
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regioselective and enantiospecific synthesis of the HSP co-inducer arimoclomol from chiral glycidyl derivatives†
Benjamin N.
Atkinson
 ,
Hannah L.
Woodward
,
James
Sipthorp
and
Paul V.
Fish
*
,
Hannah L.
Woodward
,
James
Sipthorp
and
Paul V.
Fish
*
Alzheimer's Research UK UCL Drug Discovery Institute, The Cruciform Building, University College London, Gower Street, London, WC1E 6BT, UK. E-mail: p.fish@ucl.ac.uk
First published on 21st November 2017
Abstract
A new efficient chiral synthesis of enantiopure arimoclomol (2) is reported from (R)-(−)-glycidyl nosylate (11) with complete retention of chiral integrity. Off-target pharmacology of arimoclomol (2) was evaluated against a representative set of drug targets and showed modest binding to a few kinases. Pharmacokinetic data was generated in vivo in mouse and showed a low brain![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :plasma ratio. These studies will be helpful towards a better understanding of the PK-PD relationship of 2 in disease models.
:plasma ratio. These studies will be helpful towards a better understanding of the PK-PD relationship of 2 in disease models.
Introduction
Defects in proteostasis are a common feature of various neurodegenerative diseases.1,2 Cells have a dedicated protein folding and quality control apparatus to protect against misfolding and aggregation of proteins, including the heat shock response (HSR) and the unfolded protein response (UPR). In the HSR, cell stress leads to the dissociation of heat shock proteins 90 (HSP90) and 70 (HSP70) from heat shock factor 1 (HSF1), allowing the HSPs to subsequently bind to misfolded proteins. Unbound monomeric transcription factor HSF1 forms trimers and translocates from the cytosol to the nucleus where it binds to heat shock elements on DNA, inducing transcription of HSPs.3 The HSR stabilises and refolds misfolded proteins, preventing aggregation and subsequent cellular toxicity in several models of protein aggregation induced cell death.4The small molecules bimoclomol (1) and arimoclomol (2) (Fig. 1) have been reported to be HSP co-inducers.5–9 When added to cells in the absence of cell stress, 1 and 2 do not elicit a chaperone response. In contrast, in the presence of cell stress (e.g. heat shock, HSP90 inhibition or protein aggregates), 1 and 2 potentiate the induction of HSPs and help protect cells from damage. This apparent activity-dependence makes them an attractive means of modulating this ubiquitous cellular pathway for potential therapeutic benefit.
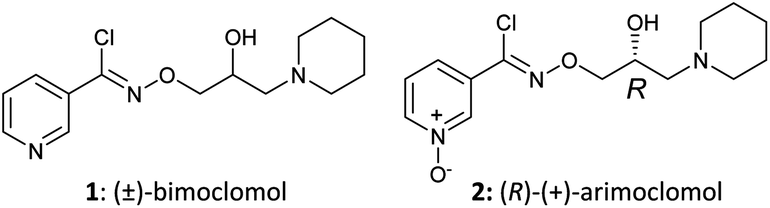 | ||
| Fig. 1 Structures of (±)-bimoclomol (1) and (R)-(+)-arimoclomol (2). | ||
Arimoclomol (2) has progressed to human clinical studies for evaluation as a potential treatment for SOD1 positive familial amyotrophic lateral sclerosis (ALS) (NCT00706147).10 This study was a phase II randomized, double-blind, placebo-controlled trial that included 36 patients with rapidly progressive ALS due to mutations in the SOD1 gene. Patients were treated with 2 at a dose of 200 mg three times per day for up to 12 months. The primary outcome was safety and tolerability; secondary outcome was efficacy as measured in terms of survival and functional decline.11 There has also been multiple reports demonstrating the efficacy of 1 and 2 in in vitro and in vivo preclinical models of neurodegeneration,5 retinal degeneration,6 sphingolipidoses,7 inclusion body myositis8 and other conditions.9 However, the molecular target(s) of 1 and 2 in these models has not yet been determined.
Herein, we disclose a new regioselective and enantiospecific synthesis of the HSP co-inducers 1 and 2 from chiral glycidyl derivatives. This new method is suitable for the synthesis of analogues to establish structure–activity-relationships (SAR) required to direct the creation of chemical probe reagents suitable for target pulldown experiments in relevant cellular models of disease. In addition, we report pharmacology and pharmacokinetic studies which will be helpful in gaining a better understanding of the pharmacokinetic-pharmacodynamic (PK-PD) relationship of 2 in disease models.
