
Dehydrogenative lactonization of diols with a platinum-loaded titanium oxide photocatalyst†
Emiko
Wada‡
a,
Akanksha
Tyagi
a,
Akira
Yamamoto
ab and
Hisao
Yoshida
 *ab
*ab
aGraduate School of Human and Environmental Studies, Kyoto University, Yoshida Nihonmatsu-cho, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: yoshida.hisao.2a@kyoto-u.ac.jp
bElemental Strategy Initiative for Catalysts and Batteries (ESICB), Kyoto University, Kyotodaigaku-katsura, Nishikyo-ku, Kyoto, 615-8520, Japan
First published on 10th October 2017
Abstract
A new catalytic route for the lactonization of diols was developed by using a metal-loaded TiO2 photocatalyst. In particular, Pt-loaded rutile TiO2 exhibited a high photocatalytic activity with high selectivity. In addition, it was found that a heterogeneous acid catalyst can accelerate this photocatalytic lactonization.
Lactones, which are cyclic esters, are important chemical intermediates or solvents in organic syntheses.1 A conventional synthesis route for lactones is the intramolecular dehydrative esterification of hydroxycarboxylic acid by a Brønsted acid.2 Recently, many research groups have reported new reaction routes to obtain lactones, such as through the reactions of epoxide or aliphatic alcohols with carbon monoxide,3 and reactions of allenol or butadiene with carbon dioxide.4 Another promising route is the dehydrogenative oxidation of diols, for which many homogeneous catalysts such as Ir- or Ru-complexes have been reported.5,6 A Cu-complex with nitroxyl radicals such as TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) efficiently catalyzes the aerobic oxidative lactonization of diols at room temperature under ambient conditions.7 Although Fujita et al. developed a reusable Ir-complex catalyst,5 homogeneous catalytic reaction systems are usually considered to have difficulties in product separation and reusing the catalyst.
On the other hand, heterogeneous catalysts for lactonization have also been reported. For example, Cu-based catalysts8 and Au-loaded TiO2 catalysts9 were reported for the lactonization of 1,4-butanediol to form γ-butyrolactone in the gas phase. CeO2-based binary oxides were developed for the gaseous phase lactonization of 1,6-hexanediol to ε-caprolactone.10 As for the liquid phase lactonization, the Au-loaded hydrotalcite catalyst was active for the lactonization of 1,4-butanediol with oxygen molecules.11 Recently, Touchy and Shimizu found a Pt-loaded SnO2 catalyst that well promotes the oxidative lactonization of various diols at 453 K under solvent-free conditions.12 In addition, the reaction route with metal oxide catalysts such as tungstic acids13 or ZrO2-supported WO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 with hydrogen peroxide has been studied for the dehydrogenative lactonization of 1,2-benzenedimethanol to form phthalide in high yield (ca. 90%) with high selectivity. Phthalide is a common chemical intermediate for pharmaceutical chemicals and other fine chemicals.13–15 Although phthalide can also be obtained through the hydrogenation of phthalic anhydride by using a heterogeneous catalyst, it requires a high temperature such as 453 K and a high pressure of hydrogen gas.16 Development of new routes for lactonization of various diols is still being studied. As a new methodology, we have examined dehydrogenative lactonization of diols under mild conditions without consuming any other reagents by using a photocatalyst.
14 with hydrogen peroxide has been studied for the dehydrogenative lactonization of 1,2-benzenedimethanol to form phthalide in high yield (ca. 90%) with high selectivity. Phthalide is a common chemical intermediate for pharmaceutical chemicals and other fine chemicals.13–15 Although phthalide can also be obtained through the hydrogenation of phthalic anhydride by using a heterogeneous catalyst, it requires a high temperature such as 453 K and a high pressure of hydrogen gas.16 Development of new routes for lactonization of various diols is still being studied. As a new methodology, we have examined dehydrogenative lactonization of diols under mild conditions without consuming any other reagents by using a photocatalyst.
