
Titania hollow spheres modified with tungstophosphoric acid with enhanced visible light absorption for the photodegradation of 4-chlorophenol†
M. Á.
Orellana
a,
L.
Osiglio
a,
P. M.
Arnal
b and
L. R.
Pizzio
*a
aCentro de Investigación y Desarrollo en Ciencias Aplicadas “Dr. Jorge J. Ronco” (CINDECA), Facultad de Ciencias Exactas, UNLP-CCT La Plata, CONICET, 47 No 257, 1900-La Plata, Argentina. E-mail: lrpizzio@química.unlp.edu.ar
bCETMIC, Centro de Tecnología de Recursos Minerales y Cerámica, CIC-CONICET Gonnet, La Plata, Argentina
First published on 3rd October 2016
Abstract
Titania hollow spheres were synthesized using silica nanospheres as the template. The core was removed using NaOH solution. They were subsequently impregnated with tungstophosphoric acid (TPA) solutions and annealed at two different temperatures (100 and 500 °C). These materials were characterized by several physicochemical techniques (XRD, BET, SEM, DRS, FT-IR, FT-Raman and 31P MAS-NMR). The 31P MAS-NMR and FT-IR characterization showed that the main species present in the samples was the [PW12O40]3− anion, which was partially transformed into the [P2W21O71]6− anion during the synthesis and drying step. 31P MAS-NMR, and FT-Raman characterization revealed the evidence of a strong interaction between the Keggin anion of TPA and TiO2 surfaces, possibly due to the formation of surface heteropolyacid-TiO2 complexes. The DRS results showed that the absorption threshold onset continuously shifted to the visible region with increased TPA concentration and calcination at 500 °C. The enhanced visible light absorption could be related to the formation of a surface complex TPA Keggin anion–TiO2. The catalytic activity of the materials in the photodegradation of 4-chlorophenol under UV and visible light irradiation increased when the TPA content and the calcination temperature of the samples were raised.
1. Introduction
The potential of heterogeneous photocatalysis over TiO2 to destroy a wide range of waterborne pollutants and microorganisms has aroused enormous interest.1,2 The preparation of titania-based photocatalysts with high catalytic performance has also attracted much attention. TiO2 has been the most widely used because it is easily available, inexpensive, nontoxic, and it shows relatively high chemical stability.3It is claimed that titania performance in the photodegradation of contaminants contained in wastes depends on its crystal structure, crystallinity, surface area, surface hydroxyl density, band gap energy, and morphology,4 among other factors. The low surface area and the fast recombination of the photoinduced electrons and holes are the main effects that can lead to a low photocatalytic activity. Additionally, titania absorbs only UV light, which is not abundant on earth's surface, representing an enormous limitation in solar-driven photocatalytic processes.
In the last few years, the synthesis of titania hollow microspheres (@TiO2) has attracted much attention due to their large surface area, low density, and highly efficient light-harvesting properties.5–9
They were successfully synthesized using pentahydrated copper sulfate,6 latex7 or silica9,10 spheres as templates. The synthesis using a template free solvothermal method has been also performed.8
Additionally, shell and core–shell particles (@TiO2 and SiO2@TiO2, respectively) may be less harmful to biological systems than TiO2 nanoparticles.10
Tungstophosphoric and tungstosilicic acid (TPA and TSA, respectively) were employed as effective homogeneous photocatalysts in the degradation of organic pollutants in water. They have also been used to modify TiO2 in order to reduce the charge recombination and enhance visible light absorption.3
The photocatalytic degradation of 4-chlorophenol (4-CP) has been investigated by many research groups and has become a standard reaction for evaluating various experimental parameters in photocatalysis.11–13
We present here, for the first time, the preparation and characterization of titania hollow microspheres modified with TPA. The influence of some preparation conditions on the catalytic activity in 4-CP photodegradation was studied.
