresponsive pNIPAM corona-forming block and a statistical copolymer of hydrophilic N,N-dimethylacrylamide (DMA) and hydrophobic n-butyl acrylate (nBA) as the core-forming block.60 These micelles underwent macroscopic precipitation upon heating, in a similar fashion to thermoresponsive homopolymers, owing to the change in solubility of the pNIPAM corona. We found that micelles with a tunable Nagg could be obtained by varying the composition of hydrophobic nBA in the core-forming block. Although the transition temperatures across the series were independent of core composition or Nagg, the degree of hysteresis was found to increase as a function of core hydrophobicity. It was postulated that these differences were in fact a result of differences in core hydration, another important factor which determines reversibility in thermoresponsive self-assemblies (vide infra).
Following on from these findings, herein we study the effects of micellar structure on the thermal hysteresis of four distinct thermoresponsive polymers, namely pNIPAM, pDEAm, poly(diethylene glycol monomethyl ether methacrylate) (pDEGMA) and poly(oligo(ethylene glycol) monomethyl ether methacrylate) (pOEGMA), the properties of which are outlined in Fig. 1. For each corona, a set of micelles with tunable Nagg was prepared using the previously studied p(nBA-co-DMA) core-forming block with varying core hydrophobicity (Schemes 1 and 2). The effect of corona hydrogen bonding ability, hydrophilicity, Tg, core composition and Nagg of the micelles (which in turn relates to coronal chain confinement) on the thermoresponsive behavior of the micelles were studied. More generally, we also demonstrate that tunable Nagg micelles can be a powerful tool for uncovering structure–property relationships in stimuli-responsive self-assemblies (Scheme 1).
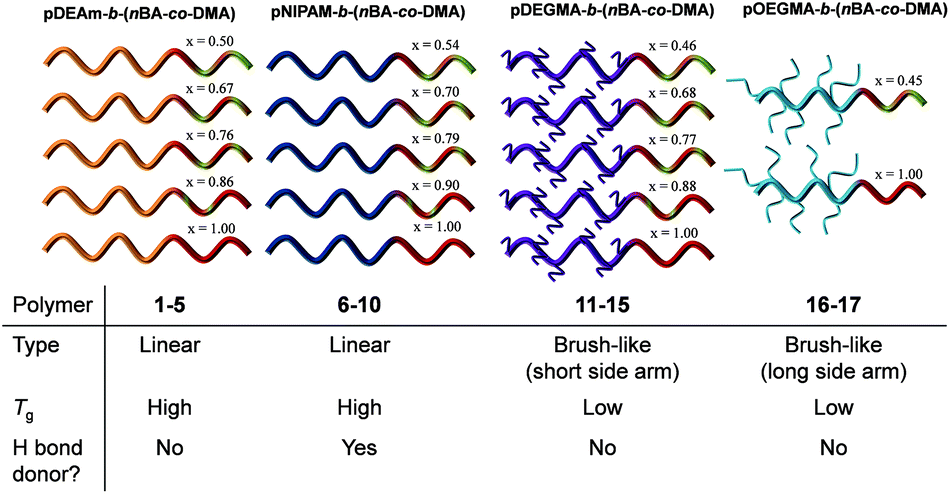 | ||
| Fig. 1 Illustration of the thermoresponsive diblock copolymers used in this study. Key: x = mol% nBA in the core-forming block. Below is a table outlining the differences in the corona blocks’ properties. | ||
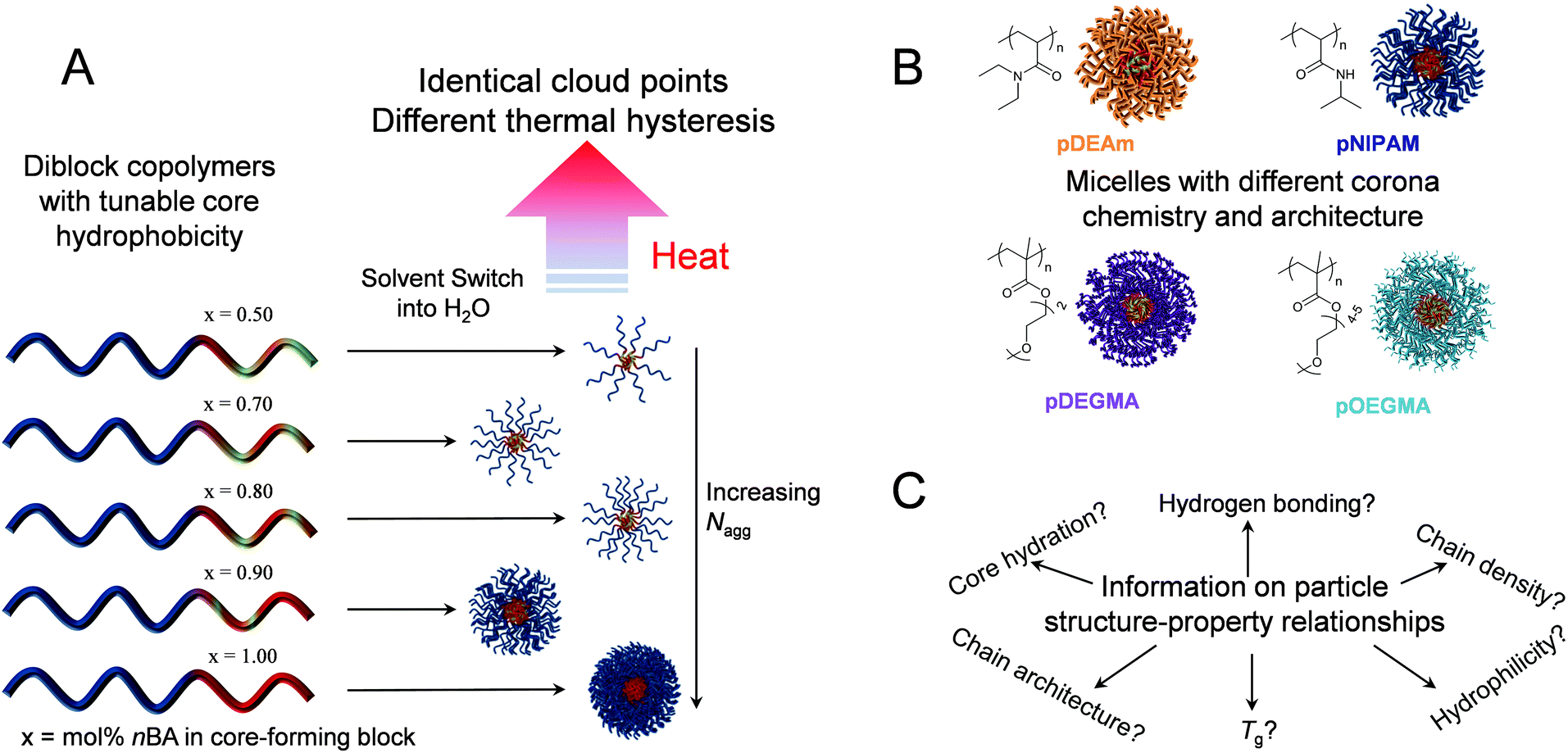 | ||
| Scheme 1 A: Schematic of one series of diblock copolymers with identical corona-forming blocks and tunable p(nBA-co-DMA) core compositions that self-assemble in water to yield micelles with a tunable Nagg. The resulting particles show identical cloud points but different thermal hysteresis when heated in solution. B: Design of four micellar series with different corona-forming blocks with distinct chemistry and architecture, but whose cores contain the same p(nBA-co-DMA) compositions. In each case, the chemical structure of the corona block is shown. C: Studying the thermoresponsive behaviors of the four micellar series gives information on the structure–property relationships regarding thermal hysteresis in thermoresponsive self-assemblies. | ||
 | ||
| Scheme 2 Synthesis of the four corona-forming macroCTA blocks (mCTA1–4) and their subsequent chain extension to yield the amphiphilic diblock copolymers (1–17). | ||
Experimental
Materials