In the present study, we found that a Pt-loaded rutile TiO2 photocatalyst can promote the lactonization for the dehydrogenative lactonization of 1,2-benzenedimethanol to phthalide (eqn (1)) with high yield and selectivity, which can also be applied to various diols for producing lactones. To the best of our knowledge, this is the first report for the dehydrogenative lactonization of diols with hydrogen evolution by heterogeneous photocatalysis around room temperature. In addition, it was demonstrated that heterogeneous acid catalysts having moderately weak acid sites such as alumina can facilitate the photocatalytic reaction further.
 | (1) |
Three kinds of TiO2 powders supplied from the Catalysis Society of Japan (JRC-TIO-8, anatase, 338 m2 g−1; JRC-TIO-6, rutile, 100 m2 g−1; and JRC-TIO-4, anatase and rutile, 50 m2 g−1) were employed as the photocatalysts, where rutile and anatase are referred to as (R) and (A), respectively. Metal loaded TiO2 (M/TiO2) samples were prepared by a conventional photodeposition method.17,18 The Pt loading amount was 0.1 wt%. The average particle size of the loaded Pt nanoparticles was determined to be 2.1 nm by a CO adsorption method.17 For each catalytic reaction test, 0.1 g of the M/TiO2 sample was used. Before the reaction, the sample was photoirradiated for 20 min by using a xenon lamp (PE300BUV) with an optical long pass filter (λ > 350 nm), where the light intensity measured at 360 nm ± 15 nm in wavelength was 27 mW cm−2. After an argon purge, the reaction mixture, 200 or 400 μmol of diol dissolved in 4 mL of a solvent, was introduced into a quartz reactor (46 cm3). The reaction tests were carried out for 1 h or more under photoirradiation. The products in the gas phase were analyzed by GC-TCD (Shimadzu, GC-8A) and those in the liquid phase were analyzed by GC-MS (Shimadzu, GCMS-QP2020). The product amounts were determined by each calibration curves, although only the amount of 2-(hydroxymethyl)benzaldehyde was determined by using the calibration curve of phthalide. As the amount of hydrogen produced was large, it could not be determined precisely.
Fig. 1 shows the time courses of the product yields in the lactonization of 1,2-benzenedimethanol (1) over the Pt/TiO2 samples consisting of an anatase or rutile TiO2 photocatalyst in an acetonitrile solution. In the initial period of the reaction over the Pt/TIO-8(A) photocatalyst, 2-(hydroxymethyl)benzaldehyde (2) was preferably formed and the yield of phthalide (3) gradually increased (Fig. 1a). Then, phthalide became the major product after 2 h with a decrease of 2-(hydroxymethyl)benzaldehyde yield. The yield of phthalide reached 60% in 3 h. This result indicates that 2-(hydroxymethyl)benzaldehyde is formed as an intermediate, and phthalide is produced by further oxidation. As for the gaseous product, only hydrogen was detected. Therefore, the lactonization of benzenedimethanol to form phthalide proceeds via two-step dehydrogenative oxidation (scheme in Table 1). However, the reaction over the Pt/TIO-8(A) photocatalyst provided a low selectivity to phthalide such as 60% after 3 h, but only 25% after 1 h (Table 1, entry 2). The carbon balance was also low i.e., 60% after 3 h, which means that the formation of other byproducts or decomposition of the substrate and products would also take place.
