
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective lysine modification of native peptides via aza-Michael addition†
Hongli
Chen
 *,
Rong
Huang
,
Zhihong
Li
,
Wei
Zhu
,
Jiakang
Chen
,
Yuexiong
Zhan
and
Biao
Jiang
*
*,
Rong
Huang
,
Zhihong
Li
,
Wei
Zhu
,
Jiakang
Chen
,
Yuexiong
Zhan
and
Biao
Jiang
*
The institute for Advanced Immunochemical Studies, ShanghaiTech University, 99 Haike Road, Pudong, Shanghai, 201210, P.R. China. E-mail: chenhl@shanghaitech.edu.cn; jiangbiao@shanghaitech.edu.cn
First published on 22nd August 2017
Abstract
A series of vinylsulfonamides were synthesized and screened for site-selective modification of the ε-amino group of lysine-bearing free α-amine residues. N-Methyl-N-phenylethenesulfonamide has emerged as an applicable reagent and has been developed for efficient and highly selective modification of the lysine residue of native peptides in the presence of a free N-terminus via aza-Michael addition. We demonstrated that functional N-phenylvinylsulfonamide derivatives with a fluorescent moiety or drug could also be conjugated to the lysine residue of octreotide and insulin with high specificity, without modifying the N-terminus. Our method provides a promising strategy for site-selective lysine functionalization in native peptides with a free N-terminus.
Introduction
The modification of native peptides at a single amino acid or specific site with synthetic moieties is a significant challenge.1–3 Among the approximately 20 primary amino acids that compose proteins, only a subset can serve as appropriate targets for bioconjugation. Many strategies have been developed to target nucleophilic natural amino acid residues, of which cysteine and lysine residues are the most popular labeling sites. Cysteine is capable of being involved in a chemoselective reaction due to its superior nucleophilic properties.4–7 However, the low abundance of free cysteine residues due to its tendency to form disulfide bonds limits its application. Lysine residues are abundant in native proteins, and methods to modify primary amines can provide versatile techniques to label lysine residues.8,9 However, lysine conjugation methods often modify the N-terminus.10,11 Site-specific modification of the N-terminus is easier to achieve because the ε-amino group of lysine is more difficult to deprotonate due to its higher pKa (∼10) compared to the N-terminal α-amino group (pKa ∼8). A number of strategies for selective N-terminal modification of native peptides or proteins have been developed.12–18 In contrast, site-specific functionalization of the ε-amino group of lysine has rarely been studied.19,20 Typically, the most common reagents for amine modification, N-hydroxysuccinimides (NHS) esters, also react with serine, tyrosine and histidine, affording heterogeneous bioconjugates.21 Although the stepwise addition of the substoichiometric amounts of an NHS ester can achieve a single lysine-selective modification, tediousness and low efficiency are the problems.22 Indeed, low reactivity and the generation of byproducts are the major problems encountered when attempting to selectively label an amine. The Michael-type addition is also an attractive strategy for protein conjugation. Michael acceptors, such as maleimides, vinyl sulfones and acrylamides, have been frequently utilized in the modification of proteins. Vinyl sulfonamides, with a similar structure to vinyl sulfones, are easier to prepare and have also received some attention in applications for bioconjugation.23–26Although the lysine ε-amino group requires a higher pH to deprotonate, it has higher intrinsic nucleophilicity. Therefore, selective lysine modification can be achieved by carefully adjusting the nucleophilicity and basicity. Herein, we report a class of vinylsulfonamides which site-specifically modify the lysine residue in native peptides rather than the N-terminus via aza-Michael addition.
Results and discussion
We first synthesized a series of vinylsulfonamides (Fig. 1, compounds 1–7). We found that activated phenylamines were the easiest to use to produce divinylsulfonamides (Fig. 1, compounds 8–10), which would be developed as efficient reagents for peptide stapling in our coming research. Then, we began to screen the abilities of different vinylsulfonamides to promote selective ε-amino group modification. Our initial study found that triethylenediamine (DABCO) buffer was a good medium for the aza-Michael addition to take place. Therefore we employed an L-lysine methyl ester (11) to react with vinylsulfonamides (1–7) in DABCO buffer and CH3CN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at pH = 7. Based on RP-HPLC analysis, compounds 1–2 did not react with Lys-OMe (ESI, Table S1,† entries 1 and 2). Compounds 3–4 showed poor selectivity (ESI, Table S1,† entries 3 and 4). Compounds 5–6 displayed good selectivity, however the conversion was poor (<20%) (ESI, Table S1,† entries 5 and 6). N-Methyl-N-phenylethenesulfonamide (compound 7) emerged as a potential reagent with good selectivity and moderate conversion (∼30%) (ESI, Table S1,† entry 7). We further screened a series of alkaline buffers at pH 7–9 for the reaction of compound 7 and Lys-OMe (11) (ESI, Table S1,† entries 8–17). Unfortunately, the selectivity or conversion became bad under all of those reaction conditions.