The solvents petroleum ether (40–60 °C), diethyl ether, dichloromethane, ethyl acetate and acetone, and the reagents cyanomethyl dodecyl trithiocarbonate, 4,4′-azobis(4-cyanovaleric acid) (ACVA) and 4-dimethylaminopyridine (DMAP) were purchased from Sigma Aldrich and used as received. N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) was purchased from Alpha Aesar and used as received. 1,4-Dioxane, and the monomers DEGMA, OEGMA (Mn = 300 g mol−1), nBA and DMA were purchased from Sigma Aldrich and passed through a column of basic alumina prior to use. NIPAM was purchased from Sigma Aldrich and recrystallized from a toluene–hexane mixture prior to use. 2,2′-Azobisisobutyronitrile (AIBN) was purchased from Sigma Aldrich and recrystallized from methanol prior to use. Dialysis membrane (MWCO = 3.5–5 kDa) was purchased from Spectra/Por. The synthesis of N,N-diethylacrylamide,61 2-cyano-2-propyl dodecyl trithiocarbonate,62 4-cyano-4-(((ethylthio)carbonothioyl)thio) pentanoic acid63 have been described previously in the literature.Small molecule and polymer analysis
SEC analyses of polymers mCTA2 and 6–10 were performed on a Varian PL-GPC 50 Plus instrument fitted with mixed C columns and an RI detector using 5 mM NH4BF4 in N,N-dimethylformamide (DMF) as the eluent. SEC analyses of polymers mCTA1, mCTA3, mCTA4, 1–5 and 11–17 were performed on a Varian PL-GPC 50 Plus instrument fitted with mixed C columns and an RI detector using tetrahydrofuran (THF) containing 2% triethylamine (TEA) as the eluent. Mn values determined by SEC were calculated using poly(methyl methacrylate) standards. Additional triple detection SEC analysis was performed on mCTA1 using 5 mM NH4BF4 in DMF as the eluent.1H NMR spectroscopy was performed at 300 MHz on a Bruker Avance III HD-300 or a Bruker Avance AV-300 spectrometer, or at 400 MHz a Bruker Avance III HD-400 spectrometer. 13C NMR was performed at 75 MHz or 100 MHz on a Bruker Avance AV-300 or a Bruker Avance III HD-400 spectrometer respectively. For 1H and 13C NMR spectroscopy, chemical shifts (δ) in parts per million (ppm) are reported relative to the residual CHCl3 solvent peak at 7.26 ppm or 77.0 ppm, respectively.
High resolution electrospray ionization time of flight mass spectrometry (HRMS (ESI-ToF)) was performed on a Bruker MaXis mass spectrometer. Fourier transform infra-red (FT-IR) spectroscopy was performed on a Perkin-Elmer Spectrum 100 FT-IR spectrometer.
Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ethyl acetate:petroleum ether) to yield a yellow solid. Yield = 855 mg (81%). 1H NMR (300 MHz, CDCl3) δ/ppm: 3.70 (3H, s, COOCH3), 3.33 (2H, q, 3JH–H = 7.4 Hz, SCH2CH3), 2.65–2.57 (2H, m, C(CN)(CH3)CH2CH2), 2.56–2.35 (2H, m, C(CN)(CH3)CH2CH2), 1.87 (3H, s, C(CN)(CH3)CH2CH2), 1.35 (3H, t, 3JH–H = 7.4 Hz, SCH2CH3). 13C NMR (75 MHz, CDCl3) δ/ppm: 216.7 (C
:3 ethyl acetate:petroleum ether) to yield a yellow solid. Yield = 855 mg (81%). 1H NMR (300 MHz, CDCl3) δ/ppm: 3.70 (3H, s, COOCH3), 3.33 (2H, q, 3JH–H = 7.4 Hz, SCH2CH3), 2.65–2.57 (2H, m, C(CN)(CH3)CH2CH2), 2.56–2.35 (2H, m, C(CN)(CH3)CH2CH2), 1.87 (3H, s, C(CN)(CH3)CH2CH2), 1.35 (3H, t, 3JH–H = 7.4 Hz, SCH2CH3). 13C NMR (75 MHz, CDCl3) δ/ppm: 216.7 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S), 171.9 (CO), 118.9 (C(CN)(CH3)CH2CH2), 52.1 (COOCH3), 46.3 (C(CN)(CH3)CH2CH2), 33.8 (SCH2CH3), 31.3 (C(CN)(CH3)CH2CH2), 29.5 (C(CN)(CH3)CH2CH2), 24.8 (C(CN)(CH3)CH2CH2), 12.7 (SCH2CH3). FT-IR (neat) ν/cm−1: 2965, 2934, 2848 (C–H stretch), 2237 (C
S), 171.9 (CO), 118.9 (C(CN)(CH3)CH2CH2), 52.1 (COOCH3), 46.3 (C(CN)(CH3)CH2CH2), 33.8 (SCH2CH3), 31.3 (C(CN)(CH3)CH2CH2), 29.5 (C(CN)(CH3)CH2CH2), 24.8 (C(CN)(CH3)CH2CH2), 12.7 (SCH2CH3). FT-IR (neat) ν/cm−1: 2965, 2934, 2848 (C–H stretch), 2237 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch), 1737 (CO stretch). HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C10H15NNaS3 300.0157; Found 300.0156.
O), 1454, 1434 (C–H bend).
N stretch), 1737 (CO stretch). HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C10H15NNaS3 300.0157; Found 300.0156.