Results and discussion
Chemistry
Our first synthesis of 2 was undertaken by reproduction of the published patent route (Scheme 1).12 Addition of NH2OH·HCl to 3-cyanopyridine-N-oxide (3) gave the amidoxime 4 which was then coupled with 2-hydroxy-4-azoniaspiro[3.5]nonane chloride (6) (derived from (±)-epichlorohydrin (5) and piperidine) to yield the O-alkylated amidoxime 7. Diazotisation of 7 followed by in situ chlorination with HCl gave racemic arimoclomol (8) in moderate yield over the 2 steps. Initially, resolution of (±)-8 using (−)-dibenzoyl-L-tartaric acid, followed by conversion to the preferred citrate salt, furnished 2 as the desired (R)-enantiomer with modest efficiency.12 Separation of (±)-8 by preparative supercritical fluid chromatography (SFC) gave (R)-(+)-arimoclomol (2) (er, R:S, 98:2) and (S)-(−)-arimoclomol (9) (er, R:S, 1:>99). The enantiomers were assigned based on comparison with an authentic sample of (R)-(+)-arimoclomol citrate (er, R:S, 99:1).13
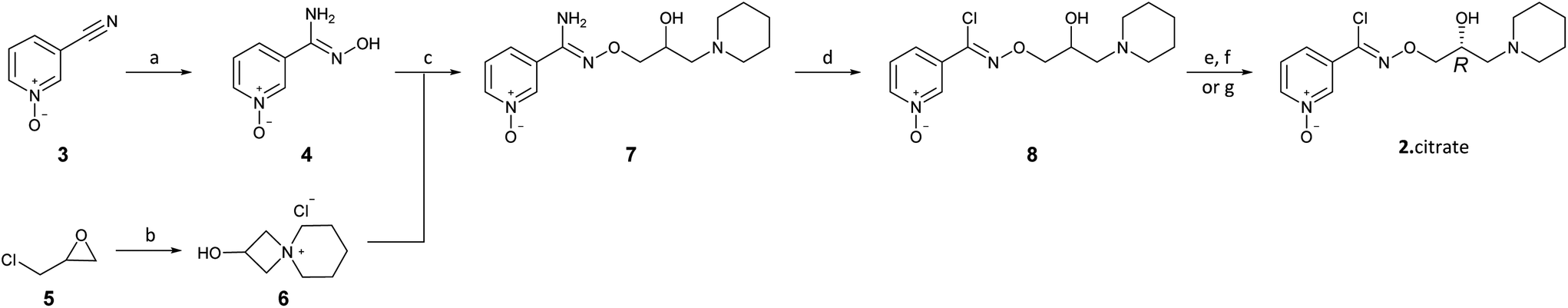 | ||
| Scheme 1 Synthesis of arimoclomol (2) by reproduction of the published patent route. Reagents and conditions: (a) NH2OH·HCl (1.2 equiv.), NaHCO3 (1.2 equiv.), H2O, rt, 18 h 91%; (b) piperidine (0.9 equiv.), MeOH, 65 °C, quant.; (c) 6, NaOH (1.3 equiv.), EtOH, H2O, 70 °C, 18 h; (d) NaNO2 (1.3 equiv.), conc. HCl, H2O, −5 °C, 2.5 h 51% over 2 steps; (e) (−)-dibenzoyl-L-tartaric acid, EtOH then NaOH, CH2Cl2; (f) citric acid (1.0 equiv.), acetone; (g) supercritical fluid chromatography. | ||
However, this linear synthetic approach, and necessity for preparative SFC to produce enantiopure compounds, would not allow for the direct preparation of homochiral analogues.14 Hence, our aim was to develop a robust, efficient and enantioselective route to novel analogues of 1 and 2, which, combined with simple late stage modifications, would allow facile exploration of the SAR.
Retrosynthetic analysis of 2 shows a disconnection into a central chiral glycidyl synthon which could be sequentially functionalised to introduce the aldoxime and amine components (Scheme 2). The stereochemical integrity at C2 would be retained by sequential and specific reaction at C1 followed by C3 of the glycidyl synthon (Scheme 2). Two complementary methods would support the development of SAR at the right-hand side (RHS) amine (path a) and left-hand side (LHS) pyridine (path b) groups from advanced intermediates by the double application of a suitable glycidyl synthon.