:1) at pH = 7. Based on RP-HPLC analysis, compounds 1–2 did not react with Lys-OMe (ESI, Table S1,† entries 1 and 2). Compounds 3–4 showed poor selectivity (ESI, Table S1,† entries 3 and 4). Compounds 5–6 displayed good selectivity, however the conversion was poor (<20%) (ESI, Table S1,† entries 5 and 6). N-Methyl-N-phenylethenesulfonamide (compound 7) emerged as a potential reagent with good selectivity and moderate conversion (∼30%) (ESI, Table S1,† entry 7). We further screened a series of alkaline buffers at pH 7–9 for the reaction of compound 7 and Lys-OMe (11) (ESI, Table S1,† entries 8–17). Unfortunately, the selectivity or conversion became bad under all of those reaction conditions.
 | ||
| Fig. 1 The structure of vinylsulfonamides 1–10. | ||
Compound 7 was allowed to react with Lys-OMe (11, 3 eq.) in the presence of trimethylamine (Et3N 6 eq.) in CH3CN. Compound 7 was consumed, and only one high yield product (12, purified yield: 87%) was obtained, which was mono-labeled based on the HRESIMS of the [M + H]+ and [M + Na]+ (ion peaks at 358.1917 and 380.1615, respectively). The attached position was the ε-amino group rather than the α-amino group based on HSQC and HMBC analysis; key correlations were observed between H-8 and C-10, C-13, C-14 (Scheme 1).
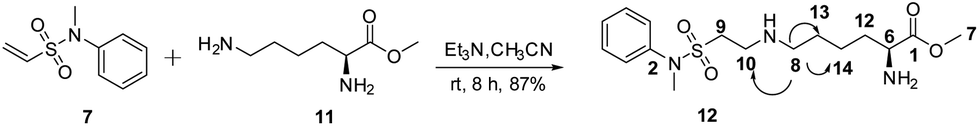 | ||
| Scheme 1 Selective modification of the ε-amino group. | ||
Having observed the high yield and good selectivity of the N-methyl-N-phenylethenesulfonamide (compound 7)-based conjugate, we next used this method to label peptides. Octreotide (13), a synthetic octapeptide with the sequence H2N-D-Phe-Cys-Phe-D-Trp-Lys-Thr-Cys-Thr-ol (disulfide bridge Cys2–Cys7) that is one of the most extensively studied somatostatin analogues, was selected to evaluate the lysine site-specific modification. Octreotides have been widely investigated for conjugation with toxic drugs for targeted delivery into SSTR-selective cancer cells. The studies have revealed that a mixture of products (modification at the N-terminus, the lysine residue, or both) was produced when octreotide was allowed to react with NHS esters.27–30 Octreotide was treated with compound 7 (3 eq.) in the presence of Et3N (6 eq.) in a CH3CN and H2O (1:1) solution at room temperature. The reaction proceeded smoothly, and octreotide was almost completely converted to a single product (14) after 4 h (Fig. 2A), as analyzed by RP-HPLC (Fig. 2B). The HRESIMS spectrum showed that product 14 was mono-labeled, with signals at 1216.4995 [M + H]+ and 608.7549 [M + 2H]2+ (Fig. 2C). After the reduction of product 14 with TCEP, peptide fragments were obtained and identified by MS2 (Fig. 2D). The peptide fragment Lys-Thr-Cys-Thr-ol with m/z 635.28 [M + H]+ carried the Michael acceptor moiety, because it corresponds to the calculated M.W. of 634.28 g mol−1. The other peptide fragment H2N-D-Phe-Cys-Phe-D-Trp with m/z 584.23 [M + H]+ was not modified (Fig. 2E). Therefore, the lysine rather than the N-terminus was selectively functionalized. Compound 14 could be further labeled with fluorescein isothiocyanate (FITC) at the N-terminus to produce compound 15 with high yield (87%). The MS and MS2 experiment for compound 15 confirmed its structure and attachment site based on an m/z of 1606.5349 [M + H]+ and 803.7729 [M + 2H]2+ and the presence of the fragment FITC-NH-D-Phe with m/z 537.11 (Fig. 2F–H).
