
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure–reactivity correlations of the abnormal Beckmann reaction of dihydrolevoglucosenone oxime†
Amani
Alhifthi
ab,
Benjamin L.
Harris
ab,
Lars
Goerigk
 a,
Jonathan M.
White
*ab and
Spencer J.
Williams
*ab
a,
Jonathan M.
White
*ab and
Spencer J.
Williams
*ab
aSchool of Chemistry, University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: sjwill@unimelb.edu.au; whitejm@unimelb.edu.au
bBio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville, Victoria 3010, Australia
First published on 14th November 2017
Abstract
A structural, spectroscopic and computational study of a series of oximes was undertaken to investigate how geometric and structural changes relevant to the reaction coordinate for the Beckmann reaction (normal Beckmann) and Beckmann fragmentation (abnormal Beckmann) manifest in the ground state. X-ray structures of a range of oximes derived from dihydrolevoglucosan (Cyrene™; which undergoes the abnormal Beckmann reaction), in which the oxygen substituent was systematically varied were determined. As the electron demand of the OR group increased, the major structural changes included lengthening of the N–OR bond distance, and a decrease in the magnitude of the C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–O bond angle, consistent with the changes seen for cyclohexanone oximes, which undergo the normal Beckmann reaction. However, unique to the Cyrene oximes, an increase in the length of the fissile C1–C2 bond was observed, which correlated with a decrease in the 13C–13C 1-bond coupling constants as the electron demand of the OR substituent increased. Computational studies of Cyrene and cyclohexanone oximes using Natural Bond Orbital analysis support an electronic structure involving n(O) → σ*C1–C2 and σC1–C2 → σ*N–O localized orbital interactions.
N–O bond angle, consistent with the changes seen for cyclohexanone oximes, which undergo the normal Beckmann reaction. However, unique to the Cyrene oximes, an increase in the length of the fissile C1–C2 bond was observed, which correlated with a decrease in the 13C–13C 1-bond coupling constants as the electron demand of the OR substituent increased. Computational studies of Cyrene and cyclohexanone oximes using Natural Bond Orbital analysis support an electronic structure involving n(O) → σ*C1–C2 and σC1–C2 → σ*N–O localized orbital interactions.
Introduction
Ketoximes undergo rearrangement reactions involving cleavage of the carbon–carbon bond adjacent to the imino carbon.1 These reactions include the Beckmann rearrangement (‘normal’ Beckmann reaction),2–4 leading to amides, and the Beckmann fragmentation (‘abnormal’ Beckmann reaction),5,6 leading to nitriles. The normal Beckman reaction involves the generation of an incipient carbocation at the carbon atom located anti to the oxime oxygen, which is intercepted by migration to nitrogen to afford a nitrilium ion intermediate, which is subsequently hydrolyzed to yield an amide (Fig. 1). The abnormal Beckmann reaction occurs in cases where the adjacent carbon can stabilize positive charge; a carbocation intermediate is formed that undergoes heterolytic fragmentation of the carbon–carbon bond anti to the leaving group to yield a nitrile.7–12 The initiating step of both the normal and abnormal Beckmann reactions involve the departure of an oxygen leaving group from nitrogen, with the propensity of a substrate to undergo one or the other transformation dictated by the nature of the anti substituent.13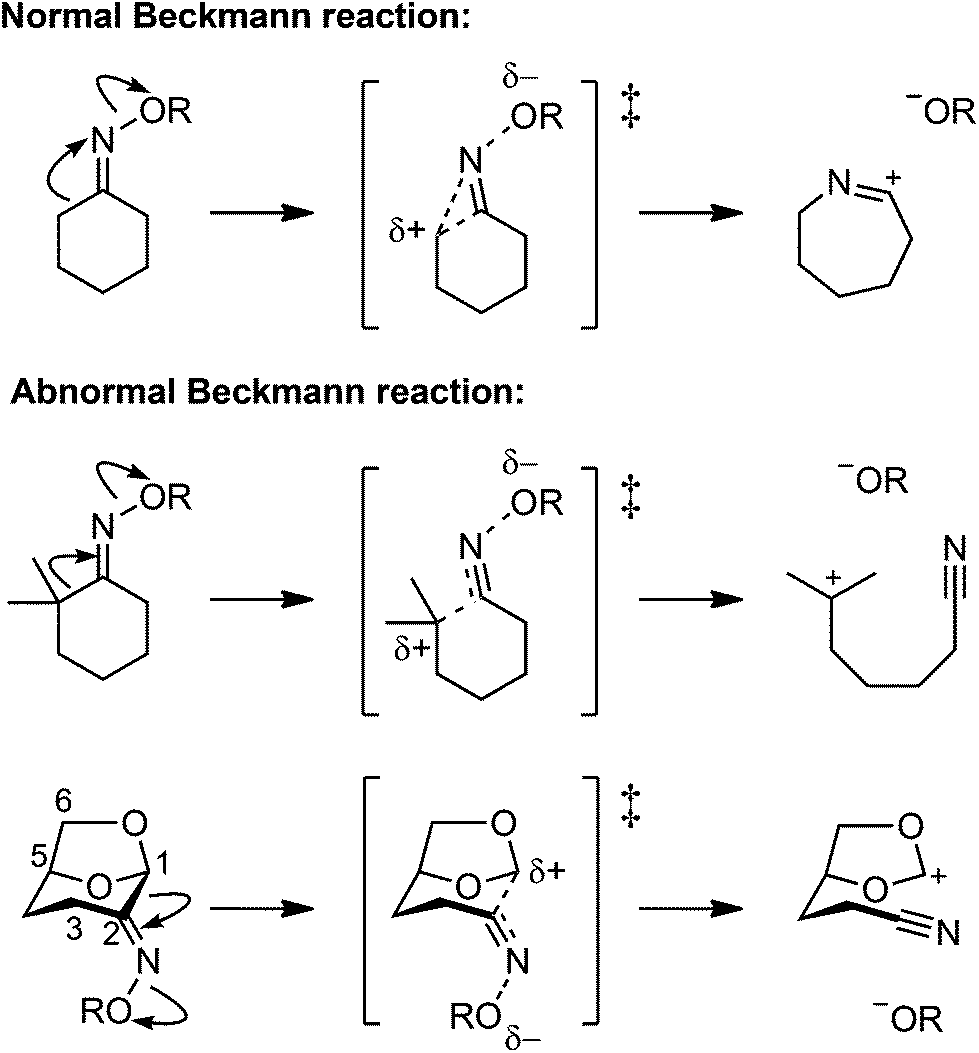 | ||
| Fig. 1 Normal Beckmann (Beckmann rearrangement) and abnormal Beckmann (Beckmann fragmentation) reactions of oximes. | ||
The structure-correlation principle emerged from early studies by Dunitz14–16 and Kirby17–19 who noted that for unimolecular reactions, changes in bond length and geometry at reactive sites in a molecule vary in a systematic manner that correlates with changes in reactivity of the substrate induced by varying its electronic nature. The structure-correlation principle can be applied in systems where the minimum energy (hereafter ‘ground state’) geometries are similar to that of the transition state. This allows application of the qualitative concept of frontier molecular orbital (FMO) interactions that facilitate the reaction to be manifested in the ground state. The structure-correlation principle is most readily applied by performing low temperature X-ray structures on substrates that are systematically varied to allow accurate measurement of geometric parameters, which are correlated with reactivity parameters such as leaving group ability. Structure–reactivity correlations of cyclohexanone oxime analogues that varied in the nature of the group OR revealed structural changes consistent with the manifestation of early stages of the normal Beckmann reaction.20 These included an increasing N–OR bond distance and a closing of the N–C1–C2 bond angle, consistent with the early stages of bond breaking and of migration of the antiperiplanar carbon onto nitrogen. Additionally, the magnitude of the 1JC–C coupling constants for the migrating bond varied in a systematic way that supported meaningful changes in bond order consistent with the normal Beckmann reaction. A similar plot for the 2,2-dimethylcyclohexanone oxime analogues, which are prone to abnormal Beckmann reaction, revealed structural effects that varied in a similar fashion but which were weaker in magnitude.
In the present work we apply the structure-correlation principle to the abnormal Beckmann reaction for an oxime derivative bearing heteroatom donors on the anti carbon. We used the ketone dihydrolevoglucosenone (Cyrene™), which has recently become readily available on large scale through the valorization of wood-waste using the Furacell process.21 Cyrene readily forms an oxime which undergoes the abnormal Beckmann reaction as a result of the effects of two donor oxygen atoms on the antiperiplanar carbon, causing a Grob fragmentation.22 Moreover, the bicyclic nature of this compound provides a rigid structure that minimizes molecular flexibility while at the same time orienting the donor oxygens so as to provide effective orbital mixing in the ground state.