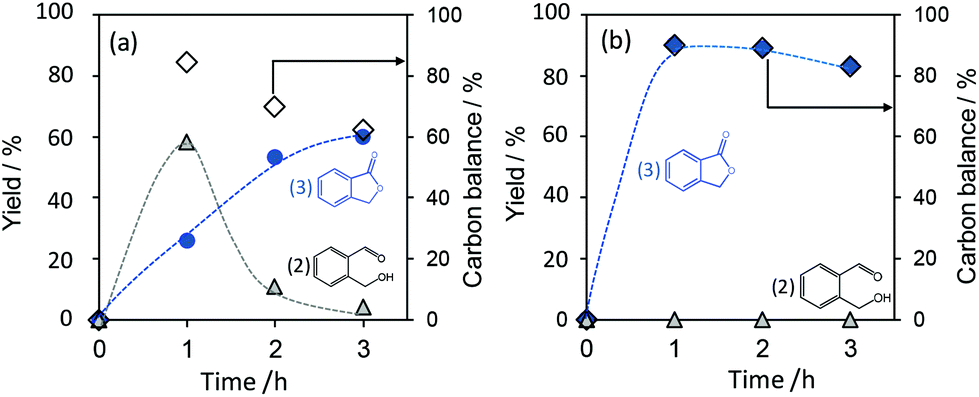 | ||
| Fig. 1 Time courses of product yields in the lactonization of 1,2-benzenedimethanol (1) over the Pt/TIO-8(A) photocatalyst (a) and the Pt/TIO-6(R) photocatalyst (b). Products were 2-(hydroxymethyl)benzaldehyde (2) and phthalide (3). Reaction conditions: 200 μmol of 1,2-benzenedimethanol, 4 mL of acetonitrile, 0.1 g of the Pt/TiO2 photocatalyst. The wavelength of the irradiation light was longer than 350 nm, and the light intensity was 27 mW cm−2 measured at 360 ± 15 nm. | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Crystal phaseb | Specific surface area/m2 g−1 | Yield/μmol | Phthalide, 3 | |||
| Phthalide 3 | Aldehyde 2 | H2c |
Yield (%) | Selectivity (%) | ||||
| a Reaction conditions were the same as those in Fig. 1. b (R) and (A) represent rutile and anatase, respectively. c Hydrogen was actually detected, but the value for the obtained amount would contain a large experimental error. d Not detected. | ||||||||
| 1 | Pt/TIO-6 | (R) | 100 | 180 | n.d.d | 200 | 90 | 90 |
| 2 | Pt/TIO-8 | (A) | 338 | 52 | 110 | 110 | 25 | 25 |
| 3 | Pt/TIO-4 | (A, R) | 50 | 31 | 12 | 90 | 19 | 15 |
| 4 | Pd/TIO-6 | (R) | 100 | 45 | 10 | 45 | 56 | 23 |
| 5 | Rh/TIO-6 | (R) | 100 | 98 | 40 | 65 | 60 | 49 |
| 6 | TIO-6 | (R) | 100 | n.d. | n.d. | n.d. | — | — |

On the other hand, the Pt/TIO-6(R) photocatalyst produced phthalide in the yield of 90% even at initial 1 h with high selectivity i.e., 90% (Fig. 1b and Table 1, entry 1), where the intermediate 2-(hydroxymethyl)benzaldehyde was not detected. That is, the Pt/TIO-6(R) sample consisting of rutile TiO2 showed 3.5 times higher yield of phthalide than the Pt/TIO-8(A) sample consisting of anatase TiO2, although the BET surface area of TIO-6(R) (100 m2 g−1) is smaller than that of TIO-8(A) (338 m2 g−1) (Table 1, entries 1 and 2). The Pt/TIO-4(A, R) photocatalyst consisting of both anatase and rutile phases with a low specific surface area did not afford much phthalide (Table 1, entry 3). These results suggest that a pure rutile phase is suitable for this lactonization.
The results of the photocatalytic reaction tests with the Pt/TiO2 samples, prepared from the calcined TIO-6(R) and TIO-8(A) samples having lower specific surface areas, are plotted in Fig. S1.† The yield of phthalide increased with an increase of the specific surface area, and the slope of the Pt/TIO-6(R) photocatalysts was clearly steeper than that of the Pt/TIO-8(A) photocatalysts. These results suggested that the rutile TiO2 of a large specific surface area would be efficient to produce phthalide in high yield.
As a co-catalyst, Pd and Rh were examined on the rutile TiO2 photocatalyst. Although the Pd/TIO-6(R) sample and the Rh/TIO-6(R) sample also promoted the reaction (Table 1, entries 4 and 5), the phthalide yield was less than that with the Pt/TIO-6(R) sample. In addition, the bare rutile TiO2 sample exhibited no activity (Table 1, entry 6). Thus, it is clear that the precious metal co-catalyst is necessary, and among them Pt drastically improves the photocatalytic activity. The deposited metal nanoparticles would enhance the electron–hole separation as an electron receiver, decrease their recombination, and promote the reduction of protons by the photoexcited electron.