O), 1454, 1434 (C–H bend).
| Polymer | Corona |
nBA:DMA feed ratio |
DP nBAb | DP DMAb | mol% nBA in coreb |
M
n NMRb (kg mol−1) |
M
n SECc (kg mol−1) |
Đ |
R
Hd (nm) |
R
coree (nm) |
|---|---|---|---|---|---|---|---|---|---|---|
| a M n calculated from conversion 1H NMR spectroscopy. b Calculated using 1H NMR spectroscopy relative to the polymer end group (mCTA2–4) or known mCTA DP (1–17) (see Experimental section for details). c Calculated from SEC analysis using 2% TEA in THF (mCTA1, mCTA3–4, 1–5 and 11–17) or 5 mM NH4BF4 in DMF (mCTA2 and 6–10) as the eluent against poly(methyl methacrylate) standards. d Calculated from multiple angle DLS analysis using the Stokes Einstein equation. e Calculated from multiple angle SLS analysis (see ESI). | ||||||||||
| mCTA1 | pDEAm | — | — | — | — | 8.6a | 7.7 | 1.11 | — | — |
| 1 | pDEAm | 1:1 |
19 | 19 | 50 | 12.9 | 12.0 | 1.16 | 7.3 | 3.4 |
| 2 | pDEAm | 7:3 |
22 | 11 | 67 | 12.5 | 12.0 | 1.14 | 10.6 | 4.5 |
| 3 | pDEAm | 4:1 |
25 | 8 | 76 | 12.6 | 13.0 | 1.16 | 11.9 | 5.0 |
| 4 | pDEAm | 9:1 |
24 | 4 | 86 | 12.1 | 12.4 | 1.14 | 11.4 | 5.1 |
| 5 | pDEAm | 1:0 |
34 | 0 | 100 | 13.0 | 13.7 | 1.16 | 14.7 | 6.3 |
| mCTA2 | pNIPAM | — | — | — | — | 8.8 | 10.0 | 1.07 | — | — |
| 6 | pNIPAM | 1:1 |
20 | 17 | 54 | 12.9 | 13.0 | 1.10 | 8.3 | 3.5 |
| 7 | pNIPAM | 7:3 |
26 | 11 | 70 | 12.8 | 12.8 | 1.11 | 12.1 | 5.6 |
| 8 | pNIPAM | 4:1 |
27 | 7 | 79 | 12.4 | 12.7 | 1.10 | 13.4 | 6.2 |
| 9 | pNIPAM | 9:1 |
37 | 4 | 90 | 13.0 | 11.2 | 1.11 | 13.2 | 7.3 |
| 10 | pNIPAM | 1:0 |
40 | 0 | 100 | 13.9 | 10.9 | 1.11 | 17.5 | 8.1 |
| mCTA3 | pDEGMA | — | — | — | — | 12.4 | 12.3 | 1.36 | — | — |
| 11 | pDEGMA | 1:1 |
16 | 19 | 46 | 16.4 | 15.8 | 1.41 | 8.4 | 2.7 |
| 12 | pDEGMA | 7:3 |
26 | 12 | 68 | 17.0 | 15.7 | 1.46 | 13.1 | 5.5 |
| 13 | pDEGMA | 4:1 |
27 | 8 | 77 | 17.0 | 18.3 | 1.37 | 13.7 | 5.6 |
| 14 | pDEGMA | 9:1 |
30 | 4 | 88 | 16.7 | 16.6 | 1.45 | 12.0 | 5.8 |
| 15 | pDEGMA | 1:0 |
40 | 0 | 100 | 17.6 | 14.3 | 1.41 | 14.6 | 6.9 |
| mCTA4 | pOEGMA | — | — | — | — | 19.8 | 15.4 | 1.23 | — | — |
| 16 | pOEGMA | 1:1 |
17 | 22 | 44 | 24.2 | 20.1 | 1.34 | 9.0 | 3.1 |
| 17 | pOEGMA | 1:0 |
40 | 0 | 100 | 24.9 | 22.0 | 1.31 | 12.5 | 4.9 |
O stretch), 1534 (N–H bend).
O), 1459 (C–H bend), 1111 (C–O stretch).
O stretch), 1454 (C–H bend), 1100 (C–O stretch).
O stretch, ester), 1638 (CO stretch, amide), 1534 (N–H bend). Owing to the overlap of the polymer peaks of polymers 1–5 with the end groups and the polymer peaks of mCTA1, the ratio of nBA to DMA for polymers 1–5 was performed using the normalized integrals of the corresponding peaks at 4.00 (pnBA) and 3.22–2.77 ppm (pDMA) after subtraction of the spectrum of mCTA1 (see ESI†). Calculation of the ratio of nBA to DMA for polymers 6–10 was determined by comparison of the integral of the peak at 3.33 ppm (end group), with the integrals at 4.00 (pNIPAM + pnBA) and 3.22–2.77 ppm (pDMA). Calculation of the ratio of nBA to DMA for polymers 11–17 was performed using the integrals of the corresponding peaks at 4.00 (pnBA) and 3.22–2.77 ppm (pDMA) respectively, with respect to the known integral of the terminal methoxy protons at around 3.36 ppm at the end of the pDEGMA or pOEGMA side chain.
Particle analysis
For each micellar system, the resulting g2(q, t) autocorrelation functions from DLS analysis for each angle were analyzed by the REPES algorithm to determine a relaxation time, τ. The τ values at each angle were plotted against the square of the scattering wave vector, q to determine the apparent diffusion coefficient D according to eqn (1).64,65 Apparent hydrodynamic radii (RH) were then calculated using the Stokes Einstein eqn (2), where η is the solvent viscosity, kB is the Boltzmann constant and T is the absolute temperature.