 | ||
| Fig. 2 Stepwise modification of the lysine residue and N-terminus of octreotide. (A) The reaction scheme. (B) RP-HPLC of the reaction. (C) The HRESIMS spectrum of product 14. (D) The MS2 spectrum of product 14. (E) Peptide fragments of compound 14. (F) The HRESIMS spectrum of product 15. (G) The MS2 spectrum of product 15. (H) Peptide fragments of compound 15. | ||
To demonstrate the general applicability of our method to selectively modify native peptides at lysine residues, we also investigated the reaction of compound 7 with lanreotide (16) and insulin (17). Lanreotide, similar to octreotide, is a somatostatin analog with the sequence H-D-2Nal-Cys(2)-Tyr-D-Trp-Lys-Val-Cys(7)-Thr-NH2. It was modified with compound 7 in a similar fashion to obtain the lysine-modified product 18 with high yield (84%) (Fig. 3A). The HRESIMS spectrum showed that product 18 was mono-labeled, with signals at 647.2653 [M + 2H]2+ (Fig. 3B). After the reduction of product 18 with TCEP (Fig. 3C), peptide fragments were obtained and identified by MS2 (Fig. 3D and E). The result showed that the lysine residue was modified.
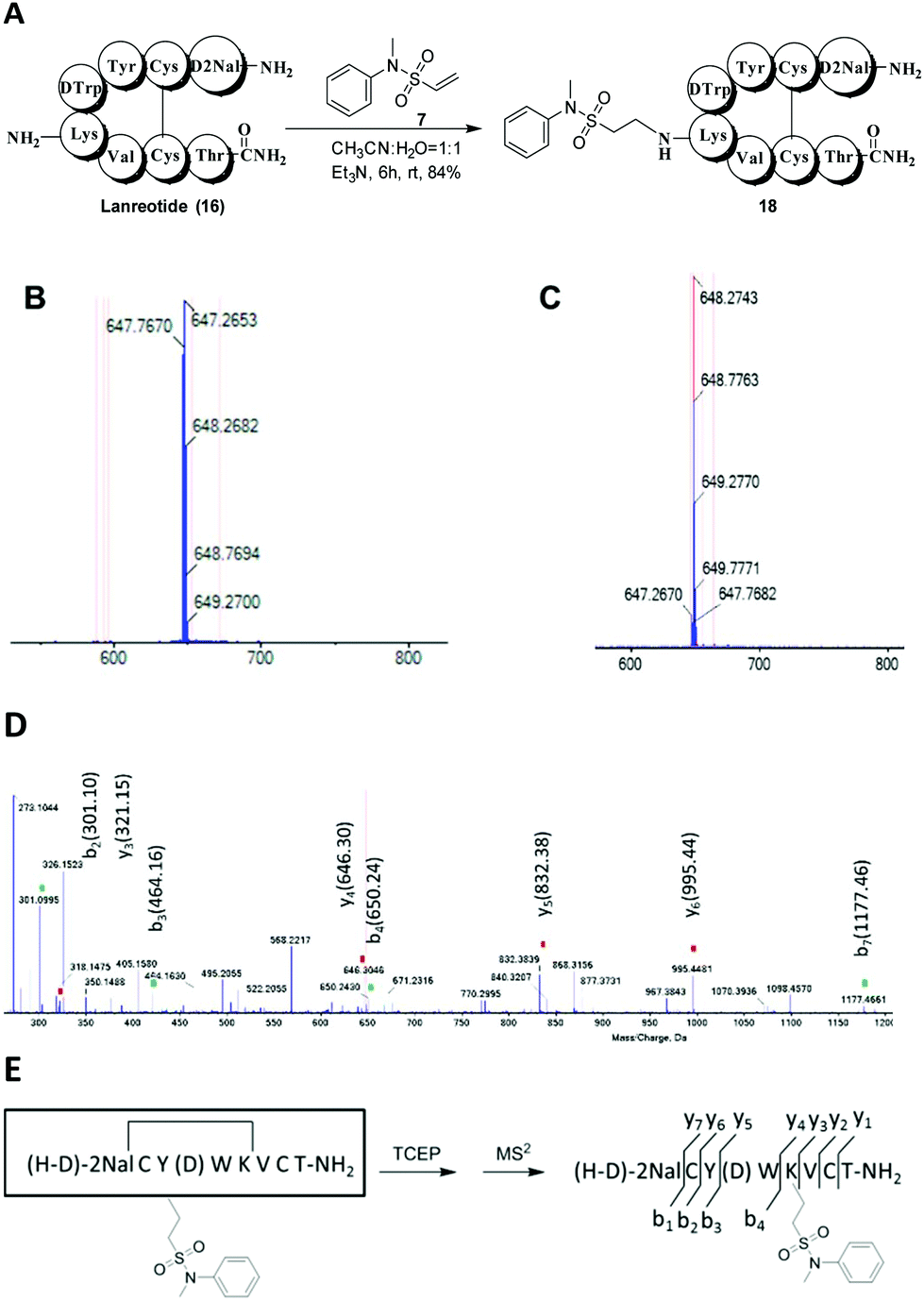 | ||
| Fig. 3 Selective lysine modification of lanreotide. (A) The reaction scheme. (B) The HRESIMS spectrum of product 18. (C) The HRESIMS spectrum of the reduced product of compound 18. (D) The MS2 spectrum of the reduced product of compound 18. (E) Peptide fragments. | ||
Insulin (17), a peptide hormone consisting of two polypeptide chains – the A chain (glycine-A1) and B chain (phenylalanine-B1) – has a lysine in the B chain (lysine-B29). The B29 position is generally tolerant with respect to the insulin receptor affinity, and B29-modified analogues such as detemir and degludec (FDA approved drugs) have demonstrated excellent safety profiles.31 B29 was found to react at a much slower rate than the N-termini of A1 and B1 when insulin was treated with FITC at pH 9.1, producing a mono-A1, mono-B1, di-A1B1, and tri-substituted A1B1B29-labeled mixture.32 Studies have revealed that NHS ester-based reagents are preferable to selectively modify B29,33,34 however, there is also a report on the difficulty to selectively attach a single group to the available amine sites.35 Here, the ability of vinylsulfonamide-based aza-Michael addition to site-specifically label insulin B29 was investigated. To strengthen the applicability of this reaction, more suitable reaction conditions were explored. First, insulin was treated with compound 7 in a buffer series containing various bases at approximately pH 7 and room temperature. The buffer adjusted with DBU was optimal at promoting the site-specific B29 modification of insulin. Although the majority of the conversion yielded a single product, the conversion of insulin was poor (<10%). The effect of pH was examined based on the DBU buffer. By screening buffers from pH 7 to pH 12, we found that the conversion of insulin was low when the pH was below 8, and the reaction became complicated when the pH was more than 10. Moderate conversion (52%) and excellent selectivity (almost a single product 19 in 49% yield and 90% yield based on the recovered insulin) were observed in pH 