The solvent also affected the activity and selectivity of the photocatalyst (Table S1†). Some solvents such as water, acetone, and THF gave undesired byproducts such as homo-coupling products of solvents or other byproducts from the reaction between 1,2-benzenedimethanol and solvents (Table S1, entries 2–4†). When acetonitrile was used as a solvent, these byproducts were not detected (Table S1, entry 1†). Thus, acetonitrile was chosen as the best solvent among the tested solvents for this lactonization reaction.
To clarify the reason for the different activities between anatase and rutile, some experiments were carried out (Table S2†). First, the adsorption amount of the substrate and the product, i.e., 1,2-benzenedimethanol and phthalide, on the catalyst surface was measured in acetonitrile in the dark. Both the Pt/TIO-6(R) and Pt/TIO-8(A) photocatalysts showed very low and similar values (Table S2, entries 1 and 2†), suggesting that the adsorption properties of these samples cannot explain the difference in the photocatalytic activity. Second, the stability of the product against the successive reaction was examined, where the Pt/TiO2 sample was photoirradiated for 1 h in the presence of 200 μmol of phthalide in the acetonitrile solution. After the photoirradiation, the amount of phthalide was not changed much in the presence of the Pt/TIO-6(R) photocatalyst (Table S2, entry 1†), whereas it decreased much in the presence of the Pt/TIO-8(A) sample and the Pt/TIO-4(A, R) sample (Table S2, entries 2 and 3†). This means that the successive reaction of the desired product hardly takes place on the Pt loaded rutile TiO2 photocatalyst. Therefore, it is one of the reasons why the Pt/TIO-6(R) photocatalyst exhibited a high yield of phthalide with high selectivity. Since the Pt/TIO-8(A) photocatalyst containing anatase TiO2 showed less selectivity even at the initial stage of the reaction test (Fig. 1a), anatase TiO2 would promote not only the successive reaction of the product but also some other reactions.
The present photocatalytic lactonization consists of the two dehydrogenation steps depicted as the scheme in Table 1. The reaction did not proceed in the dark or in the absence of the photocatalyst, indicating that the lactonization proceeds photocatalytically. Fig. S2† shows the pseudo Arrhenius plot for the phthalide formation with the Pt/TIO-6(R) photocatalyst, which was obtained from the temperature-controlled photocatalytic reaction experiments. The apparent activation energy for the formation of phthalide calculated from the plot was 4.6 kJ mol−1, which would be an acceptable value as a typical activation energy for photocatalytic reactions.19 Although it was reported in the literature12 that Pt nanoparticles can function as a metal catalyst in dehydrogenative lactonization, they did not show catalytic performance on a TiO2 support. Also in the present study, the result shows that metal catalysis did not contribute to the reaction rate under photocatalytic conditions. Thus, it was clarified that the rate-determining step of the reaction is not thermally activated catalysis but photocatalysis.
Further, the addition of a heterogeneous acid catalyst to the reaction system was examined (Table S3†). The yield of phthalide increased when the acid catalysts such as Al2O3 or protonated titanate nanotubes (TiNT)20 were introduced to the photocatalytic reaction mixture in the coexistence of the Pt/TIO-6(R) photocatalyst (Table S3, entries 3 and 4†). However, in the presence of TiNT some side reactions were also promoted, indicating that the acid catalyst having moderately strong acid sites20 is not available for this purpose. In contrast, the acid catalyst having weak acid sites such as Al2O3 can selectively accelerate the photocatalytic reaction rate without promoting the side reactions. These results suggest that the weak acid sites can improve the reaction to form phthalide. Separate experiments evidenced that the acid catalysts itself did not promote the reaction without a photocatalyst, confirming that the acid catalysis accelerates at least one step during the photocatalytic reaction, which might be the second cyclization step. Here, it is proposed that the blended catalyst consisting of the Pt/TiO2 photocatalyst and the Al2O3 acid catalyst is efficient for the photocatalytically dehydrogenative lactonization. The optimization of their combination, such as kinds of acid catalysts and the ratio of components, will further improve this reaction system. On the other hand, although TiO2 also has acid sites,21 the yield of phthalide was not increased by the addition of a bare TIO-6(R) sample to the reaction mixture including the Pt/TIO-6(R) sample, but rather decreased (Table S3, entry 2†). As mentioned above, the bare TIO-6(R) sample did not give phthalide under light irradiation (Table 1, entry 6). It is considered that the additional bare TiO2 powder could rather shade the Pt/TIO-6(R) photocatalyst from the light to reduce the photoexcitation of the Pt/TIO-6(R) photocatalyst.