Results and discussion
Synthesis and structure-determinations of Cyrene-based oximes
A range of Cyrene-based oximes of varying leaving group ability (pKa 16–2.85 for the conjugate acid) were synthesized. Reaction of Cyrene with HONH2·HCl in pyridine afforded the parent oxime 1 as a single geometrical isomer,22 a result which reflects both steric as well as electronic effects (Fig. 2a). Alternatively, reaction of Cyrene under similar conditions with PhONH2·HCl afforded the O-phenyl oxime 3. A range of acylated and alkylated oximes of varying leaving group ability were prepared directly from 1 as shown in Fig. 2b. Thus alkylation or arylation using NaH in N,N-dimethylformamide (DMF) or tetrahydrofuran (THF) afforded 2 and 4. Acylation with anhydrides or acyl halides in pyridine afforded 5–8.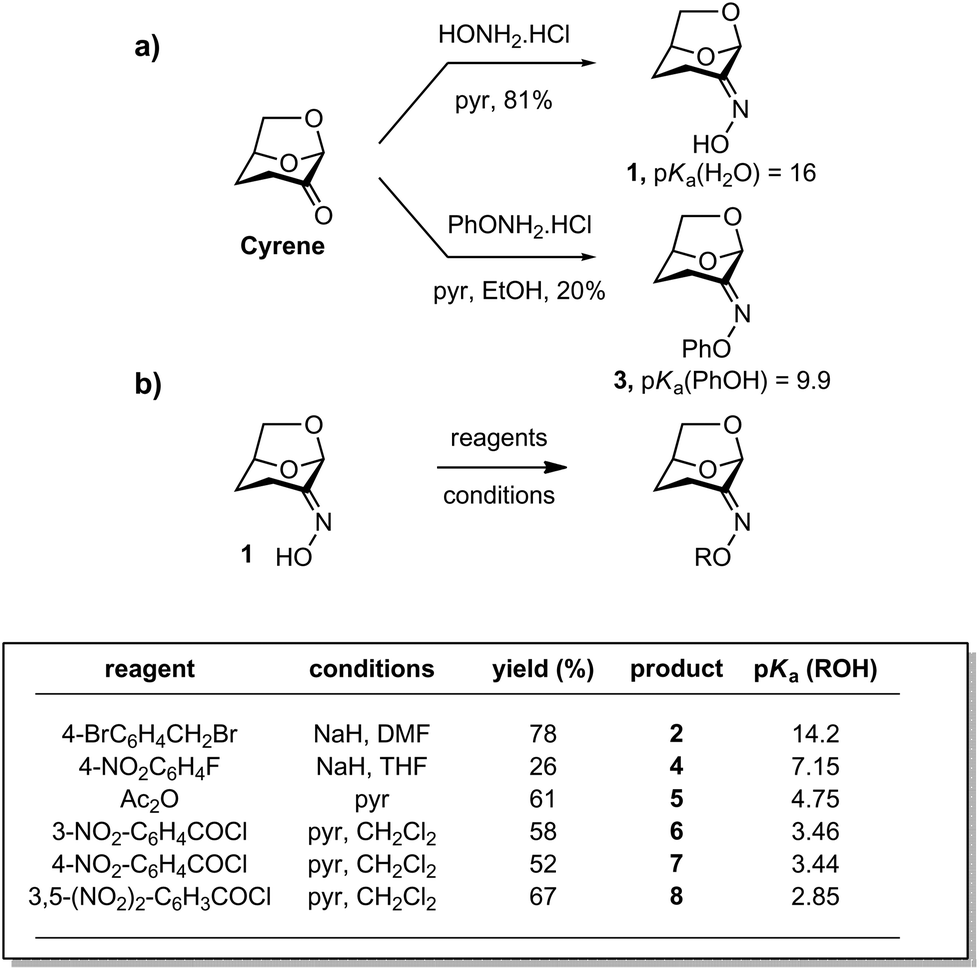 | ||
| Fig. 2 Synthesis of Cyrene oxime and derivatives. (a) Condensation reactions for preparation of compounds 1 and 3. (b) Acylation, arylation and alkylation reactions for preparation of compounds 2 and 4–8. pKa values are from ref. 23 and are for aqueous solutions. | ||
Oximes 1–8 were crystalline compounds and provided crystals suitable for single crystal X-ray structure determinations. The structures of oxime derivative 1–8 were determined at low temperature to minimise the effects of thermal libration on the measured geometric parameters. Representative thermal ellipsoid plots are shown in Fig. 3. In several cases unit cells contained non-equivalent structures (Z′ > 1), which were treated as independent in subsequent analysis. Selected bond-distances and angles are presented in ESI Table S1.†
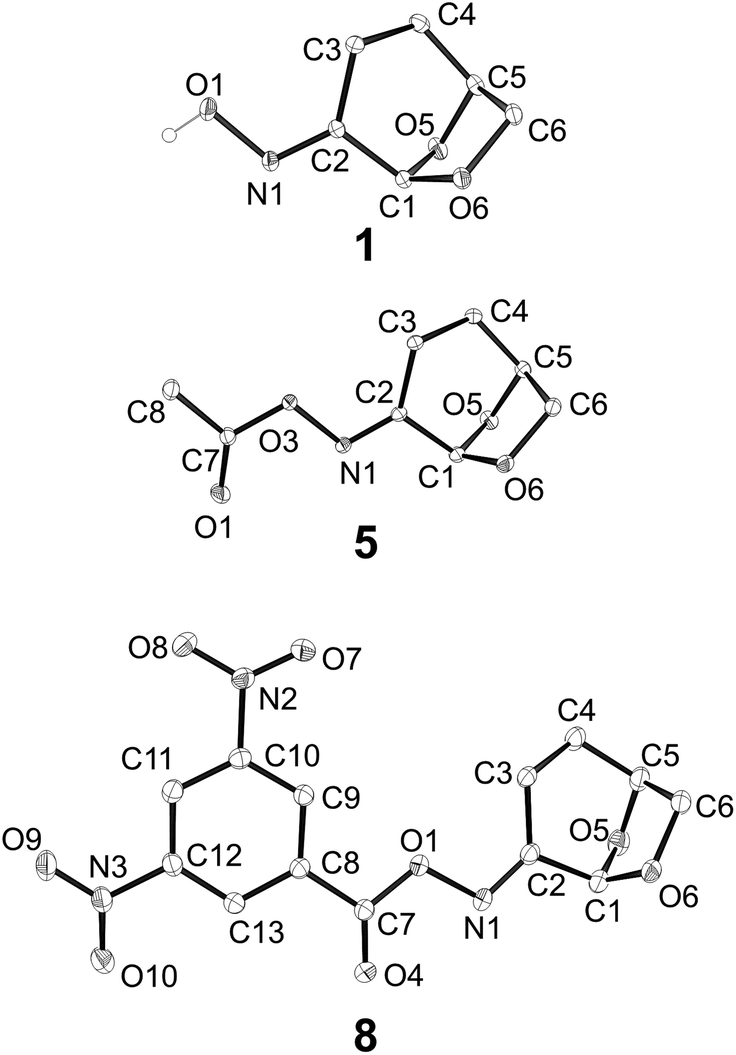 | ||
| Fig. 3 Representative X-ray structures of oxime 1, oxime acetate 5, and 3,5-dinitrophenyloxime 8. | ||
The ability of Cyrene-derived oximes to act as substrates for the abnormal Beckmann was investigated by refluxing a solution of oxime acetate 5 in acetic anhydride (Fig. 4). This led to the conversion to a mixture of nitriles 9 and 10. While the volatility of the products led to their isolation in only modest yield, thin layer chromatography of the reaction mixture suggested that these were the only products to arise from the reaction and thus that abnormal Beckmann reaction is the major reaction channel. We also prepared and unsuccessfully attempted to obtain diffraction quality crystals of the 2-nitrobenzoyl oxime 11 (for 2-nitrobenzoic acid, pKa = 2.17). Interestingly, a solution of 11 in CDCl3 rearranged smoothly to a similar mixture of nitriles 12 and 13 upon storage at room temperature for 10 days.
