
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NMR-based assignment of isoleucine vs. allo-isoleucine stereochemistry†
Zoe J.
Anderson
,
Christian
Hobson
,
Rebecca
Needley
,
Lijiang
Song
,
Michael S.
Perryman
,
Paul
Kerby
and
David J.
Fox
 *
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: d.j.fox@warwick.ac.uk
First published on 1st November 2017
Abstract
A simple 1H and 13C NMR spectrometric analysis is demonstrated that permits differentiation of isoleucine and allo-isoleucine residues by inspection of the chemical shift and coupling constants of the signals associated with the proton and carbon at the α-stereocentre. This is applied to the estimation of epimerisation during metal-free N-arylation and peptide coupling reactions.
Introduction
The assignment of the relative stereochemistry of amino-acid residues within complex natural products is normally achieved by degradation of the product to its constituent fragments, followed by reaction with a chiral derivatising agent, commonly Marfey's reagent.1 During peptide coupling, epimerisation is usually measured by HPLC separation of the diastereoisomeric products.21H, 13C and 15N NMR spectrometry can also be used to calculate the level of epimerisation, either via chiral derivatisation or with a chiral shift reagent.3 Recently, during the structural assignment4 and synthesis5 of the azolemycins, we noted that the 1H NMR spectrum of the isoleucine portion of the natural product was significantly different from its allo-isoleucine diastereoisomer.L-Isoleucine is one of the common amino-acids found in eukaryotic and prokaryotic proteins,6 but is unusual in that it possesses two stereogenic centres. It can therefore exist as four distinct stereoisomers (Fig. 1).7 Epimerisation at the α-centre can occur due to resonance stabilisation and electron-withdrawing effects from the amino- and carboxy-groups to give D-allo-isoleucine. In contrast, the β-centre is distanced from these effects and so is much more difficult to epimerise to give the less common L-allo-isoleucine stereoisomer.7a,8 This lack of reactivity allows the β-configuration at the carbon to act as a marker for epimerisation at the α-carbon. Isoleucine as a marker for the study of racemisation during peptide synthesis has been known for a number of years,9 but the final assessment of chiral purity is usually performed by hydrolysis followed by analytical chromatography.10
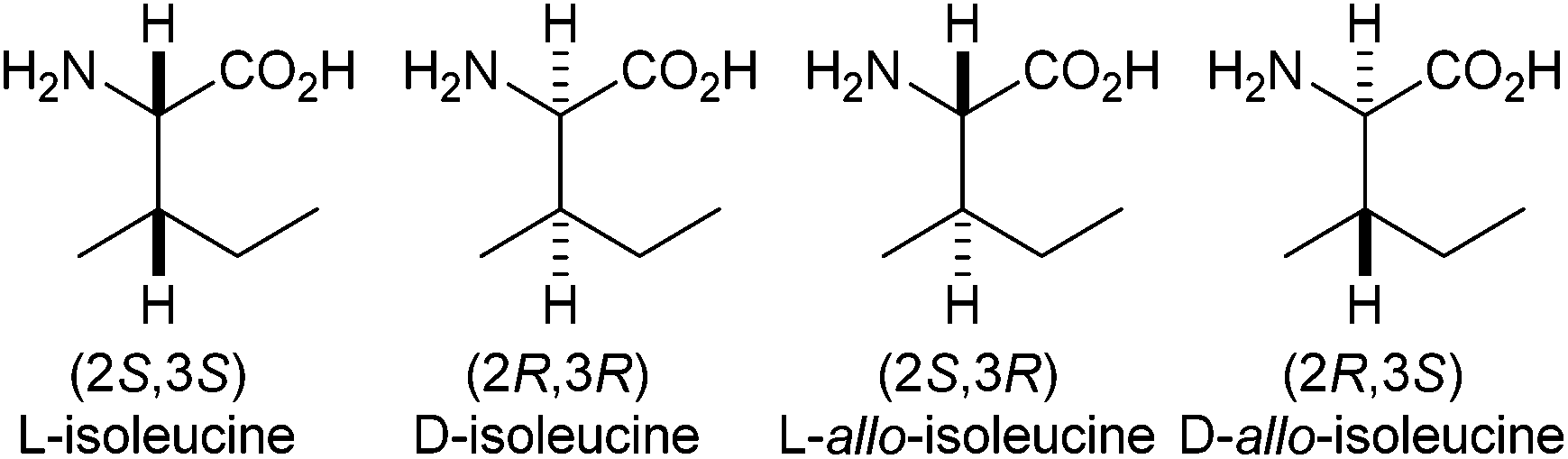 | ||
| Fig. 1 Isoleucine and allo-isoleucine stereochemistry. | ||
To our knowledge, only two reports exist describing the specific differentiation of L-isoleucine and D-allo-isoleucine by 1H NMR spectrometry. In 1983, Khatskevich et al. reported that both the chemical shift and the coupling constants for the hydrogen at the α-carbon differed for N-benzoyl-L-isoleucine methyl ester and N-benzoyl-D-allo-isoleucine methyl ester ((2S,3S) diastereoisomer, δH ppm 4.70, dd, 3JCH–CH 5.15 Hz), 3JCH–NH 8.62 Hz; ((2R,3S) diastereoisomer, δH ppm 4.90, dd, 3JCH–CH 4.30 Hz, 3JCH–NH 9.0 Hz).11 Similarly, in 2003 Biron and Kessler noted that, when hydrolysing Nα-methyl-Nα-o-NBS-L-isoleucine methyl ester, epimerisation at the α-stereocentre could be observed by the appearance of a new peak (corresponding to the D-allo-isoleucine stereoisomer α-CH) downfield from the usual peak observed for the L-isoleucine-derived stereoisomers.12 During the course of our studies into peptide bond synthesis, we decided to use this difference between L-isoleucine and its epimer, D-allo-isoleucine by 1H NMR spectrometry to easily assess the comparative levels of epimerisation during a range of reactions of molecules with a C-terminal L-isoleucine residue.