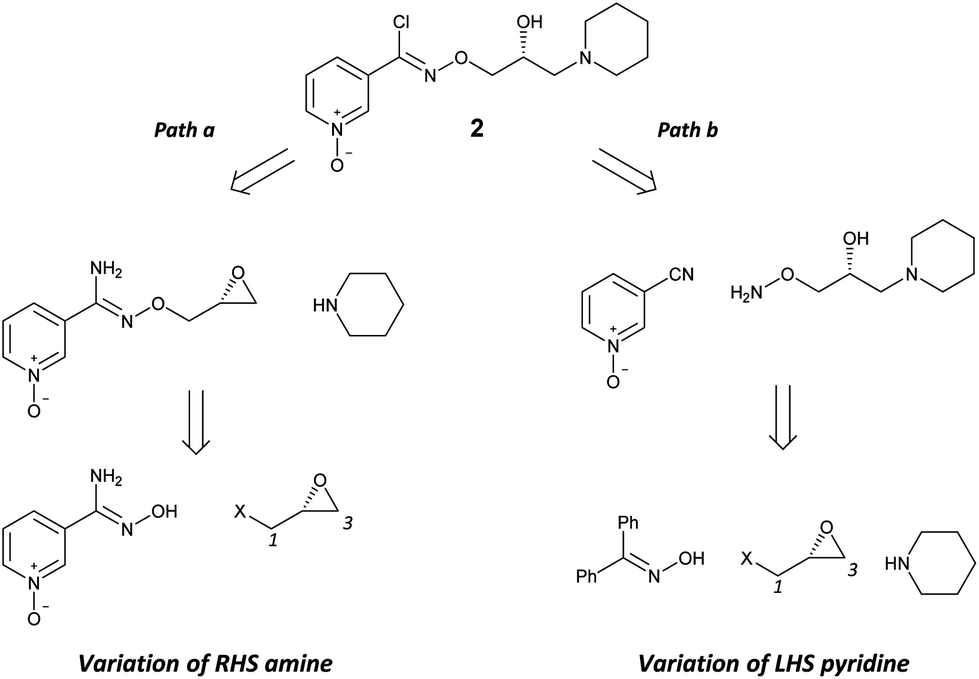 | ||
| Scheme 2 Retrosynthetic analysis of 2 towards a central chiral glycidyl synthon. | ||
Our initial experiments with (R)-(−)-epichlorohydrin (10) (ee > 97.5%)15 as a glycidyl synthon were flawed as reactions with amidoxime 4 gave poor yields of desired products and were accompanied by lack of discrimination between C1 and C3. In contrast, the use of (R)-(−)-glycidyl nosylate (11) (ee > 97.5%)15 was specific for the C1 reaction with the sodium alkoxide of 4 as the first nucleophile to yield epoxide intermediate 13, followed by C3 reaction with piperidine as the second nucleophile, to afford 12 in 71% yield (Scheme 3) (Scheme 2, path a). The regiospecific and sequential reactions of 11 with alkoxide and then amine nucleophiles is in accord with the precedent of Sharpless who developed this methodology with phenols and applied it to the preparation of β-blocker (2S)-(−)-penbutolol.16 The reaction was operationally simple and was performed as a one-pot procedure by the three-step sequential addition of NaH, 11 and then piperidine to 4 in DMF. This reaction is highly atom-efficient as 20/21 heavy atoms of 2 are installed from readily available starting materials.
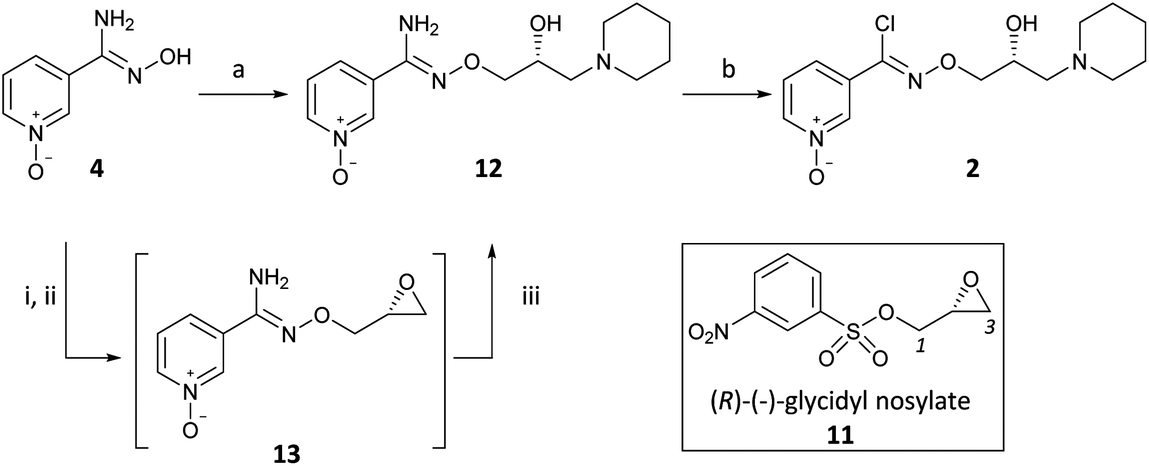 | ||
| Scheme 3 Arimoclomol (2) synthesis via chiral glycidyl nosylate synthon. Reagents and conditions: (a) (i) NaH (60% wt), DMF, 0 °C, 0.5 h; (ii) (R)-(−)-glycidyl nosylate (11) (1.06 equiv.), rt, 2 h; (iii) piperidine (1.1 equiv.), 80 °C for 4 h then rt for 18 h, 71%; (b) NaNO2 (1.3 equiv.), conc. HCl, H2O, −5 °C, 2.5 h, 73%. | ||
As before, diazotisation of 12 followed by in situ chlorination with HCl gave (R)-(2) with subsequent salt formation giving (R)-(2) citrate (Scheme 3).17 The absolute configuration of 2 was confirmed as the (R)-enantiomer by chiral SFC with an enantiomeric ratio (R:S) of >99:1 showing retention of chiral integrity of C2 throughout.13
This short sequence was also performed employing (Z)-pyridine-3-amidoxime as the amidoxime starting material giving (R)-bimoclomol (1) maleate18 with a high enantiopurity (er, R:S, 99:1).13 Furthermore, it is important to note that the epoxide intermediate 13 could be intercepted by a number of amines to give a variety of amino analogues of 2 for the development of SAR.19