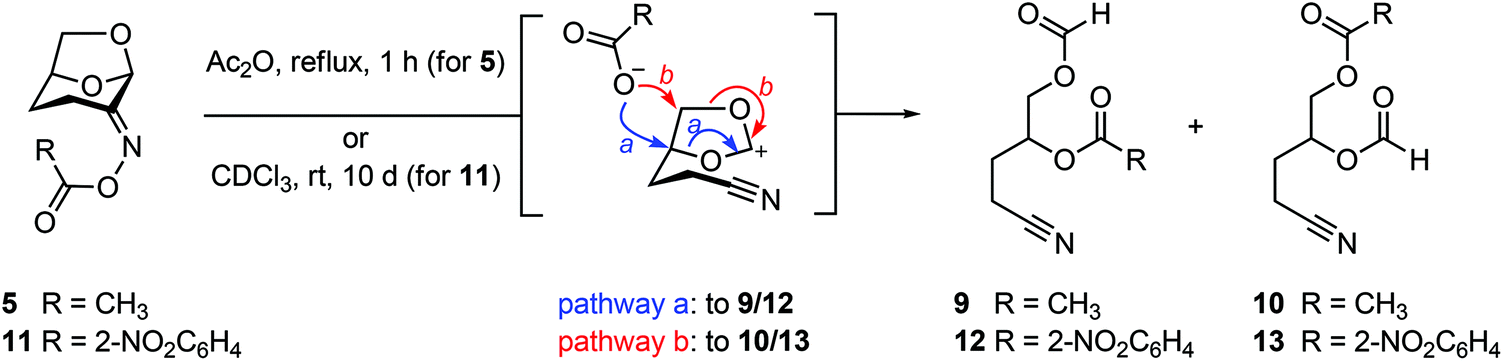 | ||
| Fig. 4 Abnormal Beckmann reactions of Cyrene oxime acetate 5 or 2-nitrobenzoate 11 affords a mixture of nitrile regioisomers. The stereochemistry was not determined. | ||
Structural correlations in Cyrene oximes
The geometrical data obtained from the low temperature X-ray structures was used to construct the plots presented in Fig. 5. The data in Fig. 5a represents an application of the variable oxygen probe, with the slope exhibiting sensitivity to the nature of the leaving group. That is, as the leaving group ability increases (as quantified based on the pKa value of the conjugate acid), the N–OR bond distance increases. This lengthening of the fissile bond can be taken to represent the early stages of the abnormal Beckmann reaction. Fig. 5b and c show the effects of the variable oxygen probe on the C1–C2–N and C2–N–O bond angles. These plots show that as leaving group ability increases, both bond angles decrease. Again, these changes can be interpreted as representing early stages of an abnormal Beckmann in which departure of the leaving group results in a change in hybridization at C2 and N from sp2 to sp; this should lead to a contraction of the C1–C2–N angle (and a concomitant expansion of the C3–C2–N angle), and contraction of the C2–N–O angle reflecting the changing hybridisation of the N2 lone pair electrons from sp2 towards sp and repulsion of the σN–O electrons (vide infra).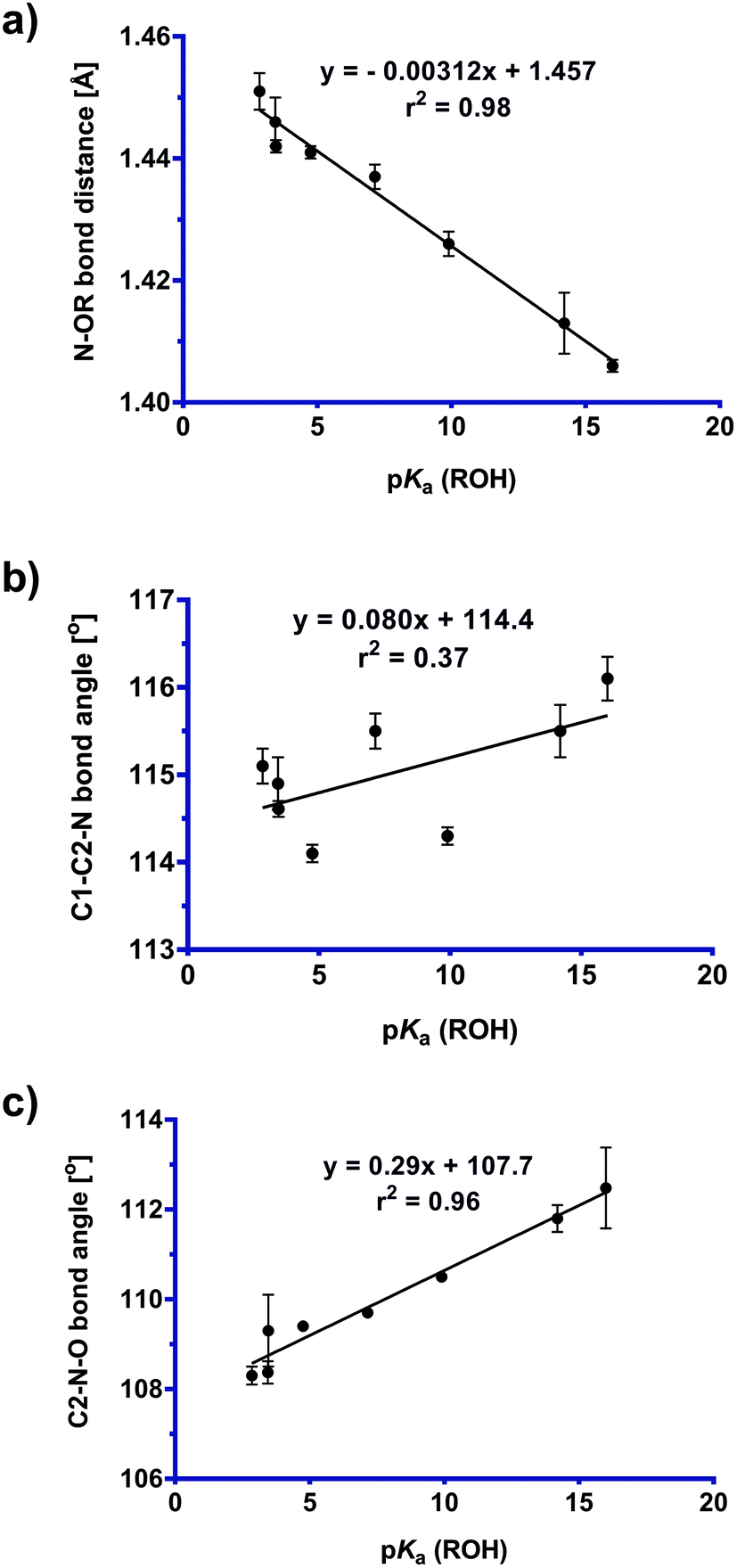 | ||
| Fig. 5 Plot of (a) N–OR bond distance, (b) C1–C2–N bond angle [°], and (c) C2–N–O bond angle [°] versus pKa (ROH) values for Cyrene oxime derivatives (1–8). See ESI Fig. S1† for a more comprehensive plot of critical bond distances and angles across this series. | ||
From an FMO perspective, the observed geometric changes can be considered to arise from σC1–C2 → σ*N–O interactions for the C1–C2 bond, which because of the presence of the two donor oxygens acts as a ‘Grob-like’ donor. Evidence for participation of the anti bond (C1–C2) in this interaction is illustrated in Fig. 6a in which the C1–C2 bond distance increases systematically with increasing leaving group ability of the OR substituent, while the C2–C3 bond (the syn bond) is essentially unchanged over the same series. Structure-correlation plots for the O5–C1 and O6–C1 bonds reveal a weak contraction of the bond distance as leaving group ability increases (Fig. 6b). The weakness of this correlation is presumably a consequence of distribution of structural effects over two bonds for the two donors, attenuating the structural consequences of electronic perturbation. Examination of the geometry of the X-ray structures reveals that O5, which is present within a tetrahydropyran ring, is geometrically disposed to provide better donation into the C1–C2 bond than O6, which is present within a tetrahydrofuran ring. An estimate for the angle of the donor lone pair can be obtained by using the AFIX 23 command within SHELXL2014,24 which was used to place hydrogens on O5 and O6, enabling measurement of the dihedral angle for the C1–C2 bond of −168° for O5 and 150° for O6 for hypothetical sp3 hybrid lone pair orbitals on each oxygen. This donor effect manifests within the X-ray structures as slightly shorter distances for the C1–O5 bonds compared to C1–O6, suggesting a stronger nO5 → σ*C1–C2 interaction (vide infra).