9 buffer adjusted with DBU (Fig. 4A). The isolated product 19 exhibited signals with multiple charges at 1001.6223 [M + 6H]6+, 1201.7446 [M + 5H]5+ and 1502.1787 [M + 4H]4+, demonstrating that it was a mono-labeled product (Fig. 4B). After the reduction of the disulphide bonds and in-solution digestion of compound 19 by chymotrypsin, peptide fragments were obtained and analyzed by MS2 (Fig. 4C). The modification site was determined as B29 (lysine) based on the fragment (GFFYTPKT+ compound 7) with m/z 579.2698 [M + 2H]2+, which corresponded well to the calculated M.W. of 1156.5263.
 | ||
| Fig. 4 Selective modification of insulin B29 lysine. (A) The reaction scheme. (B) The HRESIMS spectrum of product 19. (C) The MS2 spectrum of product 19. | ||
An effective bioconjugation method has to have the ability to attach functional groups of interest, such as fluorescent probes and drugs. Therefore, we further explored vinylsulfonamides as versatile building blocks for the site-specific preparation of more interesting bioconjugates. Coumarin- and combretastatin A-4 (CA4, a potent cytotoxic agent)-functionalized vinylsulfonamides (20 and 21) were synthesized. The synthesis of product 20 was carried out by condensation of N-(2-aminoethyl)benzeneamine (22) and coumarin-3-carboxylic acid (23) to form the amide (24), followed by treatment with 2-chloroethanesulfonyl chloride (25) to afford the desired product 20. Compound 21 was prepared in a similar manner: Conjugation of CA4 derivative 28, easily obtained from CA4 (26) according to the reported procedure,36 with compound 22 yielded compound 29, which was subsequently allowed to react with compound 25 to obtain the target product 21 (Scheme 2).
 | ||
| Scheme 2 Synthesis of functional vinylsulfonamides 20 and 21. | ||
Based on the successful incorporation of the probes into vinylsulfonamides, their selective conjugation ability for the peptides, octreotide and insulin was investigated. As previously described, octreotide was allowed to react with compounds 20 and 21 in the presence of Et3N, leading to the mono-labeled derivatives 30 and 31 with high yield and successful selectivity (Fig. 5A). For compound 30, the mass peak of m/z 709.2782 [M + 2H]2+ and 1417.5403 [M + H]+ was found in the HRESIMS spectrum, matching its calculated M.W. of 1416.5341 g mol−1 (Fig. 5B). After in-gel digestion by chymotrypsin, the MS2 spectrum of compound 30 was obtained. The degradation product (m/z 1109.4239) was detected under MS2 conditions due to the instability of the attached probe 20. Compound 20 was degraded to a residue with m/z 308.1147 when it was directly used for the MS2 experiment under the same conditions. The modified peptide fragment Lys-Thr-Cys-Thr-ol with m/z 527.1955 included a fragment from probe 20 (m/z 399.04 − 308.11 = 90.93) and the unmodified peptide fragment H2N-D-Phe-Cys-Phe-D-Trp with m/z 584.2334, indicating labeling at lysine (Fig. 5C). For compound 31, the mono-lysine-modified feature was also determined by HRESIMS with signals at m/z 801.3308 [M + 2H]2+ and 1602.6505 [M + H]+ (calculated value: 1600.6441) and MS2 with an unmodified peptide fragment H2N-D-Phe-Cys-Phe-D-Trp (m/z 584.2324) (Fig. 5D).