(R)-(−)-Glycidyl nosylate 11 was also conveniently used as the starting material to develop an advanced intermediate 16 for the exploration of the SAR of the pyridinyl chloro-oxime moiety of 2 (Scheme 4) (Scheme 2, path b). Benzophenone oxime, an N-protected hydroxylamine synthon, underwent reaction at C1 of 11 to give epoxide 14 in a good yield. Regioselective ring opening of epoxide 14 with piperidine gave 15 and subsequent acid hydrolysis of the benzophenone protecting group unmasked the required O-alkylated hydroxylamine 16 which was isolated as the mono HCl salt after treatment with the weakly basic polymeric resin Amberlyst A21.
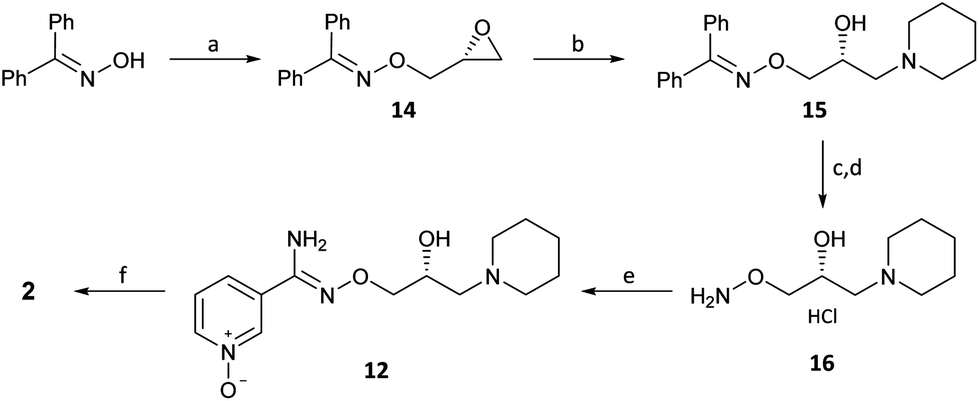 | ||
| Scheme 4 Chiral hydroxylamine route to arimoclomol (2). Reagents and conditions: (a) (i) NaH (60% wt), DMF, 0 °C, 0.5 h; (ii) (R)-(−)-glycidyl nosylate (11) (1.1 equiv.), rt, 2 h, 83%; (b) piperidine (1.05 equiv.), iPrOH, 50 °C, 18 h, quant.; (c) HCl (6 M), 95 °C, 18 h; quant.; (d) Amberlyst A21, MeOH, rt, 4 h, 98%; (e) 3-cyanopyridine-N-oxide (3) (0.8 equiv.), HSCH2CO2H (17) (1.5 equiv.), Et3N, EtOH, 85 °C, 24 h, 75%; (f) NaNO2 (1.3 equiv.), conc. HCl, H2O, −5 °C, 66%. | ||
The next step in the sequence initially proved problematic. The addition of hydroxylamine 16 into 3-cyanopyridine-N-oxide (3) was sluggish (<10% conversion) and heating at elevated temperatures caused a nitrile hydration product to form. However, the application of thioglycolic acid (17) as a nucleophilic promoter gave amidoxime 12 in good yield.20,21 Performing the reaction under identical conditions but without 17 gave <5% conversion to 12 showing the critical importance of 17 in promoting this reaction. Conversion of 12 into 2 was again achieved by standard conditions (er, R:S, >99:1).13 The analogous sequence was also performed with 3-cyanopyridine giving (R)-(+)-bimoclomol 1 with a high enantiomeric ratio (er, R:S, >99:1).13 The addition of hydroxylamine 16 to pyridine nitriles promoted by 17 proved to be successful with a number of substrates and gave access to functionalised pyridine analogues of 2.19
With (R)-(+)-bimoclomol 1 in hand, synthesis of (R)-(+)-arimoclomol 2 was completed by development of a selective method for pyridine N-oxidation of (R)-1 to 2 (Scheme 5). This method was of significant value as it would deliver a short synthesis of both 1 and 2 from common chiral intermediates. This conversion has been reported by oxidation of 1 mesylate with H2O2 in AcOH to yield 2 on multigram scale.22 When this method was repeated, albeit on smaller scale, the reaction was found to be capricious and product yields varied greatly. Although the high er was maintained, the maximum yield achieved in producing 2 from (R)-1 and was 31%.