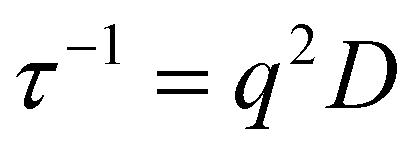 | (1) |
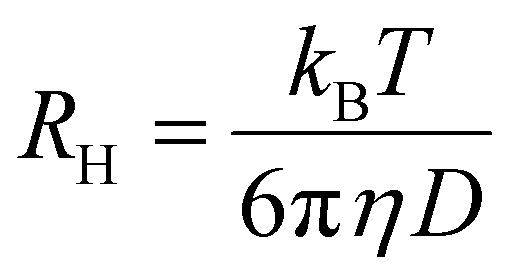 | (2) |
Using SLS analysis at the same angles, partial Zimm plots were obtained and the apparent aggregation number, Nagg for each set of micelles was calculated using eqn (3) and (4).64,65
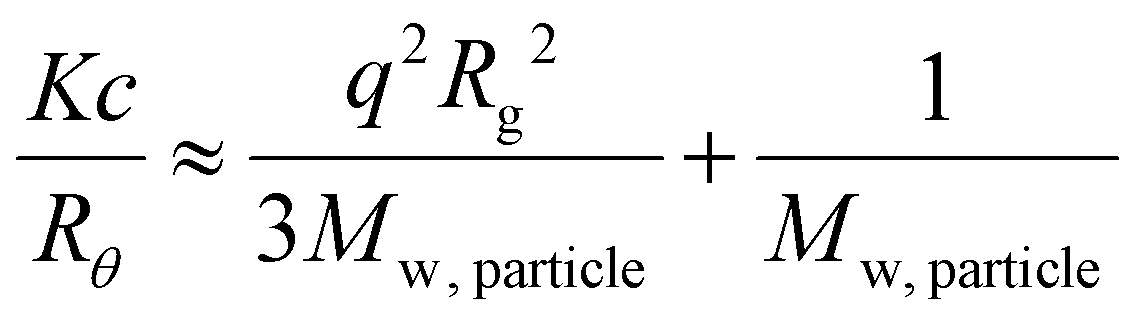 | (3) |
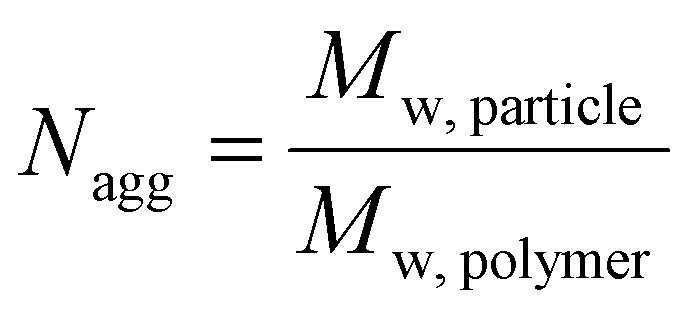 | (4) |
For SLS analysis, the intensity of the scattered light (Isample) was used to calculate Kc/Rθ for each angle, where c is the polymer concentration and K and Rθ are defined in the ESI.† It should be noted that since the radius of gyration (Rg) of these micelles was less than 20 nm, the average value of Kc/Rθ over the angles analyzed was equal to the inverse of the particles’ molecular weight (Mw, particle) and was used to calculate Nagg. In the event of two relaxation modes being present, the REPES algorithm was used to determine the relative amplitudes of the fast and slow modes (Afast and Aslow). The scattering intensity contribution from each mode was determined using eqn (5) and (6).65–67
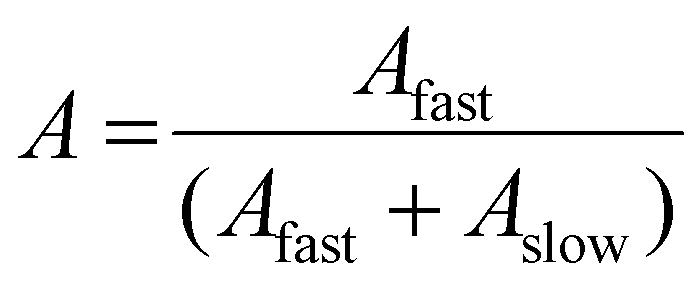 | (5) |
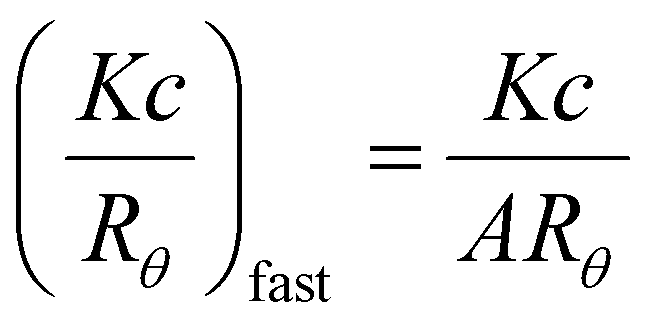 | (6) |
In the event of a data point from one observation angle falling outside of 10% error of Kc/Rθ, the point was excluded from the average in the calculation of Mw, particle. For additional calculations relating to the light scattering data, see ESI.† Polymers 1–10 and 16–17 were analyzed at 20 °C, however because of the lower transition temperature of the pDEGMA-stabilized micelles, polymers 11–15 were analyzed at 10 °C.
Results and discussion
Diblock copolymer synthesis and micelle preparation
A series of diblock copolymers with varying degrees of core hydrophobicity were synthesized by RAFT polymerization for each of the coronas investigated. In each case, the corona-forming block was synthesized first (mCTA1–4) such that each of the diblock copolymers in the series had identical corona lengths and molecular weight distributions (Table 1 and Scheme 2). For mCTA2 and mCTA3, the methylene protons adjacent to the trithiocarbonate group of the ω-end group could be observed in the 1H NMR spectrum, and so it was possible to calculate Mn by NMR spectroscopy (Mn, NMR) by comparing the integrals of the corresponding proton peaks to the integrals of the proton peaks on the polymer side chain. For mCTA4, Mn, NMR was calculated using the integrals of the four protons from the two methylene groups on the α-end group relative to the integrals of the side chain proton peaks. For mCTA1, it was not possible to recognize any peaks from the end group protons in the 1H NMR spectrum so the Mn, NMR was calculated from the conversion of monomer to polymer taken after the polymerization. This number (8600 g mol−1) is in good agreement with triple detection SEC, which calculated an Mn of 8100 g mol−1. Chain extension of the macro chain transfer agents yielded the amphiphilic diblock copolymers (1–17, Scheme 2). The degree of hydrophobicity of each of the copolymers was able to be varied by altering the monomer feed ratios of permanently hydrophilic DMA and hydrophobic nBA. It can be seen in Table 1 and Fig. S4, S7, S10 and S13† that for each of the diblock copolymers there is an increase in the Mn, SEC relative to the respective mCTAs, whilst the dispersity does not increase significantly after the extension. The percentage nBA of each of the core-forming blocks, determined by 1H NMR spectroscopy, was also found to be similar to the initial monomer feeds. As has been discussed previously, the nBA and DMA units in the core are randomly incorporated into the core-forming block as predicted by the reactivity ratios of the two monomers.68 Because of this phenomenon, we expect there to be no phase separation between the incompatible units in the core, and that the core-forming block can be thought of as having a uniform hydrophobicity determined by its composition. Once the four series of diblock copolymers were achieved, each of them were self-assembled into micellar structures using a solvent switch technique from acetone into ice cold water (see Experimental section for more details).Multi-angle light scattering analysis