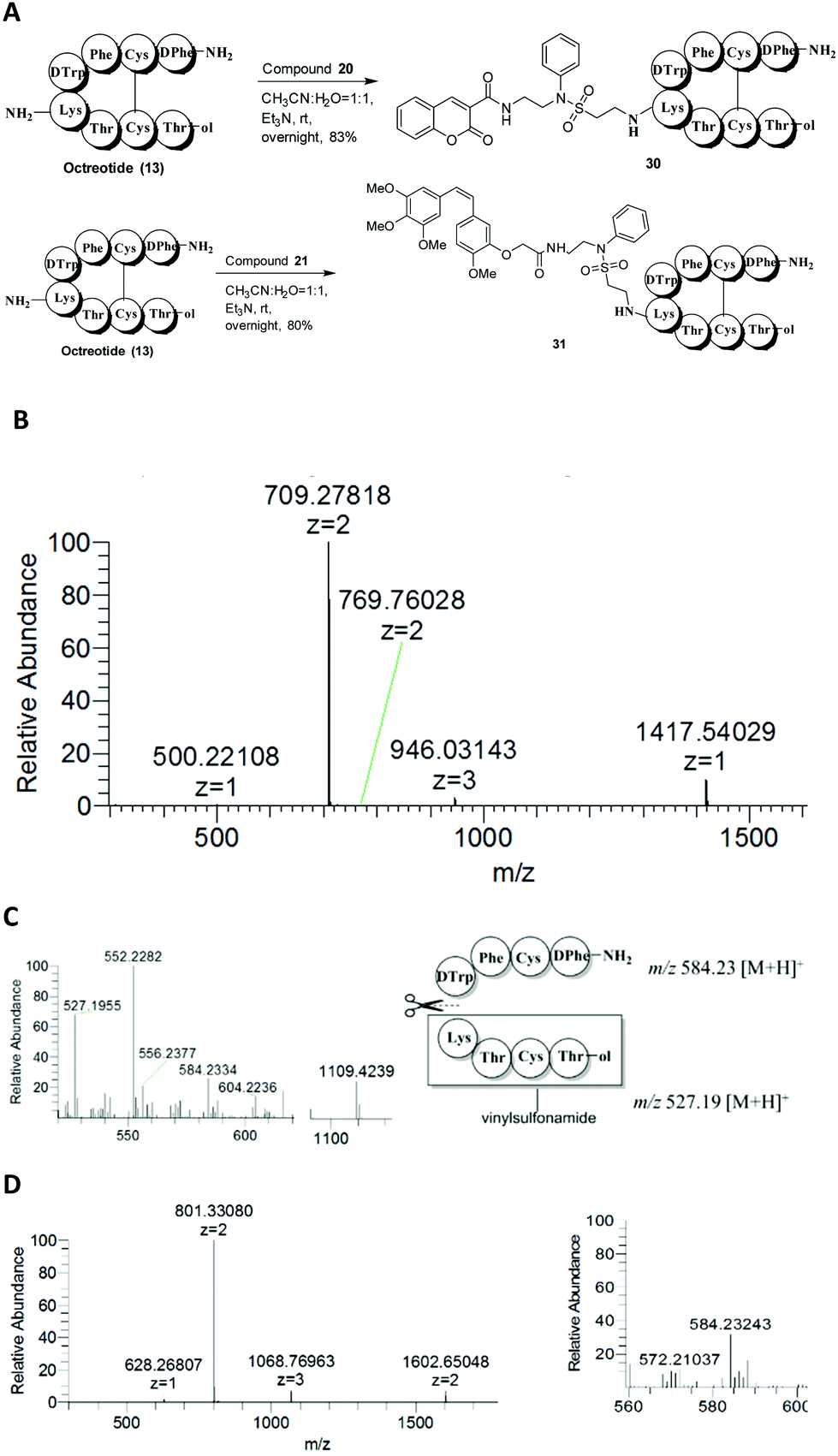 | ||
| Fig. 5 Selective lysine modification of octreotide with functional vinylsulfonamides 20 and 21. (A) The reaction scheme. (B) The HRESIMS spectrum of product 30. (C) The MS2 spectrum of product 30. (D) The HRESIMS and MS2 spectrum of product 31. | ||
In the case of insulin, to conjugate with compound 20 or 21, specific mono-modification of the B29 lysine was performed using the above optimized method with DBU buffer at pH = 9 (Fig. 6A). Products 32 and 33 were also confirmed by MS and MS2 (Fig. 6B and C). Compound 21 was an excellent coupling reagent for insulin that resulted in successful conversion (>95%), yield (92%) and selectivity (almost a single product) (Fig. 6D).