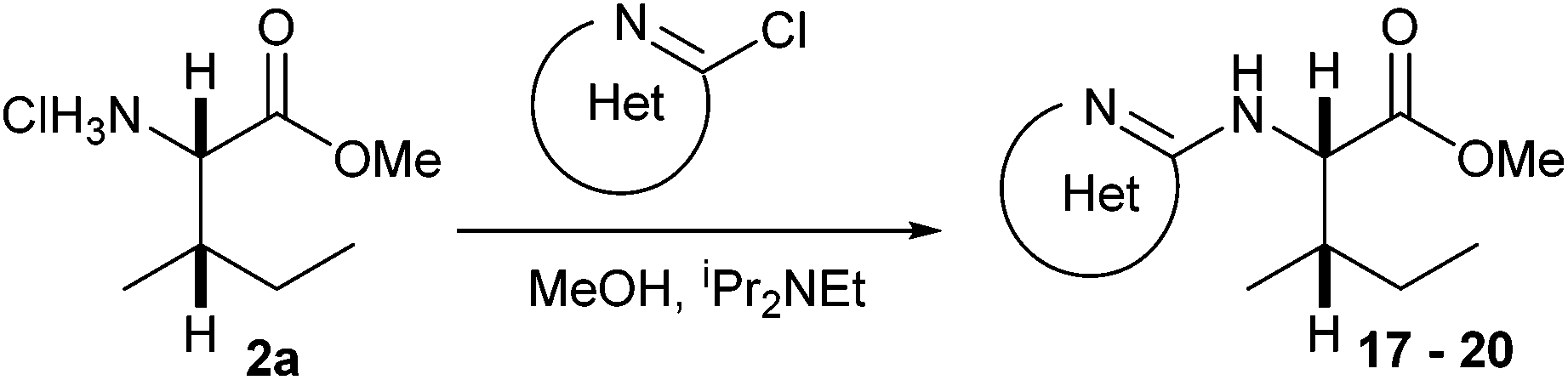
A range of C- and N-substituted isoleucine-containing molecules were synthesised using pure L-isoleucine and then repeated for comparison with a mixture of L-isoleucine and D-allo-isoleucine by standard methods (Fig. 2, see ESI† for details). N-Undec-10-enoyl 5a and N-benzoyl-L-isoleucine 6a were epimerised to a mixture of L-isoleucine and D-allo-isoleucine derivatives using the method of du Vigneaud et al.13 Compounds lacking an N-terminal acyl group were synthesised from an epimeric mixture of D-allo-isoleucine and L-isoleucine, prepared from L-isoleucine according to the method of Yamada et al.14 In addition, a model dipeptide octanoyl-glycyl-isoleucine was prepared as both the methyl ester 9 and the acid 10, again diastereoisomerically pure and as a mixture (Scheme 1).
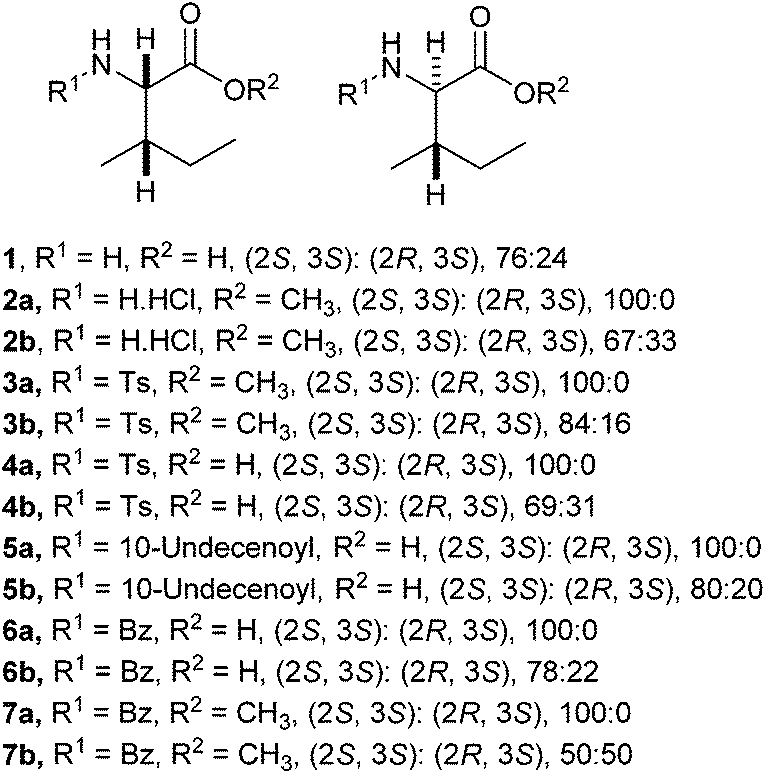 | ||
| Fig. 2 Isoleucine derivatives prepared wth L-isoleucine and a mixture of L-isoleucine and D-allo-isoleucine stereochemistry. | ||
 | ||
| Scheme 1 Reagents and conditions: (i) EDCI, HOBt, NMM, EtOH, 0 °C to rt, 17 h; (ii) LiOH, THF, H2O, 0 °C, 4 h. | ||
For the isoleucine derivatives 2 to 7, differences of L-isoleucine and D-allo-isoleucine stereochemistry due to epimerisation were readily observed by the appearance of a new peak, assignable as the proton at the α-stereocentre of D-allo-isoleucine by comparison with the equivalent compound prepared from a diastereoisomeric mixture of amino-acids. Compounds 5 and 10 were coupled to fluorenylmethane thiol 11 according to the method of Crich et al.,15 to give the corresponding thioesters 12 and 13 respectively (Scheme 2). In each case, considerable epimerisation at the L-isoleucine stereocentre of the products 12a and 13a was readily identified by the appearance of a new peak in the 1H NMR spectrum, assignable as the proton at the α-stereocentre of D-allo-isoleucine, by comparison with the equivalent products 12b and 13b prepared from the diastereoisomeric mixtures 5b and 10b. N-Propyl amides 15 and 16 were prepared by deprotection of the thioesters and reaction with the sulphonamide of propylamine 14. In each case, the products were obtained as a mixture of diastereoisomers in roughly the same proportions as the starting materials. Again, it was possible to distinguish between the L-isoleucine and D-allo-isoleucine derivatives by 1H NMR spectrometry (Scheme 3). For comparison, both 15 and 16 were also prepared using a direct PyBOP mediated peptide coupling reaction between propylamine and the equivalent acids 5a and 10a (not shown, see ESI†).
