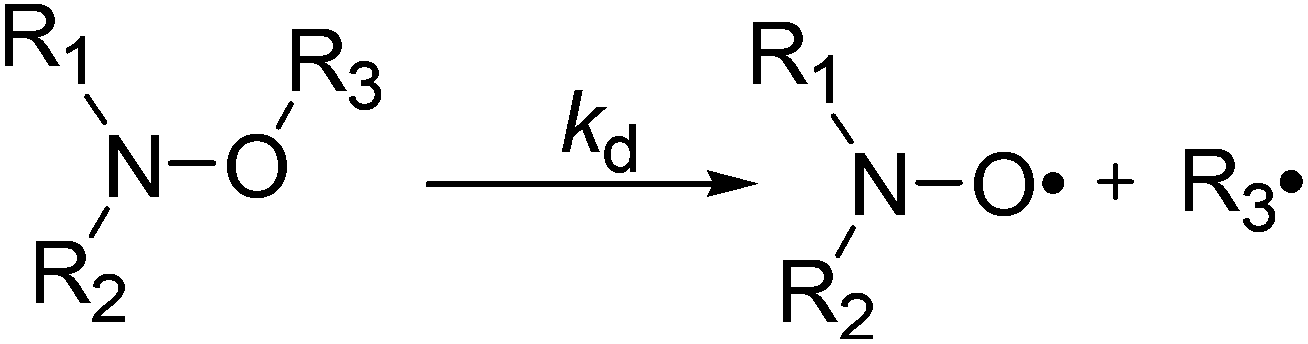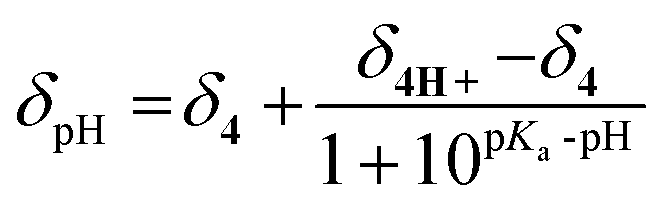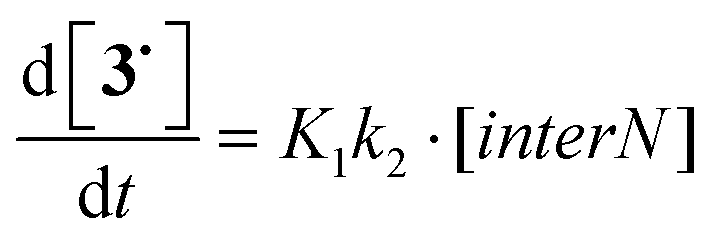How intramolecular hydrogen bonding (IHB) controls the C–ON bond homolysis in alkoxyamines†
Received
4th September 2017
, Accepted 14th September 2017
First published on 27th September 2017
Abstract
Recent amazing results (Nkolo et al., Org. Biomol. Chem., 2017, 6167) on the effect of solvents and polarity on the C–ON bond homolysis rate constants kd of alkoxyamine R1R2NOR3 led us to re-investigate the antagonistic effect of intramolecular hydrogen-bonding (IHB) on kd. Here, IHB is investigated both in the nitroxyl fragment R1R2NO and in the alkyl fragment R3, as well as between fragments, that is, the donating group on the alkyl fragment and the accepting group on the nitroxyl fragment, and conversely. It appears that IHB between fragments (inter IHB) strikingly decreases the homolysis rate constant kd, whereas IHB within the fragment (intra IHB) moderately increases kd. For one alkoxyamine, the simultaneous occurrence of IHB within the nitroxyl fragment and between fragments is reported. The protonation effect is weaker in the presence than in the absence of IHB. A moderate solvent effect is also observed.
Introduction
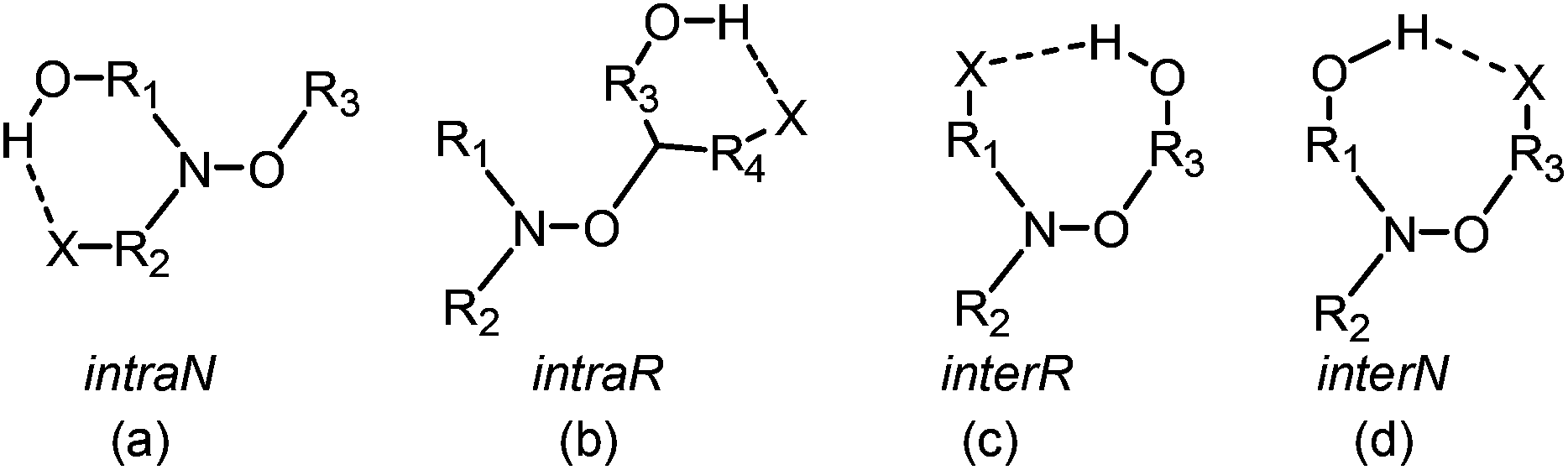
Since the pioneering work of Rizzardo and co-workers,1,2 alkoxyamines have been applied in several fields such as tin-free radical organic chemistry3,4 – as initiators for radical cyclization,5,6 1,2-radical additions,7 and several others5 – NMP (nitroxide mediated polymerization) and its variants: in situ NMP,8 ESCP,9 NMP2,10,11 SL-NMP,12 and CI-NMP,13 materials sciences – for self-healing polymers,14 optoelectronic materials,15 and encoding systems16 – and in biology,17–19 as agents for theranostics. For alkoxyamines to be used as agents for theranostics (Fig. 1), the concept of a “smart” alkoxyamine was proposed,17 that is, a highly stable alkoxyamine switching to a highly labile alkoxyamine through chemical reactions (Fig. 2). For several years, our group has been promoting the chemical alkoxyamine activation using protonation,20 oxidation,20 alkylation,20 or metal cation coordination.21,22 However, in our view, activation processes based on physico-chemical events cannot be disregarded. Indeed, very recently,23 a dramatic solvent effect on the C–ON bond homolysis rate constant kd (Scheme 1) has been reported for 1 (Fig. 3). Intramolecular hydrogen-bonding (IHB) displays very different trends, that is, IHB within the nitroxyl fragment affords an increase in kd (Fig. 4a)24–27 and IHB from the alkyl fragment to the nitroxyl fragment (interR, Fig. 4c) affords a decrease in kd spanning from weak (2–3 kJ mol−1)28 to moderate29 (ca. 10 kJ mol−1). Interestingly, other types of IHBs have not yet been investigated – IHB from the nitroxyl fragment to the alkyl fragment (interN, Fig. 4d) and IHB within the alkyl fragment (Fig. 4b) – and are the focus of this article. Several models 2–7 (Fig. 3) were prepared and their structures were determined by X-ray and NMR analysis.
 |
| | Fig. 1 Concept for the application of alkoxyamines as agents for theranostics. Reproduced with permission of the American Chemical Society (see ref. 46). | |
 |
| | Fig. 2 Concept for “smart” alkoxyamines. Reproduced with permission of the Royal Society of Chemistry (see ref. 17). | |
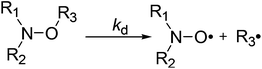 |
| | Scheme 1 C–ON bond homolysis in alkoxyamines. | |
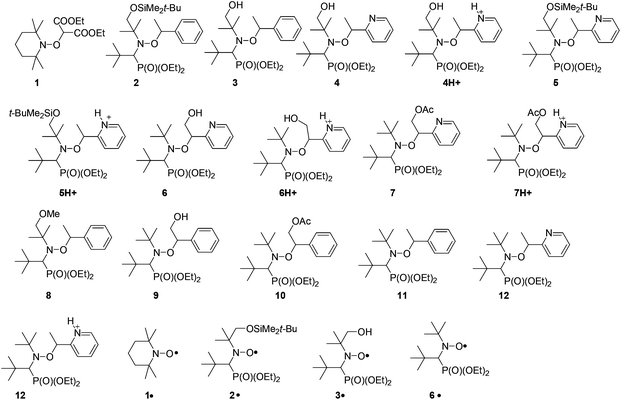 |
| | Fig. 3 Alkoxyamines and nitroxides discussed in the article. | |
 |
| | Fig. 4 Various types of IHBs: (a) within the nitroxyl fragment (intraN), (b) within the alkyl fragment (intraR), (c) from alkyl to nitroxyl fragments (interR), and (d) from nitroxyl to alkyl fragments (interN). Dotted blue lines represent IHB. | |
The occurrence and the type of IHB were determined by combining 31P and 1H NMR with DFT calculations. The influence of IHB on kd was investigated in tert-butylbenzene (t-BuPh) as a non-polar solvent and water as a polar/hydrogen bond acceptor (HBA) solvent, known to suppress IHB.30
Results
Preparation of alkoxyamines 2–7
Alkoxyamine 3 was prepared either as previously reported26 (black route in Scheme 2) or by the hydrolysis31 of 2 prepared from protected amino alcohol 2a (blue and magenta routes in Scheme 2). Alkoxyamine 2 was prepared either by the protection32 of 3 (black route in Scheme 2) or from the protected phosphorylated amino alcohol 2b (magenta route in Scheme 2).
 |
| | Scheme 2 Preparation of 2 and 3: (a) (1) 2a or 3a (1.1 eq.), t-Bu-CHO (1 eq.), pentane, 65 °C, 2 day, (2) (EtO)2P(O)H (1.1 eq.), 45 °C, 7 days, 64–78%; (b) 2b or 3b (1 eq.), Oxone® (4 eq.), Na2CO3 (6 eq.), EtOH/H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 5 h, rt, 51–56%; (c) (1) Cu(0) (1.1 eq.), CuBr (0.55 eq.), PMDETA (0.55 eq.), argon bubbled benzene, 30 min, rt, (2) 1-bromoethylbenzene (1.1 eq.), 2˙ or 3˙ (1 eq.), rt, overnight, 65–72%; (d) 3a, 3b, or 3 (1 eq.), t-BuMe2SiOTf (1.5 eq.), 2,6-lutidine (2 eq.), DCM, 4 h, rt 80–90%; (e) 2 (1 eq.) TBAF (1 M in THF) (1.2 eq.), THF, rt, 92%. :1), 5 h, rt, 51–56%; (c) (1) Cu(0) (1.1 eq.), CuBr (0.55 eq.), PMDETA (0.55 eq.), argon bubbled benzene, 30 min, rt, (2) 1-bromoethylbenzene (1.1 eq.), 2˙ or 3˙ (1 eq.), rt, overnight, 65–72%; (d) 3a, 3b, or 3 (1 eq.), t-BuMe2SiOTf (1.5 eq.), 2,6-lutidine (2 eq.), DCM, 4 h, rt 80–90%; (e) 2 (1 eq.) TBAF (1 M in THF) (1.2 eq.), THF, rt, 92%. | |
Alkoxyamine 4 was prepared using the conventional Mn(salen)2 salt procedure (Scheme 3).33 The two diastereoisomers were separated and RR/SS-4‡ was recrystallized. Then, it was protected using t-BuMe2SiCl to afford 5 in good yield (Scheme 3).34§
 |
| | Scheme 3 Preparation of 4 and 5: (a) (1) salen ligand (0.05 eq.), MnCl2 (0.05 eq.), i-PrOH, 30 min, rt; (2) 3˙ (1 eq.), 2-vinyl pyridine (1 eq.), i-PrOH; (3) NaBH4 (5 eq.), 4 h, rt, 54%; (b) (1) 4 (1eq.), pyridine (5 eq.), AgNO3 (1.5 eq.), THF, 5 min, rt; (2) t-BuMe2SiCl (1.5 eq.), overnight, rt, 59–67%. | |
Alkoxyamine 6 was prepared from the corresponding bromide 6b coupled to 6˙ using the conventional ATRA procedure.35 However, 7 was obtained as a crude mixture of diastereoisomers which cannot be separated. Then, crude 7 was hydrolyzed into crude 6 whose diastereoisomers were separated, and the diastereoisomer RS/SR‡ was crystallized. The diastereoisomers of 6 were acetylated into 7 (Scheme 4).