The resultant particles were analyzed using a combination of static and dynamic light scattering at multiple angles (SLS and DLS, respectively). DLS gave information on the particles sizes, specifically their hydrodynamic radii (RH) in solution, whereas SLS gave the average molecular weights of the particles (Mw, particle) and the radii of the particles’ cores (Rcore). The two radii for each micelle are outlined in Table 1. As can be seen in Fig. S15–S18,† for each particle, their relaxation time determined by DLS showed a q2 dependence indicative of Brownian motion and their partial Zimm plots returned a negligible slope owing to the micelles’ low expected Rg values. Additional calculations were performed on the SLS data to give Nagg (see Experimental section and ESI†). For each of the micellar series, the Nagg and therefore Rcore increased as a function of the percentage composition of nBA in the core-forming block (Fig. 2 and Table 1). This can be understood by the necessity of highly hydrophobic moieties to shield themselves from the aqueous environment, as described by the hydrophobic effect. The increase in both Nagg and Rcore with increasing hydrophobicity results in there being a lower overall number of particles in solution. This is energetically favorable as it leads to a reduction in the total interfacial area between the hydrophobic core and the solvent, and therefore a lower interfacial energy.45 The results from each of the series of micelles demonstrate that increasing the percentage composition of hydrophobic monomers in the core-forming block is a robust method for synthesizing micelles with tunable Nagg, along with other reports in the literature where variation in the polymer's ionization,67 core to corona block length ratio,45 and addition of hydrophobic homopolymer69 yield similar results.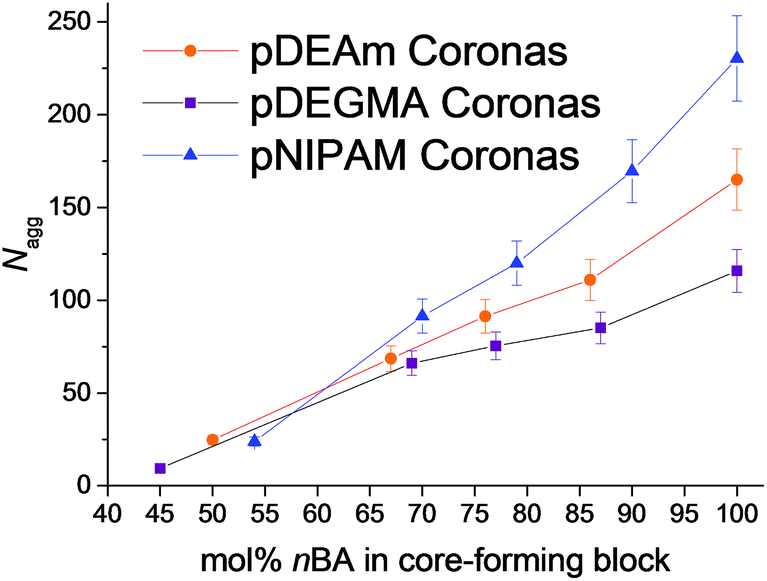 | ||
| Fig. 2 Variation in the particles’ Nagg with the molar fraction of hydrophobic nBA in the core-forming block, as determined by SLS analysis. Micelles with pDEAm (polymers 1–5, orange circles), pNIPAM (polymers 6–10, blue triangles) and pDEGMA coronas (polymers 11–15, purple squares) are shown. Error bars represent 10% error. | ||
Thermoresponsive behavior
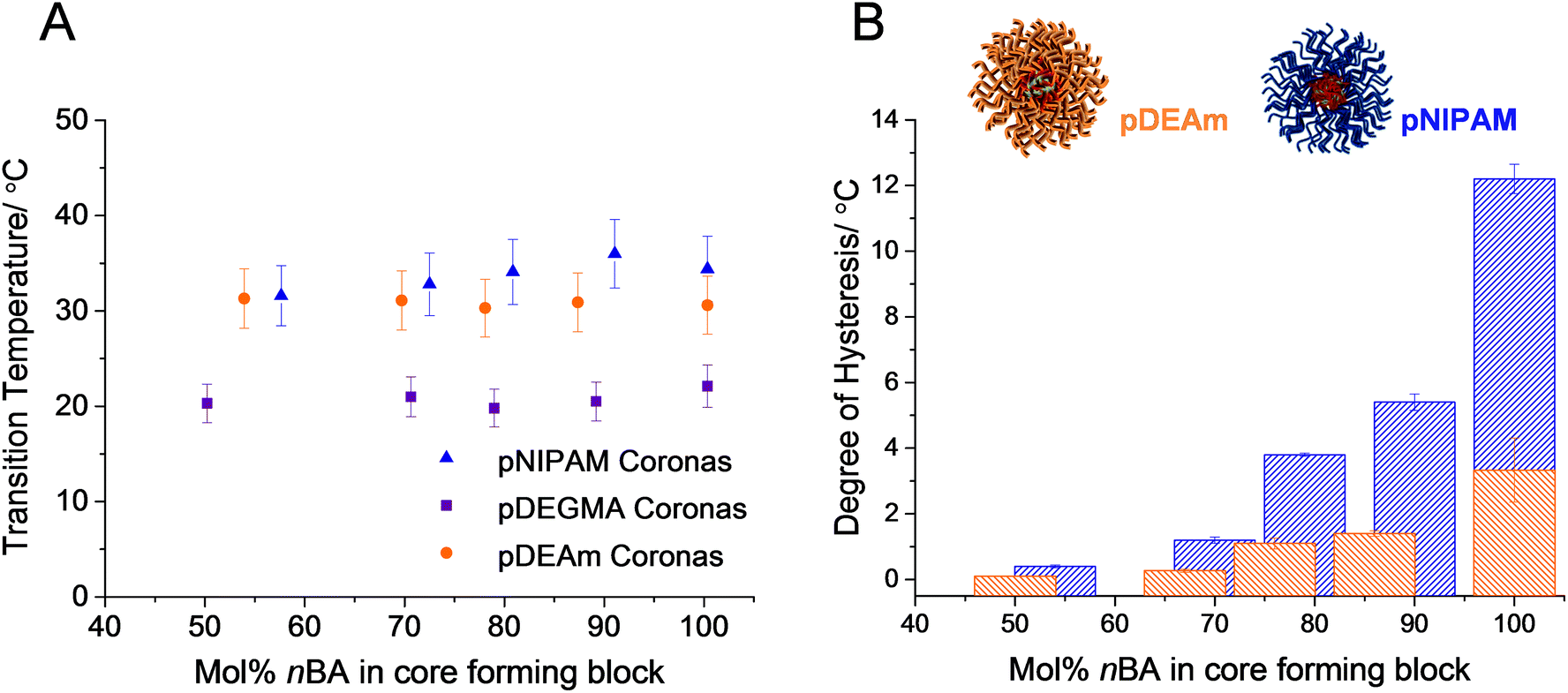 | ||
| Fig. 3 A: Variation of the cloud point transition temperatures of polymers 1–5 (orange circles), 6–10 (blue triangles) and 11–15 (purple squares) with the molar fraction of hydrophobic nBA in the core-forming block, as determined by turbidimetry. Error bars represent 10% error. B: Variation of thermal hysteresis of micelles comprised of pDEAm coronas, which cannot form hydrogen bonds between polymer chains (polymers 1–5, orange bars), and pNIPAM coronas, which can (polymers 6–10, blue bars). Values determined by turbidimetry and plotted as a function of mol% nBA in the core-forming block. Error bars represent the standard deviation across 3 repeats. | ||
| Polymer | Corona | Mol% nBA in core block | N agg | Cloud point/°C | Degree of hysteresis/°C |
|---|---|---|---|---|---|
| a Mean cloud point upon heating the micellar solutions determined using turbidimetry data across from three heating and cooling cycles. b For micelles with irreversible transitions, the cloud point from the first heating cycle is shown. c The degree of hysteresis from solutions exhibiting irreversible phase transitions was not determined as the normalized transmittance did not reach 0.5 in the cooling cycle. d Heated from 10–40 °C. e Heated from 50–95 °C. | |||||
| 11 | mCTA3 | 46 | 9 | 20.3a,d | 0.4 |
| 12 | mCTA3 | 68 | 66 | 21.0a,d | 0.5 |
| 13 | mCTA3 | 77 | 75 | 19.8a,d | 0.1 |
| 14 | mCTA3 | 88 | 85 | 20.5b,d | Irreversiblec |
| 15 | mCTA3 | 100 | 116 | 22.1b,d | Irreversiblec |
| 16 | mCTA4 | 44 | 12 | 61.1a,e | 1.1 |
| 17 | mCTA4 | 100 | 33 | 62.5b,e | Irreversiblec |