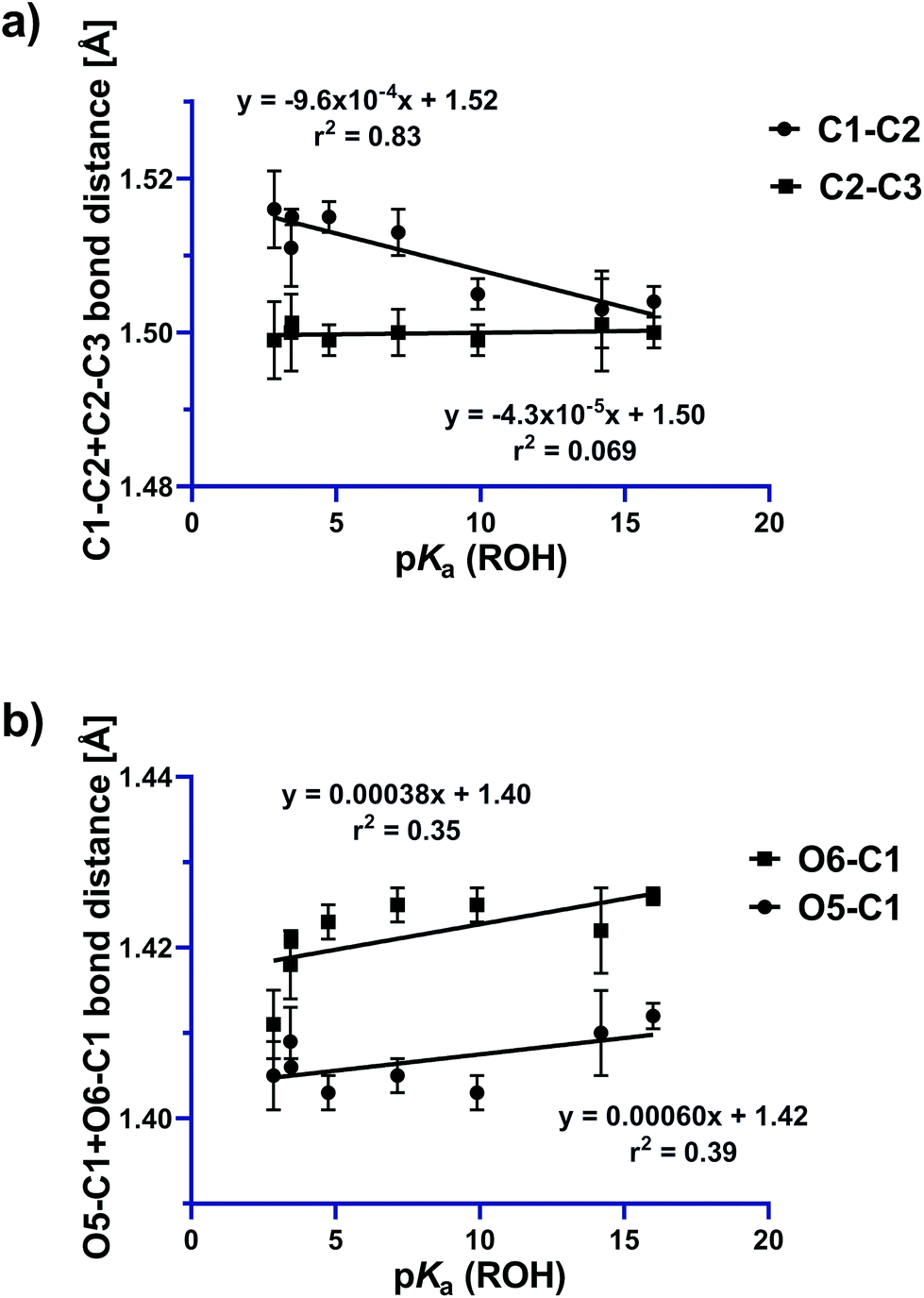 | ||
| Fig. 6 Plots showing variations in (a) C1–C2 (anti) and C2–C3 (syn) bond distances and (b) O5–C1 and O6–C1 bond distances, as a function of leaving group ability. | ||
One-bond 13C–13C coupling constants have been shown to be sensitive to the effects of hyperconjugation.25 Further insight into σC1–C2 → σ*N–O interactions in these structures, and their increasing magnitudes with leaving group ability is provided by the one-bond 13C–13C coupling constants, which were measured for selected derivatives. The one-bond 13C–13C coupling constants were determined using a 1D-INADEQUATE pulse sequence, or alternatively could be measured directly from the 1D 1H decoupled 13C NMR spectrum acquired for concentrated solutions. However, because of the poor sensitivity of these measurements, and the low solubility of several of the oxime derivatives (4 and 6), the coupling constants could only be obtained for six compounds (1–3, 5, 7 and 8). Fig. 7 shows that the 1JC1–C2 coupling constant decreases with increasing leaving group ability of the OR group, which indicates a reduction in bond order, and is consistent with a greater σC1–C2 → σ*N–O interaction across the series. Comparison of the line of least squares for that of a similar plot of cyclohexanone oximes (y = 0.240x + 42.47, r2 = 0.97), with that determined here for the Cyrene oximes (y = 0.33x + 56.09, r2 = 0.87), reveals that while they exhibit similar slopes, there is a striking difference in the Y-intercept, representing the expected coupling magnitude of C1–C2 for a leaving group with pKa value of 0 for the conjugate acid, with the former some 14 Hz greater. This difference most likely reflects a fundamental mechanistic distinction of the normal and abnormal Beckmann reactions. In the normal Beckmann reaction, alkyl migration produces a partial positive charge on the oxime carbon, whereas in the abnormal Beckmann, the Grob-like fragmentation leads to positive charge development on the adjacent carbon (C1). ESI Fig. S2† shows the trend in 13C chemical shift values for C1 and C2. C1 exhibits a decrease in chemical shift as a function of leaving group ability, while C2 exhibits an increase in chemical shift. Caution must be taken in the interpretation of these results as 13C chemical shifts are sensitive to both electron demand and hybridization effects. We interpret the steep increase in C2 as arising primarily from the inductive effect of a better leaving group, while the weak decrease in C1 presumably reflects increased electron density at C1 from the donor oxygen atoms which is more than compensating for the decrease in electron density at C1 due to the σC1–C2 → σ*N–O interaction.
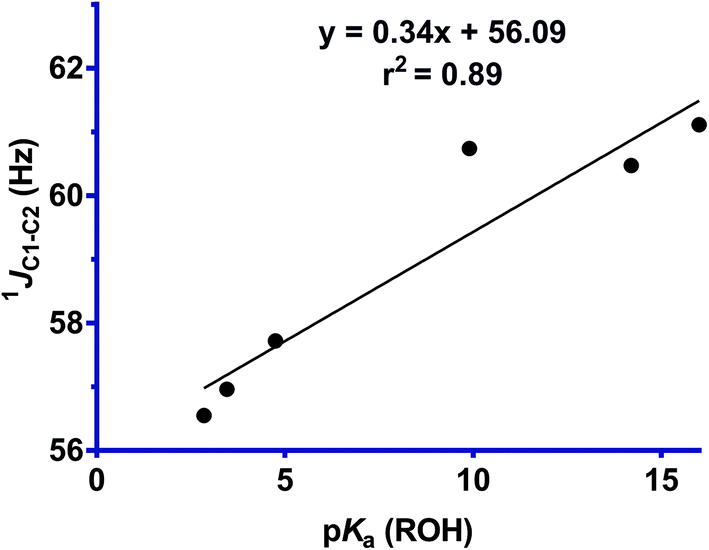 | ||
| Fig. 7 Plot of 1JC1–C2 for the antiperiplanar bond versus the pKa (ROH) value for oxime derivatives 1–3, 6–8. The poor solubility of compounds 4 and 6 prevented measurement of their coupling constants. | ||
The data presented here can be compared to published structure-correlation data for both the normal Beckmann reaction of cyclohexanone oximes, and the abnormal Beckmann reaction of 2,2-dimethylcyclohexanone oximes (Table 1). These data show that across the three series of compounds, the sensitivity of N–O bond length to increased leaving group ability is greatest for substrates that undergo the normal Beckmann reaction, with reduced sensitivity for substrates that undergo the abnormal Beckmann reaction, and especially the Cyrene oximes. This may reflect the increased ability of oxygen donors to support charge development at the remote site, leading to a reduced polarization of the C1–C2 bond. The sensitivity of the change in the NC–C bond angle is greatest for the cyclohexanone series, and weakest for the two abnormal Beckmann series and especially the Cyrene oximes. In all three cases these angular structure–reactivity correlations reflect increasing sp character at the oxime carbon as the reaction progresses to a vinyl cation or nitrile (vide infra). These differences in sensitivity indicate that structural (and presumably electronic) rearrangements towards the azacyclopropene-like transition state of the normal Beckmann reaction occur earlier in the reaction coordinate, whereas the equivalent changes for the abnormal Beckmann reaction occur later. Finally, the sensitivity of the CN–O bond angles are essentially the same for both reactions, indicating that departure of the leaving group occurs with similar timing and through a similar trajectory.
Electronic-structure correlations of normal and abnormal Beckmann reactions
Computational chemistry was used to provide qualitative insight into the electronic structure of the oximes and how this contributes to their propensity to undergo normal or abnormal Beckmann reactions. In particular, we sought to understand the key orbital interactions that contribute to their innate reactivity. We applied Natural Bond Orbital (NBO) analysis,26,27 which transforms a given wave function or Kohn–Sham determinant from Density Functional Theory (DFT) into localized NBOs that represent lone pairs and covalent bonds commensurate with a traditional Lewis-type picture. For each bonding atom within a bond or lone pair, NBO data can be expressed in terms of percentage s, p and d character, or normalized relative to s character as a natural atomic hybrid (hA). Departures from this localized Lewis-type structure through electron delocalization can subsequently be quantified using second order perturbation analysis. For each occupied Lewis-type donor NBO (i) and unoccupied non-Lewis-type acceptor NBO (j), the donor–acceptor stabilization energy E(2) associated with i → j delocalization is calculated as:| E(2) = ΔEij(2) = qiF(i,j)2/(εj − εi) |
The geometries of a series of oximes derived from Cyrene and cyclohexanone of varying leaving group ability were optimized at the TPSS-D3(BJ)/def2-TZVP level of theory (see ESI†). The electronic structures of these models were then analyzed by NBO analysis at the hybrid-DFT PW6B95/def2-TZVP level of theory. Note that this DFT approximation has shown to be one of the most robust and reliable hybrids.31Table 2 shows that both systems exhibit σC1–C2 → σ*N–O interactions, the magnitude of which, quantified by E(2), increases as the electron demand of the variable oxygen probe increases. In a similar vein this data also shows that the occupancy of the σC1–C2 donor orbital (qi) decreases with increasing electron demand, which is consistent with these structures manifesting electronic changes equivalent to early stages along the reaction coordinate for both abnormal and normal Beckmann reactions. A connection exists with the structure–reactivity correlations in which contractions of the C1–C2–N and C2–N–O angles provided qualitative evidence for a change in hybridization at C2 and N from sp2 to sp. Across the series %s at N for the lone pair rises from 45.0 to 48.7 (hA: sp1.22 to sp1.05), and %s at C2 for σC2–C3 rises more modestly from 35.2 to 35.8 (hA: sp1.83 to sp1.78) (Tables SI16 and SI17†). Simultaneously, the fissile bonds exhibit a reduction in s character at these two sites as the nucleofugacity of the leaving group OR increases; %s at N of the σN–O bond falls from 14.8 to 11.6 (hA: sp5.75 to sp7.56), and %s at C2 for σC1–C2 falls from 29.5 to 28.8 (hA: sp2.38 to sp2.46) (Tables SI11 and SI15†).