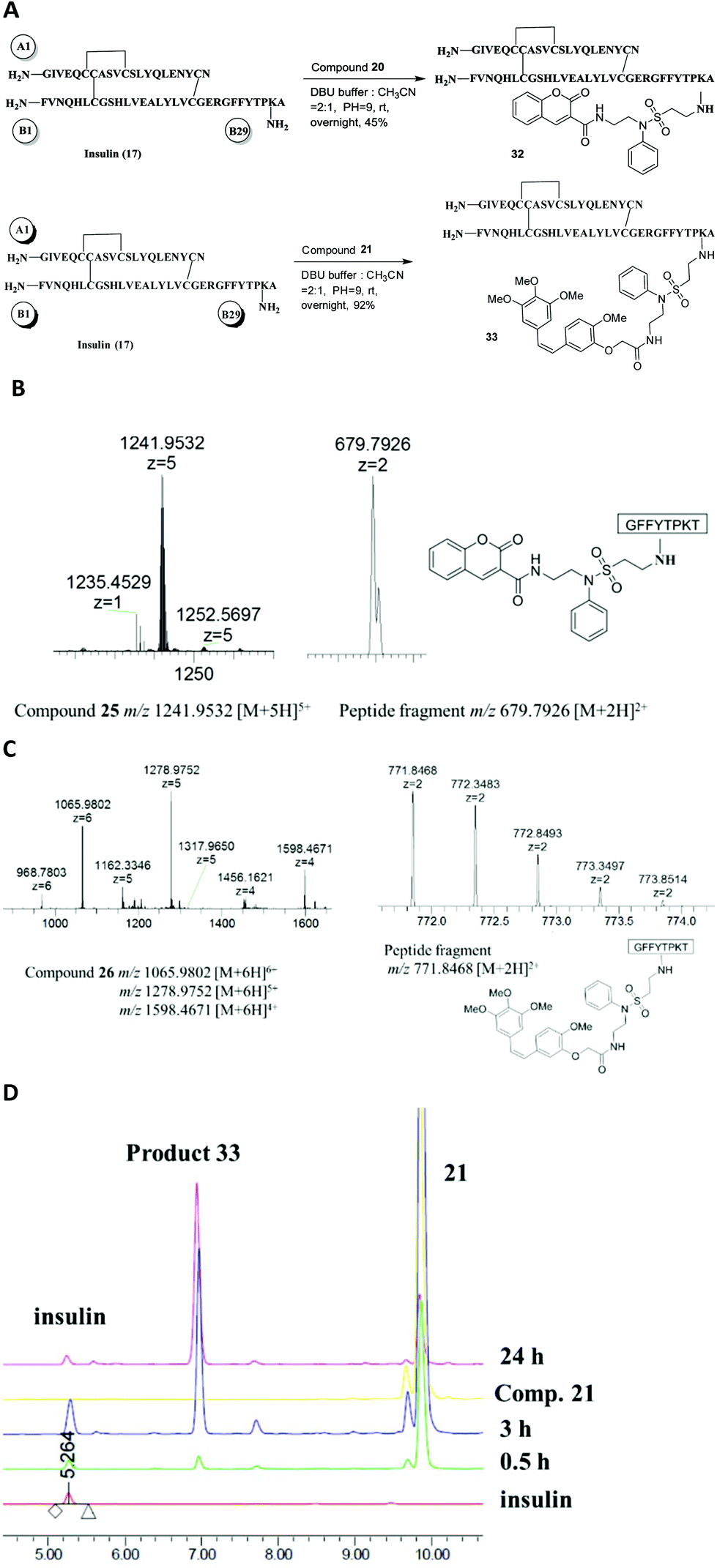 | ||
| Fig. 6 Selective lysine modification of insulin with functional vinylsulfonamides 20 and 21. (A) The reaction scheme. (B) The HRESIMS and MS2 spectrum of product 32. (C) The HRESIMS and MS2 spectrum of product 33. (D) RP-HPLC of the reaction showing that compound 21 was conjugated to insulin. | ||
Somatostatin (34) which has a free N-terminus and two lysine residues with the sequence H-Ala-Gly-Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys-OH (disulfide bridge Cys3–Cys14) was employed to investigate whether there was a preference for a single lysine residue. Somatostatin was reacted with compound 7 in DBU buffer (PH = 9) (Fig. 7A). The resulting HRESIMS spectrum revealed that a mono-labeled product 35 (9%) was obtained, with signals at 612.2921 [M + 3H]3+ and 917.9351 [M + 2H]2+ (Fig. 7B). Similarly, after the reduction of product 35 with TCEP (Fig. 3C), peptide fragments were obtained and identified by MS2 (Fig. 7D and E). The single K9 residue of somatostatin was modified. However, the conversion was low and the reaction system became complicated with the prolonged reaction time. When somatostatin (34) was reacted with compound 7 (3 eq.) in the presence of Et3N (6 eq.) in a CH3CN and H2O (1:1), a main di-labelled product (36) was yielded (63%) with signals at 678.3126 [M + 3H]3+ and 1016.9655 [M + 2H]2+ (Fig. 7F). After the reduction of product 36 with TCEP (Fig. 7G), the resulting HRESIMS spectrum showed that A1 and K9 were modified (Fig. 7H–I).