 |
| | Scheme 4 Preparation of 6 and 7: (a) 5a (1 eq.), NBS (1.1 eq.), benzoyl peroxide (0.1 eq.), CCl4, reflux, 69%; (b) (1) CuBr (0.5 eq.), Cu(0) (1 eq.), PMEDTA (0.5 eq.), benzene, Ar, 10 min, rt; (2) 6˙ (1.1 eq.), 6b (1 eq.), benzene, 12 h, rt, (3) MeOH, K2CO3, H2O, 3 days, rt, 63%; (c) (1) 6 (1 eq.), Et3N (4 eq.), CH2Cl2; (2) Ac2O (3 eq.), 3 days, rt, 65–90%. | |
X-ray analysis
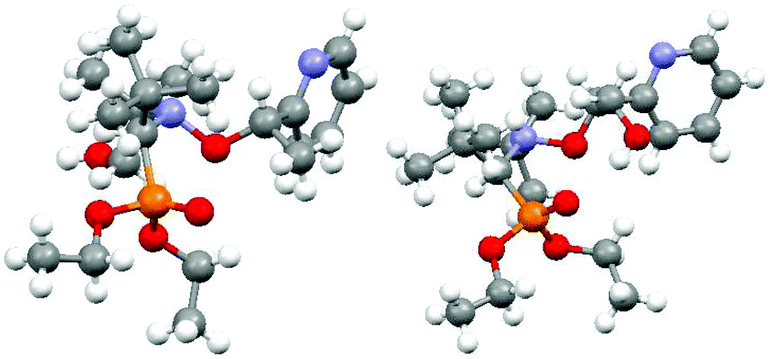
Alkoxyamines RR/SS-4 and RS/SR-6 were crystallized and analyzed by XRD (Fig. 5).‡ X-ray structures exhibit the conventional distances and angles observed for such types of molecules (see the ESI†). Dihedral angles <O5C6C7N8> are 45° and 90° larger (Fig. 6d) than the 90° required at TS for RR/SS-4 and RS/SR-6, respectively.36 However, from the X-ray structure (Fig. 5 and 6) it is clear that no IHB occurs in RR/SS-4 in the solid state although it is a good model to observe IHB of type (a) or (d) (Fig. 4) or both. By contrast, RS/SR-6, which is a good model for IHB of type (b) or (c) or both (Fig. 4), exhibits only IHB of type (c).¶37,38 The absence of IHB in the RR/SS-4 X-ray structure is in sharp contrast with its occurrence in solution (vide infra). This difference in conformation between the crystal and solution has already been observed several times for alkoxyamines27,28 and it is ascribed to the packing effect, which forces intermolecular H-bonding at the expense of IHB.
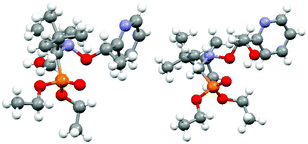 |
| | Fig. 5 X-ray structures for RR/SS-4 (left) and RS/SR-6 (right).‡ | |
 |
| | Fig. 6 Cram (a) and Newman projections through (b) N4–O5, (c) O5–C6, (d) C6–C7, and (e) C3–N4 bonds for RR/SS-4 (top) and RS/SR-6 (bottom). | |
NMR analysis
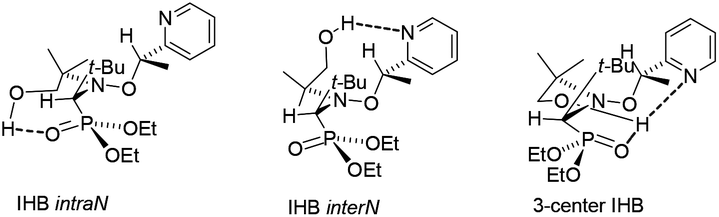
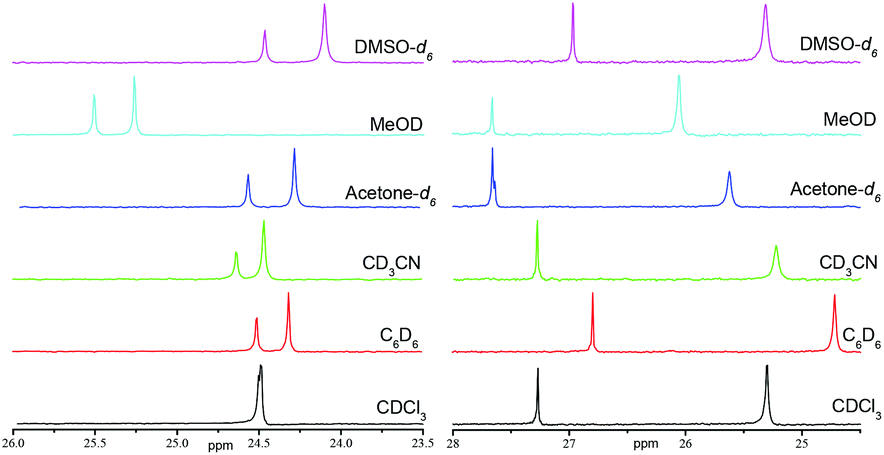
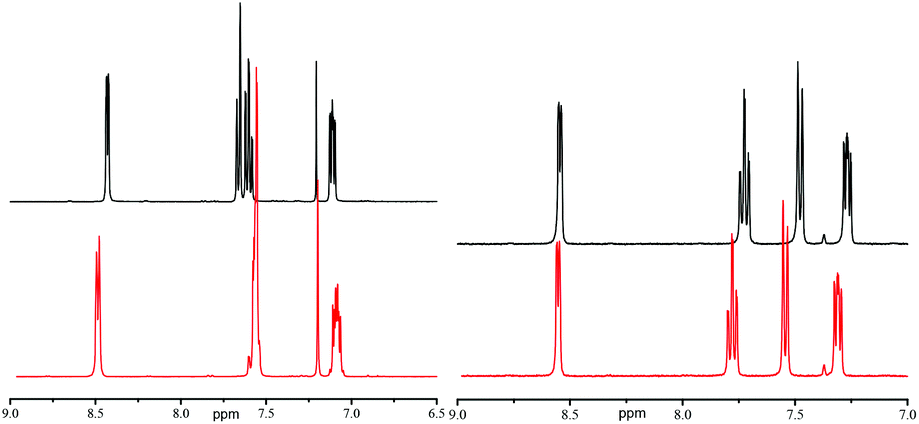
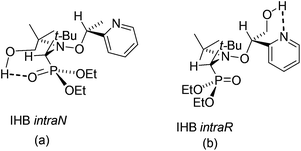
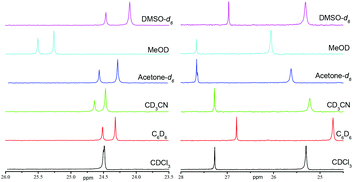
In contrast with XRD, 1H and 31P NMR spectra show that IHBs of types (a) and (d) occur at the same time for RR/SS-4.|| As already reported for 2 and 3,29 Δδ for 4 and 5 decreases from benzene-d6 to DMSO-d6 (Table 1 and Fig. 7), as expected from the increase in the parameter β30 – the hydrogen bond acceptor (HBA) property of the solvent – that is, the increase in the ability to suppress IHB. The occurrence of IHB between the diethylphosphoryl and the hydroxyl groups (intraN in Fig. 8) is supported by the difference in shifts δ and the signal pattern for the MeCH2O group observed between RR/SS-4 and RR/SS-5 in benzene-d6 (protons labelled a in Fig. 9)** and its suppression in DMSO-d6 (very similar patterns for protons labelled a–c for RR/SS-4 and RR/SS-5 in Fig. 9). However, the chemical shifts δ and the signal pattern of pyridyl protons (Fig. 9) are very different for RR/SS-4 and RR/SS-5 in benzene-d6, while they are similar in DMSO-d6. As similar observations were also reported upon protonation (or other types of activation) of the pyridyl moiety in alkoxyamines,20–22 this change in the signal is ascribed to the occurrence of IHB between the hydroxyl group and the pyridyl moiety (interN in Fig. 8). Temperature dependence shows only line broadening when the temperature is decreased down to −80 °C, meaning that the rotation around the C–N bond of the group carrying the hydroxyl function is faster than the resolution time of 1H NMR. DFT calculations (vide infra) led us to disregard the occurrence of 3-center IHB, as shown in Fig. 8.
 |
| | Fig. 7
31P NMR signals in various solvents for (a) a (2:1) mixture of RR/SS-4 (left) and RR/SS-5 (right), and (b) a (2:1) mixture of RS/SR-4 (left) and RS/SR-5 (right). | |
 |
| | Fig. 8 Possible conformations affording intraN IHB (left), interN IHB (middle), and 3-center IHB (right) for RR/SS-4. | |
 |
| | Fig. 9
1H NMR spectra of RR/SS-4 (top) and RR/SS-5 (bottom) in the range 2.5–6 ppm (top row) and 6–9 ppm (pyridyl proton zone, bottom row) in benzene-d6 (left) and DMSO-d6 (right). Labelling of protons: a–e represent MeCH2O, CHP, CHMe, OH, and CH2O, respectively. | |
Table 1 Solvent effect on diastereoisomers of 4 and 5 investigated by 31P NMR
| Solventa |
β
|
δ (ppm) |
Δδd (ppm) |
δ (ppm) |
Δδd (ppm) |
|
RR/SS-4c |
RR/SS-5c |
RS/SR-4c |
RS/SR-5c |
|
85% H3PO4 was used as an internal reference (0 ppm).
Hydrogen bond acceptor parameter β. Given in ref. 30.
Ratio 4:5 = 2:1.
Δδ = δ4 − δ5.
|
| CDCl3 |
0.10 |
27.29 |
26.11 |
1.18 |
26.13 |
25.22 |
0.91 |
| C6D6 |
0.10 |
27.35 |
25.86 |
1.49 |
26.29 |
24.95 |
1.34 |
| CD3CN |
0.40 |
27.17 |
26.25 |
0.92 |
26.57 |
25.25 |
1.32 |
| Acetone-d6 |
0.48 |
27.27 |
26.31 |
0.96 |
26.69 |
25.21 |
1.48 |
| CD3OD |
0.66 |
27.32 |
27.04 |
0.28 |
26.42 |
26.02 |
0.40 |
| DMSO-d6 |
0.76 |
26.46 |
26.15 |
0.31 |
25.61 |
24.95 |
0.66 |
The same trends are observed for Δδ (Fig. 7 and Table 1) and 1H NMR signals of EtO protons (labelled a in Fig. 1SI†) for RS/SR diastereoisomers of 4 and 5 as those for diastereoisomers RR/SS and are ascribed to the occurrence of IHB between the diethylphosphoryl and the hydroxyl groups (Fig. 10). In sharp contrast to what was observed for the diastereoisomer RR/SS, no significant differences in chemical shifts and signal patterns in the pyridyl proton zone are observed for RS/SR-4 and RS/SR-5, whatever the solvent (Fig. 1SI†). Consequently, only intraN IHB (type (a) in Fig. 4) is observed in RS/SR-4 (Fig. 10a).
 |
| | Fig. 10 Possible conformations affording intraN IHB for RS/SR-4 (a) and intraR IHB for RR/SS-6 (b). | |
The same trends are observed for Δδ (Fig. 11b and Table 2) and 1H NMR signals of EtO protons (labelled a in Fig. 2SI†) for RS/SR diastereoisomers of 6 and 7 as those for 4 and 5 (vide supra) and are ascribed to the occurrence of IHB between the diethylphosphoryl and the hydroxyl groups (Fig. 6) in RS/SR-6. On the other hand, the very similar signal patterns of protons in the aromatic zone for RS/SR-6 and RS/SR-7 (Fig. 2SI†) support the non-occurrence of intraR IHB (type b in Fig. 4 and 6).
 |
| | Fig. 11
31P NMR signals in various solvents for (a) a (2:1) mixture of RR/SS-6 (left) and RR/SS-7 (right), and (b) a (2:1) mixture of RS/SR-6 (left) and RS/SR-7 (right). | |
Table 2 Solvent effect on diastereoisomers of 6 and 7 investigated by 31P NMR
| Solventa |
β
|
δ (ppm) |
Δδd (ppm) |
δ (ppm) |
Δδd (ppm) |
|
RR/SS-6c |
RR/SS-7c |
RS/SR-6c |
RS/SR-7c |
|
85% H3PO4 was used as an internal reference (0 ppm).