2. Experimental section
2.1. Catalysts’ synthesis
Nano-spherical particles with an amorphous, solid core of SiO2 and a porous, polycrystalline shell of TiO2 were formed in a three-step procedure.10 The first step yielded silica spheres, the second step, silica spheres covered with a layer of amorphous titanium oxide (SiO2@TiO2), and in the last step titanium oxide crystallized in a shell after a mild acidic chemical treatment. Finally, the silica core was removed using a NaOH solution, yielding titania hollow spheres (@TiO2). The modification @TiO2 with tungstophosphoric acid (H3PW12O40) was realized by wet impregnation. The amount of TPA in the solution was fixed in order to obtain TPA concentrations of 10, 20 and 30% by weight in the final material. The system was kept at room temperature until dry. The solids were thermally treated at 100, and 500 °C for 2 h, and were named @TiO2TPAXXT100 and @TiO2TPAXXT500, respectively, where XX was the TPA concentration. The TPA content in the @TiO2TPAXX samples was estimated as the difference between the W amount contained in the tungstophosphoric acid water solution originally used for the impregnation and the amount of W that remained in the beaker after removing the dried samples. Finally, in order to remove the TPA weakly bound to the titania surface, the materials were brought into contact with water (typically 1 g in 1 l) for 24 h, filtered, and dried at 100 °C. The amount of W in the water solutions obtained after the ground solids were extracted was determined by atomic absorption spectrometry using a Varian AA Model 240 spectro-photometer. The calibration curve method was used with standards prepared in the laboratory. The analyses were carried out at a wavelength of 254.9 nm, bandwidth of 0.3 nm, lamp current of 15 mA, phototube amplification of 800 V, and burner height of 4 mm, and using acetylene–nitrous oxide flame (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14).
:14).
2.2. Catalyst characterization
The specific surface area of the solids was determined from N2 adsorption–desorption isotherms at liquid-nitrogen temperature. They were obtained using Micromeritics ASAP 2020 equipment. The samples were previously degassed at 100 °C for 2 h. Powder XRD patterns were obtained on Philips PW-1732 with a built-in recorder, using Cu Kα radiation, a nickel filter, 20 mA and 40 kV in the high voltage source, and a scanning angle between 5 and 60° of 2θ at a scanning rate of 1° per minute. FT-IR spectra of the supports and catalysts were obtained in the 400–4000 cm−1 wavenumber range using a Bruker IFS 66 FT-IR spectrometer and pellets in KBr. Fourier transform Raman spectra were recorded on a Raman Horiba Jobin-Yvon T 64000 instrument with an Ar+ laser source of 488 nm wavelength in a macroscopic configuration. The 31P MAS-NMR spectra were recorded using the CP/MAS 1H–31P technique with Bruker Avance II equipment. A sample holder of 4 mm diameter and 10 mm height was employed, using 5 μs pulses, and a repetition time of 4 s, working at a frequency of 121.496 MHz for 31P at room temperature. The spin rate was 8 kHz and several hundred pulse responses were collected. Phosphoric acid 85% was employed as an external reference. DRS spectra of samples were recorded using a UV-visible Lambda 35, Perkin Elmer spectrophotometer, to which a diffuse reflectance chamber with an integrating sphere of 50 mm diameter and internal Spectralon coating is attached. The range covered was 200–800 nm. The secondary electron micrographs (SEM) and the energy dispersive X-ray (EDX) patterns of the samples were obtained using Phillips 505 Model scanning electron microscopy equipment and an EDAX 9100 analyser at a working potential of 15 kV and graphite-supported samples metallized with gold.
Transmission electron microscopy (TEM) was performed on a JEOL 100 CXII microscope, working at 100 kV and at a magnification of 80000×. The samples were crushed in an agate mortar, ultrasonically dispersed in isobutanol, and deposited on a carbon-coated copper grid.