It was noted that some of the micelles in each series exhibited a thermal hysteresis, whereby the reversibility of the transition was slow. The degree of hysteresis, defined here as the difference in the cloud points upon heating and upon cooling the micelles, increased as a function of mol% nBA in the core-forming block, for each of the micellar systems 1–10 (Fig. 3B). This was attributed to differences in the hydration of the micellar cores in solution. For instance micelles of polymer 1 have a higher degree of core hydration than micelles of polymer 5, owing to the fact that there is a larger mol% of hydrophilic DMA in its core-forming block. By this virtue, upon heating the micelles, those comprised of polymer 1 form precipitates that are more hydrated than those of polymer 5. In turn, this facilitates the rapid redissolution of micelles with moderately hydrophobic cores, compared with those with a highly hydrophobic character.60 It is therefore important to note that although pDEAm has been successfully designed in the literature to yield homopolymers with an LCST close to pNIPAM but with negligible hysteresis, when incorporated into a micellar structure, the composition of the core-forming block is as important as the design of the corona-forming block when considering hysteresis.
It can be seen in Fig. 3B that this effect is exaggerated when the chains are no longer homopolymers and are instead covalently grafted from a hydrophobic micellar core. For example, the difference in hysteresis between the loosely packed micelles in each series (polymers 1 and 6) is much smaller than the difference in hysteresis between densely packed micelles with more hydrophobic cores (polymers 5 and 10). This is a result of differences in core hydration, as discussed previously. This finding demonstrates that design of polymers with a lower degree of thermal hysteresis on the whole is possible by limiting the strength of the polymer–polymer interactions through changing the hydrogen bonding ability. However, caution should be taken when designing these polymers for a specific application as the introduction of some degree of thermal hysteresis will occur in self-assemblies with very hydrophobic cores, owing to the hydration effects discussed in the previous section.
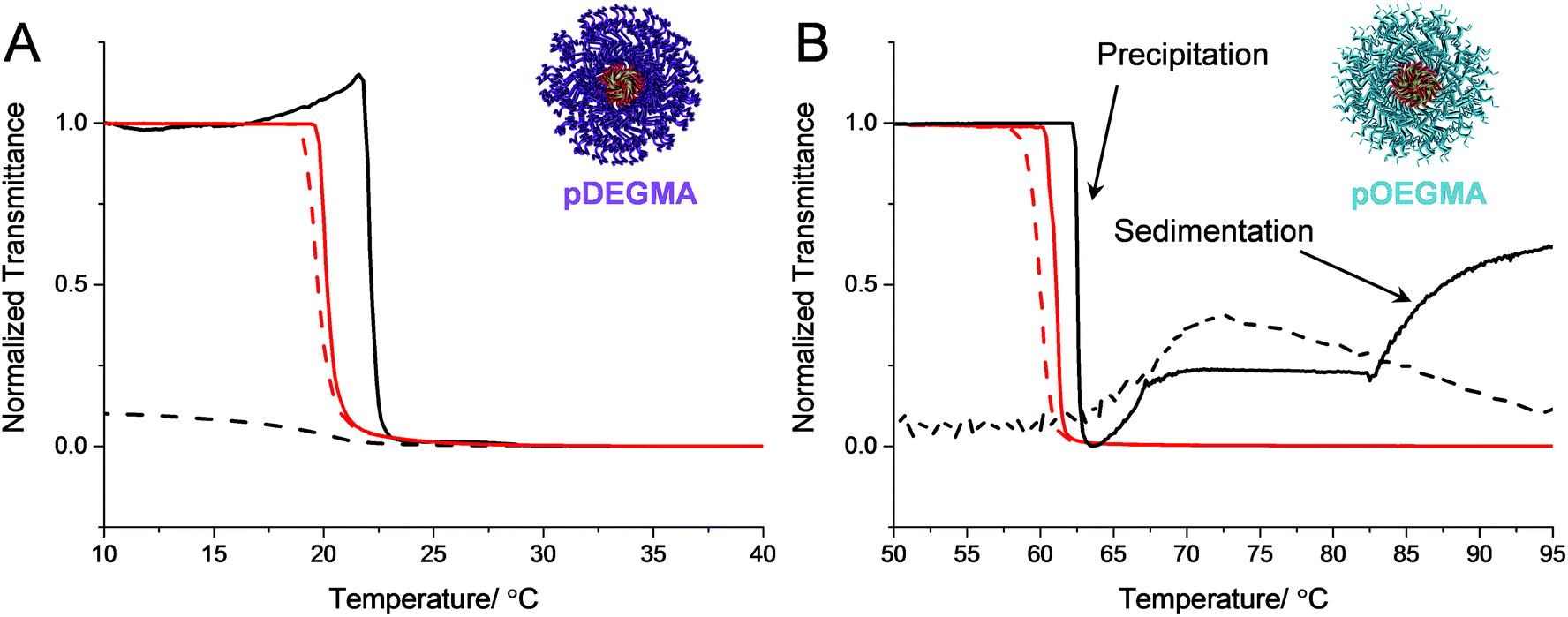 | ||
| Fig. 4 Turbidimetry analyses of micelles with pDEGMA (A) and pOEGMA coronas (B). In each case, solid lines represent heating cycles and dashed lines represent cooling cyles. A shows turbidimetry curves for polymers 11 (red) and 15 (black). B shows turbidimetry curves for polymers 16 (red) and 17 (black). For clarity, some instances of macroscopic precipitation and sedimentation have been labelled and the cooling curve for polymer 17 has been smoothed. | ||
As the cloud point measurements for polymers 16–17 were performed at a much higher temperature than polymers 11–15, it was postulated that hydrolysis of the nBA ester side-chain could have occurred, which may have led to irreversible phase separation. SEC analysis revealed that polymer 17, recovered by lyophilization, had an essentially identical molecular weight distribution before and after three heating cycles indicating that the polymer was unchanged by the turbidimetry analysis (Fig. S14†). It should be noted that upon heating micelles comprised of polymer 17 to just 70 °C, rather than 95 °C in the initial study, a reversible phase transition with a degree of hysteresis of roughly 2 °C was observed (Fig. S23†). However, when cycling the heating and cooling experiments the micelles showed poor reversibility. This finding indicated that the polymer can entangle and potentially rearrange in the precipitated bulk when heated for long periods, which leads to irreversible transitions, however the rearrangement of the chains is hindered when heated for just short periods. The fact that pOEGMA and pDEGMA have Tg values lower than their transition temperature means the chains have the mobility to achieve this in the precipitated form, in contrast to the pDEAm and pNIPAM micelles, whose coronal chains are vitrified and glassy above the transition temperature.