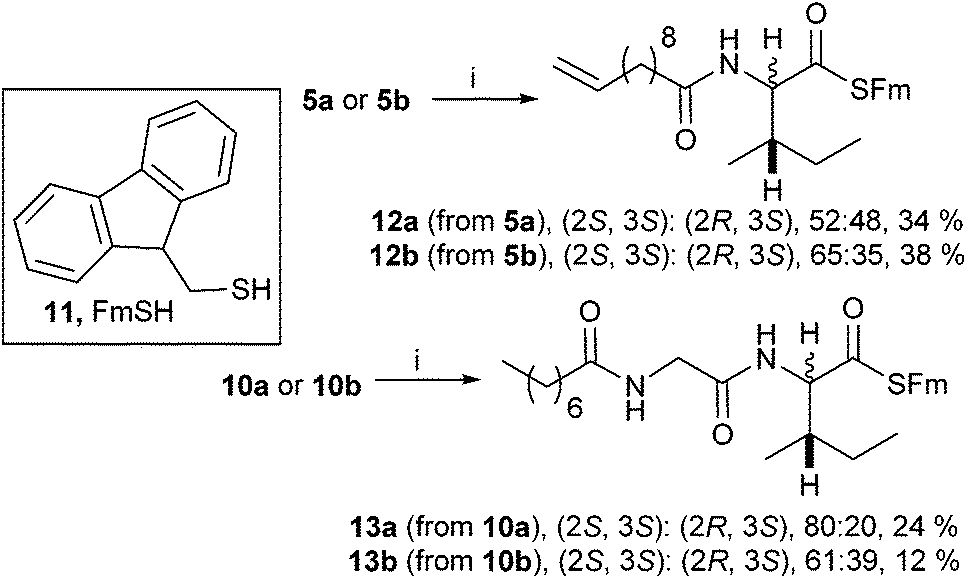 | ||
| Scheme 2 Reagents and conditions: (i) 11, PyBOP, iPr2NEt CH2Cl2, −20 °C, 4 h. | ||
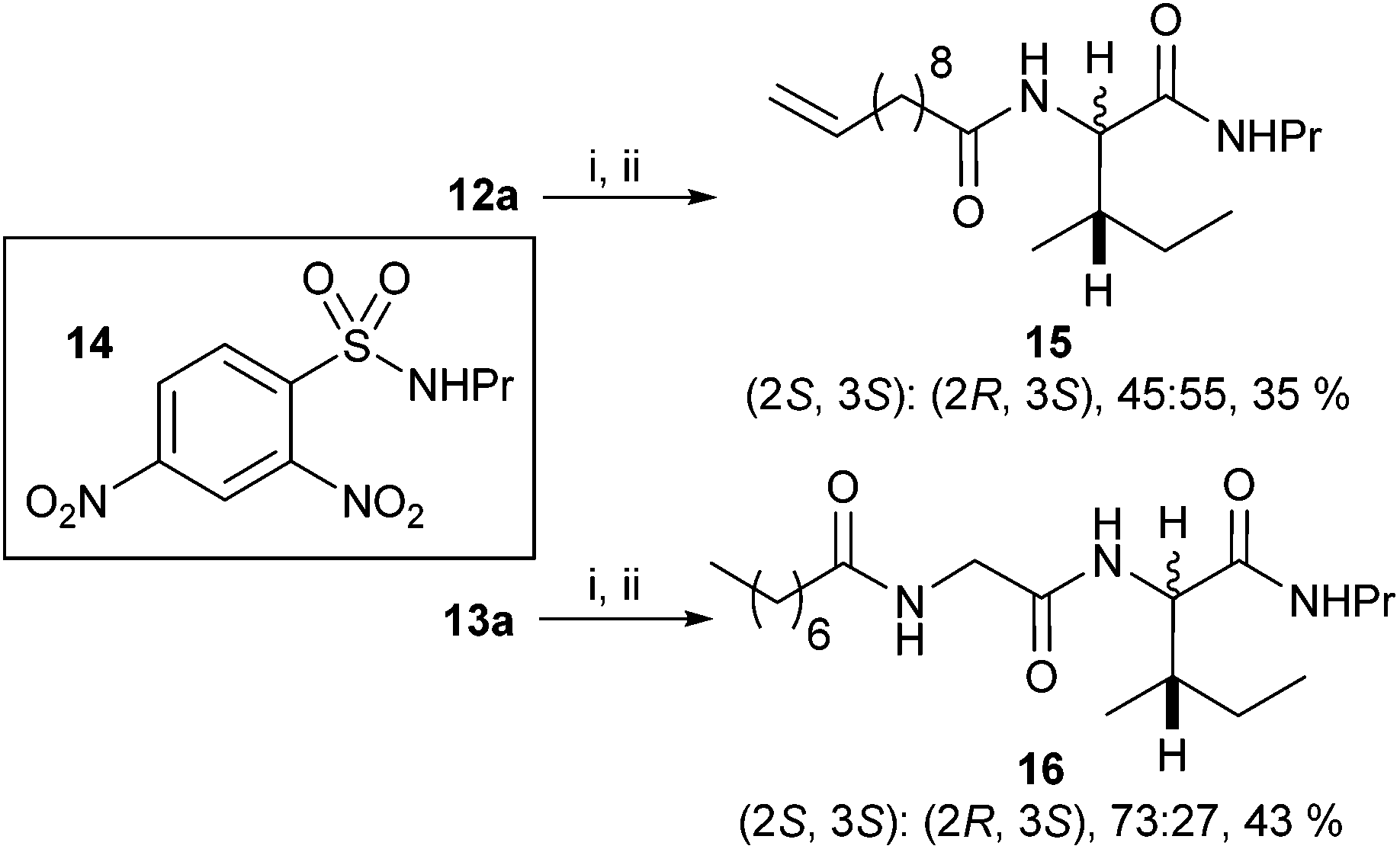 | ||
| Scheme 3 Reagents and conditions: (i) Piperidine, DMF, rt, 1.5 h; (ii) Cs2CO3, DMF, 14, rt, 1 h. | ||
The 1H NMR spectra of the isoleucine derivatives were recorded in CDCl3, CD3OD and (CD3)2SO, both as a mixture of diastereoisomers and, where possible, as the single diastereoisomer (Tables 1–3). 700 MHz 1H NMR spectra were obtained and, after calibration against the internal solvent peak, the chemical shifts and coupling constants for the α-CH hydrogen were measured. 13C NMR spectra were also obtained for most of these compounds at 100 MHz in CDCl3 only. The 13C chemical shifts corresponding to the α-CH were measured (Table 1).
