
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel sulfonamide non-classical carbenoid: a mechanistic study for the synthesis of enediynes†
Theodore O. P.
Hayes
 a,
Ben
Slater
a,
Richard A. J.
Horan
b,
Marc
Radigois
a and
Jonathan D.
Wilden
*a
a,
Ben
Slater
a,
Richard A. J.
Horan
b,
Marc
Radigois
a and
Jonathan D.
Wilden
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: j.wilden@ucl.ac.uk
bGlaxoSmithkline, Medicines Research Centre, Gunnels Wood Road, Stevenage, Hertfordshire SG1 2NY, UK
First published on 20th November 2017
Abstract
Alkynyl sulfonamides undergo sequential 1,4- then 1,2-addition/rearrangement with lithium acetylides to yield enediynes in the absence of any promoters or catalysts. Mechanistic investigations suggest that the reaction proceeds via 1,4-conjugate addition of the nucleophile to the unsaturated system to give a key alkenyl lithium species which is stabilised by an intramolecular coordination effect by a sulfonamide oxygen atom. This species can be considered a vinylidene carbenoid given the carbon atom bears both an anion (as a vinyllithium) and a leaving group (the sulfonamide). The intramolecular coordination effect serves to stabilise the vinyllithium but activates the sulfonamide motif towards nucleophilic attack by a second mole of acetylide. The resulting species can then undergo rearrangement to yield the enediyne framework in a single operation with concomitant loss of aminosulfinate.
Introduction
Enediynes are a fascinating class of anti-cancer1 and antibiotic2 molecules with a highly unusual chemical structure which has been of interest to synthetic and medicinal chemists for a number of years.3–5 Their biological mechanism of targeted DNA damage (strand cleavage via the Bergman cyclisation)6 has made the family of natural products to which they belong popular targets for manipulation and functionalisation to improve their drug-like qualities. Many synthetic approaches to enediynes and related compounds however employ toxic and/or expensive protocols based on transition or heavy metals such as tin,7 copper,8 palladium,9 or zinc.10 Research in our own laboratory has in recent years focused on transition metal free reactions where the benign properties of earth-abundant elements such as lithium,11 potassium,12 sodium and sulfur13 are exploited. In addition, electrophilic vinylidene carbenoids have long been invoked in the preparation of complex molecules and elegant structural work14a–c has proposed a structure of an electrophilic, metalated intermediate 1 where the leaving group bridges the carbon atom and the metal and retains the stereochemical information of the starting metallated alkene leading to attack at the opposite face by nucleophiles (outlined in Scheme 1).14b In some cases, full dissociation to give the metallated cationic intermediate 1a has also been suggested, although the associated model is generally preferred given the preservation of stereochemical information.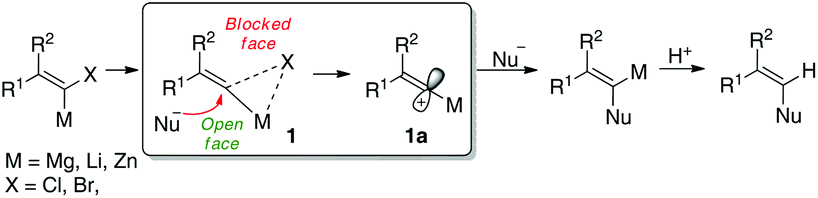 | ||
| Scheme 1 Classical electrophilic carbenoid model. | ||
We here report a protocol where unsymmetrical enediynes can be prepared via simple 1,4-addition of lithium acetylides to alkynyl sulfonamides. At first we considered that the mechanistic pathway would be similar to that described by Satoh, involving a carbenoid intermediate which undergoes a [1,2] shift to yield a diyne product (Scheme 2).15 However, mechanistic studies have revealed a distinct pathway from other work which allows control of the carbon–carbon bond forming process. In addition, our mechanistic study helps to explain and support other observations in the recent literature.
 | ||
| Scheme 2 Comparison of this and previous work. | ||
Results and discussion
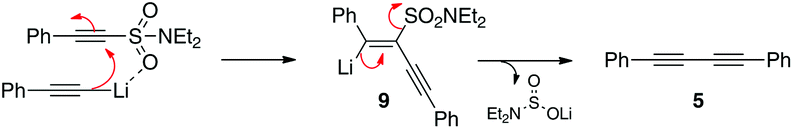
In our ongoing investigations into the chemistry of alkynes and organosulfur chemistry, we made the observation that exposure of the unsaturated sulfonamide 2 to a lithium acetylide (e.g.3) leads to three products; what appears to be the 1,4-addition product 4, diyne 5 and enediyne 6 (Scheme 3). Furthermore, increasing the temperature of the reaction improves the yield of the diyne and enediyne. Interestingly, both 4 and 6 were isolated as single geometrical isomers. It was also noticed that for enediyne 6, the incoming nucleophilic alkyne adopts the same configuration as the parent sulfonamide. | ||
| Scheme 3 Initial observations. | ||
These initial results immediately suggested initial 1,4-addition of the alkyne nucleophile to the conjugated system followed by slower conversion to the products 5 and 6 (via carbene/carbenoid pathways with loss of aminosulfinate 8). At this stage however, we did not know whether 5 and 6 were formed directly from 4 or were the result of other secondary reactions. The various pathways we initially envisaged are outlined in Scheme 4 and we then set out to explore the various possibilities.
 | ||
| Scheme 4 Possible reaction pathways: α-addition pathway in red, β-addition pathway in blue and the carbenoid pathway in green. | ||
We initially wanted to demonstrate that none of the observed products were resulting from nucleophilic addition reactions at sulfur leading to secondary reactions or sulfonyl exchange processes (Scheme 5, which is known for some sulfones16 but, to our knowledge, have not been described for sulfonamides).
 | ||
| Scheme 5 Potential exchange reactions. | ||
We therefore repeated the reaction with different lithium acetylides. Scheme 6 outlines our results with lithium m-methoxyphenylacetylide with 13, 14, and 15 being isolated. No symmetrical diynes or mixtures of enediynes were observed indicating that sulfonyl exchange is not occurring in this case and gives us confidence that the method will in due course be adaptable to the preparation of various different non-symmetrical analogues.