Hydrogen bond acceptor parameter β. Given in ref. 30.
Ratio 6 : 7 = 2:1.
Δδ = δ6 − δ7.
|
| CDCl3 |
0.10 |
24.48 |
24.48 |
0.0 |
27.27 |
25.30 |
1.97 |
| C6D6 |
0.10 |
24.52 |
24.32 |
0.20 |
26.90 |
24.82 |
2.08 |
| CD3CN |
0.40 |
24.64 |
24.47 |
0.17 |
27.25 |
25.19 |
2.06 |
| Acetone-d6 |
0.48 |
24.63 |
24.34 |
0.29 |
27.65 |
25.62 |
2.03 |
| CD3OD |
0.66 |
25.51 |
25.27 |
0.24 |
27.65 |
26.05 |
1.60 |
| DMSO-d6 |
0.76 |
24.45 |
24.08 |
0.37 |
26.97 |
25.30 |
1.67 |
No significant changes in Δδ (Table 2 and Fig. 11b) and in the signal patterns of EtO-groups (protons labelled a in Fig. 3SI†) are observed for RR/SS-6 and RR/SS-7, pointing to the absence of interR IHB (type c in Fig. 4). By contrast, significant changes in the signal patterns of aromatics are observed between RR/SS-6 and RR/SS-7 in benzene-d6, whereas very similar signal patterns for RS/SR-6 and RS/SR-7 are observed (Fig. 12) in DMSO-d6, i.e., the absence of any type of IHB, supporting the occurrence of intraR IHB in RR/SS-6 in benzene-d6 (Fig. 10b).
 |
| | Fig. 12
1H NMR spectra of RR/SS-6 (top) and RR/SS-7 (bottom) in the range 3–6 ppm (top row) and 6–9 ppm (pyridyl proton zone, bottom row) in CDCl3 (left) and DMSO-d6 (right). Labelling of protons: a–e represent MeCH2O, CHP, CHMe, OH, and CH2O, respectively. | |
Protonation of alkoxyamines
Alkoxyamines 4–7 carry a pyridyl moiety on the alkyl fragment suitable for activation by protonation. Consequently, protonation in benzene-d6 (model for tert-butylbenzene t-BuPh) in the presence of trifluoroacetic acid is confirmed by changes in 1H NMR shifts in the aromatic zone, e.g. RR/SS-4 (Fig. 4SI†). The values of pKa (Table 3)39 are estimated from the 1H NMR shift of the signal in the aromatic zone, e.g., RR/SS-4 (Fig. 13), and are given by the modified Hasselbach–Henderson equation (eqn (1), exemplified by 4/4H+, and Table 3).40,41| |  | (1) |
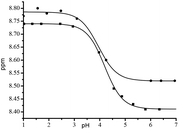 |
| | Fig. 13 Titration curves for RR/SS-4 (0.01 M, ■, inset: signal of the aromatic proton zone) and RS/SR-4 (0.01 M, ●), D2O/MeOH-d4 = 1:1. | |
Table 3 Values of pKa for 4–7
| |
4
|
5
|
6
|
7
|
|
RR/SS |
RS/SR |
RR/SS |
RS/SR |
RR/SS |
RS/SR |
RR/SS |
RS/SR |
|
All pH values measured in D2O/MeOH-d4 (1:1) were re-estimated using pH = 0.929·pH* + 0.42. pH* is the pH measured in D2O/MeOH-d4 solutions using a pH-meter calibrated with non-deuterated water. See ref. 39.
pKa = 5.89 for ortho-ethylpyridine. See ref 40.
pKa = 4.85 for ortho-2-hydroxyethylpyridine. See the ESI.
pKa values of 4.21 (RS/SR) and 3.99 (RR/SS) were reported for 12. See ref. 41.
|
| pKaa,b |
4.08c |
4.32c |
3.81c |
4.25c |
4.25c,d |
3.61c,d |
3.24c,d |
3.33c,d |
As already reported, the pKa of 12 is ca. 1.7–1.9 units lower than that reported for the ortho-ethylpyridine. It is ascribed to the presence of the nitroxyl fragment as a strong electron withdrawing group (EWG). The one unit lower pKa for the ortho-hydroxyethylpyridine is also ascribed to the presence of the OH group as an EWG. Therefore, the lower pKa values for 4–7 (Fig. 13) than those for ortho-ethylpyridine and ortho-hydroxyethylpyridine are due to the presence of the nitroxyl fragment as an EWG. It is noteworthy that alkoxyamines 4–6 exhibit pKa values very close to those for model 12, except for RR/SS-6.
The one unit lower pKa value for 7 than that for 12 is ascribed to the acylated hydroxyl group, which exhibits a higher electron withdrawing property than the OH group, the basicity of the pyridine moiety being thus decreased.
Kinetic investigations
Homolysis rate constants kd,T were measured using either the 31P NMR method27 for 4, 4H+, 5, 7 and 7H+ or the EPR method20 for 2, 2′, 2′′, 3′, 6 and 6H+, as previously reported. Differences in Ea observed between diastereoisomers for 2, 2′, 2′′, 3, 5 and 7 in t-BuPh as a solvent are in the range of 0–2 kJ mol−1 – generally observed for diastereoisomers reported in the literature – and do not deserve more comments.36 Alkoxyamines exhibiting IHB are discussed later. Except for 6, comments made for diastereoisomers in t-BuPh as a solvent hold in water, as IHB is suppressed.
DFT calculations
DFT calculations at the M062X/6-31+G(d,p) level of theory in the gas phase were performed to determine both the thermodynamics of the homolysis and the most stable conformations for the diastereoisomers of 2–4 and 6 (Table 1SI†).42 Important geometrical parameters – bond lengths, distances and angles – are reported in Table 1SI† and agree with X-ray data and those reported for molecules of the same family, except for the occurrence of IHB in RR/SS-4. Thermodynamics of 4 and 6 show the same trends as that experimentally reported (Table 1SI†). Only the 3 most stable conformations A, B and C†† are reported for 4 and 6 (Fig. 14). Valence angles, bond lengths and distances for IHB are reported in Table 5. Depending both on the valence angle α and on the distance dH⋯X between the H and X atoms implied in the H-bonding, IHB is given as strong (α > 150° and d smaller than the sum of van der Waals radii of H and X atoms), as weak (α < 120° and d closer to the sum of the van der Waals radii of H and X atoms) or as medium for other combinations.¶37,38 On these grounds, IHB for the most stable conformers is considered as strong for RS/SR-6 (conformer A) and RS/SR-9, and as medium for other diastereoisomers (Fig. 14 and Table 5). For all cases, IHB is smaller by ca. 1.8 Å and by 0.5 Å for OH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P and for OH⋯N interactions, respectively, than the respective sums of van der Waals radii¶ – dOH⋯OP = 2.61 Å and dOH⋯N = 2.64 Å – and all valence angles α are larger than 130°. Nevertheless, the occurrence of IHB is controlled mainly by the steric hindrance. As illustrated by RR/SS-9, when steric hindrance is too large, IHB does not occur, and in some cases, for example, RR/SS-4 and RR/SS-6, the most stable conformer B does not display the strongest IHB.‡‡ All other possible IHBs are disregarded due to distances dOH⋯OP and dOH⋯N much larger than the sums of the van der Waals radii, except for OH⋯NO for the conformer C of RS/SR-6. Nonetheless, the IHB observed between the hydroxyl group and the N atom of the nitroxyl moiety in the conformer C is not strong enough to balance the steric strain in C, which is less stable than A by 19 kJ mol−1 (Fig. 14).
P and for OH⋯N interactions, respectively, than the respective sums of van der Waals radii¶ – dOH⋯OP = 2.61 Å and dOH⋯N = 2.64 Å – and all valence angles α are larger than 130°. Nevertheless, the occurrence of IHB is controlled mainly by the steric hindrance. As illustrated by RR/SS-9, when steric hindrance is too large, IHB does not occur, and in some cases, for example, RR/SS-4 and RR/SS-6, the most stable conformer B does not display the strongest IHB.‡‡ All other possible IHBs are disregarded due to distances dOH⋯OP and dOH⋯N much larger than the sums of the van der Waals radii, except for OH⋯NO for the conformer C of RS/SR-6. Nonetheless, the IHB observed between the hydroxyl group and the N atom of the nitroxyl moiety in the conformer C is not strong enough to balance the steric strain in C, which is less stable than A by 19 kJ mol−1 (Fig. 14).
 |
| | Fig. 14 Most stable conformations A–C†† for RR/SS-4, RS/SR-4, RR/SS-6, and RS/SR-6. All conformers are on the same energetic scale (kJ mol−1). Dotted lines for the difference in energy between isomers and full lines for the difference in energy between conformers. | |
Discussion
Free motions in the nitroxyl fragment
Taking into account (i) NMR observations for RR/SS-4 denoting the simultaneous occurrence of intraN and interN IHBs (Fig. 4), (ii) calculations ascribing these two IHBs to two different conformers (Fig. 14 and Table 5), (iii) dramatically restricted bond rotations in the group carrying the diethylphosphoryl group attached to the nitroxyl moiety in nitroxides, and in the subsequent alkoxyamines,§§43–45 the question of the free rotation around the C–N bond for the other alkyl group attached to the nitroxyl moiety is raised. Indeed, the conformations of the nitroxyl fragment are expected to be ruled by the conformations of the nitroxide, as highlighted by the occurrence of the same IHB in 3˙26 and 3.27 Whatever the route for the preparation of 2 and 3 (Scheme 2 and ESI†), 1H NMR spectra of intermediates are the same (see the ESI†) as well as kd values within the experimental error (Table 4), supporting that for the group carrying the hydroxyl function, the rotation of the C–N bond is free, whereas the rotation of the C–N bond for the moiety carrying the diethylphosphoryl group is, in general, strongly restricted in the nitroxide,46,47 and likely in the alkoxyamine.
Table 4 C–ON bond homolysis rate constant kd,T at various temperatures T for alkoxyamines under various conditions of solvents and pH and their corresponding activation energies Ea, the homolysis rate constant kd re-estimated at 120 °C and predicted activation energies E′a
| |
Solvent |
T (°C) |
RR/SS |
RS/SR |
E′aa,b |
Referencesj |
|
k
d,Tc,d |
E
ab,e |
k
df,g |
k
d,Tc,d |
E
ab,h |
k
df,i |
RR/SS |
RS/SR |
|
Predicted values of Ea using the incremental scale.
In kJ mol−1.
In 10−4 s−1.
Given by eqn (4).
Estimated using kd values in the 4th column and the frequency factor A = 2.4 × 1014 s−1 in eqn (5). See ref. 51.
In 10−3 s−1.
Estimated using Ea values in the 5th column and the frequency factor A = 2.4 × 1014 s−1 in eqn (5). See ref. 51.
Estimated using kd values in the 7th column and the frequency factor A = 2.4 × 1014 s−1 in eqn (5). See ref. 51.
Estimated using Ea values in the 8th column and the frequency factor A = 2.4 × 1014 s−1 in eqn (5). See ref. 51.
t.w.: this work. n.d.: not determined. n.m.: not measured.
Prepared via the route 3a → 3b → 3˙ → 3 → 2.
Measured by EPR.
Used as a model: Δ11→2 = +0.4 kJ mol−1 for the diastereoisomer RR/SS and Δ11→2 = +0.9 for the diastereoisomer RS/SR.
Prepared via the route 3a → 2a → 2b → 2˙ → 2′(2) → 3′ (3).
Prepared via route 3a → 3b → 2b → 2˙ → 2′′ (2).
Used as a model: Δ11→3 = −1.5 for RR/SS and Δ11→3 = −1.2 for RS/SR.
k
d determined by 31P NMR. See ref. 20.
E′a = Ea,11 + Δ11→3 + Δ11→12.
Protonation is performed by adding 2 eq. of TFA.
Protonation expected to decrease Ea by 7.9 kJ mol−1, as highlighted by the protonation of 12 in t-BuPh. See ref. 41.
E′a = Ea,11 + Δ11→2 + Δ11→12.
E′a = Ea,11 + Δ11→9 + Δ11→12.
E′a = Ea,11 + Δ11→10 + Δ11→10.
Used as a model: Δ11→9 = −0.6 for RR/SS and Δ11→9 = +6.9 for RS/SR.
Used as a model: Δ11→10 = −1.0 for RR/SS and Δ11→10 = +0.9 for RS/SR.
Used as a reference.
Used as a model: Δ11→12 = 0 for RR/SS and Δ11→12 = +1.0 for RS/SR.
H2O:MeOH (1:1) is used as a solvent.
Used as a model: Δ11→3 = +1.1 for RR/SS and Δ11→3 = +0.2 for RS/SR.
