
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total synthesis and mass spectrometric analysis of a Mycobacterium tuberculosis phosphatidylglycerol featuring a two-step synthesis of (R)-tuberculostearic acid†
Satvika
Burugupalli
,
Mark B.
Richardson
 and
Spencer J.
Williams
*
and
Spencer J.
Williams
*
School of Chemistry and Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville, Victoria, Australia 3010. E-mail: sjwill@unimelb.edu.au
First published on 11th August 2017
Abstract
We report the total synthesis of (R)-tuberculostearic acid-containing Mycobacterium tuberculosis phosphatidylglycerol (PG). The approach features a two-step synthesis of (R)-tuberculostearic acid, involving an (S)-citronellyl bromide linchpin, and the phosphoramidite-assisted assembly of the full PG structure. Collision-induced dissociation mass spectrometry of two chemically-synthesized PG acyl regioisomers revealed diagnostic product ions formed by preferential loss of carboxylate at the secondary (sn-2) position.
Introduction
Phosphatidylglycerols are phospholipids produced by a wide range of organisms including bacteria, fungi, plants and animals.1 Within animal cells PG is mainly found in the mitochondrial membranes,2 and is believed to be released into the cellular milieu only upon cellular damage or stress. PGs are also produced by a wide range of bacteria, including important human pathogens such as Listeria, Streptococcus, and Mycobacterium tuberculosis (Mtb). PGs can be presented on several antigen presenting proteins of the cluster of differentiation 1 (CD1) class, specifically CD1b3 and CD1d,4,5 where they are recognized by natural killer T cells and lipid-reactive T cells. Thus PGs appear to be a molecular signature allowing immune recognition of bacterial infection or damaged self.In mycobacteria, PG is an intermediate in the biosynthesis of lysinylated PG,6 phosphatidylinositol,7 and cardiolipin.7,8 It has been proposed that PG is synthesized in two steps (Fig. 1). Step 1 is catalyzed by phosphatidylglycerophosphate synthetase (PgsA3), which catalyzes the phosphatidylation of glycerol-3-phosphate by CDP-diacylglycerol to afford phosphatidylglycerophosphate;7 and step 2 which involves the dephosphorylation of phosphatidylglycerophosphate to afford PG, by a phosphatidylglycerophosphatase. Mtb PG occurs as a range of lipoforms including an abundant species that bears (R)-tuberculostearic acid (R-TBSA) at the sn-1 position, and palmitic acid at the sn-2 position,4 a decoration that matches that of other phosphatidyl-containing lipids9 such as phosphatidylethanolamine,10 and phosphatidylinositol mannosides.11 Possibly because of its role as a biosynthetic intermediate, Mtb PG occurs at low levels and the challenges of its isolation4 from the natural source are compounded by the pathogenicity and low growth rate of Mtb. Recent work has shown that Mtb PG can be presented by the CD1d antigen presenting molecule to natural killer T cells that display ‘type II’ T cell receptors.4 Type II NKT cells are distinguished from their better studied type I variants, by lacking reactivity to the prototypical NKT ligand α-galactosyl ceramide, and through possessing a diverse array of αβ chains that comprise the T cell receptor.12 While most studies have focused on type I NKT cells, type II NKT cells are more abundant in humans, and are functionally distinct from type I NKT cells.13 However, studies of type II NKT cells have been limited by a lack of well-defined lipid antigens.