Here, the reaction mechanism is tentatively proposed as shown in Fig. S3.† The photoexcitation of the TiO2 photocatalyst provides an excited electron and a hole. The hydroxyl group of 1,2-benzenedimethanol is oxidized by the hole to form the radical species and proton. Then, the hydrogen radical elimination follows to form the intermediate, 2-(hydroxymethyl)benzaldehyde. On the other hand, the excited electron reduces the proton to form a hydrogen radical, which react with another hydrogen radical to form molecular hydrogen. By the second photoexcitation of the TiO2 photocatalyst, further oxidation of the hydroxyl group of the 2-(hydroxymethyl)benzaldehyde takes place, and the formed oxygen radical moiety attacks the carbonyl carbon to form the phthalide and hydrogen radical. This step might be assisted by the surface acid sites when an additional acid catalyst is used for the reaction. Molecular hydrogen is produced again from the hydrogen radicals. Thus, it is suggested that this photocatalytic lactonization is a two-photon process.
In order to expand the scope of the current reaction system, various diols were examined for lactonization with the Pt/TiO2 photocatalyst (Table 2). It was found that all the examined diols were transformed into the corresponding lactones. The Pt/TIO-6(R) photocatalyst promoted all these reactions faster than the Pt/TIO-8(A) photocatalyst (Table 2, entries 1–8). The yields of lactones in these cases were lower than that in the lactonization of 1,2-benzenedimethanol mentioned above. The lactonization of cis-1,2-cyclohexanedimethanol (4) over the Pt/TIO-6(R) photocatalyst gave dominantly the corresponding lactone compounds 6 and 7 ((3aS,7aR)-hexahydroisobenzofuran-1(3H)-one and (3aR,7aS)-hexahydroisobenzofuran-1(3H)-one, total 83 μmol, 42% yield) as the stereoretentive products, and the other lactone compounds 8 and 9 ((3aR,7aR)-hexahydroisobenzofuran-1(3H)-one and (3aS,7aS)-hexahydroisobenzofuran-1(3H)-one, total 8.0 μmol, 4.0% yield) as the minor products of the stereoinversion reaction (Table 2, entry 1). The Pt/TIO-8(A) photocatalyst gave the same products with low yields (Table 2, entry 2). The Pt/TIO-6(R) photocatalyst gave a higher stereoretention ratio, r = 11, than the Pt/TIO-8(A) photocatalyst, r = 5.2 (Table 2, entries 1 and 2). It was experimentally confirmed that the stereoinversion products, 8 and 9, were formed by a photocatalytic reaction22 and not by a catalytic keto–enol tautomerization23 or a Norrish I type photochemical reaction24 from the major products, 7 and 6, respectively, under the present conditions since the r values for the Pt/TIO-6 and Pt/TIO-8 samples did not change even after the stirring of the resulting reaction mixture in the dark with the catalyst or the photoirradiation of its filtrate without the catalyst. It should be noted that the activity of the photocatalytic epimerization depends on the properties of the TiO2 photocatalyst. The same tendency to retain the stereochemical structure was observed in the reaction of trans-1,2-cyclohexanedimethanol (5) (Table 2, entries 3 and 4), but the total yields of lactone compounds decreased in comparison with the lactonization of cis-1,2-cyclohexanedimethanol (4), which would be related to the structural torsion of the lactones 8 and 9. The lactonization of 1,4-butanediol (10) also proceeded as well as that of other diols (Table 2, entries 5 and 6), but the reaction of 1,5-pentanediol (12) hardly proceeded (Table 2, entries 7 and 8). In these cases, the high flexibility of the carbon chain structure would decrease the chance to form a cyclic structure, which decreases the yield of lactones.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Reactant | Product | Yield (%) | r | ||