| Entry | α-C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) (ppm) (ppm) |
3 J CH–CH (Hz) | 3 J CH–NH (Hz) | α-![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) H (ppm) H (ppm) |
||||
|---|---|---|---|---|---|---|---|---|
| L | D-allo | L | D-allo | L | D-allo | L | D-allo | |
| a To the nearest 0.1 Hz. | ||||||||
| 3 | 3.80 | 3.90 | 5.5 | 4.0 | 10.0 | 10.0 | 60.2 | 59.0 |
| 4 | 3.87 | 3.99 | 4.9 | 3.3 | 9.5 | 9.9 | 59.9 | 58.6 |
| 5 | 4.62 | 4.75 | 5.1 | 4.0 | 8.0 | 8.6 | 56.5 | 55.2 |
| 6 | 4.86 | 4.98 | 4.9 | 3.9 | 8.1 | 8.5 | 57.0 | 55.7 |
| 7 | 4.84 | 4.95 | 5.2 | 4.0 | 8.4 | 8.8 | 56.8 | 55.8 |
| 9 | 4.54 | 4.65 | 5.3 | 4.1 | 8.4 | 8.8 | 56.7 | 55.6 |
| 10 | 4.53 | 4.69 | 5.3 | 4.5 | 7.5 | 8.5 | 56.9 | n/a |
| 12 | 4.63 | 4.76 | 5.1 | 4.0 | 8.2 | 9.1 | 63.2 | 61.8 |
| 13 | 4.53 | 4.66 | 4.8 | 4.0 | 8.8 | 8.8 | 63.8 | 62.3 |
| 15 | 4.21 | 4.32 | 7.9 | 6.6 | 7.9 | 8.4 | 57.5 | 57.0 |
| 16 | 4.34 | 4.45 | 7.5 | 5.7 | 7.5 | 8.8 | 56.7 | 55.6 |
| Entry | α-C (ppm) |
3 J CH–CH (Hz) | ||
|---|---|---|---|---|
| L | D-allo | L | D-allo | |
| a To the nearest 0.1 Hz. | ||||
| 2 | 4.00 | 4.03 | 4.0 | 3.9 |
| 3 | 3.70 | 3.82 | 7.0 | 5.0 |
| 4 | 3.67 | 3.82 | 5.5 | 4.5 |
| 5 | 4.37 | 4.54 | 5.8 | 4.7 |
| 6 | 4.57 | 4.74 | 6.4 | 5.0 |
| 7 | 4.56 | 4.74 | 6.9 | 5.6 |
| 9 | 4.42 | 4.56 | 6.0 | 4.4 |
| 10 | 4.40 | 4.55 | 5.3 | 4.0 |
| 12 | 4.31 | 4.48 | 6.1 | 5.1 |
| 13 | 4.33 | 4.48 | 5.7 | 4.8 |
| 15 | 4.15 | 4.31 | 8.3 | 6.7 |
| 16 | 4.20 | 4.35 | 7.5 | 5.7 |
| Entry | α-C (ppm) |
3 J CH–CH (Hz) | 3 J CH–NH (Hz) | |||
|---|---|---|---|---|---|---|
| L | D-allo | L | D-allo | L | D-allo | |
| a To the nearest 0.1 Hz. b Not measurable. | ||||||
| 1 | 3.05 | 3.08 | 3.2 | 2.9 | ||
| 2 | 3.91 | 3.92 | 4.2 | 3.8 | ||
| 3 | 3.54 | 3.69 | 7.5 | 6.0 | 9.0 | 10.0 |
| 4 | 3.52 | 3.67 | 6.0 | 4.5 | 9.0 | 9.5 |
| 5 | 4.17 | 4.35 | 6.5 | 5.0 | 8.1 | 8.5 |
| 6 | 4.33 | 4.53 | 7.6 | 5.8 | 7.6 | 8.5 |
| 7 | 4.36 | 4.55 | 7.7 | 6.4 | 7.7 | 8.0 |
| 9 | 4.23 | 4.39 | 6.6 | 5.1 | 8.4 | 8.6 |
| 10 | 4.19 | 4.36 | 5.9 | 4.4 | 8.6 | 8.8 |
| 12 | 4.15 | 4.33 | 7.4 | 5.1 | 7.4 | 8.7 |
| 13 | 4.15 | 4.33 | 6.2 | 5.1 | 8.4 | 8.6 |
| 15 | 4.10 | 4.25 | 8.5 | 6.3 | 8.5 | 9.0 |
| 16 | 4.12 | 4.25 | 7.5 | 5.7 | 8.8 | 9.2 |
The general trends noted earlier were further supported by these results. In all cases we found that the α-CH chemical shift for the D-allo-isoleucine diastereoisomer occurs at a higher chemical shift than that for the L-isoleucine diastereoisomer. In addition, the 3JCH–CH coupling constants for the L-isoleucine diastereoisomer α-CH are always larger, and the 3JCH–NH coupling constants are usually smaller, than those for the D-allo-isoleucine diastereoisomer.
Generally, the difference in chemical shifts corresponding to the α-CH of L-isoleucine and D-allo-isoleucine was greatest in DMSO-d6 and smallest in CDCl3, but the α-CH 1H NMR peaks were always sufficiently distinct not to overlap at both 700 MHz and 400 MHz. This suggests they would be suitable for the approximate assignment of relative stereochemistry of isoleucine residues in compounds with unknown relative stereochemistry. The 13C NMR data also suggest a useful trend with the chemical shift corresponding to the α-H of the D-allo-isoleucine derivative always occurring at a lower chemical shift than that for the L-isoleucine derivative, in most cases by more than 1 ppm. Taken in conjunction with the 1H NMR data, the 13C NMR data are also likely to prove extremely useful for the approximate assignment of relative stereochemistry of isoleucine residues in novel natural products.
N-Arylation and peptide coupling reactions
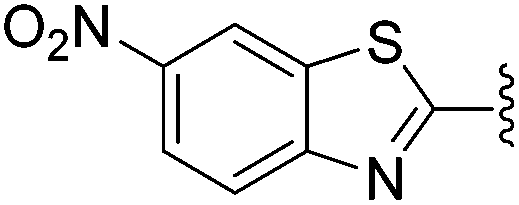
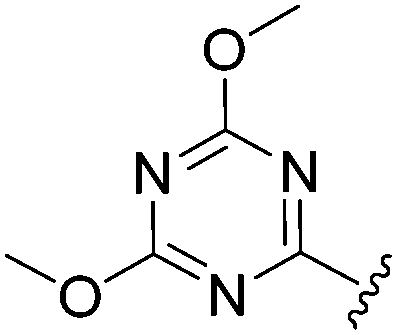
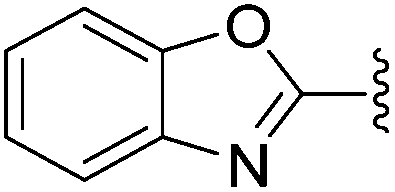
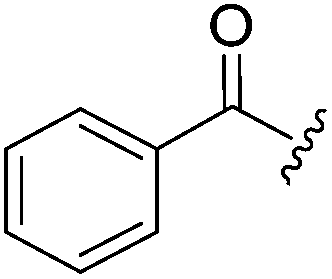