2.3. Catalytic test
The catalytic activity of the materials was evaluated in the photodegradation of 4-chlorophenol (4-CP) in water, at 25 °C. The tests were carried out employing 5 UV black light lamps Philips TLD 18 W (emission spectra: 330–400 nm and UV intensity between 300 and 400 nm: 38 W m−2) or 5 blue fluorescent lamps Philips TLD-18 W (emission spectra: 400–500 nm with UV intensity: 0.1 W m−2 and global intensity between 290 and 1100 nm: 60 W m−2). UV and global intensities were monitored with a Kipp & Zonen (CM3) power meter (Omni instruments Ltd, Dundee, UK). The catalyst was maintained in the suspension by stirring, and air was continuously bubbled. Previously, the 4-CP solution (100 ml, 1.5 10−4 mol l−1) containing 50 mg of the catalyst was magnetically stirred in the absence of light for 60 min to ensure that the adsorption–desorption equilibrium of 4-CP on the surface of the materials is attained. The pH of the experiments was 6.9. During the course of the experiments, samples were periodically withdrawn, filtered using a Millipore syringe adapter (porosity, 0.45 μm) and then analyzed. The variation of the 4-CP concentration as a function of the reaction time was determined by using a UV-visible LAMBDA 35 Perkin Elmer double-beam spectrophotometer, measuring the absorbance at 225 nm. The concentration of the released chloride ions was measured by using a selective Cl− electrode (pHoenix CLO1508) with an ion meter (Consort P903). The percentage of 4-CP mineralized was determined using the Total Organic Carbon, Method 10129 DR/4000 (HACH).3. Results and discussion
According to the SEM micrograph (Fig. 1a), the silica cores (mean diameter in the range of 650–750 nm) present a smooth surface and are well dispersed, without the formation of silica aggregates. On the other hand, the SiO2@TiO2 sample (Fig. 1b) displays a rough surface and diameters in the range of 800–900 nm. | ||
| Fig. 1 SEM micrographs of SiO2 (a), SiO2@TiO2 (b) and @TiO2 (c). | ||
For the @TiO2 material (Fig. 1c), the thickness of the titania layer estimated by TEM is around 70 nm. No significant changes in the morphology of the samples after the impregnation with TPA and the thermal treatment were detected by SEM and TEM. Analyses performed by EDX show that the silica core was successfully removed using a NaOH solution. The EDX measurements (see the ESI†) revealed the presence of the Si Kα (at 1.74 keV) and Ti Kα (4.51 keV) signals in SiO2@TiO2 and @TiO2 samples. For the SiO2@TiO2 material, the ratio between the Si Kα and the Ti Kα signal area (ASik/ATik) was 1.43. This value was significantly lower for the @TiO2 sample (ASik/ATik = 0.22) due to the almost complete silica core elimination.
The N2 adsorption–desorption isotherms of @TiO2TPAXXT100 and @TiO2TPAXXT500 samples can be classified as type IV, characteristic of mesoporous materials. For all the materials the hysteresis loop (H2 type) was very small.
The specific surface area (SBET) of the @TiO2TPAXXT100 and @TiO2TPAXXT500 samples determined from N2 adsorption–desorption isotherms using the Brunauer–Emmett–Teller (BET) method, together with the average pore diameter (Dp), are shown in Table 1. The specific surface area of the micropores (Smicro) was estimated by the t-plot method. As can be observed, all the samples are mesoporous materials with a Dp higher than 9.0 nm. The SBET decreased and the mean pore radius increased when both the TPA content and the temperature of the thermal treatment were raised.
| Samples | S BET (m2 g−1) | D p BJH (nm) | ||
|---|---|---|---|---|
| T100 | T500 | T100 | T500 | |
| @TiO2 | 77 | 46 | 9.9 | 15.2 |
| @TiO2TPA10 | 65 | 26 | 10.1 | 18.9 |
| @TiO2TPA20 | 34 | 23 | 13.8 | 19.0 |
| @TiO2TPA30 | 16 | 13 | 19.1 | 21.3 |
Taking into account the values of Smicro, less than 10% of the total surface area resulted from a microporous structure. The XRD pattern of @TiO2 calcined at 100 °C exhibited only a wide peak at 2θ = 25.2° (101) assigned to the anatase phase. The XRD pattern of @TiO2 treated at 500 °C also presents three new wide peaks at 37.9 (004), 47.8 (200) and 54.3° 2θ, due to the increment of the crystallinity (see the ESI†).
On the other hand, for the @TiO2TPA10T100 sample, no diffraction lines attributed to crystalline TPA or its decomposition products were present in the XRD pattern. However, a new set of peaks ascribed to the H3PW12O40·6H2O, (one of the more common tungstophosphoric acid hydrates) is present in the XRD pattern of the @TiO2TPA20T100 and @TiO2TPA30T100 samples.14
The patterns of the @TiO2TPA10T500, @TiO2TPA20T500 and @TiO2TPA30T500 samples only showed the characteristic peaks of the anatase phase; no diffraction lines corresponding to TPA were observed indicating that the species present are highly dispersed as a noncrystalline form or as crystallites low enough to be detected by this technique.
The leaching experiments revealed that TPA was firmly attached to the titania surface (less than 3% of the TPA content was eliminated). In the case of the @TiO2TPA10T100, @TiO2TPA10T500, and @TiO2TPA20T500 samples, the loss of TPA was practically negligible (0.90%, 0.50%, and 0.65% respectively). For all the TPA contents, the loss was lower when the samples were treated at 500 °C (for example, 2.90% and 1.75% for @TiO2TPA30T100 and @TiO2TPA30500, respectively).