| Total | 6 + 7 | 8 + 9 | |||||
| a Reaction conditions: 200 μmol of diol, 4 mL of acetonitrile, the other conditions were the same as those in Fig. 1. b The calibration curves of the starting materials were applied to determine the yields of lactones. c r = (sum of yields of products via stereoretention reactions)/(sum of yields of products via stereoinversion reactions). d Not detected. | |||||||
| 1 | Pt/TIO-6 | 4 | 6–9 | 46 | 42 | 4.0 | 11 |
| 2 | Pt/TIO-8 | 20 | 17 | 3.3 | 5.2 | ||
| 3 | Pt/TIO-6 | 5 | 6–9 | 24 | 3.1 | 21 | 6.8 |
| 4 | Pt/TIO-8 | 10 | 1.9 | 8.1 | 4.5 | ||
| 5 | Pt/TIO-6 | 10 | 11 | 20 | |||
| 6 | Pt/TIO-8 | 5.0 | |||||
| 7 | Pt/TIO-6 | 12 | 13 | 2.5 | |||
| 8 | Pt/TIO-8 | n.d.d | |||||

In conclusion, a new synthesis route for the lactones from diols with Pt/TiO2 photocatalysts was established. The photocatalytic lactonization proceeded via a two-step dehydrogenation process as a two-photon process. The Pt loaded rutile TiO2 photocatalyst exhibited a higher yield of lactones with higher selectivity than the Pt loaded anatase TiO2. The blended catalyst consisting of the Pt/TiO2 photocatalyst and the Al2O3 acid catalyst is more efficient for the photocatalytic dehydrogenative lactonization than the Pt/TiO2 photocatalyst alone.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. Wada would like to thank JSPS for the doctoral scholarship. A. Tyagi would like to thank JICA for providing the scholarship under the IIT Hyderabad-JICA Friendship project.Notes and references
- P. M. Dewick, Essentials of Organic Chemistry: For Students of Pharmacy, Medicinal Chemistry and Biological Chemistry, Wiley, 2006 Search PubMed.
- T. Mukaiyama, Angew. Chem., Int. Ed. Engl., 1979, 18, 707 CrossRef.
- S. Kreimerman, I. Ryu, S. Minakata and M. Komatsu, Org. Lett., 2000, 2, 289 CrossRef CAS; E. Yoneda, S. W. Zhang, D. Y. Zhou, K. Onitsuka and S. Takahashi, J. Org. Chem., 2003, 68, 8571 CrossRef PubMed; J. W. Kramer, E. B. Lobkovsky and G. W. Coates, Org. Lett., 2006, 8, 3709 CrossRef PubMed.
- Y. Inoue, Y. Sasaki and H. Hashimoto, Bull. Chem. Soc. Jpn., 1978, 51, 2375 CrossRef CAS; B. Pierre, D. Matt and D. Nobel, J. Am. Chem. Soc., 1988, 110, 3207 CrossRef; S. Li, B. Miao, W. Yuan and S. Ma, Org. Lett., 2013, 15, 977 CrossRef PubMed.
- K. Fujita, W. Ito and R. Yamaguchi, ChemCatChem, 2014, 6, 109 CrossRef CAS.
- T. Suzuki, K. Morita, M. Tsuchida and K. Hiroi, Org. Lett., 2002, 4, 2361 CrossRef CAS PubMed; R. Kawahara, K. Fujita and R. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 3643 CrossRef PubMed; J. Zhao and J. F. Hartwig, Organometallics, 2005, 24, 2441 CrossRef.
- J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901 CrossRef CAS PubMed; X. Xie and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 3767 CrossRef PubMed.
- N. Ichikawa, S. Sato, R. Takahashi, T. Sodesawa and K. Inui, J. Mol. Catal. A: Chem., 2004, 212, 197 CrossRef CAS; T. Hu, H. Yin, R. Zhang, H. Wu, T. Jiang and Y. Wada, Catal. Commun., 2007, 8, 193 CrossRef.