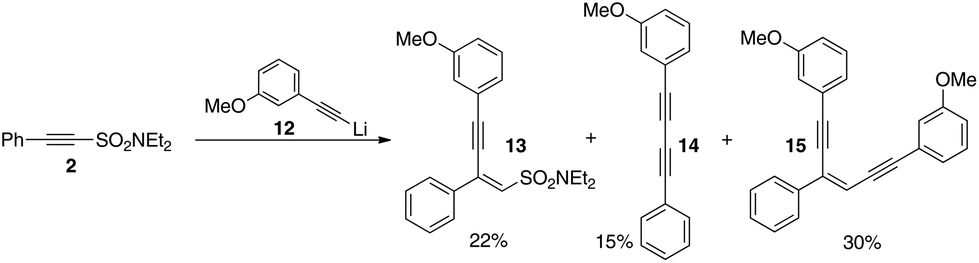 | ||
| Scheme 6 Non-symmetrical diynes and enediynes without exchange (both 13 and 15 isolated as pure Z-geometrical isomers). | ||
Satisfied that exchange reactions were not taking place we then proceeded to examine how exactly the various products were formed. Initially we suspected that classical 1,4-addition to the alkynyl sulfonamide led to the intermediate alkenyllithium 7 which then was protonated either by adventitious moisture in the reaction mixture or at the work-up stage. The stereochemistry of the addition product (an anti-carbolithiation of the alkynyl sulfonamide) would be expected based on the work of Maddaluno17 who has studied related systems and noted that carbolithiations of alkynes result in a pro-E transition state when the organolithium can coordinate to a substituent on the electrophilic alkyne (Scheme 7). This would explain our observation that only a single geometrical isomer of 4 is formed.
 | ||
| Scheme 7 Anti-carbolithiation of 2. | ||
At this stage, we strongly suspected that the enediyne was forming from addition of a second mole of lithium acetylide to 4 or from addition to the diyne 5. This we tested by isolating both components and exposing them independently to additional lithium acetylide. To our surprise no reaction was observed (Scheme 9). This also suggested to us that a vinylidene carbene might be responsible for the observed reactivity, however all of our attempts to trap such an intermediate (e.g. via the addition of excess ethyl vinyl ether) failed.
With these preliminary experiments and control reactions in hand we then proceeded to consider the mechanism in more detail. We were first drawn to the incongruous observation outlined in Scheme 8 where work-up of the reaction with D2O led to expected isotope incorporation into 13a but not into enediyne 15. We reasoned the intermediate 1,4-addition product is relatively stable and exhibits (sluggish) carbenoid behavior at low temperatures only when the reacting partner is highly reactive. The formation of a non-deuterated enediyne was initially more puzzling as via our postulated mechanism, this compound would also be expected to be deuterated as the enediyne anion appears to be the end-point for any reaction sequence. As might be expected for the formation of the enediyne, increasing the number of equivalents of the acetylide favoured its formation. When four equivalents were employed the yield of the enediyne 6 increases to 53% with an associated drop in the yields of the other two components (16% for 5 and 8% for 4). Most tellingly however, when a mixture of lithium phenylacetylide (2 eq.) and parent alkyne (2 eq.) were employed, the yield of the enediyne increased dramatically to 64% with only trace amounts of the other components detected (Scheme 10). Crucially, when the deuterated parent alkyne was employed in the same reaction, the deuterium label was identified in the enediyne product 18.
 | ||
| Scheme 8 Deuterium incorporation following anti-carbolithiation. | ||
 | ||
| Scheme 9 Ruling out secondary addition reactions. | ||
 | ||
| Scheme 10 Initial mechanistic studies via deuterium incorporation. | ||
We therefore reasoned that 7 is moderately stable and can persist in the reaction medium. Its stability is such that it is not protonated by weak proton donors such as phenylacetylene but can survive until the point where the reaction is quenched at the work-up stage. In contrast, the lithiated enediyne is highly reactive and is protonated rapidly even by trace proton sources. The addition of excess phenylacetylene provides a weak proton source that can quench this reactive anion while not interfering with any putative carbenoid or similar precursor.
These results prompted us to consider more carefully the precise nature of the reactive intermediate. We first considered that a classic electrophilic carbenoid pathway was operating (such as that outlined in Scheme 1) where the incoming nucleophile adopts the stereochemistry of the metal in the carbenoid intermediate (Scheme 1). By contrast, in our systems, the nucleophile adopts the opposite stereochemistry (Schemes 3 and 6) suggesting that a pathway distinct from both the classical carbenoid and free carbene mechanisms is in operation.
Given that the reactive intermediate in our reaction appears to exhibit no classical carbene (carbenoid) behavior, and no products of the Fritsch–Weichell–Buttenberg (FWB) rearrangement have been observed in these systems (in this or our related work) we were initially puzzled as to how the mechanistic pathway might be operating. We briefly considered a structure where the sulfonyl group fully dissociates to leave a lithiated vinyl cation (again, invoked in much of the early work on carbenoids, Scheme 1, compound 1a).14a–c Such intermediates however are known to rapidly undergo intramolecular FWB rearrangements even at low temperatures. Similarly, competitive intermolecular addition with the FWB rearrangement was reasoned to be highly unlikely, particularly given the reaction times and elevated temperatures required to maximize the conversion to the enediyne. As such, we have suggested that the sulfonyl group must stay associated with the lithiated alkene prior to the attack of the nucleophile and therefore inhibit the classical carbene/carbenoid behavior that might be expected with superior leaving groups. Since the reaction proceeds with retention of stereochemistry with respect to the sulfonamide motif, we have therefore postulated that once the 1,4-addition has taken place, the incoming nucleophile (unusually for sulfonamides) initially attacks the sulfur atom (structures 19 and 20, Scheme 11). As the temperature increases, weakening of the C–S bond then allows the alkyne nucleophile to be delivered to the cationic carbon atom (structure 21) and in doing so the stereochemistry of the alkene is preserved. We also considered that a coordination effect by the adjacent lithium atom might assist in promoting the nucleophilic attack at sulfur.