N-Heteroaryl-amine derivatives, including N-aryl-α-amino-acid derivatives, provide important building blocks for a number of biologically active products.16 We considered whether the NMR-based trends described above for distinguishing L-iso- and D-allo-isoleucine derivatives would apply to N-aryl compounds. A range of methods for N-arylation have been described, including the use of copper, palladium and nickel catalysis.16e,17N-Arylations of chiral amino-acids have been reported,17c,18 and while some methods involve harsh conditions that risk racemisation, other methods are much milder and can occur without loss of stereochemical integrity as part of amino-acid analysis.19To test these methods L-isoleucine methyl ester hydrochloride 2a was heteroarylated with four imidoylchloride-like aryl chlorides in metal free conditions (Table 4). As expected in the mildly basic reaction conditions, 1H NMR analysis indicated that arylations proceeded without significant epimerisation (17a–20a) when compared to reactions of a diastereoisomeric mixture of L-iso- and D-alloisoleucine methyl esters 17b–20b (not drawn, see ESI†). The product ratios measured by 1H NMR were confirmed by HPLC. Peptide couplings of N-arylated amino-acids have been used in the synthesis of a variety of peptidomimetic molecules, including DNA topoisomerases,20 cathepsin S. inhibitors,21 and benzoxaborales.22 We decided to extend the current study to include the synthesis of two N-aryl isoleucylglycine methyl esters, as well as the related N-benzoyl and N-tosyldipeptides for comparison. N-Aryl methyl esters 18a and 20a were hydrolysed and coupled to glycine methyl ester without isolation of the acids. Acids 4a and 6a were coupled directly (Table 5). Dipeptides 21a–23a were produced as essentially single diastereoisomers, by comparison by both NMR spectrometry and HPLC with authentic stereoisomeric mixture 21b–24b (not drawn, see ESI†). The N-benzoyl product 24a was produced as a near equal mixture of diastereoisomers. While it is well known that N-benzoyl amino acids tend to racemise during standard peptide coupling reactions,23 and that N-sulfonyl amino acids do not,24 to the best of our knowledge, there is little quantitative information regarding the levels of epimerisation of the C-terminal N-arylated amino-acids during these peptide coupling reactions. There are two main mechanisms of epimerisation during peptide coupling reactions, deprotonation of the α-carbon and 5(4H)-oxazolone formation.9c In practise, these are normally minimised by careful control of reaction temperature, pH and N-substituent on the C-terminal amino-acid. Generally, N-amido amino-acids are more prone to racemisation than N-carbamate amino-acids (e.g. N-Cbz, N-Boc or N-Fmoc amino acids) having a greater tendency to oxazolone formation.9c,25 Despite this, the benzoxazole and triazine active esters do not epimerise during peptide coupling via the related imidazolinone even though the heterocyclic nitrogens are in an analogous position to that of the amide oxygen in the benzamide (Scheme 4). Such a difference may be due to reduced nitrogen nucleophilicity because of increased steric crowding compared to the amide. Relative basicity does not correlate with nucleophilicity in this case as, even though 2-amino-4,6-dimethoxy-s-triazine is a very weak base in water,26 2-aminobenzoxazole has a pKaH(H2O) of 3.73,27 and is therefore significantly more basic than benzamide (pKaH(H2O) = −1.53).28
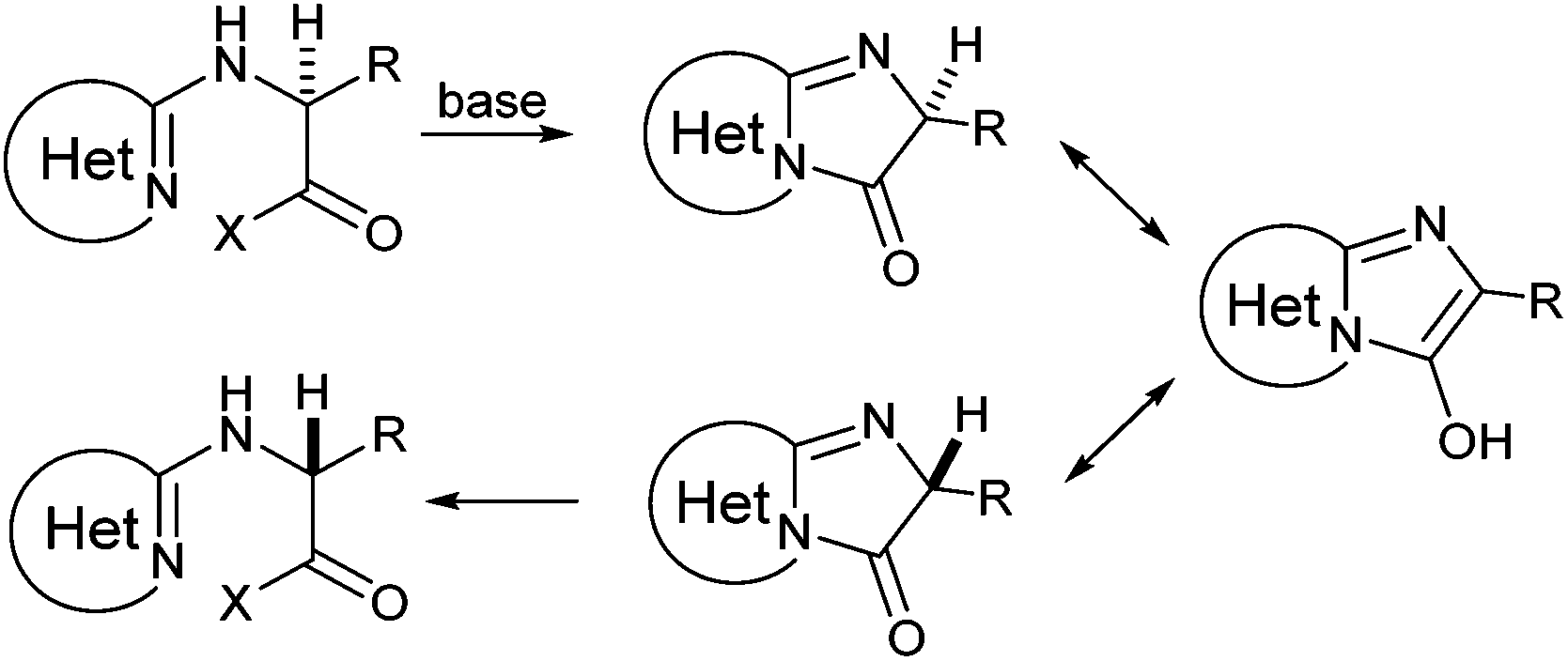 | ||
| Scheme 4 Potential racemization mechanism of N-imidoyl amino-acids during peptide coupling. | ||
|
|
||||
|---|---|---|---|---|
| Entry | R | Yield | (2S,3S)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(2R,3S) by NMR :(2R,3S) by NMR |
(2S,3S):(2R,3S) by HPLC |
| Reagents (i) LiOH H2O, THF then EDCI, HOBt, NMM, EtOH, 0 °C to rt, 17 h. | ||||
| 21a |
|
50% | >95:5 |
>99:1 |
| 22a |

|
76% | 97:3 |
>97:3 |
| 23a |

|
84% | >95:5 |
>99:1 |
| 24a |
|
54% | 44:56 |
Not measured |
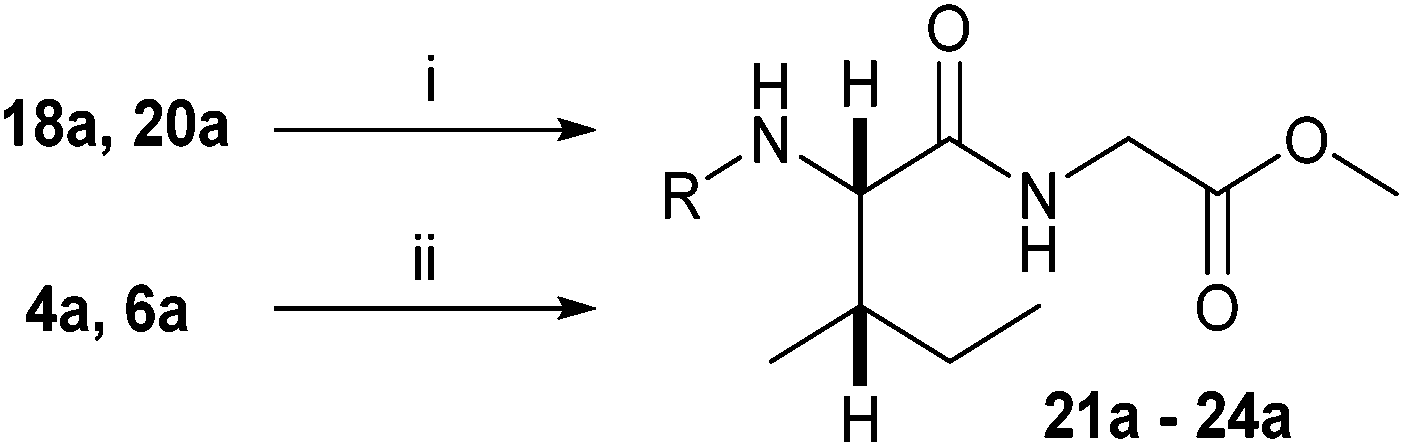
Analysis of the 1H NMR and 13C NMR data for the N-aryl esters and the dipeptides show that the trends in relative chemical shift and coupling constants for the (2S,3S)- and (2R,3S)-diastereoisomers described above apply to these compounds as well (Table 6).