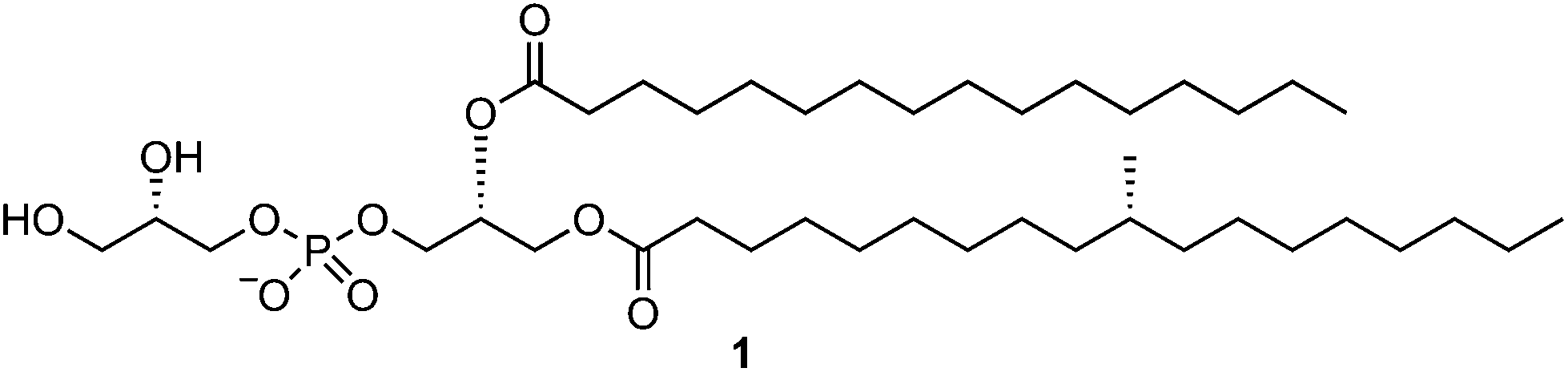 | ||
| Fig. 1 Structure of Mycobacterium tuberculosis (Mtb) TBSA-containing phosphatidylglycerol (PG). | ||
Herein we report the total synthesis of the TBSA-containing Mtb PG (1) using phosphoramidite chemistry. As part of this effort we have devised the shortest synthesis yet reported of R-TBSA (2) in just two steps from commercially available (S)-citronellyl bromide, and its incorporation into Mtb PG. Finally, we demonstrate that collision-induced dissociation mass spectrometry of chemically-synthesized PG regioisomers results in the formation of diagnostic fragment ions that have utility in the structural characterization of PGs.
Results and discussion
Synthesis of (R)-tuberculostearic acid
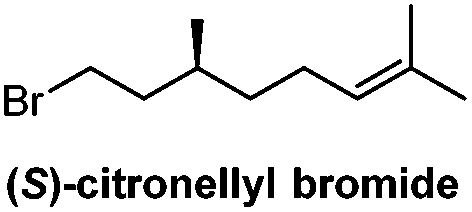
Numerous syntheses of R-TBSA have been reported, using chiral pool sources,17,19,20 chiral auxiliaries,21 catalytic enantioselective induction,10,18 and resolution methods14–16 (Table 1). The most efficient synthesis reported to date is that of Roberts and Baird who prepared TBSA in four steps from (S)-citronellyl bromide, in a route involving stepwise elongations using copper(I)-catalyzed cross-coupling and Wittig extension.22 We were inspired by this use of citronellyl bromide as a linchpin, and anticipated that diverting the cross-coupled intermediate into a Grubbs-cross-metathesis/hydrogenation process could provide further synthetic abbreviation. Thus, according to the reported method,22 copper(I)-catalyzed cross-coupling of hexyl magnesium bromide with (S)-citronellyl bromide afforded the alkene 3 (Scheme 1). Initially, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ethyl 6-heptenoate and 3 were reacted in the presence of Hoveyda–Grubbs II catalyst, affording a 58% yield of an E/Z mixture of cross-coupled alkenes. We next attempted a direct tandem cross-metathesis/hydrogenation reaction.39 Use of a 1:1 mixture of alkenes afforded a 48% yield of ethyl R-TBSA, which could be improved to 76% by using a 3:1 ratio of ethyl heptenoate/3. Saponification of ethyl R-TBSA afforded R-TBSA (2) in three steps. Encouraged by these results, we next enacted a two-step synthesis of R-TBSA by employing a 3:1 ratio of 6-heptenoic acid/3 under the same conditions, which directly afforded R-TBSA in 73% yield.
:1 mixture of ethyl 6-heptenoate and 3 were reacted in the presence of Hoveyda–Grubbs II catalyst, affording a 58% yield of an E/Z mixture of cross-coupled alkenes. We next attempted a direct tandem cross-metathesis/hydrogenation reaction.39 Use of a 1:1 mixture of alkenes afforded a 48% yield of ethyl R-TBSA, which could be improved to 76% by using a 3:1 ratio of ethyl heptenoate/3. Saponification of ethyl R-TBSA afforded R-TBSA (2) in three steps. Encouraged by these results, we next enacted a two-step synthesis of R-TBSA by employing a 3:1 ratio of 6-heptenoic acid/3 under the same conditions, which directly afforded R-TBSA in 73% yield.