Water as a solvent.
E′a = Ea,11 + Δ11→3 + Δ11→12.
Protonation expected to decrease Ea by 13.5 kJ mol−1, as highlighted by the protonation of 12 in a H2O/MeOH mixture. See ref. 41.
E′a = Ea,11 + Δ11→9 + Δ11→12.
Used as a model: Δ11→9 = −1.8 for RR/SS and Δ11→9 = +1.4 for RS/SR.
Used as a model: Δ11→12 = −2.0 for RR/SS and Δ11→12 = −1.0 for RS/SR.
|
|
2k,l |
t-BuPh |
90 |
3.0 |
124.4 |
7.1 |
3.5 |
123.9 |
8.2 |
—m |
—m |
t.w. |
|
2′l,n |
t-BuPh |
100 |
9.4 |
124.3 |
7.3 |
10.5 |
124.0 |
8.0 |
n.d. |
n.d. |
t.w. |
|
2′′l,o |
t-BuPh |
90 |
3.2 |
124.2 |
7.6 |
3.5 |
124.0 |
8.0 |
n.d. |
n.d. |
t.w. |
|
3l |
t-BuPh |
n.m. |
n.m. |
122.5 |
12.5 |
n.m. |
121.8 |
15.5 |
—p |
—p |
29
|
|
3′l,n |
t-BuPh |
100 |
14.7 |
122.9 |
11.2 |
16.7 |
122.5 |
12.7 |
n.d. |
n.d. |
t.w. |
|
4q |
t-BuPh |
80 |
4.1 |
120.1 |
26.1 |
2.4 |
121.7 |
16.0 |
122.5r |
122.8r |
t.w. |
|
4H+q |
t-BuPhs |
50 |
0.8 |
114.3 |
154.1 |
0.6 |
115.3 |
113.5 |
112.2t |
113.8t |
t.w. |
|
5q |
t-BuPh |
100 |
110.0 |
123.7 |
8.7 |
220.0 |
121.7 |
16.0 |
124.4u |
124.9u |
t.w. |
|
5H+q |
t-BuPh |
50 |
1.3 |
113.1 |
225.4 |
1.0 |
113.7 |
187.6 |
115.8t |
113.8t |
t.w. |
|
6l |
t-BuPh |
100 |
23.7 |
121.5 |
17.0 |
2.9 |
128.0 |
2.3 |
123.4v |
128.9v |
t.w. |
|
6H+l |
t-BuPhs |
83 |
11.8 |
117.9 |
51.2 |
1.2 |
124.8 |
6.2 |
113.6t |
120.1t |
t.w. |
|
7q |
t-BuPh |
83 |
2.8 |
122.2 |
13.7 |
4.4 |
120.9 |
20.4 |
123.0w |
124.9w |
t.w. |
|
7H+q |
t-BuPhs |
61 |
0.5 |
119.2 |
34.9 |
0.8 |
118.3 |
45.9 |
114.1t |
117.0t |
t.w. |
|
8
|
t-BuPh |
n.m. |
n.m. |
123.2 |
10.1 |
n.m. |
122.7 |
11.8 |
n.d. |
n.d. |
27
|
|
9
|
t-BuPh |
n.m. |
n.m. |
123.4 |
9.5 |
n.m. |
129.9 |
1.3 |
—x |
—x |
29
|
|
10
|
t-BuPh |
n.m. |
n.m. |
123.0 |
10.7 |
n.m. |
123.9 |
8.1 |
—y |
—y |
29
|
|
11
|
t-BuPh |
n.m. |
n.m. |
124.0 |
7.9 |
n.m. |
123.0 |
10.7 |
—z |
—z |
29
|
|
12
|
t-BuPh |
n.m. |
n.m. |
124.1 |
7.8 |
n.m. |
123.8 |
8.4 |
—aa |
—aa |
41
|
|
12H+
|
t-BuPh |
n.m. |
n.m. |
116.0 |
|
n.m. |
116.2 |
|
n.d. |
n.d. |
41
|
| |
|
3
|
—ab |
n.m. |
n.m. |
125.1 |
5.7 |
n.m. |
122.8 |
11.4 |
—ac |
—ac |
27
|
|
4q |
pH = 7.0ad |
80 |
8.3 |
118.0 |
49.6 |
12.8 |
116.8 |
71.7 |
121.3ae |
123.6ae |
t.w. |
|
4H+q |
pH = 1.4ad |
50 |
4.6 |
109.6 |
649.3 |
4.9 |
109.4 |
690.3 |
104.5af |
103.3af |
t.w. |
|
6l |
pH = 7.0ad |
93 |
14.9 |
120.3 |
24.6 |
5.0 |
124.1 |
7.7 |
118.4ag |
124.8ag |
t.w. |
|
6H+l |
pH = 1.4ad |
72 |
14.6 |
113.8 |
179.6 |
12.8 |
114.2 |
158.9 |
106.8af |
110.6af |
t.w. |
|
8
|
—ab |
n.m. |
n.m. |
123.8 |
8.4 |
n.m. |
123.2 |
10.1 |
n.d. |
n.d. |
29
|
|
9
|
—ab |
n.m. |
n.m. |
122.2 |
13.7 |
n.m. |
124.4 |
7.0 |
—ah |
—ah |
29
|
|
11
|
—ab |
n.m. |
n.m. |
123.0 |
10.7 |
n.m. |
124.0 |
7.9 |
—z |
—z |
29
|
|
12
|
—ab |
n.m. |
n.m. |
121.4 |
17.5 |
n.m. |
122.5 |
12.5 |
—ai |
—ai |
41
|
|
12H+
|
—ab |
n.m. |
n.m. |
108.0 |
|
n.m. |
109.0 |
|
n.d. |
n.d. |
41
|
IHB in alkoxyamines
A quick glance at 3, 4, 6 and 9 shows that several types of IHBs may occur (Fig. 4). The occurrence of interR IHB for RS/SR-9 has been reported in the literature.29 It has been observed by X-ray,29 NMR,29 and IR29 and is supported by DFT calculations and does not deserve more comments, as RS/SR-9 is used as a model. Diastereoisomer RR/SS-9 does not exhibit IHB.29 The occurrence of intraN IHB for 3 was recently studied by NMR¶¶27 and supported by DFT calculations in this work (see the ESI† and Table 5) and does not deserve more comments. X-ray data, NMR analysis, and DFT calculations support IHB of type interR, intraR, and intraN (Fig. 4, 8, 10 and 14) for RS/SR-6, RR/SS-6, and RS/SR-4, respectively (see the ESI†). In contrast with diastereoisomer RS/SR-6, no IHB is observed in diastereoisomer RR/SS-4 by X-ray analysis, although 31P NMR supports the occurrence of intraN IHB, 1H NMR supports the occurrence of interN IHB. The appealing 3-center IHB displayed in Fig. 8 is disregarded by DFT calculations (too high energy conformer).|||| On the other hand, DFT calculations show a small difference in energy of 2.5 kJ mol−1 between the stable conformer B and conformer A, more stable than C by 16 kJ mol−1, meaning that a fast equilibrium between B and A likely accounts for the simultaneous occurrence of intraN and interN IHBs observed by NMR. This fast equilibrium requires fast bond rotation around the C–N bond of the group carrying the hydroxy function. This fast rotation is nicely supported both by the temperature dependence of RR/SS-4 (not shown) and the kinetics and NMR observations reported for 2 and 3 (vide supra). The small difference in energy of 8 kJ mol−1 between diastereoisomers of 4 agrees with the small difference of 1.6 kJ mol−1 in Ea.
Table 5 Valence angles α <OHOP> and <OHN>, and distances dOH⋯OP and dOH⋯N for conformers A,aB,b and Cc of diastereoisomers of 4 and 6
| Alkoxyamine |
Conformer |
α
(°) |
Distancesd (Å) |
Δ
IHBe |
| <OHOP> |
<OHN> |
d
OH⋯OP
|
d
OH⋯N
|
|
Conformer exhibiting IHB between HO and PO functions.
Conformer exhibiting IHB between OH and N functions.
The most stable conformer with no IHB.
Bold values denote the most stable conformers.
In kJ mol−1. n.e. not eligible.
Δ
IHB = Ea,3 − Ea,8.
Not given because of too long dH⋯X forbidding any IHB. See ref. 38.
Δ
IHB = Ea,4 − Ea,5. See the ESI.
Δ
IHB = Ea,6 − Ea,7. See the ESI.
Not available.
A medium strength IHB between the HO group and the N-atom of the nitroxyl moiety is observed: dOH⋯N = 1.99 Å and <OHN> = 140°.
Δ
IHB = Ea,9 − Ea,10. See the ESI.
|
|
RR/SS-3 |
A
|
148
|
—j |
1.83 |
—e |
−0.7f |
|
RS/SR-3 |
A
|
150
|
—j |
1.83
|
—e |
−0.9f |
|
RR/SS-4 |
A
|
148
|
—g |
1.83
|
6.55 |
−3.6h |
|
B
|
—g |
149
|
6.25 |
2.06
|
n.e. |
|
C
|
—g |
—g |
6.79 |
6.87 |
n.e. |
|
RS/SR-4 |
A
|
146
|
—g |
1.85
|
6.61 |
0h |
|
B
|
—g |
161 |
4.78 |
2.13 |
n.e. |
|
C
|
—g |
—g |
6.68 |
8.05 |
n.e. |
|
RR/SS-6 |
A
|
170 |
—g |
1.82 |
5.09 |
n.e. |
|
B
|
—g |
136
|
5.16 |
2.11
|
−0.7i |
|
C
|
—g |
—g |
6.65 |
4.82 |
n.e. |
|
RS/SR-6 |
A
|
173
|
—g |
1.79
|
4.56 |
+7.1i |
|
B
|
—g |
136 |
4.99 |
2.03 |
n.e. |
|
C
|
—g |
—j,k |
4.91 |
4.84k |
n.e. |
|
RR/SS-9 |
C
|
—g |
—j |
6.00 |
—j |
n.e. |
|
RS/SR-9 |
A
|
174
|
—j |
1.79
|
—j |
+6.0l |
| |
|
RR/SS-4 |
X-ray |
—g |
—g |
6.63 |
6.91 |
n.e. |
|
RS/SR-6 |
X-ray |
164 |
—g |
1.96 |
4.29 |
n.e. |
Influence of IHB on kd
Several linear multiparameter relationships developed over the last two decades to investigate various effects involved in the changes in kd25,36 cannot account for the effect of IHB. In parallel with these relationships, the group additivity approach48 provides Ea values predicted with a good accuracy using a scale developed a decade ago.49 To use the group additivity approach, alkoxyamine 11 was selected as a benchmark, as it displays the nitroxyl fragment with the lowest functionalization in that series and a secondary aromatic alkyl fragment. Thus, estimated activation energies E′a were determined (Table 4 and ESI†). Taking into account the small difference in polarity between the alkyl fragment carrying a pyridyl (σI = 0.06)50,51 or a phenyl (σI = 0.07)52 moiety, no significant difference was reported,41 and the presence of the hydroxyl group capable of intraR IHB does not provide more than a 2-fold increase in kd, as highlighted by the couples 6/9 and 4/3. To unveil the effect of IHB on kd, the hydroxyl group was protected by silylation in 2 and 5, by methylation in 8, and by acylation in 7 and 10, to suppress the occurrence of IHB (see the ESI†). Kinetic results showed that the 4 types of IHBs can be gathered into two main families: alkyl and nitroxyl fragments exhibiting intra IHB which show no significant differences (less than a factor 2) between the alkoxyamines carrying the free hydroxyl group (3, RS/SR-4, and RR/SS-6) and those carrying the protected hydroxyl group (2, RS/SR-5, and RR/SS-7) and inter IHB between alkyl and nitroxyl fragments which shows a clear 6–9-fold decrease in kd between the alkoxyamines carrying the free hydroxyl group (diastereoisomers RS/SR of 6 and 9) and those carrying the protected hydroxyl group (diastereoisomers RS/SR of 7 and 10). Deeper insights into the geometrical parameters – the IHB valence angle α <OHX> and the IHB distance dOH⋯X – ruling the strength of the IHB and its influence on kd given by ΔIHB (see Table 5) are unveiled by the 3D plot f(ΔIHB,d,α) in Fig. 15. Indeed, intra IHB exhibits a very weak effect (−1 < ΔIHB < 0 kJ mol−1 except for intraN in RR/SS-4) associated with short H-bonds (dOH⋯X < 1.9 Å) and closed angles α (α < 150°) denoting medium strength IHB,*** whereas interR IHB exhibits a strong effect (ΔIHB > 6 kJ mol−1) associated with short H-bonds (dOH⋯X ≈ 1.8 Å) and open angles α (α ≈ 175°) denoting strong IHB. As mentioned above, diastereoisomer RR/SS-4 simultaneously displays intraN and interN IHBs and, thus, the decay of RR/SS-4 is described in Scheme 5 with K1 = k1/k−1 for the equilibrium constant between interN and intraN conformers, and k2 and k3 the rate constants for the homolysis of interN and intraN conformers, respectively. Taking into account that interN IHB makes a bond between nitroxyl and alkyl fragments as interR IHB does, the same effect is expected, i.e., an increase in Ea in regard to the homologue with no IHB. Therefore, k3 must be smaller than k2 which corresponds to intraN IHB noted to increase kd (vide supra). Hence, disregarding k3, and assuming both a fast equilibrium – supported by very close calculated energy levels (vide supra) – between conformers B (interN) and A (intraN) and the pre-equilibrium assumption (k−1 ⋙ k2 and K1 = k1/k−1), kd (considered as the apparent rate) is given as kd = k1k2/(k−1 + k2) (eqn (2) or as kd = K1k2 as in eqn (3), Scheme 5). As the decrease in Ea is in the expected range (see Table 4), the values of kd should be very close to k2, meaning that K1 ≈ 1 is in good agreement with the value estimated by DFT calculations, i.e., K1 = 0.42.†††| |  | (2) |
| |  | (3) |
 |
| | Fig. 15 A 3D plot highlighting the relationship between the energetic effect of IHB ΔIHB (kJ mol−1), IHB valence angle α (°), IHB distances dOH⋯X (Å) for the diastereoisomers of 3, 4, 6 and 9. Black and red symbols are intra and inter fragment IHB, respectively (see Fig. 4). Blue dots are for projection on the XY plane. | |
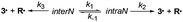 |
| | Scheme 5 Equilibrium between interN and intraN IHB and the subsequent homolysis. | |
Solvent effect on kd
The solvent effect in alkoxyamines does not increase too much interest despite the very amazing results recently reported, such as a striking 1500-fold increase in kd from t-BuPh to water as solvents or a slight but clear 5–20-fold increase in kd for homologue alkoxyamines of 12.53,54 Solvent effects for 3,279,29 and 1241 have been previously reported. Alkoxyamines 4, 6, 9 and 12 show a small ca. 1.5–3-fold increase in kd, as expected from the literature,53,54 except for RS/SR-4, RS/SR-6 and RS/SR-9. The 3-fold and 5-fold increases in kd for diastereoisomers RS/SR of 6 and 9 are ascribed to the suppression of IHB from t-BuPh to water/MeOH as solvents. The slight 4.5-fold increase in kd observed for RS/SR-4, from t-BuPh to water/MeOH as solvents, might be ascribed to the suppression of stabilizing IHB which balances the steric strain due to the configuration. The unexpected slight ca. 2-fold decrease in kd for 3 is ascribed to a better solvation of the alkoxyamine, and hence a better stabilization, and higher Ea from t-BuPh to water/MeOH as solvents than for the other alkoxyamines investigated.