| Cyrene oximes | Cyclohexanone oximes | ||||||
|---|---|---|---|---|---|---|---|
| R | pKa | E(2) (kcal mol−1) | ΔEij (au) | q i | E(2) (kcal mol−1) | ΔEij (au) | q i |
| H | 16 | 5.65 | 0.64 | 1.97376 | 5.60 | 0.61 | 1.97090 |
| C6H5 | 9.9 | 6.00 | 0.64 | 1.97219 | 6.00 | 0.61 | 1.96928 |
| CH3CO | 4.75 | 6.43 | 0.62 | 1.97017 | 6.34 | 0.59 | 1.96779 |
| 3-NO2C6H4CO | 3.46 | 6.53 | 0.61 | 1.96894 | 6.36 | 0.60 | 1.96759 |
| 3,5-(NO2)2C6H3CO | 2.85 | 6.79 | 0.60 | 1.96750 | 6.60 | 0.59 | 1.96629 |
| CF3CO | 0.50 | 7.20 | 0.59 | 1.96574 | 7.11 | 0.57 | 1.96344 |
NBO analysis was used to investigate the nature of remote donor effects by the endocyclic oxygens O5 and O6. The data in Table 3 reveals that donation by lone pairs on these remote oxygens increases as the leaving group ability increases, exhibiting the donor interactions necessary for the abnormal Beckmann reaction in the ground state for this system. While the X-ray structures suggest that O5, within the tetrahydropyran ring, is geometrically better disposed to act as a more effective donor than O6, within the tetrahydrofuran ring, NBO analysis does not provide evidence that the two oxygens are in fact significantly different in donor ability. To rule out any artefacts stemming from the chosen DFT approximation, extensive NBO calculations at five different levels of theory with the general gradient approximation (GGA) method revPBE, the meta-GGA TPSS, and the hybrid methods PW6B95, B3LYP, and PBE0 were conducted (see ESI Tables S3–10†). None of these provided clear evidence for superior donation by either oxygen.
| LP O5 | LP O6 | ||||||
|---|---|---|---|---|---|---|---|
| R | pKa | E(2) (kcal mol−1) | ΔEij (au) | q i | E(2) (kcal mol−1) | ΔEij (au) | q i |
| H | 16 | 3.30 | 0.75 | 1.90644 | 3.50 | 0.74 | 1.91418 |
| C6H5 | 9.9 | 3.37 | 0.74 | 1.91333 | 3.54 | 0.74 | 1.83150 |
| CH3CO | 4.75 | 3.45 | 0.74 | 1.90456 | 3.61 | 0.73 | 1.91275 |
| 3-NO2C6H4CO | 3.46 | 3.55 | 0.74 | 1.90366 | 3.64 | 0.73 | 1.91189 |
| 3,5-(NO2)2C6H3CO | 2.85 | 3.62 | 0.73 | 1.90300 | 3.79 | 0.73 | 1.91114 |
| CF3CO | 0.50 | 3.63 | 0.73 | 1.90239 | 3.79 | 0.73 | 1.91076 |
Conclusions
The structure–reactivity correlations of Cyrene oxime and derivatives show changes in bond-length and angles that are consistent with the early stages of the abnormal Beckmann reaction: lengthening of the fissile N–OR bond, and contraction of the CN–O and C1–C2N bond angles. More modest changes in the O5–C1 and O6–C1 bond distances are consistent with donor orbital contributions shared across these two donor atoms. While modest lengthening of the fissile C1–C2 bond distance was evident, more compelling evidence for electronic changes of this bond were revealed by measurement of one-bond 13C–13C coupling constants by NMR spectroscopy, indicating a reduction in electron density as leaving group ability increases. These experimental conclusions are supported by computational studies using NBO analysis, which revealed an increasing donor–acceptor stabilization energy E(2), and a reduction in bond order (qi), of the C1–C2 bond with increasing leaving group ability, and an increase in n → σ*C1–C2 interactions of the adjacent oxygen donors. Collectively, the structural and electronic data are consistent with the major orbital interactions in the Cyrene oxime structures involving n → σ*C1–C2 and σC1–C2 → σ*N–O (Fig. 8).
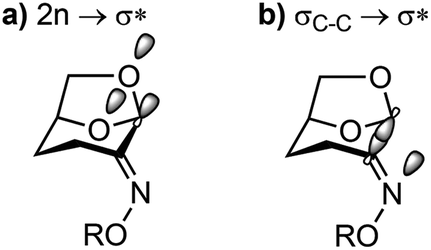 | ||
| Fig. 8 Major orbital interactions in Cyrene oximes. | ||
This work shows the interplay between structural and electronic reorganization in the ground state32 and provides glimpses of the orbital and geometrical changes that occur upon O⋯N bond scission. We extend early observations of Kirby et al.33,34 for the structure–reactivity correlations of oximes in the Beckmann rearrangement and more recent work by White et al.20 describing structure–reactivity correlations for the abnormal Beckmann rearrangement of 2,2-dimethylcyclohexane oximes.20 Broadly, the sensitivity of most of the Cyrene oxime correlations are similar to that for related plots of cyclohexanone oximes and 2,2-dimethylcyclohexanone oximes, which undergo normal and abnormal Beckmann reactions respectively. However, uniquely, the Cyrene oximes show a lengthening of the C1–C2 bond upon increased leaving group ability, and a greater magnitude of one-bond 13C–13C coupling constants, supporting divergence of mechanism for the normal and abnormal Beckmann reactions.
Experimental
Synthesis
Proton (1H NMR, 400 or 600 MHz) and proton-decoupled carbon-13 (13C NMR, 100 or 150 MHz) nuclear magnetic resonance spectra were obtained in deuterochloroform or acetone-d6 with residual protonated solvent or solvent carbon signals as internal standards. Abbreviations for multiplicity are s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Flash chromatography was carried out on silica gel 60 according to the procedure of Still et al.35 Analytical thin layer chromatography (t.l.c.) was conducted on aluminium-backed 2 mm thick silica gel 60 F254 and chromatograms were visualized with UV light or ceric ammonium molybdate (Hanessian's stain). High resolution mass spectra (HRMS) were obtained using an ESI-TOF-MS; all samples were run using 0.1% formic acid. Dry CH2Cl2, THF, and Et2O were obtained from a dry solvent apparatus (Glass Contour of SG Water, Nashua, USA) as per the procedure of Pangborn et al.36 Dry DMF was dried over 4 Å molecular sieves, pyridine was dried over KOH. Pet. spirits refers to petroleum ether, boiling range 40–60 °C.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 petroleum ether/EtOAc to afford the oxime 1 as white needles (8.85 g, 81%), mp = 104–110 °C; [α]20D −185.9 (c 0.7, CHCl3; lit.22 [α]20D −100.5); 1H NMR (400 MHz, CDCl3) δ 7.71 (1 H, s, OH), 5.52 (1 H, s, H1), 4.69–4.63 (1 H, m, H5), 3.93 (1 H, d, J = 7.2 Hz, H6), 3.88 (1 H, t, J = 5.7 Hz, H6′), 3.07 (1 H, dd, J = 16.9, 7.3 Hz, H3), 2.25 (1 H, ddd, J = 16.9, 11.6, 8.2 Hz, H3′), 2.13–2.01 (1 H, m, H4), 1.76 (1 H, dd, J = 13.8, 8.2 Hz, H4′); 13C NMR (150 MHz, CDCl3) δ 154.1 (C2), 100.2 (C1), 73.3 (C5), 67.0 (C6), 27.5 (C4), 16.0 (C3); IR (thin film) ν 689, 741, 883, 950, 1105, 1170, 1428, 1449, 2900, 3299 cm−1.