It is worthy of mention that the peaks in the XRD patterns of the @TiO2TPA20T100 and @TiO2TPA30T100 samples assigned to the H3PW12O40·6H2O disappear as a result of leaching. We can suggest that the loosely bound TPA eliminated during the leaching experiments was mainly the H3PW12O40·6H2O hexahydrate. In this case, a hydrogen bond type interaction between oxygen atoms [W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and W–O–W] of TPA species and hydroxyl groups [Ti–OH] of the titania matrix could take place.
O and W–O–W] of TPA species and hydroxyl groups [Ti–OH] of the titania matrix could take place.
However, when TPA is firmly attached to the titania surface the interaction can be of the electrostatic type due to transfer of protons to Ti–OH according to:15
| Ti–OH + H3PW12O40 → [Ti–OH2+] [H2PW12O40−]. |
Taking into account Lefebvre's report,16 we can assume that the elimination of water from [Ti–OH2+] during the thermal treatment should involve direct bonding of the polyanion to titania [Ti–O–W].
Li et al. have suggested that TPA could interact with titania surfaces through Ti–O–W covalent bonds formed by the interaction of WO or W–O–W bonds with Ti–OH species.17,18
31P MAS-NMR spectra corresponding to the @TiO2TPAXXT100 (Fig. 2) and @TiO2TPAXXT500 materials show two signals; the more intense at −14.2 ppm attributed to the [PW12O40]3− anion and the other at −12.4 ppm, to the dimer [P2W21O71]6−.19 The transformation of the [PW12O40]3− anion into the dimer is due to the limited stability range of the Keggin anion in solution. At pH 1.5–2, it is reversibly and quickly transformed according to the scheme: [PW12O40]3− ⇔ [P2W21O71]6− ⇔ [PW11O39]7− when the hydroxyl concentration is increased.20
 | ||
| Fig. 2 31P MAS-NMR spectra of @TiO2TPAXXT100 samples. | ||
The downfield shift and slight increase of the line width observed, compared to the bulk compound (−15.3 ppm), can be attributed to the interaction between the anion and the titania matrix21 such as [Ti–OH2+][H3−XPW12O40]X−.
The 31P MAS-NMR spectra of the @TiO2TPAXXT500 samples show that the ratio between the intensity of the signal attributed to the [PW12O40]3− Keggin anion and the one assigned to the dimer [P2W21O71]6− increases with the increment of the calcination temperature. We have reported3 that a noticeable transformation of the [PW12O40]3− anion into the dimeric one takes place when the thermal treatment is performed at temperatures higher than 400 °C. Nevertheless, the line assigned to the [PW12O40]3− anion is the most intense in all the samples. Additionally, the calcination at 500 °C produces a slight increment of the line at −14.2 ppm that could be related with the direct bonding of the polyanion to titania [Ti–O–W].
The FT-IR spectrum of TPA (Fig. 3) shows bands at 1081, 982, 888, 793, 595, and 524 cm−1, in agreement with those reported in the literature for the H3PW12O40 acid.22 They are assigned to the stretching vibrations P–Oa, W–Od, W–Ob–W, and W–Oc–W, and to the bending vibration Oa–P–Oa, respectively. In the FT-IR spectra of the @TiO2TPAXXT100 samples, the P–Oa, W–Od, and W–Ob–W stretching vibrations of the [PW12O40]3− anion are overlapped to the TiO2 wide band. The main FT-IR bands assigned to the stretching vibrations P–Oa (1095 and 1085 cm−1), W–Od (972 cm−1), and W–O–W (890 and 790 cm−1) of the dimer [P2W21O71]6− appear at wavenumber values similar to those of the [PW12O40]3− Keggin anion.23 For the @TiO2TPA20T100 and @TiO2TPA30T100 samples the band at 1080 cm−1 displays a shoulder (at 1094 cm−1) assignable to the presence of the [P2W21O71]6− anion.
 | ||
| Fig. 3 FT-IR spectra of @TiO2, TPA, and @TiO2TPAXXT100 samples. | ||
From the FT-IR and 31P MAS-NMR results, we can state that the main species present in the samples is the [PW12O40]3− Keggin anion. However, it was partially transformed into the [P2W21O71]6− dimer during the synthesis and drying steps.