| Entry | α-C (ppm) |
3 J CH–CH (Hz) | 3 J CH–NH (Hz) | α-H (ppm) |
||||
|---|---|---|---|---|---|---|---|---|
| L | D-allo | L | D-allo | L | D-allo | L | D-allo | |
| a To the nearest 0.5 Hz. b Value is the centre of an undefined multiplet. c Not measurable. | ||||||||
| 17 | 4.76b | 4.93b | 58.5 | 57.4 | ||||
| 18 | 4.58 | 4.70 | 5.5 | 4.5 | 8.5 | 9.0 | 60.4 | 59.4 |
| 19 | 4.63b | 4.79b | 8.5 | 9.0 | 62.1 | 60.5 | ||
| 20 | 4.59 | 4.73 | 5.0 | 4.5 | 8.5 | 9.5 | 57.9 | 56.8 |
| 21 | 4.30b | 4.48b | 61.7 | 60.8 | ||||
| 22 | 4.47 | 4.64 | 7.5 | 5.0 | 7.5 | 8.5 | 59.3 | 58.1 |
| 23 | 3.55 | 3.67 | 4.0 | 5.5 | 8.5 | 8.5 | 61.5 | 60.2 |
| 24 | 4.57 | 4.69 | 7.0 | 5.5 | 8.5 | 8.5 | 57.9 | 57.1 |
While L-isoleucine is the most common diastereoisomer in natural products, D-allo-isoleucine is also fairly common in microbial peptides, including the sporidemolides, stendomycin, angolide, actinomycin C, calpinactam and peptidolipin N.A.29 In addition, D-allo-isoleucine has been identified in peptides isolated from the skin of the yellow bellied toad, Bombina variegate.30 Unsurprisingly, considering the comparative stability of the β-C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) of isoleucine peptides containing L-allo-isoleucine and D-isoleucine are far less common, though a few examples do exist.31 Alteration of relative stereochemistry of isoleucine within a peptide can have significant effects of its structure and folding, and potentially its biological activity.31d,32 The ability to distinguish between, and potentially predict, the stereochemistry isoleucine diastereoisomers by a non-destructive method such as NMR, instead of peptide hydrolysis, is valuable, particularly for complex natural proteins, which are often only obtained in minute quantities. While in this study we cannot distinguish between L-allo-isoleucine and D-allo-isoleucine, and between the L- and D-isoleucine, the comparative rarity of the D- and L-allo forms means that this method will prove extremely useful. This method can be applied to the structure of the antibiotic peptide teixobactin.33 By hydrolysis, and derivatisation using Marfey's method, teixobactin was found to contain three L-isoleucines and one D-allo-isoleucine. It is worth noting that the full assignments of the NMR spectra of the peptide in deuteriated DMSO show that in the 1H NMR spectrum the peak corresponding to the α-CH hydrogen of the D-allo-isoleucine residue has a higher chemical shift (4.36 ppm) than the peaks due to the α-CH hydrogens of the three L-isoleucine residues (4.29, 4.12 and 4.03 ppm), and that in the 13C NMR spectrum the peak corresponding to the α-CH carbon of the D-allo-isoleucine has a lower chemical shift (56.8 ppm) than those corresponding to α-CH carbons of the three L-isoleucine residues (57.3, 57.8 and 57.9 ppm), in agreement with the trends observed in Tables 1–3 and 6.
of isoleucine peptides containing L-allo-isoleucine and D-isoleucine are far less common, though a few examples do exist.31 Alteration of relative stereochemistry of isoleucine within a peptide can have significant effects of its structure and folding, and potentially its biological activity.31d,32 The ability to distinguish between, and potentially predict, the stereochemistry isoleucine diastereoisomers by a non-destructive method such as NMR, instead of peptide hydrolysis, is valuable, particularly for complex natural proteins, which are often only obtained in minute quantities. While in this study we cannot distinguish between L-allo-isoleucine and D-allo-isoleucine, and between the L- and D-isoleucine, the comparative rarity of the D- and L-allo forms means that this method will prove extremely useful. This method can be applied to the structure of the antibiotic peptide teixobactin.33 By hydrolysis, and derivatisation using Marfey's method, teixobactin was found to contain three L-isoleucines and one D-allo-isoleucine. It is worth noting that the full assignments of the NMR spectra of the peptide in deuteriated DMSO show that in the 1H NMR spectrum the peak corresponding to the α-CH hydrogen of the D-allo-isoleucine residue has a higher chemical shift (4.36 ppm) than the peaks due to the α-CH hydrogens of the three L-isoleucine residues (4.29, 4.12 and 4.03 ppm), and that in the 13C NMR spectrum the peak corresponding to the α-CH carbon of the D-allo-isoleucine has a lower chemical shift (56.8 ppm) than those corresponding to α-CH carbons of the three L-isoleucine residues (57.3, 57.8 and 57.9 ppm), in agreement with the trends observed in Tables 1–3 and 6.