:70) of the residue afforded a colourless solid, which was recrystallized from MeOH/pet. spirits to give compound 2 as white needles (685 mg, 78%), mp = 72–76 °C; [α]20D −82.6 (c 0.66, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.47(2 H, app. d, J = 8.3 Hz, Ar), 7.20 (2 H, app. d, J = 8.2 Hz, Ar), 5.51 (1 H, s, H1), 5.00 (2 H, s, CH2), 4.66–4.62 (1 H, m, H5), 3.91 (1 H, d, J = 7.0 Hz, H6), 3.87 (1 H, t, J = 6.2 Hz, H6′), 3.01 (1 H, dd, J = 17.0, 7.3 Hz, H3), 2.25 (1 H, ddd, J = 17.0, 11.5, 8.2 Hz, H3′), 2.09–2.00 (1 H, m, H4), 1.73 (1 H, dd, J = 14.1, 8.5 Hz, H4′); 13C NMR (150 MHz, CDCl3) δ 153.4 (C2), 100.2 (C1), 73.2 (C5), 67.2 (C6), 27.5 (C4), 16.7 (C3), 75.1 (CH2Ar), 121.8, 129.7, 131.5, 136.6 (6 C, Ar); IR (thin film) ν 685, 722, 782, 797, 870, 885, 983, 1103, 1107, 1339, 1588, 2891, 2935, 2969 cm−1.
:70) of the residue afforded a solid that was recrystallized from CH2Cl2/Et2O to give the oxime as beige blocks (250 mg, 26%), mp = 125–128 °C; [α]20D −116.6 (c 0.64, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.21 (2 H, app. d, J = 9.3 Hz, Ar), 7.27 (2 H, app. d, J = 9.5 Hz, Ar), 5.70 (1 H, s, H1), 4.73 (1 H, br s, H5), 4.0 (1 H, d, J = 7.3 Hz, H6), 3.95 (1 H, t, J = 6.3 Hz, H6′), 3.24 (1 H, dd, J = 17.1, 7.2 Hz, H3), 2.48 (1 H, ddd, J = 17.1, 11.6, 8.3 Hz, H3′), 2.21–2.12 (1 H, m, H4), 1.86 (1 H, dd, J = 13.7, 8.5 Hz, H4′);13C NMR (150 MHz, CDCl3) δ 157.9 (C2), 142.5, 114.4, 125.6, 163.71 (6 C, Ar), 99.6 (C1), 73.2 (C5), 67.5 (C6), 27.7 (C4), 17.5 (C3); IR (thin film) ν 701, 749, 883, 1105, 1231, 1338, 1484, 1509, 1587, 1608, 1725, 2854, 2924 cm−1.
:1 petroleum ether/EtOAc to afford compound 5 as white needles (400 mg, 61%), mp = 40–46 °C; [α]20D −292.7 (c 1.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.71 (1 H, s, H1), 4.69 (1 H, br s, H5), 3.97 (1 H, d, J = 7.3 Hz, H6), 3.91 (1 H, t, J = 6.9 Hz, H6′), 3.06 (1 H, dd, J = 17.0, 7.2 Hz, H3), 2.39 (1 H, ddd, J = 17.0, 11.7, 8.2 Hz, H3′), 2.16–2.07 (1 H, m, H4), 1.181 (1 H, dd, J = 14.0, 8.2 Hz, H4′); 13C NMR (150 MHz, d6-acetone) δ 168.6 (CO), 159.7 (C2), 99.6 (C1), 73.1 (C5), 67.5 (C6), 27.7 (C4), 17.9 (C3), 19.5 (CH3); IR (thin film) ν 677, 796, 864, 921, 966, 988, 1110, 1166, 1369, 1432, 1654, 1760, 2910, 2980 cm−1.
:1 MeOH/CH2Cl2 to afford compound 6 as colorless blocks (471 mg, 58%), mp = 79–83 °C; [α]20D −135.2 (c 0.73, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.82 (1 H, s, Ar), 8.46 (1 H, d, J = 8.2 Hz, Ar), 8.39 (1 H, d, J = 8.9 Hz, Ar), 7.70 (1 H, t, J = 8.0 Hz, Ar), 5.82 (1 H, s, H1), 4.74 (1 H, br s, H5), 4.01 (1 H, d, J = 7.4 Hz, H6), 3.95 (1 H, t, J = 6.3 Hz, H6′), 3.21 (1 H, dd, J = 17.0, 7.2 Hz, H3), 2.60 (1 H, ddd, J = 17.1, 11.6, 8.3 Hz, H3′), 2.26–2.15 (1 H, m, H4), 1.90 (1 H, dd, J = 13.7, 8.5 Hz, H4′); 13C NMR (150 MHz, CDCl3) δ 161.82, 161.78 (C2, CO), 148.3, 135.4, 130.5, 130.1, 127.9, 124.4 (6 C, Ar), 99.3 (C1), 73.2 (C5), 67.6 (C6), 27.7 (C4), 18.4 (C3); IR (thin film) ν 677, 795, 864, 890, 921, 966, 988, 1110, 1167, 1186, 1203, 1222, 1344, 1369, 1432, 1653, 1761, 2909, 2982 cm−1.
:1 petroleum ether/EtOAc to afford compound 7 as pale yellow blocks (105 mg, 52%), mp = 119–123 °C; [α]20D −65.7 (c 0.25, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.32 (2 H, app. d, J = 8.9 Hz, Ar), 8.22 (2 H, app. d, J = 8.9 Hz, Ar), 5.88 (1 H, s, H1), 4.75 (1 H, br s, H5), 4.00 (1 H, d, J = 7.3 Hz, H6), 3.96 (1 H, t, J = 6.2 Hz, H6′), 3.2 (1 H, dd, J = 17.0, 6.5 Hz, H3), 2.57 (1 H, ddd, J = 17.1, 11.6, 8.3 Hz, H3′), 2.2–2.1 (1 H, m, H4), 1.98 (1 H, dd, J = 13.8, 8.3 Hz, H4′); 13C NMR (150 MHz, CDCl3) δ 161.8, 161.6 (C2, CO), 150.7, 130.7, 134.3, 123.7 (6C, Ar), 99.3 (C1), 73.1 (C5), 67.6 (C6), 27.8 (C4), 18.3 (C3); IR (thin film) ν 659, 709, 864, 858, 961, 983, 1010, 1069, 1165, 1237, 1342, 1520, 1740, 1754, 2904, 2975, 3113 cm−1.
:1 Et2O/CH2Cl2 to afford compound 8 as white fine crystals (777 mg, 67%), mp = 73–77 °C; [α]20D −116.4 (c 0.56, CHCl3); 1H NMR (400 MHz, CDCl3) δ 9.27 (1 H, app. d, J = 1.7 Hz, Ar), 9.15 (2 H, app. t, J = 3.4 Hz, Ar), 5.83 (1 H, s, H1), 4.77 (1 H, br s, H5), 4.03 (1 H, d, J = 7.5 Hz, H6), 3.97 (1 H, t, J = 7.2 Hz, H6′), 3.20 (1 H, dd, J = 17.0, 7.2 Hz, H3), 2.64 (1 H, ddd, J = 19.4, 10.6, 8.8 Hz, H3′), 2.29–2.18 (1 H, m, H4), 1.94 (1 H, dd, J = 14.0, 8.2 Hz, H4′); 13C NMR (150 MHz, CDCl3) δ 162.8, 159.8 (C2, CO), 148.8, 132.6, 129.8, 122.8 (6C, Ar), 99.1 (C1), 73.1 (C5), 67.7 (C6), 27.8 (C4), 18.5 (C3); IR (thin film) ν 716, 830, 870, 910, 920, 1074, 1094, 1110, 1133, 1183, 1250, 1345, 1416, 1471, 1540, 1597, 1630, 1705, 1757, 3094 cm−1.
:1 petroleum ether/EtOAc to afford the oxime 1 as white needles (8.85 g, 81%), mp = 104–110 °C; [α]20D −185.9 (c 0.7, CHCl3; lit.22 [α]20D −100.5); 1H NMR (400 MHz, CDCl3) δ 7.71 (1 H, s, OH), 5.52 (1 H, s, H1), 4.69–4.63 (1 H, m, H5), 3.93 (1 H, d, J = 7.2 Hz, H6), 3.88 (1 H, t, J = 5.7 Hz, H6′), 3.07 (1 H, dd, J = 16.9, 7.3 Hz, H3), 2.25 (1 H, ddd, J = 16.9, 11.6, 8.2 Hz, H3′), 2.13–2.01 (1 H, m, H4), 1.76 (1 H, dd, J = 13.8, 8.2 Hz, H4′); 13C NMR (150 MHz, CDCl3) δ 154.1 (C2), 100.2 (C1), 73.3 (C5), 67.0 (C6), 27.5 (C4), 16.0 (C3); IR (thin film) ν 689, 741, 883, 950, 1105, 1170, 1428, 1449, 2900, 3299 cm−1.