 | ||
| Fig. 7 Modification of somatostatin 34. (A) The reaction scheme. (B) The HRESIMS spectrum of product 35. (C) The HRESIMS spectrum of the reduced product of compound 35. (D) The MS2 spectrum of the reduced product of compound 35. (E) Peptide fragments. (F) The HRESIMS spectrum of product 36. (G) The HRESIMS spectrum of the reduced product of compound 36. (H) The MS2 spectrum of the reduced product of compound 36. (I) Peptide fragments. | ||
Conclusions
Selective and effective modification of a lysine residue in the presence of an N-terminus is anticipated to be difficult due to the lower pKa of the N-terminal position. Here, a useful and efficient method for utilizing vinylsulfonamide-based aza-Michael addition to selectively modify lysine instead of the N-terminus on peptides was developed. Furthermore, we demonstrated that functional vinylsulfonamide derivatives could also be produced via reaction with a fluorescent moiety or drug and achieve the selective bioconjugation. The CA4-conjugated probe showed excellent reactivity and selectivity to modify the B29 lysine of insulin. This is a promising method for native peptide modification where the N-terminus is crucial for peptide bioactivity. However, peptides that contain multiple lysine residues will result in a complicated situation. And our method is more beneficial for peptides that lack an accessible free Cys residue which has superior reactivity. Efforts to develop more useful vinylsulfonamide reagents and application of this method to modify peptides or proteins are currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by ShanghaiTech university.Notes and references
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. Bernardes, Nat. Chem., 2016, 8, 103–113 CAS.
- O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495–5551 RSC.
- O. Boutureira and G. J. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- A. Abbas, B. Xing and T. P. Loh, Angew. Chem., Int. Ed., 2014, 53, 7491–7494 CrossRef CAS PubMed.
- I. M. Serafimova, M. A. Pufall, S. Krishnan, K. Duda, M. S. Cohen, R. L. Maglathlin, J. M. McFarland, R. M. Miller, M. Frodin and J. Taunton, Nat. Chem. Biol., 2012, 8, 471–476 CrossRef CAS PubMed.
- D. Kalia, P. V. Malekar and M. Parthasarathy, Angew. Chem., Int. Ed., 2016, 55, 1432–1435 CrossRef CAS PubMed.
- S. B. Gunnoo and A. Madder, ChemBioChem, 2016, 17, 529–553 CrossRef CAS PubMed.
- H. G. Lee, G. Lautrette, B. L. Pentelute and S. L. Buchwald, Angew. Chem., Int. Ed., 2017, 56, 3177–3181 CrossRef CAS PubMed.
- C. L. Tung, C. T. Wong, E. Y. Fung and X. Li, Org. Lett., 2016, 18, 2600–2603 CrossRef CAS PubMed.
- S. T. Larda, D. Pichugin and R. S. Prosser, Bioconjugate Chem., 2015, 26, 2376–2383 CrossRef CAS PubMed.
- S. Diethelm, M. A. Schafroth and E. M. Carreira, Org. Lett., 2014, 16, 3908–3911 CrossRef CAS PubMed.
- D. Chen, M. M. Disotuar, X. Xiong, Y. Wang and D. H.-C. Chou, Chem. Sci., 2017, 8, 2717–2722 RSC.
- J. I. MacDonald, H. K. Munch, T. Moore and M. B. Francis, Nat. Chem. Biol., 2015, 11, 326–331 CrossRef CAS PubMed.
- Z. W. Lai, A. Petrera and O. Schilling, Curr. Opin. Chem. Biol., 2015, 24, 71–79 CrossRef CAS PubMed.