It is unclear at this point as to whether the differences observed are due to purely stereochemical differences in amino-acid derivatives with otherwise similar backbone conformations, or if the stereochemical differences cause a significant change in backbone conformation which is itself the reason for the observed differences in the NMR spectra.34 Differentiation between these two possibilities may be effected by studying pairs of stereoisomers with defined conformations e.g. involving cyclic peptides of various sizes. Conformational analysis would be supported with molecular modelling. While the analysis of the spectra from teixobactin,33 which contains both linear and cyclic isoleucine-containing regions, indicates that the trends presented in this paper may be applicable to more complex peptides, the issues of conformation and additional stereochemistry are the subjects of future work in our laboratory and results will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to thank the EPSRC (DTA studentship for ZJA) and the University of Warwick (PhD studentships for MSP and PK, URSS funding for CH) for funding, and Prof. G Challis for useful discussions. Data are available at http://wrap.warwick.ac.uk/74515.Notes and references
- P. Marfey, Carlsberg Res. Commun., 1984, 49, 591–596 CrossRef CAS.
- R. Bhushan and H. Brückner, Amino Acids, 2004, 27, 231–247 CrossRef CAS PubMed.
- (a) E. D. Gómez and H. Duddeck, Magn. Reson. Chem., 2009, 47, 222–227 CrossRef PubMed; (b) H. R. Kricheldorf and W. E. Hull, Org. Magn. Reson., 1979, 12, 607–611 CrossRef CAS; (c) B. Weinstein and A. E. Pritchard, J. Chem. Soc., Perkin Trans. I, 1972, 1015–1020 RSC.
- N. Liu, L. Song, M. Liu, F. Shang, Z. Anderson, D. J. Fox, G. L. Challis and Y. Huang, Chem. Sci., 2016, 7, 482–488 RSC.
- Z. J. Anderson and D. J. Fox, Org. Biomol. Chem., 2016, 14, 1450–1454 CAS.
- B. Hao, W. Gong, T. K. Ferguson, C. M. James, J. A. Krzycki and M. K. Chan, Science, 2002, 296, 1462–1466 CrossRef CAS PubMed.
- (a) J. L. Bada, M. Zhao, S. Steinberg and E. Ruth, Nature, 1986, 319, 314–316 CrossRef CAS; (b) B. Dang, R. Shen, T. Kubota, K. Mandal, F. Bezanilla, B. Roux and S. B. H. Kent, Angew. Chem., Int. Ed., 2017, 56, 3324–3328 CrossRef CAS PubMed; (c) B. Dang, S. Chhabra, M. W. Pennington, R. S. Norton and S. B. H. Kent, J. Biol. Chem., 2017, 292, 12599–12605 CrossRef CAS PubMed.
- (a) M. Bodanszky and D. Perlman, Nature, 1968, 291–292 CrossRef CAS; (b) Q. Li, X. Qin, J. Liu, C. Gui, B. Wang, J. Li and J. Ju, J. Am. Chem. Soc., 2016, 138, 408–415 CrossRef CAS PubMed.
- (a) M. Bodanszky and L. E. Conklin, Chem. Commun., 1967, 773–774 RSC; (b) M. Itoh, Bull. Chem. Soc. Jpn., 1973, 46, 2219–2221 CrossRef CAS; (c) E. Gross and J. Meienhofer, The Peptides: Major Methods of Peptide Bond Formation, Academic Press, 1979 Search PubMed.
- D. H. Spackman, W. H. Stein and S. Moore, Anal. Chem., 1958, 30, 1190–1206 CrossRef CAS.
- I. É. Khatskevich, I. K. Kalnin, E. I. Karpeiskaya and E. I. Klabunovskii, Russ. Chem. Bull., 1983, 32, 323–329 CrossRef.
- E. Biron and H. Kessler, J. Org. Chem., 2005, 70, 5183–5189 CrossRef CAS PubMed.
- V. du Vigneaud and C. E. Meyer, J. Biol. Chem., 1932, 98, 295–308 CAS.
- S. Yamada, C. Hongo, R. Yoshioka and I. Chibata, J. Org. Chem., 1983, 48, 843–846 CrossRef CAS.
- D. Crich, K. Sana and S. Guo, Org. Lett., 2007, 9, 4423–4426 CrossRef CAS PubMed.
- (a) R. Nagata, N. Ae and N. Tanno, Bioorg. Med. Chem. Lett., 1995, 5, 1527–1532 CrossRef CAS; (b) W. H. Miller, T. W. Ku, F. E. Ali, W. E. Bondinell, R. R. Calvo, L. D. Davis, K. F. Erhard, L. B. Hall, W. F. Huffman, R. M. Keenan, C. Kwon, K. A. Newlander, S. T. Ross, J. M. Samanen, D. T. Takata and C.-K. Yuan, Tetrahedron Lett., 1995, 36, 9433–9436 CrossRef CAS; (c) Y. Endo, M. Ohno, M. Hirano, A. Itai and K. Shudo, J. Am. Chem. Soc., 1996, 118, 1841–1855 CrossRef CAS; (d) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed; (e) C. Fischer and B. Koenig, Beilstein J. Org. Chem., 2011, 7, 59–74 CrossRef CAS PubMed; (f) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC; (g) T. R. M. Rauws and B. U. W. Maes, Chem. Soc. Rev., 2012, 41, 2463–2497 RSC; (h) J. Quick, B. Saha and P. E. Driedger, Tetrahedron Lett., 1994, 35, 8549–8552 CrossRef CAS.
- (a) G. Lemen and J. Wolfe, in Amination and Formation of sp2 C-N Bonds, ed. M. Taillefer and D. Ma, Springer, Berlin Heidelberg, 2013, vol. 46, pp. 1–53 Search PubMed; (b) M. Islam, S. Mondal, P. Mondal, A. Roy, K. Tuhina, N. Salam, S. Paul, D. Hossain and M. Mobarok, Transition Met. Chem., 2011, 36, 447–458 CrossRef CAS; (c) H. Hammoud, M. Schmitt, E. Blaise, F. Bihel and J.-J. Bourguignon, J. Org. Chem., 2013, 78, 7930–7937 CrossRef CAS PubMed; (d) H.-J. Cristau, P. P. Cellier, J.-F. Spindler and M. Taillefer, Eur. J. Org. Chem., 2004, 695–709 CrossRef CAS; (e) J. Bariwal and E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 9283–9303 RSC; (f) A. P. Combs, S. Tadesse, M. Rafalski, T. S. Haque and P. Y. S. Lam, J. Comb. Chem., 2002, 4, 179–182 CrossRef CAS PubMed; (g) D. S. Raghuvanshi, A. K. Gupta and K. N. Singh, Org. Lett., 2012, 14, 4326–4329 CrossRef CAS PubMed; (h) D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC; (i) D. M. T. Chan, K. L. Monaco, R. Li, D. Bonne, C. G. Clark and P. Y. S. Lam, Tetrahedron Lett., 2003, 44, 3863–3865 CrossRef CAS.