:70) of the residue afforded a colourless solid, which was recrystallized from MeOH/pet. spirits to give compound 2 as white needles (685 mg, 78%), mp = 72–76 °C; [α]20D −82.6 (c 0.66, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.47(2 H, app. d, J = 8.3 Hz, Ar), 7.20 (2 H, app. d, J = 8.2 Hz, Ar), 5.51 (1 H, s, H1), 5.00 (2 H, s, CH2), 4.66–4.62 (1 H, m, H5), 3.91 (1 H, d, J = 7.0 Hz, H6), 3.87 (1 H, t, J = 6.2 Hz, H6′), 3.01 (1 H, dd, J = 17.0, 7.3 Hz, H3), 2.25 (1 H, ddd, J = 17.0, 11.5, 8.2 Hz, H3′), 2.09–2.00 (1 H, m, H4), 1.73 (1 H, dd, J = 14.1, 8.5 Hz, H4′); 13C NMR (150 MHz, CDCl3) δ 153.4 (C2), 100.2 (C1), 73.2 (C5), 67.2 (C6), 27.5 (C4), 16.7 (C3), 75.1 (CH2Ar), 121.8, 129.7, 131.5, 136.6 (6 C, Ar); IR (thin film) ν 685, 722, 782, 797, 870, 885, 983, 1103, 1107, 1339, 1588, 2891, 2935, 2969 cm−1.
:70) of the residue afforded a solid that was recrystallized from CH2Cl2/Et2O to give the oxime as beige blocks (250 mg, 26%), mp = 125–128 °C; [α]20D −116.6 (c 0.64, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.21 (2 H, app. d, J = 9.3 Hz, Ar), 7.27 (2 H, app. d, J = 9.5 Hz, Ar), 5.70 (1 H, s, H1), 4.73 (1 H, br s, H5), 4.0 (1 H, d, J = 7.3 Hz, H6), 3.95 (1 H, t, J = 6.3 Hz, H6′), 3.24 (1 H, dd, J = 17.1, 7.2 Hz, H3), 2.48 (1 H, ddd, J = 17.1, 11.6, 8.3 Hz, H3′), 2.21–2.12 (1 H, m, H4), 1.86 (1 H, dd, J = 13.7, 8.5 Hz, H4′);13C NMR (150 MHz, CDCl3) δ 157.9 (C2), 142.5, 114.4, 125.6, 163.71 (6 C, Ar), 99.6 (C1), 73.2 (C5), 67.5 (C6), 27.7 (C4), 17.5 (C3); IR (thin film) ν 701, 749, 883, 1105, 1231, 1338, 1484, 1509, 1587, 1608, 1725, 2854, 2924 cm−1.
:1 petroleum ether/EtOAc to afford compound 5 as white needles (400 mg, 61%), mp = 40–46 °C; [α]20D −292.7 (c 1.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.71 (1 H, s, H1), 4.69 (1 H, br s, H5), 3.97 (1 H, d, J = 7.3 Hz, H6), 3.91 (1 H, t, J = 6.9 Hz, H6′), 3.06 (1 H, dd, J = 17.0, 7.2 Hz, H3), 2.39 (1 H, ddd, J = 17.0, 11.7, 8.2 Hz, H3′), 2.16–2.07 (1 H, m, H4), 1.181 (1 H, dd, J = 14.0, 8.2 Hz, H4′); 13C NMR (150 MHz, d6-acetone) δ 168.6 (CO), 159.7 (C2), 99.6 (C1), 73.1 (C5), 67.5 (C6), 27.7 (C4), 17.9 (C3), 19.5 (CH3); IR (thin film) ν 677, 796, 864, 921, 966, 988, 1110, 1166, 1369, 1432, 1654, 1760, 2910, 2980 cm−1.
:1 MeOH/CH2Cl2 to afford compound 6 as colorless blocks (471 mg, 58%), mp = 79–83 °C; [α]20D −135.2 (c 0.73, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.82 (1 H, s, Ar), 8.46 (1 H, d, J = 8.2 Hz, Ar), 8.39 (1 H, d, J = 8.9 Hz, Ar), 7.70 (1 H, t, J = 8.0 Hz, Ar), 5.82 (1 H, s, H1), 4.74 (1 H, br s, H5), 4.01 (1 H, d, J = 7.4 Hz, H6), 3.95 (1 H, t, J = 6.3 Hz, H6′), 3.21 (1 H, dd, J = 17.0, 7.2 Hz, H3), 2.60 (1 H, ddd, J = 17.1, 11.6, 8.3 Hz, H3′), 2.26–2.15 (1 H, m, H4), 1.90 (1 H, dd, J = 13.7, 8.5 Hz, H4′); 13C NMR (150 MHz, CDCl3) δ 161.82, 161.78 (C2, CO), 148.3, 135.4, 130.5, 130.1, 127.9, 124.4 (6 C, Ar), 99.3 (C1), 73.2 (C5), 67.6 (C6), 27.7 (C4), 18.4 (C3); IR (thin film) ν 677, 795, 864, 890, 921, 966, 988, 1110, 1167, 1186, 1203, 1222, 1344, 1369, 1432, 1653, 1761, 2909, 2982 cm−1.
:1 petroleum ether/EtOAc to afford compound 7 as pale yellow blocks (105 mg, 52%), mp = 119–123 °C; [α]20D −65.7 (c 0.25, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.32 (2 H, app. d, J = 8.9 Hz, Ar), 8.22 (2 H, app. d, J = 8.9 Hz, Ar), 5.88 (1 H, s, H1), 4.75 (1 H, br s, H5), 4.00 (1 H, d, J = 7.3 Hz, H6), 3.96 (1 H, t, J = 6.2 Hz, H6′), 3.2 (1 H, dd, J = 17.0, 6.5 Hz, H3), 2.57 (1 H, ddd, J = 17.1, 11.6, 8.3 Hz, H3′), 2.2–2.1 (1 H, m, H4), 1.98 (1 H, dd, J = 13.8, 8.3 Hz, H4′); 13C NMR (150 MHz, CDCl3) δ 161.8, 161.6 (C2, CO), 150.7, 130.7, 134.3, 123.7 (6C, Ar), 99.3 (C1), 73.1 (C5), 67.6 (C6), 27.8 (C4), 18.3 (C3); IR (thin film) ν 659, 709, 864, 858, 961, 983, 1010, 1069, 1165, 1237, 1342, 1520, 1740, 1754, 2904, 2975, 3113 cm−1.
:1 Et2O/CH2Cl2 to afford compound 8 as white fine crystals (777 mg, 67%), mp = 73–77 °C; [α]20D −116.4 (c 0.56, CHCl3); 1H NMR (400 MHz, CDCl3) δ 9.27 (1 H, app. d, J = 1.7 Hz, Ar), 9.15 (2 H, app. t, J = 3.4 Hz, Ar), 5.83 (1 H, s, H1), 4.77 (1 H, br s, H5), 4.03 (1 H, d, J = 7.5 Hz, H6), 3.97 (1 H, t, J = 7.2 Hz, H6′), 3.20 (1 H, dd, J = 17.0, 7.2 Hz, H3), 2.64 (1 H, ddd, J = 19.4, 10.6, 8.8 Hz, H3′), 2.29–2.18 (1 H, m, H4), 1.94 (1 H, dd, J = 14.0, 8.2 Hz, H4′); 13C NMR (150 MHz, CDCl3) δ 162.8, 159.8 (C2, CO), 148.8, 132.6, 129.8, 122.8 (6C, Ar), 99.1 (C1), 73.1 (C5), 67.7 (C6), 27.8 (C4), 18.5 (C3); IR (thin film) ν 716, 830, 870, 910, 920, 1074, 1094, 1110, 1133, 1183, 1250, 1345, 1416, 1471, 1540, 1597, 1630, 1705, 1757, 3094 cm−1.
Major compound: 1H NMR (600 MHz, CDCl3) δ 8.09 (1 H, s, OCOH), 5.28–5.24 (1 H, m, H5), 4.28 (1 H, dd, J = 12.1, 3.7, H6), 4.10 (1 H, dd, J = 12.3, 5.3 Hz, H'6), 2.44 (2 H, ddd, J = 11.7, 7.5, 4.1 Hz, H3), 2.07 (3 H, s, CH3), 2.02 (2 H, t, J = 7.4 Hz, H4); 13C NMR (151 MHz, CDCl3) δ 170.4, 160.0, 118.4, 69.4, 63.8, 26.7, 20.6, 13.5.
Minor compound: 1H NMR (600 MHz, CDCl3) δ 8.06 (1 H, s, OCOH), 5.18–5.09 (1 H, m, H5), 4.37 (1 H, dd, J = 12.1, 3.7, H6), 4.17 (1 H, dd, J = 12.3, 5.3 Hz, H6′), 2.44 (2 H, ddd, J = 11.7, 7.5, 4.1 Hz, H3), 2.10 (3 H, s, CH3), 2.02 (2 H, t, J = 7.4 Hz, H4); 13C NMR (151 MHz, CDCl3) δ 170.2, 160.2, 118.5, 69.5, 63.4, 26.7, 20.8, 13.6; HRMS (ESI)+m/z 203.1027 [C8H15N2O4+ (M + NH4)+ requires 203.1026].
IR (thin film) ν 752.8, 1050, 1161, 1231.5, 1370, 1428.7, 1722.3, 2245.8, 2923.6.