For protonated alkoxyamines 4H+, 6H+ and 12H+, a 3–12-fold increase in kd is observed and is ascribed to intimate ion pair dissociation and solvation effects,55 except for RS/SR-6, for which a 25-fold increase in kd is observed due to the combination of IHB suppression and intimate ion pair dissociation.
Effect of protonation on kd
A ca. 10-fold and 60-fold increase in kd for 12 in t-BuPh and water/MeOH as solvents was reported upon protonation. Amazingly, the protonation effect for 4, i.e., a ca. 6-fold and 12-fold increase in t-BuPh and water/MeOH as solvents, respectively, and that for 6, i.e., a 3-fold and ca. 8-fold increase (for the RR/SS diastereoisomer) in t-BuPh and water/MeOH as solvents, respectively, are less strong than that for 12, although 6 and 4 exhibit structures very similar to that of 12! Suppressing IHB in water/MeOH for RS/SR-6H+ affords a 20-fold increase in kd upon protonation. This surprising lower effect of protonation for 4 and 6 is ascribed to the better solvation of the alkoxyamine for 4 and 6 than that for 12, due to the presence of the hydroxyl group affording the better stabilization of the starting alkoxyamine, which partly balances the polar effect due to protonation.
Experimental section
Alkoxyamines 3 were prepared as previously reported,26 with the modification of the first step. All solvents and reactants for the preparation of alkoxyamines were used as received. Routine reaction monitoring was performed using silica gel 60 F254 TLC plates; spots were visualized upon exposure to UV light and a phosphomolybdic acid solution in EtOH as a stain revealed by heating. Purifications were performed on a Reveleris® X2 flash chromatography system (BUCHI, Switzerland); a solvent delivery system: high pressure HPLC pumps; pump flow rate: 1–200 mL min−1; maximum pressure: 200 psi; gradients: linear (see Fig. 5SI† for the profile); UV wavelength range: 200–500 nm; flash Reveleris® and GraceResolv™ cartridges: silica 40 μm, silica weight (g): 4, 12, 24, 48, 80 and 120. 1H, 13C, and 31P NMR spectra were recorded in CDCl3 on a 300 or 400 MHz spectrometer. Chemical shifts (δ) in ppm were reported using residual non-deuterated solvents as the internal reference for 1H and 13C-NMR spectra, and as an internal capillary filled with 85% H3PO4 for 31P-NMR spectra. High-resolution mass spectra (HRMS) were recorded on a SYNAPT G2 HDMS (Waters) spectrometer equipped with a pneumatically assisted atmospheric pressure ionization source (API). Positive mode electrospray ionization was used on samples: electrospray voltage (ISV): 2800 V; opening voltage (OR): 20 V; nebulizer gas pressure (nitrogen): 800 L h−1. Low resolution mass spectra were recorded on an ion trap AB SCIEX 3200 QTRAP instrument equipped with an electrospray source. The parent ion [M + H]+ is quoted.
Diethyl (1-((1-hydroxy-2-methylpropan-2-yl)amino)-2,2-dimethylpropyl)phosphonate (3b)
Under N2, 2-methyl-2-aminopropanol 3a (6.8 g, 1.1 eq., 76.7 mmol) was added dropwise to a solution of pivalaldehyde (6.0 g, 1 eq., 69.7 mmol) in pentane (50 mL). The solution was heated at 65 °C with a Dean–Stark device for 2 days. Then, pentane was evaporated and diethylphosphite (10.6 g, 1.1 eq., 76.7 mmol) was added at r.t., and the mixture was heated at 45 °C for seven days. The solution was acidified with 1 M HCl (50 mL), and washed with dichloromethane (2 × 30 ml). The aqueous layer was basified with NaHCO3 and then extracted with dichloromethane (2 × 20 ml), the organic layer was dried (MgSO4), and the solvent was evaporated to yield aminophosphonate 3b (16.1 g, 78%).26
Diethyl (1-((1-((tert-butyldimethylsilyl)oxy)-2-methylpropan-2-yl)amino)-2,2-dimethylpropyl) phosphonate (2b)
The same procedure as that for 3b was applied to 2a. 2a (3 g, 1.1 eq., 16.23 mmol), pivalaldehyde (1.27 g, 1 eq., 14.75 mmol), pentane (20 mL) and diethylphosphite (2.2 g, 1.1 eq., 16.23 mmol). After flash-chromatography (gradient petroleum ether (PE)/AcOEt: 0% to 100% of AcOEt), aminophosphonate 2b was isolated (3.8 g, 64%, colourless oil). 1H NMR (300 MHz, CDCl3) δ: 4.09 (qd, J = 7.1, 1.3 Hz, 4H), 3.32 (s, 2H), 2.81 (d, JH–P = 17.0 Hz, 1H), 1.68 (s, 1H), 1.30 (t, J = 7.1 Hz, 6H), 1.06 (s, 3H), 1.04 (s, 9H), 0.99 (s, 3H), 0.90 (s, 9H), 0.03 (s, 6H). 13C NMR (75 MHz, CDCl3) δ: 72.8 (d, JC–P = 1.8 Hz), 61.4 (d, JC–P = 7.4 Hz), 61.0 (d, JC–P = 7.6 Hz), 58.9 (d, JC–P = 138.7 Hz), 54.2 (d, JC–P = 1.6 Hz), 35.2, 35.1, 27.9 (d, JC–P = 6.2 Hz, 2C), 25.9 (3C), 25.1 (d, JC–P = 1.5 Hz), 24.2, 18.2, 16.5 (d, JC–P = 4.6 Hz), 16.4 (d, JC–P = 5.0 Hz), −5.5 (2C). 31P NMR (162 MHz, CDCl3) δ: 30.09. HRMS m/z (ESI) calcd for C19H45NO4PSi [M + H]+ 410.2850, found: 410.2852.
(1-(Diethoxyphosphoryl)-2,2-dimethylpropyl)-(2-((tert-butyldimethylsilyl)oxy)-1,1-dimethylethyl)amino-N-oxyl radical (2˙)
Aminophosphonate 2b (1.0 g, 1 eq., 2.4 mmol) was dissolved in a mixture of EtOH/H2O (3:1) (15 mL) and Na2CO3 (1.55 g, 6 eq., 14.6 mmol). Then, Oxone® (3.0 g, 4 eq., 9.8 mmol) was added in small portions at r.t. within 2 h under vigorous stirring. After completion, Et2O (10 ml) was added, and the precipitate was filtered off. Et2O was removed under vacuum. The aqueous phase was extracted with dichloromethane (2 × 15 ml), and dried over MgSO4 and the solvents were evaporated under reduced pressure. The crude was purified by flash chromatography (gradient PE/AcOEt: 0% to 100% of AcOEt) and nitroxide 2˙ was isolated (529 mg, 51%). HRMS m/z (ESI) calcd for C19H44NO5PSi· [M + H]+ 425.2721, found: 425.2720.
Diethyl (1-((1-((tert-butyldimethylsilyl)oxy)-2-methylpropan-2-yl)(1-phenylethoxy)amino)-2,2-dimethylpropyl)phosphonate (2)
To a suspension of CuBr (55 mg, 0.55 eq., 0.388 mmol) and Cu powder (49 mg, 1.1 eq., 0.78 mmol) in degassed benzene (argon bubbling for one hour) (3 mL) was added N,N,N′,N′,N′′-pentamethyldiethylenetriamine (80 μL, 0.55 eq., 0.39 mmol). After stirring for 10 min, a solution of nitroxide 2˙ (300 mg, 1 eq., 0.71 mmol) and (1-bromoethyl)benzene (103 μL, 1.1 eq., 0.78 mmol) in degassed benzene (3 mL) was transferred to the first solution. The mixture was allowed to stir for 12 h. The solution was diluted with Et2O, quenched with a NH4Cl sat. solution, washed with water and brine, and dried over MgSO4. The solvents were evaporated under reduced pressure to give the crude product as a 1:1 mixture of diastereoisomers (31P-NMR ratio). The crude product was purified by automatic flash-chromatography (gradient PE/AcOEt: 0% to 100% of AcOEt) to yield alkoxyamines RR/SS-2 (pale yellow oil, 119 mg, 32%) and RS/SR-2 (pale yellow oil 123 mg, 33%). RS/SR-21H NMR (300 MHz, CDCl3) δ: 7.37 (d, J = 7.3 Hz, 2H), 7.17 (m, 3H), 5.18 (q, J = 6.4 Hz, 1H), 3.86 (m, 2H), 3.69 (d, JH–P = 26.6 Hz, 1H), 3.58 (s, 2H), 3.38 (m, 1H), 3.21 (m, 1H), 1.48 (d, J = 6.5 Hz, 3H), 1.15 (m, 18H), 0.86 (m, 12H), 0.00 (s, 6H). 13C NMR (75 MHz, CDCl3) δ: 143.2, 127.9 (2C), 127.7 (2C), 127.3, 78.5, 70.0 (d, JC–P = 139.2 Hz), 69.0, 65.1, 61.6 (d, JC–P = 6.4 Hz), 58.5 (d, JC–P = 7.5 Hz), 35.4 (d, JC–P = 5.3 Hz), 30.5 (d, JC–P = 6.0 Hz, 2C), 25.9 (3C), 23.4, 22.6, 21.2, 18.2, 16.3 (d, JC–P = 5.6 Hz), 16.1 (d, JC–P = 6.9 Hz), −5.4, −5.5. 1P NMR (121 MHz, CDCl3) δ: 24.98. RR/SS-21H NMR (300 MHz, CDCl3) δ: 7.45 (m, 5H), 5.13 (d, J = 6.8 Hz, 1H), 4.53 (m, 1H), 4.22 (m, 3H), 3.86 (d, JH–P = 26.5 Hz, 1H), 3.24 (q, J = 9.9 Hz, 2H), 1.74 (d, J = 6.7 Hz, 3H), 1.49 (m, 6H), 1.40 (s, 9H), 1.06 (s, 3H), 1.03 (s, 3H), 0.98 (s, 9H), 0.00 (s, 6H). 13C NMR (75 MHz, CDCl3) δ: 145.5, 128.2 (2C), 127.1, 126.8 (2C), 85.4, 69.7 (d, J = 138.1 Hz), 69.6 (d, J = 1.6 Hz), 65.3, 61.6 (d, J = 6.3 Hz), 58.8 (d, J = 7.5 Hz), 35.7 (d, J = 6.1 Hz), 29.9 (d, J = 5.8 Hz, 2C), 25.8 (3C), 24.5, 23.7, 22.1, 18.0, 16.8 (d, J = 5.4 Hz), 16.2 (d, J = 6.7 Hz), −5.5, −5.6. 31P NMR (121 MHz, CDCl3) δ: 26.19. HRMS for RR/SS-2 and RS/SR-2 mixture m/z (ESI) calcd for C27H53NO5PSi [M + H]+ 530.3425, found: 530.3425.