- (a) S. Röttger, P. J. R. Sjöberg and M. Larhed, J. Comb. Chem., 2007, 9, 204–209 CrossRef PubMed; (b) N. Narendar and S. Velmathi, Tetrahedron Lett., 2009, 50, 5159–5161 CrossRef CAS; (c) J. R. Porter, S. C. Archibald, J. A. Brown, K. Childs, D. Critchley, J. C. Head, B. Hutchinson, T. A. H. Parton, M. K. Robinson, A. Shock, G. J. Warrellow and A. Zomaya, Bioorg. Med. Chem. Lett., 2002, 12, 1591–1594 CrossRef CAS PubMed; (d) W. Du, J. P. Jewell, L. S. Lin, V. J. Colandrea, J. C. Xiao, J. Lao, C.-P. Shen, T. J. Bateman, V. B. G. Reddy, S. N. Ha, S. K. Shah, T. M. Fong, J. J. Hale and W. K. Hagmann, Bioorg. Med. Chem. Lett., 2009, 19, 5195–5199 CrossRef CAS PubMed; (e) P. M. Chandrika, T. Yakaiah, A. R. R. Rao, B. Narsaiah, N. C. Reddy, V. Sridhar and J. V. Rao, Eur. J. Med. Chem., 2008, 43, 846–852 CrossRef CAS PubMed; (f) Y. Zou, E. Zhang, T. Xu, W. Wu, Y. Chen, M. Yuan, W. Wei and X. Zhang, RSC Adv., 2013, 3, 6545–6552 RSC.
- (a) F. Sanger, Biochem. J., 1945, 39, 507–515 CrossRef CAS PubMed; (b) S. Chen, Amino Acids, 2004, 27, 277–284 CrossRef CAS PubMed; (c) K.-i. Harada, K. Fujii, T. Mayumi, Y. Hibino, M. Suzuki, Y. Ikai and H. Oka, Tetrahedron Lett., 1995, 36, 1515–1518 CrossRef CAS; (d) R. Bhushan and R. Kumar, J. Chromatogr. A, 2009, 1216, 2592–2596 CrossRef CAS PubMed; (e) R. Bhushan and H. Brückner, J. Chromatogr. B: Biomed. Appl., 2011, 879, 3148–3161 CrossRef CAS PubMed.
- J. Rudolph, H. Theis, R. Hanke, R. Endermann, L. Johannsen and F.-U. Geschke, J. Med. Chem., 2001, 44, 619–626 CrossRef CAS PubMed.
- (a) P. B. Alper, H. Liu, A. K. Chatterjee, K. T. Nguyen, D. C. Tully, C. Tumanut, J. Li, J. L. Harris, T. Tuntland, J. Chang, P. Gordon, T. Hollenbeck and D. S. Karanewsky, Bioorg. Med. Chem. Lett., 2006, 16, 1486–1490 CrossRef CAS PubMed; (b) D. C. Tully, H. Liu, P. B. Alper, A. K. Chatterjee, R. Epple, M. J. Roberts, J. A. Williams, K. T. Nguyen, D. H. Woodmansee, C. Tumanut, J. Li, G. Spraggon, J. Chang, T. Tuntland, J. L. Harris and D. S. Karanewsky, Bioorg. Med. Chem. Lett., 2006, 16, 1975–1980 CrossRef CAS PubMed; (c) D. C. Tully, H. Liu, A. K. Chatterjee, P. B. Alper, R. Epple, J. A. Williams, M. J. Roberts, D. H. Woodmansee, B. T. Masick, C. Tumanut, J. Li, G. Spraggon, M. Hornsby, J. Chang, T. Tuntland, T. Hollenbeck, P. Gordon, J. L. Harris and D. S. Karanewsky, Bioorg. Med. Chem. Lett., 2006, 16, 5112–5117 CrossRef CAS PubMed.
- Z. Fu, J. He, A. Tong, Y. Xie and Y. Wei, Synthesis, 2013, 2843–2852 CrossRef CAS.
- G. W. Anderson and F. M. Callahan, J. Am. Chem. Soc., 1958, 80, 2902–2903 CrossRef CAS.
- (a) S. C. Miller and T. S. Scanlan, J. Am. Chem. Soc., 1998, 120, 2690–2691 CrossRef CAS; (b) A. Leggio, M. L. Di Gioia, F. Perri and A. Liguori, Tetrahedron, 2007, 63, 8164–8173 CrossRef CAS.
- B. Halpern, L. F. Chew and B. Weinstein, J. Am. Chem. Soc., 1967, 89, 5051–5052 CrossRef CAS PubMed.
- J. R. Dudley, J. Am. Chem. Soc., 1951, 73, 3007–3008 CrossRef CAS.
- A. Albert, R. Goldacre and J. Phillips, J. Chem. Soc., 1948, 2240–2249 RSC.
- J. F. Bunnett and F. P. Olsen, Can. J. Chem., 1966, 44, 1899–1916 CrossRef CAS.
- (a) M. Bodanszky and D. Periman, Science, 1969, 163, 352–358 CAS; (b) N. Koyama, S. Kojima, K. Nonaka, R. Masuma, M. Matsumoto, S. Omura and H. Tomoda, J. Antibiot., 2010, 63, 183–186 CrossRef CAS PubMed; (c) N. Koyama, S. Kojima, T. Fukuda, T. Nagamitsu, T. Yasuhara, S. Ōmura and H. Tomoda, Org. Lett., 2010, 12, 432–435 CrossRef CAS PubMed.
- G. Mignogna, M. Simmaco, G. Kreil and D. Barra, EMBO J., 1993, 12, 4829–4832 CAS.
- (a) J. Claesen and M. Bibb, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16297–16302 CrossRef CAS PubMed; (b) K. Bevan, J. S. Davies, C. H. Hassall and D. A. S. Phillips, J. Chem. Soc., Chem. Commun., 1969, 1246a–1246a RSC; (c) T. Yajima, K. Mason and E. Katz, Antimicrob. Agents Chemother., 1976, 9, 224–232 CrossRef CAS PubMed; (d) T. Yajima, K. T. Mason and E. Kaltz, Antimicrob. Agents Chemother., 1975, 7, 773–780 CrossRef CAS.
- (a) J. E. Tudor, M. W. Pennington and R. S. Norton, Eur. J. Biochem., 1998, 251, 133–141 CAS; (b) C. Beeton, B. J. Smith, J. K. Sabo, G. Crossley, D. Nugent, I. Khaytin, V. Chi, K. G. Chandy, M. W. Pennington and R. S. Norton, J. Biol. Chem., 2008, 283, 988–997 CrossRef CAS PubMed; (c) T. Yoshida and K. Katagiri, J. Bacteriol., 1967, 93, 1327–1331 CAS.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schaberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- D. S. Wishart, B. D. Sykes and F. M. Richards, Biochemistry, 1992, 31, 1647–1651 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis details, NMR spectra and chiral HPLC chromatograms. See DOI: 10.1039/c7ob01995e |
| This journal is © The Royal Society of Chemistry 2017 |