 | ||
| Scheme 11 Cooperative reactivity via nucleophilic attack at sulfur. | ||
Further support for a process following this route was found when first-principles molecular dynamics simulations were performed. These suggested that the C–S bond in compound 19 was extremely robust, varying from 1.73 Å ± 0.12 Å over a 5000 fs run at 350 K. It is therefore highly unlikely to spontaenously cleave to yield a metallated cationic intermediate such as 1a. This data, combined with our other observations would suggest a pathway where the sulfur atom directs the incoming nucleophile to obtain the observed enediyne product is more likely. Such a pathway would also support recent literature observations where phosphonium salts have been employed in a similar fashion; specifically the ‘attack@P’ mechanism proposed by Radosevich where an alkynyl phosphonium was employed with a lithium acetylide to give enediynes.18
In light of the fact that we had established that 4 did not react with lithium acetylides, we realized that our hypothesis could be tested if 4 could be lithiated independently. As such a sample of 4 was independently exposed to one equivalent of lithium tetramethylpiperidine in order to form 7. This was then exposed to the anion derived from phenylacetylene followed by aqueous work-up. This resulted in the enediyne 6 in moderate yield, the remainder of the mixture being recovered starting material (Scheme 12). In addition to this, β-diphenyldiethylvinyl sulfonamide 23 could also be lithiated with lithium tetramethylpiperidine (LiTMP) and reacts with lithium phenylacetylide to give the enyne 24 in good yield (Scheme 13).
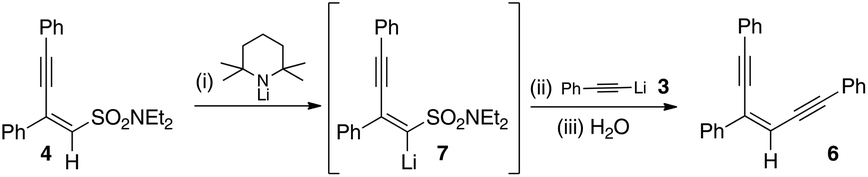 | ||
| Scheme 12 Independent lithiation and reaction of 7. | ||
 | ||
| Scheme 13 Alternative substrate. | ||
Furthermore, we were able to demonstrate the basic character of the intermediate vinyllithium by preparing the 2-methylphenylacetylinic sulfonamide 25 as depicted in Scheme 14. Exposure of this material to lithium phenylacetylide 3 in the same manner as previously would necessarily allow the labile C–H bonds of the benzylic methyl group to come into close contact with the carbenoid centre 26.
 | ||
| Scheme 14 Basicity of the carbenoid centre. | ||
A reactive intermediate exhibiting classical carbene reactivity would be expected to undergo (at least in part) C–H insertion reactions to yield cyclic products. In fact, no cyclic or enediyne products were observed. The major product of the reaction was the monoaddition product which presumably results from initial 1,4-addition (via the pro-E TS) to give the vinyllithium which is then rapidly protonated by the pendant benzylic CH3 group yielding the benzylic anion 27. Protonation on work-up then yields the observed product 28.
At this juncture, we were curious as to the effect the sulfonamide motif was having on this reaction and whether other sulfonyl acetylenes would participate in similar reactions. We were also interested to see if other metal cations might promote or attenuate the reaction. As the sulfonamide unit is well-known as only weakly activating in 1,4-addition reactions,19 we reasoned that replacing this unit with the trifluoromethanesulfonyl moiety would result in a more facile reaction. To our surprise, heating the alkynyl trifluoromethylsulfone 29 (as shown in Scheme 15) with lithium phenylacetylide resulted in none of compounds 30 or 31 being produced, even with extended reaction times and elevated temperatures. Only the starting compound 29 was recovered. Considering that the triflyl group is one of the most powerfully electron withdrawing group commonly employed in organic synthesis, this was somewhat unexpected. Similarly, generating the magnesium, sodium or potassium salts of the acetylide and reacting with 2 also resulted in no reaction occurring with the persistence of starting materials even after several hours at elevated temperatures (60 °C) (Scheme 12).
 | ||
| Scheme 15 Unreactivity of other sulfonyl species and other metal cations. | ||
These unexpected results have led us to conclude that a unique combination of the properties of the lithium cation and the sulfonamide motif were allowing this unusual reactivity to occur. We therefore turned to computational methods to assist us in understanding the reactivity of the intermediate vinyl sulfonyl lithium species as the precursor to the carbene. Quantum chemical density functional based approaches suggested that when the cation is lithium there is a strong interaction between the metal ion and the sulfonyl group oxygen(s) with bond lengths of ca. 2 Å for both the Li–O and Li–C bonds (Li–C was calculated to have a bond distance of 1.99 Å, Li–O 1.90 Å). We reason that this interaction, facilitated by the small size of the lithium ion and high effective charge density, stabilizes the initial addition product, favouring the equilibrium to a degree that would not be expected simply by considering the relative stability of the alkenyl and acetylinic anions (Scheme 16, Fig. 1). Presumably, the sulfonamide nitrogen atom serves to increase the electron density on the oxygen atoms, strengthening the Li–O interaction compared to other sulfonyl systems. This four-membered coordination complex is strongly reminiscent of the structures proposed by Biellmann,20 Durst,21 and Chassaing and Marquet22 in their studies on lithiated sulfoxides; although their work focused on subsequent reactions with electrophiles. When the alkynyl triflone is employed, the significant electron withdrawing effect of the CF3 group appears to lower the electron density on oxygen which is likely to disrupt the Li–O interaction.