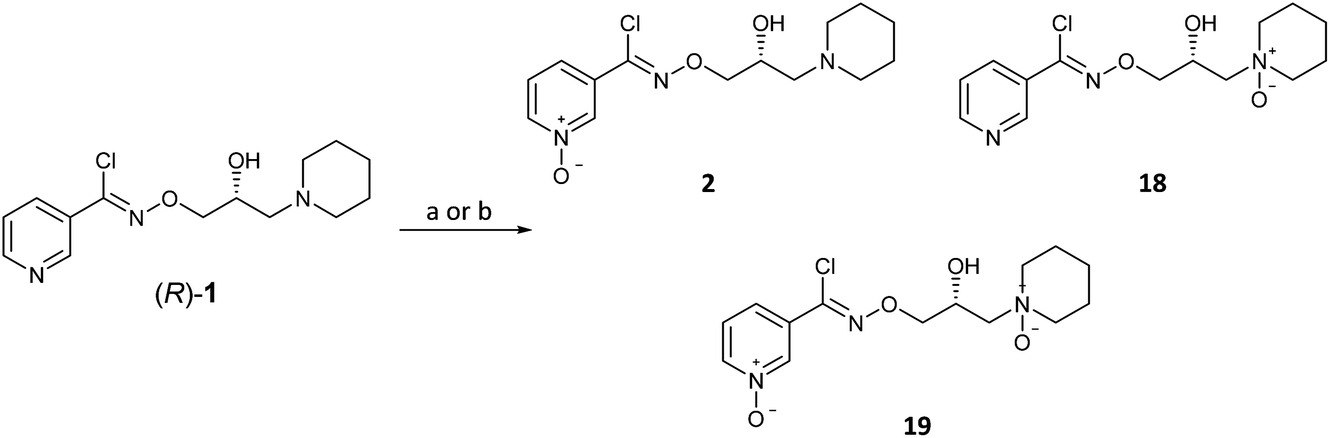 | ||
| Scheme 5 Oxidation of bimoclomol (1) to arimoclomol (2) and other N-oxide derivatives. Reagents and conditions: (a) MeSO3H (1.0 equiv.), AcOH, then 30% H2O2 (4.0 equiv.), 60 °C, 24 h, 2 isolated in 31%; (b) (i) PhSO3H (1.3 equiv.), acetone, rt, 1.5 h; (ii) H2NC(O)NH2·H2O2, (2.0 equiv.), (CF3CO)2O (2.0 equiv.), MeCN, −5 °C → rt, 18 h, 2 isolated in 55–60%. | ||
We began exploring a number of procedures for the selective N-oxidation of a pyridine in the presence of a sec-alcohol and/or tert-amine. Initially we used mCPBA in the presence of MsOH, the rationale being that the organic acid weakens the nucleophilicity of the tert-amine, preventing oxidation. However only the piperidine N-oxide 18 was produced in 31% yield under these conditions. Subsequently, it was found that pre-forming the mono-besylate salt of (R)-1 followed by oxidation with urea-hydrogen peroxide adduct with trifluoroacetic anhydride in MeCN gave a selectivity for 2 over 18.23,24 Pyridine N-oxide 2 was isolated from the reaction mixture in typically 55–60% yield. The high er from (R)-1 (er, R:S, 98:2) was retained in 2 (er, R:S, 98:2) after the oxidation.13
General pharmacology of arimoclomol
The use of target-aligned small molecule tools in cell-based screens requires an investigation of their associated pharmacology to understand the selectivity window over any off-target activities. For phenotypic screening of small molecule hits, where the primary target(s) has not been identified, general pharmacology screening is important to establish the degree of likely selectivity (or promiscuity) against a set of representative traditional drug targets.Arimoclomol (2) was screened for off-target pharmacology against a panel of 50 representative classical receptors, transporters and ion channels, and a panel of 50 representative kinases, and was found to have modest binding affinity for the FGR kinases along with a few other weaker affinities (Table 1).25 Hence, we recommend screening of 2 in cellular assays should be restricted to concentrations below 10 μM to avoid acting through promiscuous undefined polypharmacology which could confound the interpretation of biological results.