General procedure for TBS protection of alcohols 3a, 3b and 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) 32,56
32,56
In a flask under N2, alcohols 3a, 3b or 3 (1.0 eq.), tert-butyldimethylsilyl trifluoromethanesulfonate (1.5 eq.) and 2,6-lutidine (2 eq.) were stirred in dichloromethane at room temperature for 4 hours. A saturated NaHCO3 solution was added and the layers were separated. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. Purification was performed using flash chromatography, affording 2a, 2b, or 2 as an oil.
Hydrolysis31 of silylated compound 2
In a flask under N2, RR/SS-2 or RS/SR-2 (30 mg, 1 eq., 0.06 mmol) was dissolved in THF (2 mL), and tetra-n-butylammonium fluoride (1 M in THF) was added (68 μL, 1.2 eq., 0.07 mmol). After stirring for 1 hour at room temperature, THF was evaporated under reduced pressure and the crude was purified by automatic flash-chromatography (gradient PE/AcOEt: 0% to 100% of AcOEt) to yield alkoxyamine RR/SS-3′ or RS/SR-3′ with a yield higher than 90%.
Diethyl-(1-((1-hydroxy-2-methylpropan-2-yl)(1-(pyridin-2-yl)ethoxy)amino)-2,2-dimethylpropyl) phosphonate (4)
In an open flask, MnCl2 (70 mg, 0.05 eq., 0.35 mmol) was added to a stirred solution of salen ligand (130 mg, 0.05 eq., 0.35 mmol) in i-PrOH. After 30 minutes of stirring at room temperature, a solution of 3˙ (2.2 g, 1 eq., 7.1 mmol) and 2-vinylpyridine (1.15 mL, 1.5 eq., 10.6 mmol) in i-PrOH was added first, and then solid NaBH4 (1.07 g, 4 eq., 28.4 mmol) was added in small portions. The resulting suspension was stirred at room temperature for 4 h. It was then diluted with EtOAc and 1 M aq. HCl was carefully added. Solid NaHCO3 was then added until neutralization. The layers were separated, and the organic phase was washed with water and brine, and dried over Na2SO4. The solvent was evaporated to give the crude product as a 1:2 mixture of diastereoisomers (31P-NMR ratio). The diastereomers were separated by automatic flash column chromatography (gradient PE/AcOEt: 0% to 100% of AcOEt) to afford RR/SS-4 (pale yellow crystal 560 mg, 19%) and RS/SR-4 (pale yellow oil 1.01 g 35%). RS/SR-41H NMR (300 MHz, CDCl3) δ: 8.44 (dd, J = 5.1, 1.7 Hz, 1H), 7.59 (td, J = 7.7, 1.8 Hz, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.07 (ddd, J = 7.6, 4.8, 1.2 Hz, 1H), 5.20 (q, J = 6.5 Hz, 1H), 3.89 (d, J = 12.7 Hz, 1H), 3.67 (m, 4H), 3.57 (d, JH–P = 26.5 Hz, 1H), 3.44 (d, J = 12.6 Hz, 1H), 1.57 (d, J = 6.6 Hz, 3H), 1.12 (s, 3H), 1.10 (s, 9H), 1.09 (s, 3H), 1.04 (t, J = 7.1 Hz, 3H), 0.98 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 162.1, 148.6, 136.9, 122.3, 121.6, 78.7, 70.5 (d, JC–P = 136.3 Hz), 70.0, 64.9, 61.7 (d, JC–P = 6.6 Hz), 60.1 (d, JC–P = 7.9 Hz), 35.3 (d, JC–P = 4.0 Hz), 30.6 (d, JC–P = 5.7 Hz, 3C), 26.9, 22.0, 20.4, 16.4 (d, JC–P = 5.7 Hz), 15.9 (d, JC–P = 6.8 Hz). 31P NMR (121 MHz, CDCl3) δ: 26.13. HRMS m/z (ESI) calcd for C20H38N2O5P [M + H]+ 417.2513, found: 417.2513. RR/SS-41H NMR (400 MHz, CDCl3) δ: 8.49 (m, 1H), 7.66 (td, J = 7.7, 1.8 Hz, 1H), 7.43 (d, J = 7.9 Hz, 1H), 7.15 (ddd, J = 7.6, 4.9, 1.2 Hz, 1H), 5.09 (q, J = 6.7 Hz, 1H), 4.64 (s, 1H), 4.17 (m, 4H), 3.62 (d, JH–P = 26.4 Hz, 1H), 3.49 (d, J = 12.3 Hz, 1H), 3.04 (d, J = 12.4 Hz, 1H), 1.54 (d, J = 6.7 Hz, 3H), 1.33 (t, J = 7.1 Hz, 6H), 1.18 (s, 9H), 1.02 (s, 3H), 0.68 (s, 3H). 13C NMR (101 MHz, CDCl3) δ: 164.2, 148.7, 136.7, 122.4, 121.7, 86.3, 69.6 (d, JC–P = 136.1 Hz), 67.5, 65.1, 62.0 (d, JC–P = 6.6 Hz), 60.6 (d, JC–P = 7.8 Hz), 35.8 (d, JC–P = 4.6 Hz), 30.1 (d, JC–P = 5.7 Hz, 3C), 26.8, 23.6, 23.2, 16.6 (d, JC–P = 5.7 Hz), 16.4 (d, JC–P = 6.6 Hz). 31P NMR (162 MHz, CDCl3) δ: 27.29. HRMS m/z (ESI) calcd for C20H38N2O5P [M + H]+ 417.2513, found: 417.2515.
Diethyl ((R)-1-((1-hydroxy-2-methylpropan-2-yl)((S)-1-(pyridin-2-yl)ethoxy)amino)-2,2-dimethylpropyl)phosphonate (RS/SR-5)
Alkoxyamine RS/SR-4 (400 mg, 1 eq., 0.96 mmol) was dissolved in 20 mL THF, and pyridine was added (388 μL, 5 eq., 4.80 mmol). Silver nitrate (244 mg, 1.5 eq., 1.44 mmol) was added and after stirring for 5 minutes, t-butyldimethylsilyl chloride (217 mg, 1.5 eq., 1.44 mmol) was added and stirring was continued at room temperature overnight. At the end of the reaction period, the solution was washed with 10% NaHCO3 solution and extracted with DCM. The combined organic phases were dried with MgSO4 and the solvent was evaporated. The crude product was purified by flash chromatography (petroleum ether/AcOEt 7:3) to afford RS/SR-5 (pale yellow oil, 341 mg, 67%).1H NMR (400 MHz, CDCl3) δ: 8.52 (ddd, J = 4.9, 1.8, 0.9 Hz, 1H), 7.60 (m, 2H), 7.11 (ddd, J = 7.3, 4.8, 1.5 Hz, 1H), 5.26 (q, J = 6.6 Hz, 1H), 3.87 (m, 2H), 3.77 (d, JH–P = 26.7 Hz, 1H), 3.71 (m, 1H), 3.63 (s, 2H), 3.57 (m, 1H), 1.58 (d, J = 6.6 Hz, 3H), 1.17 (m, 18H), 1.03 (t, J = 7.1 Hz, 3H), 0.91 (s, 9H), 0.04 (2s, 6H). 13C NMR (101 MHz, CDCl3) δ: 162.5, 148.6, 136.1, 122.5, 122.1, 79.9, 69.8 (d, JC–P = 137.0 Hz), 69.2, 65.4, 61.3 (d, JC–P = 6.4 Hz), 59.1 (d, JC–P = 7.6 Hz), 35.5 (d, JC–P = 5.1 Hz), 30.3 (d, JC–P = 5.9 Hz), 25.9 (5C), 23.2, 22.8, 20.2, 18.3, 16.4 (d, JC–P = 5.8 Hz), 16.1 (d, JC–P = 7.0 Hz), −5.4 (2C). 31P NMR (162 MHz, CDCl3) δ: 25.22. HRMS m/z (ESI) calcd for C26H52N2O5PSi [M + H]+ 531.3378, found: 531.3380.
Diethyl((R)-1-((1-hydroxy-2-methylpropan-2-yl)((R)-1-(pyridin-2-yl)ethoxy)amino)-2,2-dimethylpropyl)phosphonate (RR/SS-5)
The same procedure as that for RS/SR-5 was applied to RR/SS-4. RR/SS-4 (210 mg, 1 eq., 0.5 mmol), 10 mL THF, pyridine (203 μL, 5 eq., 2.52 mmol), silver nitrate (128 mg, 1.5 eq., 0.76 mmol), t-butyldimethylsilyl chloride (114 mg, 1.5 eq., 0.76 mmol). RR/SS-5 (pale yellow oil, 157 mg, 59%). 1H NMR (400 MHz, CDCl3) δ: 8.53 (ddd, J = 4.9, 1.8, 0.9 Hz, 1H), 7.66 (td, J = 7.7, 1.8 Hz, 1H), 7.34 (d, J = 7.8 Hz, 1H), 7.15 (ddd, J = 7.5, 4.9, 1.2 Hz, 1H), 5.13 (q, J = 6.8 Hz, 1H), 4.37 (m, 1H), 4.09 (m, 3H), 3.73 (d, JH–P = 26.2 Hz, 1H), 3.12 (s, 2H), 1.59 (d, J = 6.8 Hz, 3H), 1.36 (t, J = 7.1 Hz, 3H), 1.30 (t, J = 7.1 Hz, 3H), 1.23 (s, 9H), 0.88 (s, 3H), 0.83 (s, 3H), 0.81 (s, 9H), −0.14 (2s, 6H). 13C NMR (75 MHz, CDCl3) δ: 164.5, 148.8, 136.2, 122.1, 121.7, 86.4, 69.8 (d, JC–P = 138.3 Hz), 69.3 (d, JC–P = 1.4 Hz), 65.4, 61.6 (d, JC–P = 6.4 Hz), 58.9 (d, JC–P = 7.5 Hz), 35.8 (d, JC–P = 6.0 Hz), 29.8 (d, JC–P = 5.8 Hz), 25.8 (3C), 23.3, 23.0, 22.6, 18.1, 16.8 (d, JC–P = 5.5 Hz), 16.2 (d, JC–P = 6.8 Hz), −5.6 (2C). 31P NMR (162 MHz, CDCl3) δ: 26.11. HRMS m/z (ESI) calcd for C26H52N2O5PSi [M + H]+ 531.3378, found: 531.3380.
2-Bromo-2-(pyridin-2-yl)ethyl acetate (6b)
2-(Pyridin-2-yl)ethyl acetate 6a (2.0 g, 1 eq., 12.0 mmol), N-bromosuccinimide (2.4 g, 1.1 eq., 13.2 mmol) and benzoyl peroxide (0.03 g, 0.01 eq., 0.12 mmol) were mixed with CCl4 (100 mL). An argon bubbled solution was refluxed (77 °C). The reaction was monitored by thin layer chromatography (TLC) after until complete disappearance of 6a. The solution was then quenched and washed with NaHCO3 sat., and the aqueous phases were extracted with CH2Cl2. The combined organic layers were washed with water, dried over MgSO4 and concentrated under reduced pressure. The crude product was subjected to automatic flash-chromatography (gradient PE/AcOEt: 0% to 100% of AcOEt) to yield bromide 6b (2.02 g, 69%). 1H NMR (400 MHz, CDCl3) δ: 8.59 (d, J = 5.6 Hz, 1H), 7.70 (t, J = 8.6 Hz, 1H), 7.42 (d, J = 7.9 Hz, 1H), 7.28–7.20 (m, 1H), 5.18 (t, J = 7.1 Hz, 1H), 4.73 (d, J = 7.1 Hz, 2H), 2.02 (s, 3H). 13C NMR (101 MHz, CDCl3) δ: 170.3, 156.7, 149.4, 137.3, 123.6, 123.1, 66.3, 49.1, 20.7. HRMS (ESI) calc. for C9H11NO2Br [M + H]+: 243.9968; found: 243.9967.