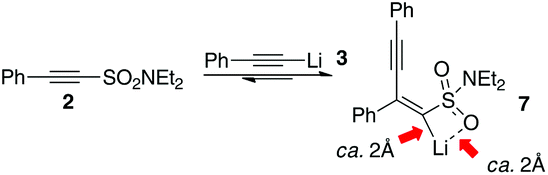 | ||
| Scheme 16 Proposed stabilised vinyllithium intermediate indicating the close interaction of carbon, lithium and oxygen atoms. | ||
 | ||
| Fig. 1 Minimum energy configuration of the proposed intermediate (the dimethylamino sulfonamide was used for the MD calculations). Lithium is displayed in pink, oxygen in red, sulfur in yellow, nitrogen in blue, carbon in cyan and hydrogen in white. | ||
The model here exhibits a distinct mode of reactivity when compared to the classical carbenoid systems described by Schleyer and others.14a–c The unique cooperativity between the lithium atom and sulfonamide group which essentially results in the displacement of a (relatively poor) leaving group at an sp2 centre is brought about by a threefold combination of properties: (i) Li–sulfonamide coordination facillitating the initial 1,4-addition reaction giving a geometrically defined addition product as depicted in Scheme 7 (ii) the formation of a sulfonamide–Li coordination complex, favouring the equilibrium shown in Scheme 16 by stabilising the addition product and (iii) activation of the sulfonyl group to nucleophilic attack by intramolecular coordination in a Lewis-acid manner as outlined in Scheme 11. As an additional note, it has occurred to us that such a coordinating effect may explain why PPh(o-An)2 proved to be a superior mediator compared to other phosphines in Radosevich's synthesis18 of enediynes since an additional coordination effect is available to stabilise the initial 1,4-addition product (32, Fig. 2).
 | ||
| Fig. 2 Potential stabilising effect of o-anisyl group in the synthesis of enediynes. | ||
Although focusing on the synthesis of enediynes we were also keen to understand the mechanism by which the minor diyne products were being formed. In addition, having invoked a carbenoid intermediate we were aware that diyne 6 could be formed by intramolecular 1,2-FWB rearrangements and we therefore chose to explore this further.
In considering the likelihood of a 1,2-FWB type rearrangement in these systems we first had to consider the relative migratory aptitude of the aryl group compared to the alkyne. Fortunately, Tykwinski et al. have demonstrated that the migratory aptitude of alkynes in FWB rearrangements is poor in coordinating solvents.23 We therefore concluded that any FWB rearrangement would most likely involve migration of the aryl group and we prepared three substrates to examine any putative rearrangements occurring. By altering the electron demand of the aryl substituent we hoped to gain some insight as to whether a 1,2-rearrangement was competing with enediyne formation. The results are outlined in Scheme 17.
 | ||
| Scheme 17 Competing reaction investigation. | ||
Clearly, increasing the electron withdrawing capability of the aromatic ring on the sulfonamide increases the proportion of diyne formed. This observation is exactly the opposite of what would be expected if a FWB rearrangement were operating. This leads us to conclude that the diyne product is most likely formed by simple 1,3-addition to the unsaturated sulfonamide, again, directed by the strong coordinating effect of the sulfonamide oxygen atoms (Scheme 18).
 | ||
| Scheme 18 Competing addition–elimination pathway. | ||
Such an addition with a strong coordinating effect will almost certainly yield the trans lithiated sulfonamide which will rapidly eliminate due to the favourable orbital overlap. This is in sharp contrast to our other work24 where less coordinating cations lead to cis anions where the elimination is much slower and consequently the anion can be trapped prior to elimination. The 1,3-addition–elimination reaction of nucleophiles with alkynyl sulfonyl compounds has been well documented for both carbon nucleophiles (Truce, Ruano)25,26 and heteroatoms (our previous work)24 however it is interesting to see that the propensity for this reaction with alkynes is much lower than for other carbon nucleophiles as these have not been reported either by Ruano or Truce's original paper where sulfones have been employed.
As a final note, it is clear that we have only presented diethyl sulfonamides as reactive partners in this study. We have also explored similar reactions with different amino groups attached to sulfur. Although we predicted that increasing the electron withdrawing capacity of the groups attached to nitrogen would facilitate the reaction by improving the group's ability to dissociate, in fact we found very little correlation on the efficiency of the reaction over a wide range of sulfonamides prepared from different amines (morpholine, N,O-dimethylhydroxylamine, piperidine). At the present time it is unclear to us why this is the case. Perhaps only a small increase in electron density on the sulfonyl oxygen atoms is required to ‘switch on’ the observed reactivity which can be provided by many amines, even less nucleophilic variants.