| Target | % inhibition |
|---|---|
| a Eurofins Cerep ExpresS Profile. Single point concentration at 10 μM; binding assays; n = 2. b Life Technologies SelectScreen Kinase Profiling. Single point concentration at 1 μM; n = 2. | |
| h.H2 (ant)a | 34% at 10 μM |
| h.5-HT2B (ag)a | 30% at 10 μM |
| FGR1b | 47% at 1 μM |
| FGR2b | 50% at 1 μM |
| MAPKAPK2b | 43% at 1 μM |
| PRKACAb | 38% at 1 μM |
| GSK3βb | 37% at 1 μM |
Pharmacokinetics of arimoclomol
Pharmacokinetic data for arimoclomol (2) was generated in vivo in mouse (Table 2). The route of administration and dose were selected to most closely match a number of relevant published mouse disease model studies.5,26 Following single intraperitoneal (i.p.) injection of 10 mg kg−1, plasma clearance was high relative to liver blood flow resulting in a plasma elimination half-life of 0.71 hours.a
| PK parameter | Plasma | Brain |
|---|---|---|
| a Pharmidex Pharmaceutical Services Ltd, UK. Male CD1 mouse; solution formulation in 10% DMSO, 10% PEG200, 80% saline; n = 3 per time point; terminal blood and brain levels measured at seven time points: 0.16, 0.50, 1, 2, 4, 8 and 24 h. | ||
| T 1/2 | 0.71 h | 0.61 h |
| T max | 0.16 h | 0.50 h |
| C max | 1600 ng mL−1 | 140 ng g−1 |
| AUC0–t | 990 h ng mL−1 | 140 h ng g−1 |
| AUC0–∞ | 1000 h ng mL−1 | 160 h ng g−1 |
Compound 2 has a low brain:plasma ratio in mouse of just 16% based on AUCinf, however, these studies are somewhat confounded by the high clearance of 2 which means steady state is unlikely to have been achieved with single acute dosing. The plasma half-life and brain penetration values from these mouse PK experiments are consistent with published preclinical PK data in rat and dog where a relatively short half-life was observed in both species (T1/2 1 and 2 h respectively).27 These data will contribute to the understanding of drug levels of 2 to overlay with appropriate in vivo efficacy endpoints, i.e. the PK-PD relationship.
Conclusions
A new efficient chiral synthesis of enantiopure 1 and 2 is reported from (R)-(−)-glycidyl nosylate (11) with retention of chiral integrity. This short 4-step sequence from simple chiral commercial starting materials has enabled the exploration of SAR. Arimoclomol (2) was then evaluated in general pharmacology assays to assess selectivity over a representative set of drug targets and pharmacokinetic data was generated in vivo in mouse; these studies will be helpful towards a better understanding of the PK-PD relationship of 2 in disease models.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The ARUK UCL Drug Discovery Institute is core funded by Alzheimer's Research UK (ARUK). Alzheimer's Research UK is a registered charity (No. 1077089 and SC042474). We thank Reach Separations Ltd for chiral chromatographic analysis and separation, the UCL Chemistry Department for spectroscopic and analytical services. In addition, we thank Professor Linda Greensmith and Dr Bernadett Kalmar (UCL Institute of Neurology) for the provision of an authentic sample of arimoclomol citrate.Notes and references
- J. J. Yerbury, L. Ooi, A. Dillin, D. N. Saunders, D. M. Hatters, P. M. Beart, N. R. Cashman, M. R. Wilson and H. Ecroyd, J. Neurochem., 2016, 137, 489–505 CrossRef CAS PubMed.
- P. Sweeney, H. Park, M. Baumann, J. Dunlop, J. Frydman, R. Kopito, A. McCampbell, G. Leblanc, A. Venkateswaran, A. Nurmi and R. Hodgson, Transl. Neurodegener., 2017, 6, 6 CrossRef PubMed.
- J. Zou, Y. Guo, T. Guettouche, D. F. Smith and R. Voellmy, Cell, 1998, 94, 471–480 CrossRef CAS PubMed.
- A. Hishiya and S. Takayama, Oncogene, 2008, 27, 6489–6506 CrossRef CAS PubMed.