Diethyl-(1-(tert-butyl-(2-hydroxy-1-(pyridin-2-yl)ethoxy)amino)-2,2-dimethylpropyl) phosphonate (6)
To a suspension of CuBr (660 mg, 0.55 eq., 4.6 mmol) and Cu powder (580 mg, 1.1 eq., 9.1 mmol) in degassed benzene (argon bubbling for one hour) (30 mL) was added N,N,N′,N′,N′′-pentamethyldiethylenetriamine (1 mL, 0.55 eq., 4.6 mmol). After stirring for 10 min, a solution of 6˙ (2.68 g, 1.1 eq., 9.1 mmol) and bromide 6b (2.0 g, 1 eq., 8.3 mmol) in degassed benzene (30 mL) was cannulated into the first solution. The mixture was allowed to stir for 12 h. The solution was diluted with EtOAc, quenched and washed with 50% (v/v) aq. ammonia solution and NaHCO3 saturated solution, and dried over MgSO4. The solvents were evaporated under reduced pressure. The crude product (1.1 g, 1 eq., 2.4 mmol) with a 1:4 diastereomeric ratio (31P NMR ratio) was dissolved in MeOH (7 mL), and a solution of K2CO3 (663 mg, 2 eq., 4.8 mmol) in H2O (7 mL) was added at once to the flask. The solution was allowed to stir for 3 days and then quenched with water. The aqueous phase was extracted with CH2Cl2 and dried over MgSO4, and the solvents were evaporated under reduced pressure. The crude product was subjected to automatic flash-column chromatography (PE/acetone 7:3) to afford RS/SR-6 (144 mg) and RR/SS-6 (490 mg) as colourless oils, corresponding to a total yield of 634 mg (63%). RS/SR-61H NMR (400 MHz, CDCl3) δ: 8.53 (d, J = 4.8 Hz, 1H), 7.64 (t, J = 7.7 Hz, 1H), 7.37 (d, J = 7.7 Hz, 1H), 7.18–7.09 (m, 1H), 5.93 (t, J = 6.9 Hz, 1H), 5.11 (s, 1H), 4.44 (dd, J = 16.5, 7.7 Hz, 1H), 4.28 (dd, J = 16.7, 9.4 Hz, 1H), 4.21–4.09 (m, 2H), 4.02 (dd, J = 15.8, 7.9 Hz, 1H), 3.84 (dd, J = 15.1, 8.5 Hz, 1H), 3.43 (d, JH–P = 27.6 Hz, 1H), 1.38 (t, J = 7.8 Hz, 3H), 1.34 (t, J = 7.6 Hz, 3H), 1.25 (s, 9H), 0.92 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 160.8, 149.0, 136.0, 122.7, 122.3, 91.0, 68.8 (d, JC–P = 139.6 Hz), 65.0, 62.1, 62.1, 59.8 (d, J = 7.6 Hz), 35.8 (d, J = 5.1 Hz), 30.9 (d, J = 5.8 Hz, 2C), 28.1 (3C), 16.7 (d, J = 5.5 Hz), 16.24 (d, J = 7.0 Hz). 31P NMR (162 MHz, CDCl3) δ: 27.16. HRMS (ESI) calc for C20H38N2O5P+: 417.2513 [M + H]+; found: 417.2512. RR/SS-61H NMR (400 MHz, CDCl3) δ: 8.48 (d, J = 5.5 Hz, 1H), 7.73 (s, 1H), 7.65 (t, J = 8.5 Hz, 1H), 7.20–7.13 (m, 1H), 5.29 (t, J = 5.6 Hz, 1H), 4.72 (s, 1H), 4.34 (dd, J = 11.2, 5.3 Hz, 1H), 4.01 (dd, J = 11.2, 6.0 Hz, 1H), 3.96–3.83 (m, 2H), 3.73–3.60 (m, 2H), 3.46 (d, JH–P = 26.7 Hz, 1H), 1.25 (s, 9H), 1.21 (s, 9H), 1.13 (t, J = 7.1 Hz, 3H), 1.03 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ: 159.7, 148.2, 136.4, 123.3, 122.6, 80.5, 70.2 (d, JC–P = 138.8 Hz), 64.7, 62.2, 61.3 (d, J = 6.7 Hz), 59.8 (d, J = 7.7 Hz), 35.4, 35.3, 30.8 (d, J = 5.9 Hz, 2C), 28.1 (3C), 16.3 (d, J = 6.0 Hz), 16.0 (d, J = 6.9 Hz). 31P NMR (162 MHz, CDCl3) δ: 24.48. HRMS (ESI) calc for C20H38N2O5P+: 417.2513 [M + H]+; found: 417.2511.
(S)-2-((tert-Butyl((R)-1-(diethoxyphosphoryl)-2,2-dimethylpropyl)amino)oxy)-2-(pyridin-2-yl)ethyl acetate (RS/SR-7)
RS/SR-6 (144 mg, 1 eq., 0.35 mmol) was diluted in CH2Cl2 (5 mL) and triethylamine (0.2 mL, 4 eq., 1.4 mmol) was added. After 2 minutes, acetic anhydride (0.1 mL, 3 eq., 1.1 mmol) was added slowly via a syringe. The reaction was allowed to stir for 3 days and was then quenched with a saturated aqueous solution of NaHCO3. The solution was extracted with CH2Cl2. The combined organic extracts were dried with MgSO4, filtered, and concentrated under vacuum. The crude was purified by automatic flash-column chromatography (petroleum ether/acetone 9:1) to yield RS/SR-7 (140 mg, 90%) as a white solid. 1H NMR (300 MHz, CDCl3) δ: 8.54 (d, J = 4.7 Hz, 1H), 7.66 (t, J = 7.7 Hz, 1H), 7.36 (d, J = 7.8 Hz, 1H), 7.21–7.12 (m, 1H), 5.30 (dd, J = 7.7, 4.2 Hz, 1H), 4.84 (dd, J = 10.9, 4.2 Hz, 1H), 4.69–4.59 (m, 1H), 4.48–4.33 (m, 1H), 4.21 (td, J = 16.6, 8.9 Hz, 1H), 4.03 (m, J = 17.2, 12.7, 7.2 Hz, 2H), 3.35 (d, JH–P = 26.1 Hz, 1H), 1.79 (s, 3H), 1.36 (t, J = 6.4 Hz, 3H), 1.31 (t, J = 7.0 Hz, 3H), 1.23 (s, 9H), 0.87 (s, 9H). 13C NMR (101 MHz, CDCl3) δ: 170.1, 160.1, 148.8, 135.9, 123.3, 122.6, 87.5, 69.5 (d, JC–P = 138.9 Hz), 65.7, 61.8 (2C), 61.7 (d, J = 6.4 Hz), 59.3 (d, J = 7.4 Hz), 35.8 (d, J = 5.4 Hz), 30.1 (d, J = 5.8 Hz, 2C), 28.4 (3C), 20.7, 16.8 (d, J = 5.4 Hz), 16.3 (d, J = 6.8 Hz). 31P NMR (121 MHz, CDCl3) δ: 25.27. HRMS (ESI) calc for C22H40N2O6P+: 459.2619 [M + H]+; found: 459.2624.
(R)-2-((tert-Butyl((R)-1-(diethoxyphosphoryl)-2,2-dimethylpropyl)amino)oxy)-2-(pyridin-2-yl)ethyl acetate (RR/SS-7)
The same procedure as that for RS/SR-7 was applied to RR/SS-6. RR/SS-6 (490 mg, 1 eq., 1.18 mmol), CH2Cl2 (10 mL), triethylamine (0.5 mL, 4 eq., 3.5 mmol), acetic anhydride (0.3 mL, 3 eq., 3 mmol) flash-column chromatography (PE/acetone 9:1) RR/SS-7 (343 mg, 65%) as a colorless solid. 1H NMR (300 MHz, CDCl3) δ: 8.55 (d, J = 4.8 Hz, 1H), 7.69–7.57 (m, 2H), 7.20–7.08 (m, 1H), 5.35 (dd, J = 6.5, 3.6 Hz, 1H), 4.76 (m, 2H), 3.88 (p, J = 7.2 Hz, 2H), 3.74–3.50 (m, 2H), 3.44 (d, JH–P = 27.1 Hz, 1H), 1.86 (s, 3H), 1.23 (s, 9H), 1.19 (t, J = 7.1 Hz, 3H), 1.15 (s, 9H), 1.00 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ: 170.6, 158.1, 148.7, 135.8, 124.6, 122.7, 80.8, 69.6 (d, JC–P = 138.6 Hz), 64.1, 62.0, 61.4 (d, J = 6.6 Hz), 59.5 (d, J = 7.7 Hz), 35.3 (d, J = 4.4 Hz), 30.7 (d, J = 5.9 Hz, 2C), 28.0 (3C), 20.7, 16.3 (d, J = 5.9 Hz), 16.1 (d, J = 6.8 Hz). 31P NMR (162 MHz, CDCl3) δ: 24.27. HRMS (ESI) calc for C22H40N2O6P+: 459.2619 [M + H]+; found: 459.2623.
Kinetic measurements
The values of the homolysis rate constant kd were determined by monitoring either the concentration of nitroxide by EPR or the concentration of alkoxyamine by 31P NMR. For EPR, a sample tube filled with a solution of 10−4 M of each diastereoisomer in tert-butylbenzene or in H2O/MeOH (1:1) was set in the EPR cavity. EPR signals were recorded. The temperature was controlled by using a BVT2000 temperature controlling unit. Measurements of kd by 31P NMR required the use of TEMPO as an alkyl radical scavenger. The NMR tubes were filled with a stock solution of 0.02 M of alkoxyamine in tert-butylbenzene or in H2O/MeOH with 2 equiv. of TEMPO. Buffer solutions were used for specific pH conditions instead of H2O. kd values were given by eqn (4). Activation energies Ea were estimated using eqn (5) and the average frequency factor A = 2.4 × 1014 s−1. The values of kd and Ea are listed in Table 4.| |  | (4) |
Conclusion
Different types of IHBs exhibiting very different effects are highlighted. These effects depend on the strength of the IHB, which in turn is straightforwardly related to its geometric parameters – dOH⋯X the distance for IHB and the IHB valence angle α. That is, strong interR IHBs are expected for α larger than 160° and dOH⋯X smaller than 1.8 Å affording an increase in Ea (Tables 4 and 5). As geometric parameters – α ≈ 150° and dOH⋯X ≈ 2.1 Å – underline a weak IHB, interN IHB does not exhibit the same effect as that of interR IHB. Indeed, this interN IHB is weak enough that it is in equilibrium with the intraN IHB which, in turn, favours slightly the C–ON bond homolysis, i.e., decreasing Ea. Nevertheless, a strong interN IHB is expected to afford the same effect as that of a strong interR IHB. IntraN and intraR IHBs exhibit features of IHB of intermediate strength – α < 150° and dOH⋯X > 1.8 Å – affording a decrease in Ea. However, the effect of intraR and intraN depends a lot on the strength of these IHBs at TS, that is, on the stereoelectronic requirement for the C–ON bond homolysis and on the stabilization of the products, i.e., partly due to the strength of IHB. Hence, stronger IHBs in products than in starting materials stabilize TS and decrease Ea. This assumption is supported by the stronger IHB in 3˙ (α = 178° and dOH⋯X = 1.57 Å) than in 3 and 4 (Table 5).‡‡‡57
As a rule of thumb, one may assume that IHB between alkyl and nitroxyl fragments (interN or interR IHB) affords an increase in Ea which might be pictured as the cleavage of two bonds – C–ON bond and IHB – and that IHB inside each fragment (intraN or intraR IHB) affords a slight decrease in Ea provided the stabilization due to IHB is larger for the released radicals than that for the starting material. Otherwise, an increase in Ea might be expected.
This work highlights the combination of several effects – IHB, solvents, intimate ion pairs, and protonation – to strikingly decrease the alkoxyamine half-life time t1/2, as highlighted by t1/2 = 123 days for 6 in t-BuPh as a solvent at 37 °C and by t1/2 = 14 hours for 6H+ in water/MeOH as a solvent at 37 °C, that is, a 210-fold increase in kd. These results nicely highlight the potential of such alkoxyamines as switches for applications in biology.17
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
SRAM, PB, VR and GA thank Aix-Marseille Université and CNRS for support. SRAM and ME are grateful to the Russian Science Foundation (grant 15-13-20020) for supporting this work and to the Multi-Access Chemical Service Center SB RAS for spectral and analytical measurements. SRAM, VR, GA and JPJ are grateful to ANR for funding this project (ANR-14-CE16-0023-01). PN thanks the Ministère de la Recherche du Gabon.