Conclusions
This preliminary study has uncovered some fascinating aspects of the reactivity of vinyl sulfonamides and how they might be exploited in synthesis. The reactive intermediates in this work present a new class of carbenoid distinct from those described previously with a unique reactivity profile mediated by the pendant sulfonamide motif. Most remarkably, this reactivity manifests itself in a sulfonamide group behaving as a leaving group at an sp2 centre only when the carbon atom to which it is attached has been deprotonated. Both of these observations are counterintuitive in fundamental organic chemistry. Furthermore, our mechanistic work also helps to support and understand other observations with alternative p-block species that have recently been disclosed in the literature.14,17 Although our work has focused on simultaneous 1,4-addition of alkynyl nucleophiles to alkynyl sulfonamides, we recognize that the scope for exploiting this new class of tamed carbenoid is vast and research is already underway in our laboratory to optimize its formation and explore its applications.Experimental
General procedure for the treatment of alkynyl sulfonamides with aromatic acetylene to produce vinylsulfonamides, diynes and enediynes
A 100 mL flame-dried flask was charged with a solution of aromatic acetylene (1.1 eq.) in distilled THF (0.01 M) under argon. The solution was cooled to 0 °C and nBuLi (2.5 M in hexanes, 1.1 eq.) was added dropwise. The mixture was allowed to warm to RT and stirred for a further 10 min. An additional, 100 mL flame-dried flask was charged with a solution of alkynyl sulfonamide (0.15–0.2 mmol, 1.0 eq.) in distilled THF (0.1 M) under argon. The solution was heated to 60 °C and the previously formed lithiated aromatic acetylene solution was added dropwise (addition rate of 0.0025 mmol min−1) with constant stirring. The reaction mixture was diluted with CH2Cl2 (200 mL), washed with H2O (100 mL) then brine (100 mL), dried over MgSO4 and concentrated in vacuo to yield the crude mixture. Separation via flash column chromatography (petroleum ether/ethyl acetate) was carried out to yield the purified products.(Z)-N,N-Diethyl-2,4-diphenylbut-1-en-3-yne-1-sulfonamide (4)
Yellow oil. Rf = 0.30 (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 EA:PE); 1H-NMR (500 MHz, CDCl3) δH 7.72 (m, 2 H, ArH), 7.62 (m, 2 H, ArH), 7.44 (m, 3 H, ArH), 7.39 (m, 3 H, ArH), 6.88 (s, 1 H, C
:80 EA:PE); 1H-NMR (500 MHz, CDCl3) δH 7.72 (m, 2 H, ArH), 7.62 (m, 2 H, ArH), 7.44 (m, 3 H, ArH), 7.39 (m, 3 H, ArH), 6.88 (s, 1 H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 3.43 (q, J = 7.2 Hz, 4 H, NCH2), 1.24 (t, J = 7.6 Hz, 6 H, NCH2CH3); 13C-NMR (500 MHz, CDCl3) δc 136.2 (Cq), 132.7 (CH), 132.2 (CH), 131.1 (Cq), 130.3 (CH), 129.7 (CH), 128.9 (CH), 128.6 (CH), 127.2 (CH), 122.3 (Cq), 103.6 (Cq), 84.9 (Cq), 41.9 (CH2), 14.5 (CH3); νmax/cm−1 3059, 2973, 2929, 2874, 2252, 2211, 1730, 1680, 1598, 1555, 1489, 1444, 1334, 1266, 1200, 1183, 1145, 1017, 934, 910, 815, 758, 735, 692, 648, 653; LRMS (ESI+) m/z (%) 868 (2), 763 (1), 701 (100), 644 (1), 460 (2), 340 (58), 267 (1); HRMS (ESI+) calc'd for C20H22NO2S (M + H)+ 340.1366, found 340.1370.
CH), 3.43 (q, J = 7.2 Hz, 4 H, NCH2), 1.24 (t, J = 7.6 Hz, 6 H, NCH2CH3); 13C-NMR (500 MHz, CDCl3) δc 136.2 (Cq), 132.7 (CH), 132.2 (CH), 131.1 (Cq), 130.3 (CH), 129.7 (CH), 128.9 (CH), 128.6 (CH), 127.2 (CH), 122.3 (Cq), 103.6 (Cq), 84.9 (Cq), 41.9 (CH2), 14.5 (CH3); νmax/cm−1 3059, 2973, 2929, 2874, 2252, 2211, 1730, 1680, 1598, 1555, 1489, 1444, 1334, 1266, 1200, 1183, 1145, 1017, 934, 910, 815, 758, 735, 692, 648, 653; LRMS (ESI+) m/z (%) 868 (2), 763 (1), 701 (100), 644 (1), 460 (2), 340 (58), 267 (1); HRMS (ESI+) calc'd for C20H22NO2S (M + H)+ 340.1366, found 340.1370.
1,4-Diphenylbuta-1,3-diyne (5)
White solid. M.p. 83–87 °C; Rf = 0.57 (20:80 EA:PE); 1H-NMR (500 MHz, CDCl3) δH 7.54 (d, J = 7.6 Hz, 4 H, ArH), 7.36 (m, 6 H, ArH); 13C-NMR (500 MHz, CDCl3) δc 132.6 (CH), 129.3 (CH), 128.6 (CH), 121.9 (Cq), 81.6 (Cq), 74.0 (Cq); νmax/cm−1 3047, 2148, 1949, 1879, 1750, 1667, 1591, 1568, 1483, 1438, 1175, 1156, 1066, 1023, 997, 964, 914, 848, 825, 751, 682, 523, 462; LRMS (EI) m/z (%) 204 (3), 203 (9), 202 (100), 200 (17), 150 (3), 101 (6); HRMS (EI) calc'd for C16H10 (M+) 202.0777, found 202.0780.
(Z)-Hexa-3-en-1,5-diyne-1,3,6-triyltribenzene (6)
Brown oil. Rf = 0.56 (20:80 EA:PE); 1H-NMR (500 MHz, CDCl3) δH 7.75 (d, J = 7.5 Hz, 2 H, ArH), 7.61 (m, 2 H, ArH), 7.54 (m, 2 H, ArH), 7.41 (t, J = 7.5 Hz, 2 H, ArH), 7.36 (m, 7 H, ArH), 6.58 (s, 1 H, CCH); 13C-NMR (500 MHz, CDCl3) δc 136.9 (Cq), 133.5 (CH), 131.9 (CH), 131.7 (CH), 128.9 (CH), 128.8 (CH), 128.7 (CH), 128.6 (CH), 128.5 (CH), 128.5 (CH), 126.2 (CH), 123.5 (Cq), 123.2 (Cq), 113.7 (Cq), 98.5 (Cq), 98.4 (Cq), 89.1 (Cq), 87.7 (Cq); νmax/cm−1 3058, 3031, 2920, 2847, 2198, 2171, 1719, 1676, 1596, 1488, 1443, 1362, 1176, 1069, 914, 843, 756, 690, 529; LRMS (EI) m/z (%) 305 (23), 304 (100), 302 (44), 300 (14), 276 (4), 226 (6), 202 (5), 178 (4), 150 (5); HRMS (EI) calc'd for C24H16 (M+) 304.1247, found 304.1246.