The main Raman vibration bands of bulk TPA appear at 1010, 990 and 930 cm−1.24 They present an important broadening when TPA is supported on @TiO2. Additionally, in the spectra of the @TiO2TPAXXT500 samples, the main Raman scattering peak of TiO2 at 141 cm−1 exhibits a strong blue shift and broadening. In agreement with Li et al.25 we consider that the shift and broadening of the Raman vibration modes of TPA and TiO2 could be associated with a strong interaction between the TiO2 network and TPA.
The UV-vis-DRS spectrum of bulk TPA presented two absorption bands in the range of 200–450 nm, assigned to the charge transfer from bridging or terminal O 2p to W 5d (W–O–W and W–Od, respectively).3 On the other hand, the @TiO2 sample displayed intense absorption in the range of 200–390 nm, corresponding to charge transfer from the valence band (O 2p) to the conduction band (Ti 3d).26
The UV-vis-DRS spectra of the @TiO2TPAXXT100 and @TiO2TPAXXT500 samples (Fig. 4a and b respectively) show an absorption threshold onset that continuously shifts to the visible region with the increment of the TPA content. The DRS results also reveal that the visible light absorption increases when the calcination temperature is raised up to 500 °C. So, the TPA–TiO2 surface complex could be responsible for the visible light absorption and spectroscopic changes observed.
 | ||
| Fig. 4 DRS spectra of @TiO2, and @TiO2TPAXX samples calcined at 100 (a) and 500 °C (b). | ||
We evaluated the activity of the synthesized materials in the photocatalytic degradation of 4-chlorophenol at the pH of the suspension obtained by addition of the catalyst to 4-CP water solution. The initial and the final pH of the solutions were in the range of 6.9–5.7.
The point of zero charge (pHPZC) of the @TiO2TPAXXT100 samples (estimated using the mass titration method proposed by Noh and Schwarz27) decreases in the following order: @TiO2TPA10T100 (5.4) > @TiO2TPA20T100 (4.9) > @TiO2TPA30T100 (4.3) due to the incorporation of TPA. In the case of the samples treated at 500 °C, the behaviour is the same: @TiO2TPA10T500 (5.2) > @TiO2TPA20T500 (4.1) > @TiO2TPA30T500 (2.7). However, the values are slightly lower than those of the @TiO2TPAXXT100 samples due to the elimination of water from [Ti–OH2+] during the thermal treatment. So, under our experimental conditions, the surface of the @TiO2TPAXX samples was mostly negatively charged. As a result, 4-CP is not significantly adsorbed on the catalyst surface.
Experiments made without the catalyst (under UV-A and visible irradiation), showed negligible 4-CP degradation after 240 min of irradiation.
The variation of the 4-CP concentration as a function of time using the @TiO2TPAXXT100 and @TiO2TPAXXT500 catalysts is shown in Fig. 5.
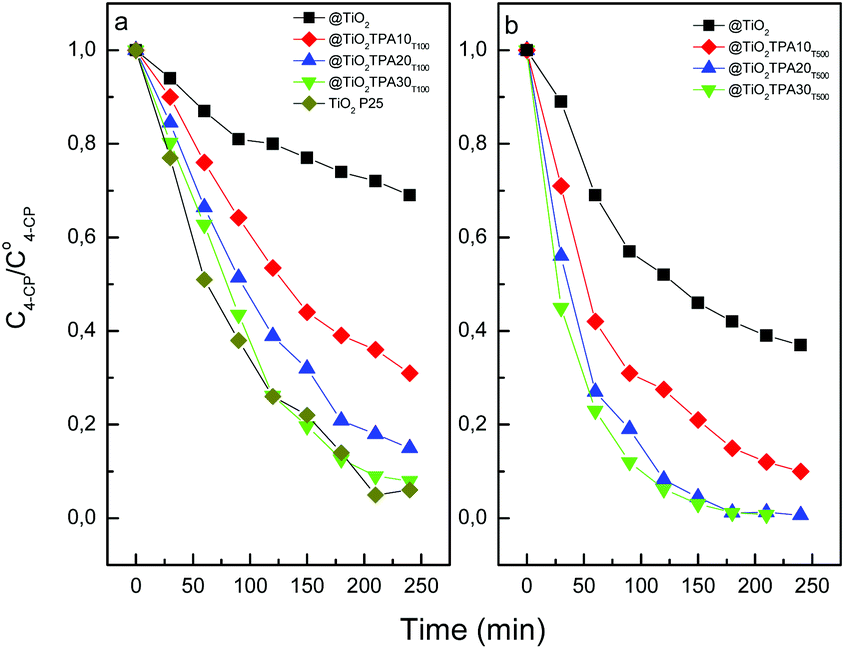 | ||
| Fig. 5 Photocatalytic degradation of 4-CP as a function of the time (under UV-A irradiation) for @TiO2, and @TiO2TPAXX samples treated at 100 °C (a), 500 °C (b), and TiO2 Evonik P25. | ||
The reduction in the 4-CP concentration was rather low (31%) for the titania hollow spheres treated at 100 °C after 240 min of irradiation. However, 4-CP degradation significantly increases in parallel with the increment of tungstophosphoric acid: @TiO2TPA10T100 (69%) < @TiO2TPA20T100 (85%) < @TiO2TPA30T100 (92%). This behavior is due to the fact that TPA on the TiO2 surface acts as an effective trap of photoinduced electrons. Electrons can be easily transferred from the TiO2 conduction band to TPA Keggin anions on the surface, decreasing the fast e−/h+ recombination28 and leaving more valence band holes (h+VB) available to react with water molecules to yield ˙OH radicals.