Notes and references
-
D. H. Solomon, E. Rizzardo and P. Cacioli, Eur. Pat. Appl135280, 1985 Search PubMed;
D. H. Solomon, E. Rizzardo and P. Cacioli, US Patent4581429, 1986 Search PubMed;
Chem. Abstr., 1985, 102, 221335q Search PubMed.
-
Nitroxide Mediated Polymerization: From Fundamentals to Applications in Materials Sciences, ed. D. Gigmes, RSC Polymer Chemistry Series 19, Royal Society of Chemistry, London, 2016; and 513 references cited therein Search PubMed.
- A. Studer, Chem. Soc. Rev., 2004, 33, 267–273 RSC.
- L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034–5068 CrossRef CAS PubMed.
- A. Studer, Angew. Chem., Int. Ed., 2000, 39, 1108–1111 CrossRef CAS PubMed.
- J. Xu, E. J. E. Caro-Diaz, L. Trzoss and E. A. Theodorakis, J. Am. Chem. Soc., 2012, 134, 5072–5075 CrossRef CAS PubMed.
- C. Wetter, K. Jantos, K. Woithe and A. Studer, Org. Lett., 2003, 5, 2899–2902 CrossRef CAS PubMed.
- V. Sciannamea, R. Jérôme and C. Detrembleur, Chem. Rev., 2008, 108, 1104–1126 CrossRef CAS PubMed.
- E. H. H. Wong, M. H. Stenzel, T. Junkers and C. Barner-Kowollik, Macromolecules, 2010, 43, 3785–3793 CrossRef CAS.
- Y. Guillaneuf, D.-L. Versace, D. Bertin, J. Lalevée, D. Gigmes and J. P. Fouassier, Macromol. Rapid Commun., 2010, 31, 1909–1913 CrossRef CAS PubMed.
- E. Yoshida, Colloid Polym. Sci., 2010, 288, 1639–1643 CAS.
- G. Audran, E. G. Bagryanskaya, P. Brémond, M. V. Edeleva, S. R. A. Marque, D. A. Parkhomenko, O. Y. Rogozhnikova, V. M. Tormyshev, E. V. Tretyakov, D. V. Trukhin and S. I. Zhivetyeva, Polym. Chem., 2016, 7, 6490–6499 RSC.
- G. Audran, E. G. Bagryanskaya, M. V. Edeleva, S. R. A. Marque, D. A. Parkhomenko, E. V. Tretyakov and S. I. Zhivetyeva, J. Polym. Sci.: Part A: Polym. Chem., 2017 Search PubMed , submitted.
- M. Q. Zhang and M. Z. Rong, Polym. Chem., 2013, 4, 4878 RSC.
- B. Schulte, M. Tsotsalas, M. Becker, A. Studer and L. De Cola, Angew. Chem., Int. Ed., 2010, 49, 6881–6884 CrossRef CAS PubMed.
- R. K. Roy, A. Meszynska, C. E. Laure, L. Charles, C. Verchin and J.-F. Lutz, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- G. Audran, P. Brémond, J.-M. Franconi, S. R. A. Marque, P. Massot, P. Mellet, E. Parzy and E. Thiaudière, Org. Biomol. Chem., 2014, 12, 719–723 CAS.
- D. Moncelet, P. Voisin, N. Koonjoo, V. Bouchaud, P. Massot, E. Parzy, G. Audran, J.-M. Franconi, E. Thiaudière, S. R. A. Marque, P. Brémond and P. Mellet, Mol. Pharmaceutics, 2014, 11, 2412–2419 CrossRef CAS PubMed.
- N. A. Popova, G. M. Sysoeva, V. P. Nicolin, V. I. Kaledin, E. V. Tretyakov, M. V. Edeleva, S. M. Balakhnin, E. L. Louschnikova, G. Audran and S. Marque, Bull. Exper. Biol. Med., 2017, 164(7), 61–65 Search PubMed.
- P. Brémond, A. Koïta, S. R. A. Marque, V. Pesce, V. Roubaud and D. Siri, Org. Lett., 2012, 14(1), 358–361 CrossRef PubMed.
- G. Audran, E. Bagryanskaya, I. Bagryanskaya, P. Brmond, M. Edeleva, S. R. A. Marque, D. Parkhomenko, E. Tretyakov and S. Zhivetyeva, Inorg. Chem. Front., 2016, 3, 1464–1472 RSC.
- G. Audran, E. Bagryanskaya, I. Bagryanskaya, M. Edeleva, S. R. A. Marque, D. Parkhomenko, E. Tretyakov and S. Zhivetyeva, ChemistrySelect, 2017, 2, 3584–3593 CrossRef CAS.
- P. Nkolo, G. Audran, P. Brémond, R. Bikanga, S. R. A. Marque and V. Roubaud, Org. Biomol. Chem., 2017, 15, 6167–6176 CAS.
- K. Matyjaszewski, S. Gaynor, D. Greszta, D. Mardare and T. Shigemoto, J. Phys. Org. Chem., 1995, 8, 306–315 CrossRef CAS.
- S. Marque, H. Fischer, E. Baier and A. Studer, J. Org. Chem., 2001, 66, 1146–1156 CrossRef CAS PubMed.
- S. Acerbis, D. Bertin, B. Boutevin and D. Gigmes, Helv. Chim. Acta, 2006, 89, 2119–2132 CrossRef CAS.
- G. Audran, P. Brémond, S. R. A. Marque and T. Yamasaki, J. Org. Chem., 2016, 81, 1981–1988 CrossRef CAS PubMed.
- E. G. Bagryanskaya, P. Brémond, T. Butscher, S. R. A. Marque, D. Parkhomenko, V. Roubaud, D. Siri and S. Viel, Macromol. Chem. Phys., 2015, 216(5), 475–488 CrossRef CAS.
- P. Brémond, T. Butscher, V. Roubaud, D. Siri and S. Viel, J. Org. Chem., 2013, 78, 10524–10529 CrossRef PubMed.
-
C. Reichardt and T. Welton, Solvent and Solvent Effect in Organic Chemistry, Wiley-VCH, Weinheim, 2011 Search PubMed.
- E. J. Corey and A. Venkateswarlu, J. Am. Chem. Soc., 1972, 94, 6190 CrossRef CAS.
- E. J. Corey, H. Cho, C. Rücker and D. H. Hua, Tetrahedron Lett., 1981, 22, 3455 CrossRef CAS.
- R. Braslau, N. Naik and H. Zipse, J. Am. Chem. Soc., 2000, 122, 8421–8434 CrossRef CAS.
- K. K. Ogilvie, G. H. Hakimelahi, Z. A. Proba and D. P. C. McGee, Tetrahedron Lett., 1982, 23((19), 1997–2000 CrossRef.
- K. Matyjaszewski, B. E. Woodworth, X. Zhang, S. G. Gaynor and Z. Metzner, Macromolecules, 1998, 31, 5955–5957 CrossRef CAS.
-
E. G. Bagryanskaya and S. R. A. Marque, in Kinetic Aspects of Nitroxide-Mediated Polymerization, RSC Polymer Chemistry Series, n° = 19, Nitroxide Mediated Polymerization: From Fundamentals to Applications in Materials Sciences, ed. D. Gigmes, Royal Society of Chemistry, 2016, ch. 2, pp. 45–113 Search PubMed.
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS.
-
G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer, Berlin, Heidelberg, 1994 Search PubMed.
- A. Krȩżel and W. Bal, J. Inorg. Biochem., 2004, 98, 161–166 CrossRef.
- R. J. L. Andon, J. D. Cox and E. F. G. Herington, Trans. Faraday Soc., 1954, 50, 918–910 RSC.
- G. Audran, M. Bim Batsiandzy Ibanou, P. Brémond, S. R. A. Marque, V. Roubaud and D. Siri, Org. Biomol. Chem., 2013, 11, 7738–7750 CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision A.02), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- S. Marque, J. Org. Chem., 2003, 68, 7582–7590 CrossRef CAS PubMed.
- S. Acerbis, E. Beaudoin, D. Bertin, D. Gigmes, S. Marque and P. Tordo, Macromol. Chem. Phys., 2004, 205, 973–978 CrossRef CAS.
- A. Studer, K. Harms, C. Knoop, C. Müller and T. Schulte, Macromolecules, 2004, 37, 27–34 CrossRef CAS.
- G. Audran, R. Bikanga, P. Brémond, J.-P. Joly, S. R. A. Marque and P. Nkolo, J. Org. Chem., 2017, 82, 5702–5709 CrossRef CAS PubMed.
- P. Nkolo, G. Audran, P. Brémond, R. Bikanga, S. R. A. Marque and V. Roubaud, Tetrahedron, 2017, 73, 3188–3201 CrossRef CAS.
-
S. W. Benson, Thermochemical Kinetics. Methods for the Estimation of Thermochemical Data and Rate Parameters, John Wiley & Sons, Inc., New York, 1968 Search PubMed.
- E. Beaudoin, D. Bertin, D. Gigmes, S. R. A. Marque, D. Siri and P. Tordo, Eur. J. Org. Chem., 2006, 1755–1768 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- D. Bertin, D. Gigmes, S. R. A. Marque and P. Tordo, Macromolecules, 2005, 38, 2638–2650 CrossRef CAS.
- M. Charton, Prog. Phys. Org. Chem., 1981, 13, 119–251 CAS.
- G. Audran, P. Brémond, S. R. A. Marque and G. Obame, Polym. Chem., 2012, 3, 2901–2908 RSC.
- G. Audran, P. Brémond, S. R. A. Marque and G. Obame, J. Org. Chem., 2012, 77(21), 9634–9640 CrossRef CAS PubMed.
- G. Audran, P. Brémond, S. R. A. Marque and G. Obame, J. Org. Chem., 2013, 78, 7754–7757 CrossRef CAS PubMed.
- T. J. Barker and E. R. Jarvo, Angew. Chem., Int. Ed., 2011, 50, 8325–8328 CrossRef CAS PubMed.
- M. C. Foti, R. Amorati, G. F. Pedulli, C. Daquino, D. A. Pratt and K. U. Ingold, J. Org. Chem., 2010, 75, 4434–4440 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR and HRMS spectra of 2–7 and 2˙. DFT calculations for the most stable conformers of 4 and 6. A gradient profile for flash chromatography. The titration curve for 2-hydroxyethylpyridine, and XRD for RR/SS-4 and RS/SR-6. CCDC 1550955 and 1550956. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ob02223a |
| ‡ CCDC: 1550955 for RR/SS-4 and 1550956 for RS/SR-6.† |
| § The procedure described in Scheme 2 was not used because it afforded the isomerization of the pure diastereoisomer. |
| ¶ dPO⋯HO = 1.96 Å and <O1H15O14> = 164°. The bond radii of H, N and O are rH = 1.09 Å, rN = 1.55 Å and rO = 1.52 Å, respectively. See ref. 37. |
| || RR/SS-5 exhibiting no IHB was chosen as a model for 4 and very similar solvent effects were expected. |
| ** Protons from CH2OH (labelled e in Fig. 9) are expected to be sensitive to the silylation of the hydroxyl functions, whereas protons labelled b and c are not expected to be very sensitive. |
| †† Conformer A exhibiting IHB between HO and PO functions, conformer B exhibiting IHB between the OH function and N atom, and C the most stable conformer with no IHB. |
| ‡‡ Indeed, conformer A exhibits geometrical parameters (Table 5) featuring stronger IHB than in B as highlighted by the shorter dOH⋯OP and flatter angle α for RR/SS-6. |
| §§ When the t-Bu group attached to the nitroxyl moiety is replaced by a bulkier group CMe2R, no difference in kd is observed, meaning that the nitroxyl fragment adopts a conformation which forces the R group to be in such a position that its bulkiness is cancelled. This is due to the levelled steric effect. See ref. 43–46. |
| ¶¶ In ref. 29, no intraN IHB is observed in X-ray data. |
| |||| Indeed, a 3-center IHB (Fig. 8) would require a conformation for the aromatic ring exhibiting a 1,3-syn allylic destabilizing interaction with H10, whereas such an interaction does not occur for conformers A and B. |
| *** The weak effect of IHB in RR/SS-6 is due to a weak IHB (the weakest of those investigated), as highlighted by its longest dOH⋯X and its closed angle α. |
| ††† For such molecules, a difference of 2.5 kJ mol−1 in energy is in the limit of accuracy and reliability of the method. |
| ‡‡‡ A similar effect on remote IHB has already been reported in ref. 57. The authors would like to thank the reviewer for pointing this issue at TS. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  c,
Jean-Patrick
Joly
a,
Sylvain R. A.
Marque
c,
Jean-Patrick
Joly
a,
Sylvain R. A.
Marque