(Z)-N,N-Diethyl-4-(3-methoxyphenyl)-2-phenylbut-1-en-3-yne-1-sulfonamide (13)
Yellow oil. Rf = 0.24 (20:80 EA:PE); 1H-NMR (500 MHz, CDCl3) δH 7.71 (m, 2 H, ArH), 7.44 (m, 3 H, ArH), 7.29 (t, J = 7.8 Hz, 1 H, ArH), 7.21 (d, J = 7.6 Hz, 1 H, ArH), 7.13 (m, 1 H, ArH), 6.96 (m, 1 H, ArH), 6.88 (s, 1 H, CCH), 3.83 (s, 3 H, CH3), 3.42 (q, J = 7.2 Hz, 4 H, NCH2) 1.23 (t, J = 7.1 Hz, 6 H, NCH2CH3); 13C-NMR (500 MHz, CDCl3) δc 159.5 (Cq), 136.1 (Cq), 132.6 (Cq), 131.2 (CH), 130.3 (CH), 129.7 (CH), 128.9 (CH), 127.2 (CH), 124.7 (CH), 123.2 (Cq), 116.7 (CH), 116.4 (CH), 103.5 (Cq), 84.6 (Cq), 55.5 (CH3), 41.9 (CH2), 14.5 (CH3); νmax/cm−1 3063, 2928, 2872, 2853, 2204, 1727, 1668, 1596, 1486, 1464, 1429, 1324, 1286, 1262, 1201, 1143, 1040, 1017, 937, 782, 757, 686, 561, 522, 461; LRMS (ES+) m/z (%) 371 (5), 370 (100); HRMS (ES+) calc'd for (C21H24NO3S) (M + H)+ 370.1477, found 370.1479.
1-Methoxy-3-(phenylbuta-1,3-diyn-1-yl)benzene (14)
Yellow oil. Rf = 0.50 (20:80 EA:PE); 1H-NMR (500 MHz, CDCl3) δH 7.53 (dt, J = 6.5, 1.7 Hz, 2 H, ArH), 7.35 (m, 3 H, ArH), 7.25 (m, 1 H, ArH), 7.13 (m, 1 H, ArH), 7.05 (s, 1 H, ArH), 6.93 (m, 1 H, ArH), 3.81 (s, 3 H, CH3); 13C-NMR (500 MHz, CDCl3) δc 159.3 (Cq), 132.6 (CH), 129.6 (CH), 129.3 (CH), 128.5 (CH), 125.2 (CH), 122.8 (Cq), 121.8 (Cq), 117.1 (CH), 116.1 (CH), 83.7 (Cq), 81.5 (Cq), 73.9 (Cq), 73.8 (Cq), 55.4 (CH3); νmax/cm−1 3060, 2998, 2956, 2924, 2851, 2217, 2189, 2145, 1727, 1670, 1592, 1573, 1485, 1463, 1425, 1342, 1315, 1284, 1251, 1168, 1081, 1042, 993, 916, 870, 855, 780, 755, 685, 580, 563, 526, 467; LRMS (CI) m/z (%) 252 (6), 251 (14), 250 (100), 232 (9); HRMS (CI) calc'd for C17H12O (M+) 232.0883, found 232.0884.
(Z)-3,3′-(3-Phenylhexa-3-en-1,5-diyne-1,6-diyl)bis(methoxybenzene) (15)
Yellow oil (4.3 mg). Rf = 0.34 (20:80 EA:PE); 1H-NMR (500 MHz, CDCl3) δH 7.74 (d, J = 7.3 Hz, 2 H, ArH), 7.40 (m, 4 H, ArH), 7.23 (t, J = 7.8 Hz, 2 H, ArH), 7.14 (m, 2 H, ArH), 7.07 (m, 1 H, ArH), 6.91 (m, 2 H, ArH), 6.57 (s, 1 H, CCH), 3.79 (s, 3 H, CH3), 3.77 (s, 3 H, CH3); 13C-NMR (500 MHz, CDCl3) δc 159.4 (Cq), 159.4 (Cq), 138.6 (Cq), 133.7 (Cq), 129.6 (CH), 129.5 (CH), 129.0 (CH), 128.7 (CH), 126.2 (CH), 124.4 (Cq), 124.3 (Cq), 124.1 (CH), 116.4 (CH), 116.2 (CH), 115.6 (CH), 115.5 (CH), 113.8 (CH), 98.4 (Cq), 96.4 (Cq), 95.2 (Cq), 88.7 (Cq), 55.4 (CH3), 55.3 (CH3); νmax/cm−1 3061, 3002, 2958, 2922, 2849, 2835, 2200, 2189, 1724, 1685, 1595, 1575, 1486, 1463, 1450, 1428, 1318, 1285, 1263, 1211, 1175, 1040, 855, 781, 761, 737, 686, 565, 518, 468; LRMS (CI) m/z (%) 383 (3), 382 (12), 367 (14), 366 (24), 365 (100); HRMS (CI) calc'd for C26H21O2 (M + H)+ 365.1536, found 365.1537.
(E)-N,N-Diethyl-4-phenyl-2-(o-tolyl)but-1-en-3-yne-1-sulfonamide (28)
Colourless oil. Rf = 0.31 (20:80 EA:PE); 1H-NMR (600 MHz, CDCl3) δH 7.52 (d, J = 7.1 Hz, 2 H, ArH), 7.35 (m, 3 H, ArH), 7.30 (m, 1 H, ArH), 7.26 (m, 3 H, ArH), 6.48 (s, 1 H, CCH), 3.43 (q, J = 7.2 Hz, 4 H, NCH2), 2.49 (s, 3 H, ArCH3), 1.26 (t, J = 7.1 Hz, 6 H, NCH2CH3); 13C-NMR (600 MHz, CDCl3) δc 137.6 (Cq), 135.7 (Cq), 134.6 (CH), 133.7 (Cq), 132.2 (CH), 131.0 (CH), 129.6 (CH), 129.2 (CH), 128.6 (CH), 128.5 (CH), 126.4 (CH), 122.4 (Cq), 104.1 (Cq), 85.4 (Cq), 41.9 (CH2), 20.3 (CH3), 14.6 (CH3); νmax/cm−1 3048, 2972, 2933, 2873, 2207, 1598, 1562, 1488, 1456, 1443, 1382, 1352, 1332, 1199, 1142, 1069, 1048, 1015, 994, 932, 879, 826, 755, 724, 688, 569, 555, 529, 507, 462, 429; LRMS (ES+) m/z (%) 408 (3), 378 (3), 377 (12), 376 (45), 356 (9), 355 (27), 354 (100); HRMS (ES+) calc'd for C21H24NO2S (M + H)+ 354.1528, found 354.1507.