- D. Kieran, B. Kalmar, J. R. T. Dick, J. Riddoch-Contreras, G. Burnstock and L. Greensmith, Nat. Med., 2004, 10, 402–405 CrossRef CAS PubMed.
- D. A. Parfitt, M. Aguila, C. H. McCulley, D. Bevilacqua, H. F. Mendes, D. Athanasiou, S. S. Novoselov, N. Kanuga, P. M. Munro, P. J. Coffey, B. Kalmar, L. Greensmith and M. E. Cheetam, Cell Death Dis., 2014, 5, e1236 CrossRef CAS PubMed.
- T. Kirkegaard, J. Gray, D. A. Priestman, K.-L. Wallom, J. Atkins, O. D. Olsen, A. Klein, S. Drndarski, N. H. T. Petersen, L. Ingemann, D. A. Smith, L. Morris, C. Bornæs, S. H. Jorgensen, I. Williams, A. Hinsby, C. Arenz, D. Begley, M. Jäättelä and F. M. Platt, Sci. Transl. Med., 2016, 8, 355ra118 CrossRef PubMed.
- M. Ahmed, P. M. Machado, A. Miller, C. Spicer, L. Herbelin, J. He, J. Noel, Y. Wang, A. L. McVey, M. Pasnoor, P. Gallagher, J. Statland, C.-H. Lu, B. Kalmar, S. Brady, H. Sethi, G. Samandouras, M. Parton, J. L. Holton, A. Weston, L. Collinson, J. P. Taylor, G. Schiavo, M. G. Hanna, R. J. Barohn, M. M. Dimachkie and L. Greensmith, Sci. Transl. Med., 2016, 8, 331ra41 CrossRef PubMed.
- K. Hesselink, J. Pain Relief, 2017, 6, 1 Search PubMed.
- ClinicalTrials.gov Identifier: NCT00706147.Phase II/III Randomized, Placebo-controlled Trial of Arimoclomol in SOD1 Positive Familial Amyotrophic Lateral Sclerosis. See: https://clinicaltrials.gov/ct2/show/NCT00706147 (accessed August 11, 2017).
- University of Miami and Orphazyme APS announce successful phase II trial of arimoclomol in ALS patients. Orphazme press release, December 9, 2016. See: https://www.orphazyme.com/news-feed/2017/2/17/press-release-2 (accessed August 11, 2017).
- L. Ürögdi, Z. Jegesné-Csákai, L. Gruber, L. Ötvös, J. Tóth, I. Tömösközi, A. Szakácsné-Schmidt, F. Reider and M. Schneiderné-Barlay, PCT Int. Appl, WO2001/79174, 2001 Search PubMed.
- See ESI† for all methods, conditions, retention times and SFC traces. R and S assignment based on comparison with an authentic sample of 2 and optical rotations.
- For achiral methods, see: (a) A. Kocak, S. Kurbanh and S. Malkondu, Synth. Commun., 2007, 37, 1155–1165 CrossRef CAS; (b) Y. Zhou, K. Vu, Y. Chen, J. Pham, T. Brady, G. Liu, J. Chen, J. Nam, P. S. Murali Mohan Reddy, Q. Au, I. S. Yoon, M.-H. Tremblay, G. Yip, C. Cher, B. Zhang, J. R. Barber and S. C. Ng, Bioorg. Med. Chem. Lett., 2009, 19, 3128–3135 CrossRef CAS PubMed; (c) A. Zhang, P. Fu, Z. Zhang, H. Chen and P. Yu, J. Pharm. Biomed. Sci., 2016, 6, 30–37 Search PubMed . For a chiral chemo-enzymatic approach, see: L. Banoth, T. K. Narayan, B. Pujala, A. K. Chakraborti and U. C. Banerjee, Tetrahedron: Asymmetry, 2012, 23, 1564–1570 Search PubMed.
- (R)-(−)-Epichlorohydrin (10) (ee > 97.5%) was purchased from Sigma Aldrich [cat no. 540072] and used as supplied. (R)-(−)-Glycidyl nosylate (11) (ee > 97.5%) was purchased from Fluorochem [cat no. 212269] and used as supplied.
- J. M. Klunder, T. Onami and K. B. Sharpless, J. Org. Chem., 1989, 54, 1295–1304 CrossRef CAS.