- J. Huang, W. L. Dai, H. Li and K. Fan, J. Catal., 2007, 252, 69 CrossRef CAS.
- T. Akashi, S. Sato, R. Takahashi, T. Sodesawa and K. Inui, Catal. Commun., 2003, 4, 411 CrossRef CAS.
- T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Green Chem., 2009, 11, 793 RSC.
- A. S. Touchy and K. Shimizu, RSC Adv., 2015, 5, 29072 RSC.
- Q. Zhu, X. Chu, Z. Zhang, W. L. Dai and K. Fan, Green Chem., 2010, 12, 205 RSC.
- Q. Zhu, X. Chu, Z. Zhang, W. L. Dai and K. Fan, Appl. Catal., A, 2012, 435, 141 CrossRef.
- Z. Zhang, Q. Zhu, J. Ding, X. Liu and W. L. Dai, Appl. Catal., A, 2014, 482, 171 CrossRef CAS.
- Y. X. Liu, T. F. Xing, Z. J. Wei, X. N. Li and W. Yan, Catal. Commun., 2009, 10, 2023 CrossRef CAS.
- E. Wada, T. Takeuchi, Y. Fujimura, A. Tyagi, T. Kato and H. Yoshida, Catal. Sci. Technol., 2017, 7, 2457 CAS.
- H. Yoshida, Y. Fujimura, H. Yuzawa, J. Kumagai and T. Yoshida, Chem. Commun., 2013, 49, 3793 RSC; A. Tyagi, T. Matsumoto, T. Kato and H. Yoshida, Catal. Sci. Technol., 2016, 6, 4577 Search PubMed; A. Tyagi, A. Yamamoto, T. Kato and H. Yoshida, Catal. Sci. Technol., 2017, 7, 2616 Search PubMed.
- R. J. Davis, J. L. Gainer, G. O'Neal and I. W. Wu, Water Environ. Res., 1994, 66, 50 CrossRef CAS; A. V. Vorontsov, E. N. Savinov, G. B. Barannik, V. N. Froitsky and V. N. Parmon, Catal. Today, 1997, 39, 207 CrossRef; T. Hisatomi, K. Miyazaki, K. Takanabe, K. Maeda, J. Kubota, Y. Sakata and K. Domen, Chem. Phys. Lett., 2010, 486, 144 CrossRef; K. Shimura, K. Maeda and H. Yoshida, J. Phys. Chem. C, 2011, 115, 9041 Search PubMed.
- M. Kitano, K. Nakajima, J. N. Kondo and M. Hara, J. Am. Chem. Soc., 2010, 132, 6622 CrossRef CAS PubMed; M. Kitano, E. Wada, K. Nakajima, S. Hayashi, S. Miyazaki, H. Kobayashi and M. Hara, Chem. Mater., 2013, 25, 385 CrossRef; E. Wada, M. Kitano, K. Nakajima and M. Hara, J. Mater. Chem. A, 2013, 1, 12768 Search PubMed.
- K. Tanabe, H. Htattori, T. Sumiyoshi, K. Tamaru and T. Kondo, J. Catal., 1978, 53, 1 CrossRef CAS; K. Hadjiivanov, Appl. Surf. Sci., 1998, 135, 331 CrossRef.
- The detailed mechanism for the production of the minor products has not been clarified. Further study is necessary to clarify it.
- H. Seto, E. Nomura, S. Fujioka, H. Koshino, T. Suenaga and S. Yoshida, Biosci., Biotechnol., Biochem., 1999, 63, 361 CrossRef CAS PubMed; G. G. Tsantali, J. Dimtsas, C. A. Tsoleridis and I. M. Takakis, Eur. J. Org. Chem., 2007, 258 CrossRef.
- N. C. Yang and R. H.-K. Chen, J. Am. Chem. Soc., 1971, 93, 530 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7pp00258k |
| ‡ Research Fellow of the Japan Society for the Promotion of Science. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2017 |