1-Methoxy-4-(phenylbuta-1,3-diyn-1-yl)benzene (35)
Colourless oil. Rf = 0.49 (20:80 EA:PE); 1H-NMR (600 MHz, CDCl3) δH 7.53 (d, J = 6.6 Hz, 2 H, ArH), 7.48 (d, J = 8.6 Hz, 2 H, ArH), 7.35 (m, 3 H, ArH), 6.87 (d, J = 8.7 Hz, 2 H, ArH), 3.83 (s, 3 H, OCH3); 13C-NMR (600 MHz, CDCl3) δc 160.5 (Cq), 134.3 (CH), 132.6 (CH), 129.2 (CH), 128.5 (CH), 122.1 (Cq), 114.3 (CH), 113.8 (Cq), 81.9 (Cq), 81.1 (Cq), 74.3 (Cq), 72.8 (Cq), 55.5 (CH3); νmax/cm−1 3074, 2953, 2923, 2842, 2541, 2216, 2139, 1976, 1887, 1759, 1599, 1566, 1506, 1487, 1457, 1439, 1344, 1290, 1246, 1170, 1106, 1070, 1026, 952, 939, 918, 827, 757, 732, 689, 643, 616, 532, 491, 443; LRMS (CI) m/z (%) 251 (19), 250 (100), 234 (12), 233 (62); HRMS (CI) calc'd for C17H13O (M + H)+ 233.0961, found 233.0960.
(Z)-(3-(4-Methoxyphenyl)hexa-3-en-1,5-diyne-1,6-diyl)dibenzene (36)
Brown oil. Rf = 0.44 (20:80 EA:PE); 1H-NMR (600 MHz, CDCl3) δH 7.70 (d, J = 8.6 Hz, 2 H, ArH), 7.61 (m, 2 H, ArH), 7.53 (m, 2 H, ArH), 7.38 (m, 3 H, ArH), 7.34 (m, 3 H, ArH), 6.94 (d, J = 8.7 Hz, 2 H, ArH), 6.48 (s, 1 H, CCH), 3.86 (s, 3 H, OCH3); 13C-NMR (600 MHz, CDCl3) δc 160.4 (Cq), 133.0 (Cq), 131.9 (CH), 131.7 (CH), 129.5 (Cq), 128.8 (CH), 128.6 (CH), 128.5 (CH), 128.5 (CH), 127.6 (CH), 123.7 (Cq), 123.3 (Cq), 114.1 (CH), 111.7 (CH), 98.2 (Cq), 97.9 (Cq), 89.4 (Cq), 87.8 (Cq), 55.5 (CH3); νmax/cm−1 3052, 2954, 2926, 2836, 2199, 2179, 1880, 1726, 1669, 1603, 1577, 1509, 1487, 1460, 1441, 1362, 1304, 1287, 1250, 1177, 1114, 1068, 1030, 913, 823, 754, 689, 605, 527, 448; LRMS (CI) m/z (%) 336 (27), 335 (100); HRMS (CI) calc'd for C25H19O (M + H)+ 335.1430, found 335.1431.
1-(Phenylbuta-1,3-diyn-1-yl)-4-(trifluoromethyl)benzene (37)
Colourless oil. Rf = 0.66 (20:80 EA:PE); 1H-NMR (600 MHz, CDCl3) δH 7.55 (d, J = 7.0 Hz, 2 H, ArH), 7.63 (m, 7 H, ArH); 13C-NMR (600 MHz, CDCl3) δc 132.8 (CH), 132.7 (CH), 130.9 (q, J = 33.1 Hz, Cq), 129.7 (CH), 128.6 (CH), 125.8 (Cq), 125.5 (q, J = 3.8 Hz, CH), 123.9 (q, J = 272.6 Hz, Cq), 121.5 (Cq), 83.0 (Cq), 79.9 (Cq), 76.3 (Cq), 73.5 (Cq); νmax/cm−1 2955, 2924, 2853, 2256, 2213, 1916, 1796, 1667, 1612, 1570, 1488, 1462, 1443, 1405, 1317, 1167, 1104, 1064, 1012, 912, 834, 753, 686, 593, 519; LRMS (CI) m/z (%) 540 (8), 371 (11), 370 (44), 342 (9), 288 (5), 271 (19), 270 (100), 260 (10), 221 (8).
(Z)-(3-(4-(Trifluoromethyl)phenyl)hexa-3-en-1,5-diyne-1,6-diyl)dibenzene (38)
Yellow oil. Rf = 0.57 (20:80 EA:PE); 1H-NMR (600 MHz, CDCl3) δH 7.85 (d, J = 8.2 Hz, 2 H, ArH), 7.67 (d, J = 8.2 Hz, 2 H, ArH), 7.61 (m, 2 H, ArH), 7.55 (m, 2 H, ArH), 7.40 (m, 3 H, ArH), 7.37 (m, 3 H, ArH), 6.64 (s, 1 H, CCH); 13C-NMR (600 MHz, CDCl3) δc 132.1 (Cq), 131.9 (CH), 131.8 (CH), 131.8 (CH), 131.7 (CH), 130.3 (q, J = 31.4 Hz, Cq), 129.1 (CH), 129.0 (CH), 128.6 (CH), 128.6 (CH), 125.7 (q, J = 3.4 Hz, CH), 125.0 (Cq), 124.1 (q, J = 273.6 Hz, Cq), 123.2 (Cq), 116.1 (Cq), 99.9 (Cq), 99.1 (Cq), 88.7 (Cq), 87.0 (Cq); νmax/cm−1 3079, 3060, 3023, 2954, 2923, 2853, 2183, 1632, 1616, 1597, 1490, 1461, 1443, 1410, 1377, 1324, 1167, 1125, 1069, 1016, 914, 843, 831, 755, 689, 620, 605, 529; LRMS (CI) m/z (%) 392 (3), 391 (11), 390 (33), 378 (5), 375 (8), 374 (28), 373 (100), 372 (12), 370 (5), 353 (4), 350 (9), 345 (26), 322 (9); HRMS (CI) calc'd for C25H16F3 (M + H)+ 373.1199, found 373.1199.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge GSK for supporting a PhD studentship (to TOPH) and the financial support of EPSRC (grant no. EP/M02220X/1). The contributions of the UCL Mass Spectrometry service (Dr K. Karu) and NMR Service (Dr A. Aliev) are gratefully acknowledged. The authors also acknowledge the work of Giwrgos Lefkaritis and Joana Wong. The helpful advice of Dr Richard Fitzmaurice is also acknowledged.Notes and references
- A. L. Smith and K. C. Nicolaou, J. Med. Chem., 1996, 39, 2103–2117 CrossRef CAS PubMed.