To the best of our knowledge, the reversibility of the thermal transition of block copolymer micelles with pOEGMA or pDEGMA coronas has not been widely reported. However, the results are in concordance with Armes and Zheng et al. who showed that polypyrrole nanoparticles stabilized using a pOEGMA corona exhibited irreversible phase transitions upon either heating the solution, or irradiating the nanoparticles with near infra-red light to induce photothermal aggregation.70 Here, the irreversible transition was attributed to the high Hamaker constant of the polypyrrole cores, indicative of strong van der Waals’ interactions between the cores in the particles’ aggregated state. This may explain to some extent why this behavior is only observed in highly hydrophobic micellar cores. However, the fact that micelles comprised of polymers with linear coronas (1–10) exhibited reversible phase transitions for all of the core compositions investigated indicates that the brush-like structure of pOEGMA and pDEGMA, coupled with their low Tg, plays a role in limiting the reversibility of micelles comprised of polymers 11–17.
This important finding highlights that the micellar tethering of the corona chains leads to behavior that is drastically different from that of unimeric homopolymers. Although poly(ethylene glycol (meth)acrylate) based thermoresponsive polymers have been hailed as tunable smart materials with low thermal hysteresis, this reversible behavior may be limited to non- or loosely assembled micellar structures with low degrees of chain entanglement. Indeed, for certain applications, an irreversible phase transition is actually the desired outcome, for instance when designing polymers to permanently block blood vessels at the site of a tumor, or for sensors or logic gates with a memory of their thermal history. As such, the design and consideration of the behavior of an entire formulation is critical as they typically display behavior very different from their constituent parts.
Conclusions
Well-defined, responsive, amphiphilic block copolymers containing four different thermoresponsive corona blocks were synthesized by RAFT polymerization and assembled into micellar structures in aqueous media. Multi-angle DLS and SLS revealed that the micelles from each series had tunable Nagg and this variation was used to probe the effects of altering the corona chemistry, chain confinement and core hydrophobicity on the thermoresponsive behavior, specifically the degree of hysteresis, of the micelles. Turbidimetry analysis revealed that higher core hydrophobicities led to a higher degree of hysteresis across each micellar series, owing to differences in core hydration. As expected, linear corona chains that could form polymer–polymer hydrogen bonding interactions (pNIPAM) showed a greater hysteresis than those that could not (pDEAm). Finally brush-like coronas (pDEGMA and pOEGMA) showed irreversible phase transitions at high core hydrophobicity and Nagg. It was postulated that this was owing to increased coronal chain entanglements or rearrangement, which prevented micellar redissolution. These results highlight the complexity of hysteresis in thermoresponsive polymer systems, and that responsive self-assembled polymer structures do not necessarily exhibit behavior analogous to the sum of their respective unimer chains. Therefore, it is important to consider the effects of the chemistry and the architecture of individual polymer chains, as well as the structure of the resultant assembled particle when predicting the ultimate phase behavior of a given formulation, and when designing these formulations for a desired application.Acknowledgements
We gratefully acknowledge Mr Robert Keogh and Dr Craig Bell at the University of Warwick for the synthesis of 2-cyano-2-propyl dodecyl trithiocarbonate and 4-cyano-4-(((ethylthio)carbonothioyl)thio) pentanoic acid respectively. Dr Lijang Song at the University of Warwick is also acknowledged for carrying out HRMS analysis. We thank the EPSRC and the ERC (638661 and 615142) for financial support.Notes and references
- J. Akimoto, M. Nakayama and T. Okano, J. Controlled Release, 2014, 193, 2–8 CrossRef CAS PubMed.
- B. P. Bastakoti, S. Guragain, K. Nakashima and Y. Yamauchi, Macromol. Chem. Phys., 2015, 216, 287–291 CrossRef CAS.
- P. Bawa, V. Pillay, Y. E. Choonara and L. C. du Toit, Biomed. Mater., 2009, 4, 022001 CrossRef PubMed.
- W.-F. Lai and H. C. Shum, Nanoscale, 2016, 8, 517–528 RSC.
- B. Louage, Q. Zhang, N. Vanparijs, L. Voorhaar, S. Vande Casteele, Y. Shi, W. E. Hennink, J. Van Bocxlaer, R. Hoogenboom and B. G. De Geest, Biomacromolecules, 2015, 16, 336–350 CrossRef CAS PubMed.
- H. Mohapatra, H. Kim and S. T. Phillips, J. Am. Chem. Soc., 2015, 137, 12498–12501 CrossRef CAS PubMed.
- E. G. Kelley, J. N. L. Albert, M. O. Sullivan and T. H. Epps III, Chem. Soc. Rev., 2013, 42, 7057–7071 RSC.
- D. J. Phillips, I. Prokes, G.-L. Davies and M. I. Gibson, ACS Macro Lett., 2014, 3, 1225–1229 CrossRef CAS.
- T. M. Reineke, ACS Macro Lett., 2016, 5, 14–18 CrossRef CAS.
- Y. Peng, X. Jiang, S. Chen, Q. Wu, J. Shen and W. Wu, Polym. Chem., 2015, 6, 8331–8342 RSC.
- D. J. Phillips, G.-L. Davies and M. I. Gibson, J. Mater. Chem. B, 2015, 3, 270–275 RSC.
- P.-F. Cao, J. D. Mangadlao and R. C. Advincula, Polym. Rev., 2015, 55, 706–733 CrossRef CAS.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- E. Cabane, X. Zhang, K. Langowska, C. Palivan and W. Meier, Biointerphases, 2012, 7, 1–27 CrossRef PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- P. Theato, B. S. Sumerlin, R. K. O'Reilly and T. H. Epps III, Chem. Soc. Rev., 2013, 42, 7055–7056 RSC.