Additionally, the increment of the catalytic activity could be due to the direct participation of TPA in the degradation of the organic substrate.3 TPA absorb strongly in the UV region of the light spectrum (λ < 400 nm). This absorption corresponds to the ligand-to-metal charge-transfer band and it can generate strongly oxidizing excited state TPA*. They can perform the oxidation of organic substrates directly via charge transfer or indirectly through the production of ˙OH reactive species that participates in the degradation of the organic substrate.29,30 After that, the corresponding reduced TPA− are usually reoxidized to their original oxidation state by an electron acceptor such as dioxygen.
The degradation profile of 4-CP using TiO2 Evonik P25 (commonly used as reference) was very similar to that of the @TiO2TPA30T100 sample.
Fig. 5b shows that the 4-CP amount degraded after 240 min of UV irradiation increased when the calcination temperature was raised up to 500 °C. For any TPA content considered, we observed that the 4-CP degraded amount increases with the increment of the calcination temperature (i.e., @TiO2TPA10T500 (90%) > @TiO2TPA10T100 (69%)). This can be due to an increase in the crystallinity of the @TiO2, resulting in a decrease in the number of defects that act as recombination centers for photogenerated electrons and holes.
For the @TiO2TPAXXT100 and @TiO2TPAXXT500 samples, we observed that the ratio between the amount of degraded 4-CP and the amount of released chloride ions as a function of the irradiation time remained practically equal to 1 during the course of the degradation. These results are in accordance with literature reports that evidence that the first step during the photocatalytic degradation of 4-CP is the Cl–aryl bond cleavage.
On the other hand, we found that the 4-CP amount degraded under blue-light irradiation (at 240 min), using titania hollow spheres modified with TPA as the catalyst, varied in the following way: @TiO2TPA10T500 (20%) < @TiO2TPA20T500 (27%) ≅ @TiO2TPA30T500 (29%). On the contrary, the catalytic activities of Evonik P25 and @TiO2 were negligible. These results show the beneficial effect of TPA on the photocatalytic activity also under visible light radiation.
Under visible light irradiation the TPA anchored to the titania surfaces through Ti–O–W covalent bonds (responsible for the visible light absorption) would be excited yielding a TPA*–TiO2 complex. The TPA*–TiO2 complex can inject a photoexcited electron to TiO2 CB.31–33 Electron injection from TPA in the excited state (TPA*) to the CB of TiO2 was clearly observed using the two-color two-laser flash-photolysis technique.33
The electron injected to TiO2 CB can react with molecular oxygen yielding a ˙O2− radical. It is well known that in aqueous media this radical can undergo disproportion reactions producing mainly hydrogen peroxide.34 H2O2 can also be reduced by CB electrons producing ˙OH radicals that are also able to oxidize organic pollutants.
Nevertheless, the generation of hydrogen peroxide by means of disproportion of ˙O2− radicals is very low in irradiated TiO2 surfaces,35,36 which should account for the low degradation of 4-CP.
It is worthy of mention that the @TiO2TPA samples can be easily separated from the resulting suspension by decantation. On the contrary, Evonik P25 can be recovered only by filtration using Millipore filters. Taking into account the obtained results, @TiO2TPA30T500 was chosen to test its reusability. To this end, after each photocatalytic experiment, the catalyst was easily separated from the resulting suspension by decantation, washed with distilled water, dried at 70 °C and reused four times (under UV-A irradiation).