- (R,Z)-3-(N′-(2-Hydroxy-3-(piperidine-1-yl)propoxy)carboximi-oylchloride)pyridine-1-oxide citrate (2-citrate, arimoclomol citrate) was prepared as an off-white amorphous solid (164 mg): m.p. 161–162 °C; [α]20D +8.0° (c = 1, H2O); IR νmax (neat): 3423, 3228, 2949, 2868, 1722, 1589, 1483, 1433, 1307, 1128, 972, 829 cm−1; 1H NMR (600 MHz, d6-DMSO) δ: 8.54 (t, J = 1.5 Hz, 1H), 8.39–8.35 (m, 1H), 7.72–7.68 (m, 1H), 7.55 (dd, J = 8.0, 6.5 Hz, 1H), 4.28 (ddd, J = 17.6, 13.3, 7.4 Hz, 3H), 3.35 (br. s, 2H), 3.13–2.74 (m, 6H), 2.59 (d, J = 15.2 Hz, 2H), 2.56–2.51 (m, 2H), 1.77–1.61 (m, 4H), 1.48 (s, 2H); 13C NMR (151 MHz, d6-DMSO) δ: 176.6, 171.3, 140.5, 136.4, 132.7, 131.5, 126.8, 123.3, 77.8, 71.4, 63.8, 58.7, 53.1, 44.0, 30.7, 23.0, 21.9; HRMS (m/z TOF MS ES+) for C14H20ClN3O3 [M + H]+ calc. 314.1271, observed 314.1263; SFC er purity R:S, >99:1.
- (R,Z)-3-(N′-(2-Hydroxy-3-(piperidine-1-yl)propoxy)carboximi-oylchloride)pyridine maleate ((R)-1-maleate, bimoclomol maleate) was prepared as an off-white amorphous solid (70 mg): m.p. 137–138 °C; [α]20D +6.0° (c = 1, MeOH); IR νmax (neat): 3269, 2937, 1577, 1477, 1440, 1348, 1205, 1070, 981, 864 cm−1; 1H NMR (600 MHz, d6-DMSO) δ: 9.09 (s, 1H), 9.01–8.98 (m, 1H), 8.73 (dd, J = 4.8, 1.5 Hz, 1H), 8.24–8.06 (m, 1H), 7.57 (ddd, J = 8.1, 4.8, 0.6 Hz, 1H), 6.02 (d, J = 4.0 Hz, 2H), 5.93 (s, 1H), 4.40–4.21 (m, 3H), 3.60–3.28 (m, 3H), 3.20 (d, J = 11.8 Hz, 1H), 3.12–3.05 (m, 1H), 3.03–2.83 (m, 2H), 1.84–1.55 (m, 5H), 1.38 (s, 1H); 13C NMR (151 MHz, d6-DMSO) δ: 167.1, 151.7, 147.4, 136.0, 135.1, 134.6, 127.9, 123.9, 77.2, 63.1, 58.0, 54.1, 51.1, 22.2, 21.3; HRMS (m/z TOF MS ES+) for C14H20ClN3O2 [M + H]+ calc. 298.1322, observed 298.1319; SFC er purity R:S, 98:2.
- Details for the preparation of analogues of 1 and 2 and the SAR will be reported separately.
- Z. Li, M. Zancanella, C. Yu, L. C. Setti, H. Sham, Q. Xu, C. Yee and M. Yu, PCT Int. Appl, WO2016/201052, 2016 Search PubMed.
- F. Al-Saffar, S. Berlin, T. Musil and S. Sivadasan, PCT Int. Appl, WO2006/090153A1, 2006 Search PubMed.
- M. Kürthy, K. Bíró, K. Nagy, L. Ürögdi, Z. Csákai, J. Szilbereky, T. Mogyorósi, M. Török, A. Komáromi, E. Márványos, M. Barabás, M. Kardos, Z. Nagy, L. Korányi and M. Nagy, US Pat. Appl, US6649628B1, 2003 Search PubMed.
- (a) J. Y. L. Chung, R. J. Cvetovich, F.-R. Tsay, P. G. Dormer, L. DiMichele, D. J. Mathre, J. R. Chilenski, B. Mao and R. Wenslow, J. Org. Chem., 2003, 68, 8838–8846 CrossRef CAS PubMed; (b) S. Caron, N. M. Do and J. E. Sieser, Tetrahedron Lett., 2000, 41, 2299–2302 CrossRef CAS.
- The bis-N-oxide product 19 was noted to form by LCMS of the crude reaction mixture but not in significant amounts and was not isolated.
- See ESI† for the complete off-target pharmacology data generated during these studies.
- B. Kalmar, S. Novoselov, A. Gray, M. E. Cheetham, B. Margulis and L. Greensmith, J. Neurochem., 2008, 107, 339–350 CrossRef CAS PubMed.
- V. Lanka, S. Wieland, J. Barber and M. Cudkowicz, Expert Opin. Invest. Drugs, 2009, 18, 1907–1918 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and data for all new compounds including traces of chiral SFC. See DOI: 10.1039/c7ob02578e |
| This journal is © The Royal Society of Chemistry 2017 |