- M. I. Gibson and R. K. O'Reilly, Chem. Soc. Rev., 2013, 42, 7204–7213 RSC.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- J. Seuring and S. Agarwal, Macromol. Rapid Commun., 2012, 33, 1898–1920 CrossRef CAS PubMed.
- J. Akimoto, M. Nakayama, K. Sakai and T. Okano, Biomacromolecules, 2009, 10, 1331–1336 CrossRef CAS PubMed.
- M. A. Ward and T. K. Georgiou, Polymer, 2011, 3, 1215–1242 CAS.
- B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2002, 54, 37–51 CrossRef CAS PubMed.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- J.-F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- R. Hoogenboom, H. M. L. Thijs, M. J. H. C. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760 RSC.
- H. Okamura, T. Mori, K. Minagawa, S. Masuda and M. Tanaka, Polymer, 2002, 43, 3825–3828 CrossRef CAS.
- S. Samanta, D. R. Bogdanowicz, H. H. Lu and J. T. Koberstein, Macromolecules, 2016, 49, 1858–1864 CrossRef CAS.
- N. S. Ieong, M. Redhead, C. Bosquillon, C. Alexander, M. Kelland and R. K. O'Reilly, Macromolecules, 2011, 44, 886–893 CrossRef CAS.
- H. Cheng, L. Shen and C. Wu, Macromolecules, 2006, 39, 2325–2329 CrossRef CAS.
- C. Wu and X. Wang, Phys. Rev. Lett., 1998, 80, 4092–4094 CrossRef CAS.
- Y. Ding, X. Ye and G. Zhang, Macromolecules, 2005, 38, 904–908 CrossRef CAS.
- Y. Lu, K. Zhou, Y. Ding, G. Zhang and C. Wu, Phys. Chem. Chem. Phys., 2010, 12, 3188–3194 RSC.
- V. Aseyev, H. Tenhu and F. M. Winnik, in Self Organized Nanostructures of Amphiphilic Block Copolymers II, ed. H. E. A. Müller and O. Borisov, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 29–89 Search PubMed.
- K. Van Durme, G. Van Assche and B. Van Mele, Macromolecules, 2004, 37, 9596–9605 CrossRef CAS.
- J. Seuring and S. Agarwal, Macromolecules, 2012, 45, 3910–3918 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- E. R. G. Moad and S. Thang, Macromolecules, 1998, 31, 5559 CrossRef.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- N. S. Cameron, M. K. Corbierre and A. Eisenberg, Can. J. Chem., 1999, 77, 1311–1326 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- Q. Yan, R. Zhou, C. Fu, H. Zhang, Y. Yin and J. Yuan, Angew. Chem., Int. Ed., 2011, 50, 4923–4927 CrossRef CAS PubMed.
- R. Tamate, T. Ueki and R. Yoshida, Adv. Mater., 2014, 27, 837–842 CrossRef PubMed.
- F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS PubMed.
- K. E. B. Doncom, C. F. Hansell, P. Theato and R. K. O'Reilly, Polym. Chem., 2012, 3, 3007–3015 RSC.
- A. Feng, C. Zhan, Q. Yan, B. Liu and J. Yuan, Chem. Commun., 2014, 50, 8958–8961 RSC.
- Y. Cai, K. B. Aubrecht and R. B. Grubbs, J. Am. Chem. Soc., 2011, 133, 1058–1065 CrossRef CAS PubMed.
- A. Sundararaman, T. Stephan and R. B. Grubbs, J. Am. Chem. Soc., 2008, 130, 12264–12265 CrossRef CAS PubMed.
- H. A. Zayas, A. Lu, D. Valade, F. Amir, Z. Jia, R. K. O'Reilly and M. J. Monteiro, ACS Macro Lett., 2013, 2, 327–331 CrossRef CAS.
- A. O. Moughton and R. K. O'Reilly, Chem. Commun., 2010, 46, 1091–1093 RSC.
- A. O. Moughton, J. P. Patterson and R. K. O'Reilly, Chem. Commun., 2011, 47, 355–357 RSC.
- K. Wei, L. Su, G. Chen and M. Jiang, Polymer, 2011, 52, 3647–3654 CrossRef CAS.
- X. Wang and L. Li, Polymer, 2016, 88, 123–132 CrossRef CAS.
- W. Wang, C. Gao, Y. Qu, Z. Song and W. Zhang, Macromolecules, 2016, 49, 2772–2781 CrossRef CAS.
- L. D. Blackman, D. B. Wright, M. P. Robin, M. I. Gibson and R. K. O'Reilly, ACS Macro Lett., 2015, 4, 1210–1214 CrossRef CAS.
- I. Idziak, D. Avoce, D. Lessard, D. Gravel and X. X. Zhu, Macromolecules, 1999, 32, 1260–1263 CrossRef CAS.
- B. A. Abel and C. L. McCormick, Macromolecules, 2016, 49, 465–474 CrossRef CAS.
- A. J. Convertine, D. S. W. Benoit, C. L. Duvall, A. S. Hoffman and P. S. Stayton, J. Controlled Release, 2009, 133, 221–229 CrossRef CAS PubMed.
- W. Schärtl, Light Scattering from Polymer Solutions and Nanoparticle Dispersions, Springer, 2007 Search PubMed.
- J. P. Patterson, M. P. Robin, C. Chassenieux, O. Colombani and R. K. O'Reilly, Chem. Soc. Rev., 2014, 43, 2412–2425 RSC.
- J. P. Patterson, E. G. Kelley, R. P. Murphy, A. O. Moughton, M. P. Robin, A. Lu, O. Colombani, C. Chassenieux, D. Cheung, M. O. Sullivan, T. H. Epps and R. K. O'Reilly, Macromolecules, 2013, 46, 6319–6325 CrossRef CAS PubMed.
- D. B. Wright, J. P. Patterson, A. Pitto-Barry, P. Cotanda, C. Chassenieux, O. Colombani and R. K. O'Reilly, Polym. Chem., 2015, 6, 2761–2768 RSC.
- D. Neugebauer and K. Matyjaszewski, Macromolecules, 2003, 36, 2598–2603 CrossRef CAS.
- L. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168–3181 CrossRef CAS.
- K. M. Au, M. Chen, S. P. Armes and N. Zheng, Chem. Commun., 2013, 49, 10525–10527 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of small molecules and polymers, SEC chromatograms of the polymers, DLS, SLS and turbidimetry data for the micelles, a discussion of the chain density of micelles 11–15, additional calculations regarding the core composition of polymers 1–5 and definitions and calculations related to the light scattering data. See DOI: 10.1039/c6py01191h |
| This journal is © The Royal Society of Chemistry 2017 |