The percentage of 4-CP degraded decreased slightly during the first and the second reuse (95% and 91% respectively), and then kept constant. The decrease could be assigned to the TPA solubilization (less than 1% of the TPA content), as was established by atomic absorption spectrometry. On the other hand, the mineralization degree was slightly lower than the amount of degraded 4-CP (78% and 70% for the first and the second reuse respectively) as a result of the formation of organic intermediates such as benzoquinone and hydroquinone.37
The activity of the @TiO2TPA solids in the degradation of 4-chlorophenol aqueous solutions under UV and visible light irradiation increases with the TPA content and the thermal treatment temperature. The sample containing 30% TPA and annealed at 500 °C showed the highest photocatalytic activity. Furthermore, the reused catalysts showed only a slight decrease in the degradation and mineralization degree, thus being promissory materials to aid in the photocatalytic treatment of wastewater containing chlorinated phenols.
4. Conclusions
Titania hollow spheres were synthesized by the sol–gel method using silica nanospheres as the template. The direct modification of @TiO2 with tungstophosphoric acid enables obtaining visible-light-absorbing materials. This fact could be due to the generation of surface complexes between TPA anions and TiO2 through the formation of Ti–O–W bonds. Due to the formation of these bonds the main Raman scattering of TPA and TiO2 displayed a strong blue shift and broadening.The FT-IR and 31P MAS-NMR results indicated that the main species present in the samples was the [PW12O40]3− anion, which was partially, transformed into the [P2W21O71]6− dimeric anion during the synthesis and thermal treatment steps.
@TiO2TPA materials exhibit higher photocatalytic activity than @TiO2 under UV-A irradiation, since the TPA acts as a sink of conduction band photo-induced electrons decreasing the e−/h+ recombination leaving available more valence band holes to either directly oxidize malachite green molecules or produce ˙OH radicals.
The visible light photocatalytic activity of these materials in the 4-CP degradation could be explained by the fact that the TPA–TiO2 complex may be excited by visible light absorption and injection of an electron into the TiO2 conduction band, inducing the indirect formation of ˙OH radicals.
Finally, the direct modification of @TiO2 with TPA is a good method to obtain catalysts with photocatalytic activity in the 4-chlorophenol degradation under UV-A and visible light irradiation.
Acknowledgements
Authors thank the financial support from CONICET (PIP 628) and Universidad Nacional de La Plata (UNLP) (Project number X638) and the technical assistance of M. Theiller (SEM/EDX analysis) and P. Fetsis (BET).References
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- T. Ochiai and A. Fujishima, J. Photochem. Photobiol., C, 2012, 13, 247–262 CrossRef CAS.
- M. N. Blanco and L. R. Pizzio, Appl. Catal., A, 2011, 405, 69–78 CrossRef CAS.
- K. B. Kyoko, S. Kazuhiro, K. Hitoshi, O. Kiyomi and A. Hironori, Appl. Catal., A, 1997, 165, 391–409 CrossRef.
- J. G. Yu, W. Liu and H. G. Yu, Cryst. Growth Des., 2008, 8, 930 CAS.
- C. Jia, Y. Cao and P. Yang, Funct. Mater. Lett., 2013, 6, 1350025–1350026 CrossRef.
- C. Liu, H. Yin, L. Shi, A. Wang, Y. Feng, L. Shen, Z. Wu, G. Wu and T. Jiang, J. Nanosci. Nanotechnol., 2014, 14, 7072–7078 CrossRef CAS PubMed.
- L. Yan, X. He, Y. Wang, J. Li and D. Wang, J. Mater. Sci.: Mater. Electron., 2016, 27, 4068–4073 CrossRef CAS.
- L. Li, S. Bai, W. Yin, S. Li, Y. Zhang and Z. Li, Int. J. Hydrogen Energy, 2016, 41, 1627–1634 CrossRef CAS.
- A. L. Di Virgilio, I. Maisuls, F. Kleitz and P. M. Arnal, J. Colloid Interface Sci., 2013, 394, 147–156 CrossRef CAS PubMed.
- C. Castañeda, F. Tzompantzi, R. Gómez and H. Rojas, J. Chem. Technol. Biotechnol., 2016, 91, 2170–2178 CrossRef.