- M. Gredičak and I. Jerić, Acta Pharm., 2007, 57, 133–150 CrossRef.
- (a) K. C. Nicolaou, G. Zuccarello, C. Riemer, V. A. Estevez and W. M. Dai, J. Am. Chem. Soc., 1992, 114, 7360–7371 CrossRef CAS; (b) K. C. Nicolaou and W. M. Dai, Angew. Chem., Int. Ed. Engl., 1991, 30, 1387–1416 CrossRef.
- P. A. Magriotis, M. E. Scott and K. D. Kim, Tetrahedron Lett., 1991, 32, 6085–6088 CrossRef CAS.
- K. C. Nicolaou, Y. Wang, M. Lu, D. Mandal, M. R. Pattanayak, R. Yu, A. A. Shah, J. S. Chen, H. Zhang, J. J. Crawford, L. Pasunoori, Y. B. Poudel, N. S. Chowdari, C. Pan, A. Nazeer, S. Gangwar, G. Vite and E. N. Pitsinos, J. Am. Chem. Soc., 2016, 138, 8235–8246 CrossRef CAS PubMed.
- K. C. Nicolaou, A. L. Smith and E. W. Yue, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5881–5888 CrossRef CAS.
- Z. Wang and K. K. Wanh, J. Org. Chem., 1994, 59, 4738–4742 CrossRef CAS.
- J. H. Ryan and P. J. Stang, J. Org. Chem., 1996, 61, 6162–6165 CrossRef CAS PubMed.
- There are many examples of Pd catalyzed synthetic approaches. For recent examples see: (a) G. W. Kabalka, G. Dong and B. Venkataiah, Tetrahedron Lett., 2005, 46, 763–765 CrossRef CAS; (b) Y. Liu and H. Gao, Org. Lett., 2006, 8, 309–311 CrossRef CAS PubMed.
- M. J. Dabdoub, V. B. Dabdoub and J. P. Marino, Tetrahedron Lett., 2000, 41, 437–440 CrossRef CAS.
- J. Cuthbertson and J. D. Wilden, Tetrahedron, 2015, 71, 4385–4392 CrossRef CAS.
- R. M. Chowdhury and J. D. Wilden, Org. Biomol. Chem., 2015, 13, 5859–5861 CAS.
- V. J. Gray, J. Cuthbertson and J. D. Wilden, Chem. Commun., 2014, 79, 5869–5874 CAS.
- (a) A. Maercker, Angew. Chem., Int. Ed. Engl., 1993, 32, 1023–1025 CrossRef; (b) P. v. R. Schleyer, T. Clark, A. J. Kos, G. W. Spitznagel, C. Rohde, D. Arad, K. N. Houk and N. G. Rondan, J. Am. Chem. Soc., 1984, 106, 6467–6475 CrossRef CAS; (c) M. Braun, Angew. Chem., Int. Ed., 1998, 37, 430–451 CrossRef.
- T. Kimura, Y. Nishimura, N. Ishida, H. Momochi, H. Yamashita and T. Satoh, Tetrahedron Lett., 2013, 54, 1049–1051 CrossRef CAS.
- T. S. Chou and L. J. Chang, J. Org. Chem., 1985, 50, 4998–5000 CrossRef CAS.
- C. Fressigné, R. Lhermet, A.-L. Girard, M. Durandetti and J. Maddaluno, J. Org. Chem., 2013, 78, 9659–9669 CrossRef PubMed.
- K. D. Reichl and A. T. Radosevich, Chem. Commun., 2014, 50, 9302–9305 RSC.
- J. J. Reddick, J. Cheng and W. R. Roush, Org. Lett., 2003, 5, 1967–1970 CrossRef CAS PubMed.
- (a) J. F. Biellmann and J. J. Vicens, Tetrahedron Lett., 1974, 15, 2915–2918 CrossRef; (b) J. F. Biellmann and J. J. Vicens, Tetrahedron Lett., 1978, 19, 467–470 CrossRef.
- T. Durst and T. Molin, Tetrahedron Lett., 1975, 16, 63–66 CrossRef.
- G. Chassaing, R. Lett and A. Marquet, Tetrahedron Lett., 1978, 19, 471–474 CrossRef.
- S. Eisler, N. Chahal, R. McDonald and R. R. Tykwinski, Chem. – Eur. J., 2003, 9, 2542–2550 CrossRef CAS PubMed.
- V. J. Gray, B. Slater and J. D. Wilden, Chem. – Eur. J., 2012, 18, 15582–15585 CrossRef CAS PubMed.
- R. L. Smorada and W. E. Truce, J. Org. Chem., 1979, 44, 3444–3445 CrossRef CAS.
- (a) J. L. G. Ruano, J. Alemán, L. Marzo, C. Alvarado, M. Tortosa, S. Díaz-Tendero and A. Fraile, Chem. – Eur. J., 2012, 18, 8414–8422 CrossRef PubMed; (b) J. L. G. Ruano, J. Alemán, L. Marzo, C. Alvarado, M. Tortosa, S. Díaz-Tendero and A. Fraile, Angew. Chem., Int. Ed., 2012, 51, 2712–2716 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental detail and characterisation data. See DOI: 10.1039/c7ob02437a |
| This journal is © The Royal Society of Chemistry 2017 |