Crystallography
Intensity data for 1–7 were collected with an Oxford Diffraction SuperNova CCD diffractometer using either Mo-Kα or Cu-Kα radiation, the temperature during all data collections was maintained at 130.0(1) using an Oxford Cryosystems cooling device. The data for 8 was collected on the MX1 beamline at the Australian Synchrotron at 100 K.37 The structures were solved by direct methods and difference Fourier synthesis.24 Thermal ellipsoid plots were generated using the program ORTEP-338 integrated within the WINGX suite of programs.39005 reflections measured, 7762 independent reflections (Rint = 0.034) the final R = 0.0408 [I > 2σ(I), 6894 data] and wR(F2) = 0.1076 (all data) GOOF = 1.087. Absolute structure parameter 0.1(2). CCDC 1578343.†
227 reflections measured, 10861 independent reflections (Rint = 0.0312) the final R = 0.0471 [I > 2σ(I), 8361 data] and wR(F2) = 0.1228 (all data) GOOF = 1.049. Absolute structure parameter −0.027(7). CCDC 1578344.†
018 reflections measured, 4053 independent reflections (Rint = 0.0256) the final R = 0.0395 [I > 2σ(I), 3573 data] and wR(F2) = 0.1102 (all data) GOOF = 1.068. Absolute structure parameter −0.3(3). CCDC 1578348.†
600 reflections measured, 8050 independent reflections (Rint = 0.0271) the final R = 0.0379 [I > 2σ(I), 7008 data] and wR(F2) = 0.1085 (all data) GOOF = 1.074. Absolute structure parameter 0.0(2). CCDC 1578347.†
391 reflections measured, 12230 independent reflections (Rint = 0.0306) the final R = 0.0495 [I > 2σ(I), 9425 data] and wR(F2) = 0.1225 (all data) GOOF = 1.022. Absolute structure parameter 0.1(6). CCDC 1578349.†
414 reflections measured, 3333 independent reflections (Rint = 0.0338) the final R = 0.0339 [I > 2σ(I), 3118 data] and wR(F2) = 0.0906 (all data) GOOF = 1.080. Absolute structure parameter −0.1(2). CCDC 1578350.†
Computational methods
All geometry optimizations and single-point energy calculations were done using the ORCA 4.0.0 program.40 Geometry optimizations of all structures were performed at the dispersion-corrected TPSS41-D3(BJ)42,43/def2-TZVP44,45 level of theory together with the resolution of the identity approximation to the Coulomb integral evaluation (RI-J).46 The ORCA numerical quadrature grid option “4” was chosen and a self-consistent-field (SCF) convergence criterion of 10−7Eh. After obtaining converged MOs in single-point calculations, NBO analyses were performed with the NBO6.047 program based on five different DFT approximations and the def2-TZVP basis set: PW6B95,48 TPSS, revPBE,49 B3LYP,50,51 and PBE0.52,53 These DFT approximations were chosen either for their known accuracy (PW6B95, TPSS, revPBE) or their popularity (B3LYP, PBE0).31 Note that an additive dispersion correction of the DFT-D3 type does not influence the MOs and their energies, hence usage of DFT-D3(BJ) was not necessary for this part of the analysis. The numerical quadrature grid and SCF convergence options in all single-point calculations were the same as for the geometry optimizations. All (meta-)GGA calculations were again carried out with the RI-J approximation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Australian Research Council (ARC) for financial support. SJW is an ARC-funded Future Fellow (FT130100103), and LG received support as an ARC DECRA Fellow (DE140100550). AA is grateful to Jazan University for financial support and thanks the Saudi Arabian Cultural Mission (SACM). LG is also grateful for generous allocation of computer time and technical support by The University of Melbourne, Melbourne Bioinformatics (project RA0005), and the National Computational Infrastructure Facility in Canberra through the National Computational Merit Allocation Scheme (project fk5). We thank Circa Group for a generous gift of Cyrene.References
- R. E. Gawley, Org. React., 1988, 35, 14–24 Search PubMed.
- E. Beckmann, Ber., 1886, 19, 988–993 CrossRef.
- A. H. Blatt, Chem. Rev., 1933, 12, 215–260 CrossRef CAS.
- B. Jones, Chem. Rev., 1944, 35, 335–350 CrossRef CAS.
- O. Wallach, Justus Liebigs Ann. Chem., 1899, 309, 1–31 CrossRef CAS.
- C. A. Grob, Angew. Chem., Int. Ed. Engl., 1969, 8, 535–546 CrossRef CAS.
- A. F. Ferris, J. Org. Chem., 1960, 25, 12–18 CrossRef CAS.
- R. K. Hill, B. G. McKinnie, R. T. Conley, P. S. Darby, H. van Halbeek and E. M. Holt, Tetrahedron, 1988, 44, 3405–3412 CrossRef CAS.
- H. Fujioka, H. Yamamoto, M. Miyazaki, T. Yamanaka, K. Takuma and Y. Kita, Tetrahedron Lett., 1991, 32, 5367–5368 CrossRef CAS.
- C. A. Grob, H. P. Fischer, H. Link and E. Renk, Helv. Chim. Acta, 1963, 46, 1190–1206 CrossRef CAS.
- R. P. Bakale, M. A. Scialdone and C. R. Johnson, J. Am. Chem. Soc., 1990, 112, 6729–6731 CrossRef CAS.
- P. A. Petukhov and A. V. Tkachev, Tetrahedron, 1997, 53, 2535–2550 CrossRef CAS.
- I. V. Alabugin, in Stereoelectronic Effects, John Wiley & Sons, Ltd, 2016, pp. 183–213, DOI:10.1002/9781118906378.ch7.
- H. B. Burgi, J. D. Dunitz, J. M. Lehn and G. Wipff, Tetrahedron, 1974, 30, 1563–1572 CrossRef.
- H. B. Burgi, J. D. Dunitz and E. Shefter, J. Am. Chem. Soc., 1973, 95, 5065–5067 CrossRef CAS.
- P. Murrayrust, H. B. Burgi and J. D. Dunitz, J. Am. Chem. Soc., 1975, 97, 921–922 CrossRef CAS.
- F. H. Allen and A. J. Kirby, J. Am. Chem. Soc., 1984, 106, 6197–6200 CrossRef CAS.
- A. J. Briggs, R. Glenn, P. G. Jones, A. J. Kirby and P. Ramaswamy, J. Am. Chem. Soc., 1984, 106, 6200–6206 CrossRef CAS.
- P. G. Jones and A. J. Kirby, J. Am. Chem. Soc., 1984, 106, 6207–6212 CrossRef CAS.
- S. D. Yeoh, B. L. Harris, T. J. Simons and J. M. White, Aust. J. Chem., 2012, 65, 905–917 CrossRef CAS.
- J. Sherwood, M. De bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650–9652 RSC.
- F. A. Valeev, E. V. Gorobets, I. P. Tsypysheva, G. S. Singizova, L. K. Kalimullina, M. G. Safarov, O. V. Shitikova and M. S. Miftakhov, Chem. Nat. Compd., 2003, 39, 563–568 CrossRef CAS.
- J. A. Dean, Lange's Handbook of Chemistry, McGraw-Hill Inc, New York, 1999 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- G. R. Jones, S. Caldarelli and P. Vogel, Helv. Chim. Acta, 1997, 80, 59–64 CrossRef CAS.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 1–42 CrossRef CAS.
- I. V. Alabugin, K. M. Gilmore and P. W. Peterson, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 109–141 CrossRef CAS.
- I. V. Alabugin and M. Manoharan, J. Org. Chem., 2004, 69, 9011–9024 CrossRef CAS PubMed.
- I. V. Alabugin and T. A. Zeidan, J. Am. Chem. Soc., 2002, 124, 3175–3185 CrossRef CAS PubMed.
- (a) L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC; (b) L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi and S. Grimme, Phys. Chem. Chem. Phys., 2017 10.1039/c7cp04913g.
- I. V. Alabugin, S. Bresch and G. dos Passos Gomes, J. Phys. Org. Chem., 2015, 28, 147–162 CrossRef CAS.
- P. G. Jones, M. R. Edwards and A. J. Kirby, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 1230–1232 CrossRef.
- M. R. Edwards, A. J. Kirby, P. R. Raithby and P. G. Jones, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 300–303 CrossRef.
- W. C. Still, M. Kahn and A. M. Mitra, J. Org. Chem., 1978, 43, 2923–2925 CrossRef CAS.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- N. P. Cowieson, D. Aragao, M. Clift, D. J. Ericsson, C. Gee, S. J. Harrop, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Williamson and T. Caradoc-Davies, J. Synchrotron Radiat., 2015, 22, 187–190 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, 2013.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2005, 109, 5656–5667 CrossRef CAS PubMed.
- Y. Zhang and W. Yang, Phys. Rev. Lett., 1998, 80, 890–890 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Chem. Phys., 1994, 98, 11623–11627 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1578343–1578350. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ob02499a |
| This journal is © The Royal Society of Chemistry 2017 |