- M. Wang, G. Fang, P. Liu, D. Zhou, C. Ma, D. Zhang and J. Zhan, Appl. Catal., B, 2016, 188, 113–122 CrossRef CAS.
- Y. Yao, L. Jiao, N. Yu, J. Zhu and X. Chen, Russ. J. Electrochem., 2016, 52, 348–354 CrossRef CAS.
- J. B. Mioc, R. Z. Dimitrijevi, M. Davidovic, Z. P. Nedic, M. M. Mitrovic and P. H. Colomban, J. Mater. Sci., 1994, 29, 3705–3718 CrossRef.
- V. M. Fuchs, E. L. Soto, M. N. Blanco and L. R. Pizzio, J. Colloid Interface Sci., 2008, 327, 403–411 CrossRef CAS PubMed.
- F. Lefebvre, J. Chem. Soc., Chem. Commun., 1992, 10, 756–757 RSC.
- K. Li, Y. Guo, F. Ma, H. Li, L. Chen and Y. Guo, Catal. Commun., 2010, 11, 839–843 CrossRef CAS.
- K. Li, X. Yang, Y. Guo, F. Ma, H. Li, L. Chen and Y. Guo, Appl. Catal., B, 2010, 99, 364–375 CrossRef CAS.
- M. T. Pope, Heteropoly and Isopolyoxometalates, Springer-Verlag, Heidelberg, 1983, p. 58 Search PubMed.
- R. Massart, R. Contant, J. Fruchart, J. Ciabrini and M. Fournier, Inorg. Chem., 1977, 16, 2916 CrossRef CAS.
- V. M. Mastikhin, S. M. Kulikov, A. V. Nosov, I. V. Kozhevnikov, I. L. Mudrakovsky and M. N. Timofeeva, J. Mol. Catal. A: Chem., 1990, 60, 65–70 CrossRef CAS.
- C. Rocchiccioli-Deltcheff, R. Thouvenot and R. Franck, Spectrochim. Acta, Part A, 1976, 32, 587–597 CrossRef.
- R. Contant, Can. J. Chem., 1978, 65, 568–573 CrossRef.
- I. Holclajtner-Antunovic, D. Bajuk-Bogdanovic, A. Popa and S. Uskokovic-Markovic, Inorg. Chim. Acta, 2012, 383, 26–32 CrossRef CAS.
- J. Li, W. Kang, X. Yang, X. Yu, L. Xu, Y. Guo, H. Fang and S. Zhang, Desalination, 2010, 255, 107–116 CrossRef CAS.
- J. T. Yates, Surf. Sci., 2009, 603, 1605–1612 CrossRef CAS.
- J. S. Noh and J. A. Schwarz, J. Colloid Interface Sci., 1988, 130, 157–164 CrossRef.
- H. Park and W. Choi, J. Phys. Chem. B, 2003, 107, 3885 CrossRef CAS.
- R. R. Ozer and J. L. Ferry, J. Phys. Chem. B, 2000, 104, 9444 CrossRef CAS.
- S. Antonaraki, T. M. Triantis, E. Papaconstantinou and A. Hiskia, Catal. Today, 2010, 151, 119–124 CrossRef CAS.
- R. Sivakumar, J. Thomas and M. Yoon, J. Photochem. Photobiol., C, 2012, 13, 277–298 CrossRef CAS.
- J. A. Rengifo-Herrera, M. N. Blanco and L. R. Pizzio, Mater. Res. Bull., 2014, 49, 618–624 CrossRef CAS.
- T. Tachikawa, S. Tojo, M. Fujitsutka and T. Majima, Chem. – Eur. J., 2006, 12, 3124–3131 CrossRef CAS PubMed.
- D. T. Sawyer and J. S. Valentine, Chem. Res., 1981, 14, 393–400 CrossRef CAS.
- Y. Nosaka, Y. Yamashita and H. Fukuyama, J. Phys. Chem. B, 1997, 101, 5822–5827 CrossRef CAS.
- H. Sakai, R. Baba, K. Hashimoto, A. Fujishima and A. Heller, J. Phys. Chem. B, 1995, 99, 11896–11900 CrossRef CAS.
- A. Mylonas and E. Papaconstantinou, J. Photochem. Photobiol., A, 1996, 94, 77–82 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6pp00175k |
| This journal is © The Royal Society of Chemistry and Owner Societies 2017 |