- A. C. Obermeyer, J. B. Jarman and M. B. Francis, J. Am. Chem. Soc., 2014, 136, 9572–9579 CrossRef CAS PubMed.
- A. O. Chan, C. M. Ho, H. C. Chong, Y. C. Leung, J. S. Huang, M. K. Wong and C. M. Che, J. Am. Chem. Soc., 2012, 134, 2589–2598 CrossRef CAS PubMed.
- G. Xu, S. B. Shin and S. R. Jaffrey, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 19310–19315 CrossRef CAS PubMed.
- J. M. Gilmore, R. A. Scheck, A. P. Esser-Kahn, N. S. Joshi and M. B. Francis, Angew. Chem., Int. Ed., 2006, 45, 5307–5311 CrossRef CAS PubMed.
- M. Chilamari, L. Purushottam and V. Rai, Chem. – Eur. J., 2017, 23, 3819–3823 CrossRef CAS PubMed.
- L. Purushottam, S. R. Adusumalli, M. Chilamari and V. Rai, Chem. Commun., 2017, 53, 959–962 RSC.
- N. Abello, H. A. Kerstjens, D. S. Postma and R. Bischoff, J. Proteome Res., 2007, 6, 4770–4776 CrossRef CAS PubMed.
- X. Chen, K. Muthoosamy, A. Pfisterer, B. Neumann and T. Weil, Bioconjugate Chem., 2012, 23, 500–508 CrossRef CAS PubMed.
- J. Dadova, M. Vrabel, M. Adamik, M. Brazdova, R. Pohl, M. Fojta and M. Hocek, Chem. – Eur. J., 2015, 21, 16091–16102 CrossRef CAS PubMed.
- J. L. Furman, M. Kang, S. Choi, Y. Cao, E. D. Wold, S. B. Sun, V. V. Smider, P. G. Schultz and C. H. Kim, J. Am. Chem. Soc., 2014, 136, 8411–8417 CrossRef CAS PubMed.
- E. H. Suh, Y. Liu, S. Connelly, J. C. Genereux, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2013, 135, 17869–17880 CrossRef CAS PubMed.
- J. Dadova, P. Orsag, R. Pohl, M. Brazdova, M. Fojta and M. Hocek, Angew. Chem., Int. Ed., 2013, 52, 10515–10518 CrossRef CAS PubMed.
- H. Y. Zhang, W. Q. Xu, Y. W. Wang, E. Omari-Siaw, Y. Wang, Y. Y. Zheng, X. Cao, S. S. Tong, J. N. Yu and X. M. Xu, Int. J. Pharm., 2016, 502, 98–106 CrossRef CAS PubMed.
- M. Huo, Q. Zhu, Q. Wu, T. Yin, L. Wang, L. Yin and J. Zhou, J. Pharm. Sci., 2015, 104, 2018–2028 CrossRef CAS PubMed.
- D. H. Na, K. C. Lee and P. P. DeLuca, Pharm. Res., 2005, 22, 743–749 CrossRef CAS PubMed.
- T. Yin, Q. Wu, L. Wang, L. Yin, J. Zhou and M. Huo, Mol. Pharm., 2015, 12, 3020–3031 CrossRef CAS PubMed.
- A. N. Zaykov, J. P. Mayer and R. D. DiMarchi, Nat. Rev. Drug Discovery, 2016, 15, 425–439 CrossRef CAS PubMed.
- D. Jacob, M. Joan Taylor, P. Tomlins and T. S. Sahota, J. Fluoresc., 2016, 26, 617–629 CrossRef CAS PubMed.
- D. H. Chou, M. J. Webber, B. C. Tang, A. B. Lin, L. S. Thapa, D. Deng, J. V. Truong, A. B. Cortinas, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2401–2406 CrossRef CAS PubMed.
- T. H. Jensen, S. Havelund, P. K. Nielsen and J. Markussen, J. Am. Chem. Soc., 2005, 127, 6158–6159 CrossRef PubMed.
- K. Thibaudeau, R. Leger, X. Huang, M. Robitaille, O. Quraishi, C. L. Soucy, N. B. Gagnon, P. V. Wyk, V. Paradis, J. P. Castaigne and D. Bridon, Bioconjugate Chem., 2005, 16, 1000–1008 CrossRef CAS PubMed.
- A. Kamal, A. Mallareddy, M. Janaki Ramaiah, S. N. Pushpavalli, P. Suresh, C. Kishor, J. N. Murty, N. S. Rao, S. Ghosh, A. Addlagatta and M. Pal-Bhadra, Eur. J. Med. Chem., 2012, 56, 166–178 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob01866e |
| This journal is © The Royal Society of Chemistry 2017 |