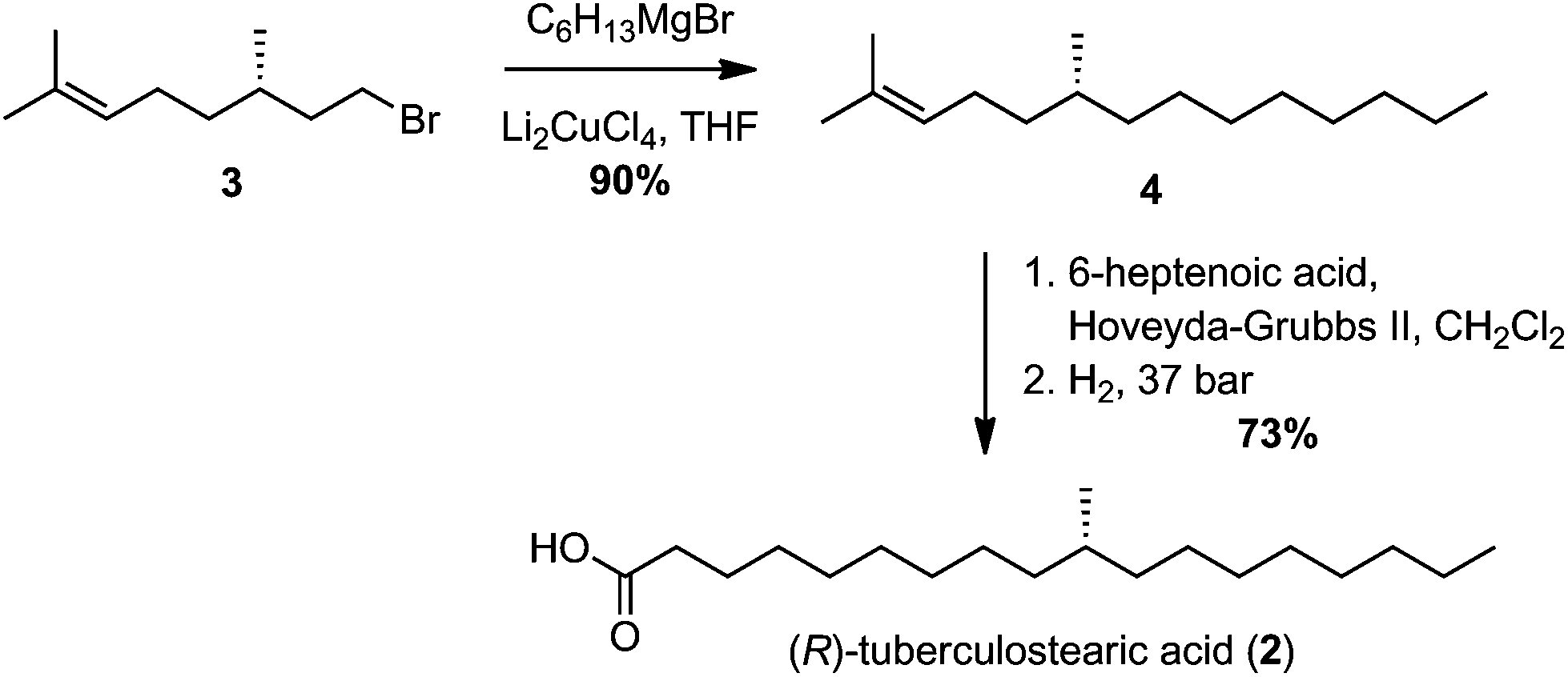 | ||
| Scheme 1 Two-step synthesis of (R)-tuberculostearic acid. | ||
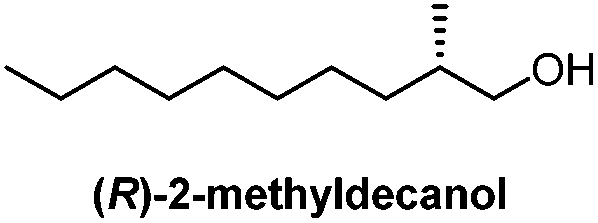
| Starting material/key intermediate | Approach/steps | Ref. | Starting material/key intermediate | Approach/steps | Ref. |
|---|---|---|---|---|---|
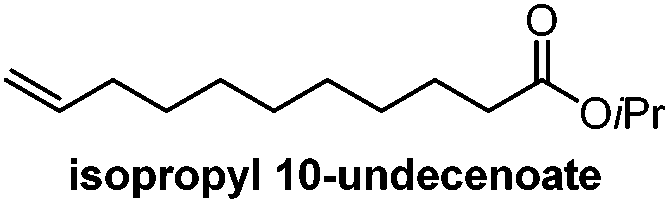
| a (R)-2-Methyldecanol was synthesized from bromooctane in 5 steps by resolution of an intermediate brucine salt. b Isopropyl 10-undecenoate can be synthesized in one step from commercially-available undecylenic acid. | |||||
|
Chiral resolution/10 stepsa | Prout, 194814 |
|
Chiral resolution/7 steps | Hahn, 201515 |
|
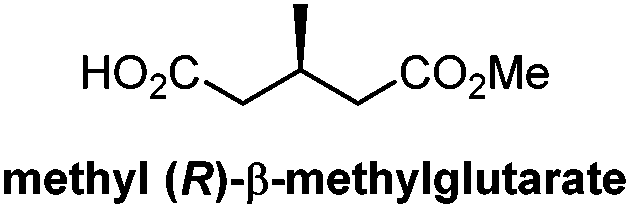
Chiral resolution/9 steps | Linstead, 195116 |
|
Catalytic enantioselective induction/6 stepsb | Feringa, Minnaard, 201010 |
|
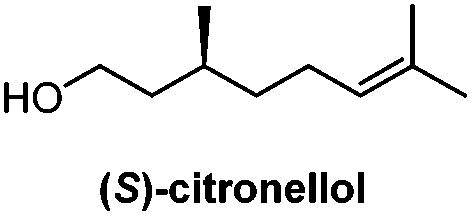
Chiral pool/8 steps | Larsen, 200717 |
|
Catalytic enantioselective induction/5 steps | Minnaard, 201318 |
|
Chiral pool/7 steps | Seeberger, 200619 |
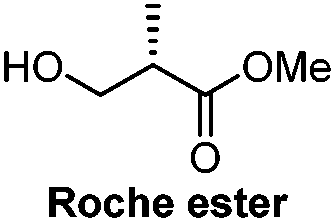
|
Chiral pool/5 steps | Hung, 201520 |
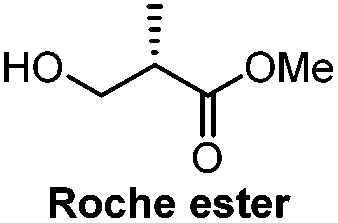
|
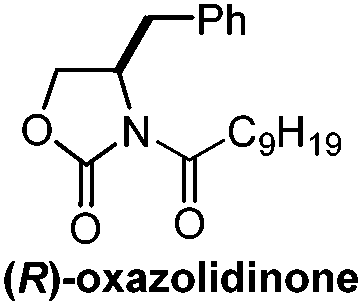
Chiral auxiliary/7 steps | Williams, 201321 |
|
Chiral pool/4 steps | Baird, 200622 |
Synthesis of phosphatidylglycerols
Reported methods for the synthesis of PGs include chemoenzymatic methods,23 and purely chemical approaches;24 however, the reported chemical methods typically employ highly moisture sensitive POCl3 as a phosphorylation reagent and do not allow regioselective introduction of two different acyl groups. In devising an approach to Mtb PG, we elected to utilize contemporary phosphoramidite chemistry, which has been widely applied in the synthesis of oligonucleotides,25 phosphatidylinositols,26 and cardiolipin.27We chose to prepare 1, and analogues lacking the 10-methyl group with alternate arrangements of the fatty acyl groups. For the construction of the requisite diacylglycerols, we employed the approach of Minnaard, which proceeds with high regioselectivity and without acyl group migration.18 Accordingly, opening of benzyl (R)-glycidyl ether (5) with palmitic acid, stearic acid, or R-TBSA using Jacobsen pre-catalyst C1,28 followed by esterification with a second fatty acid and DCC/DMAP, gave the diesters 6a–c (Scheme 2). Hydrogenolysis of the benzyl ether afforded the diacylglycerols 7a–c, which were used immediately without purification in the next step.
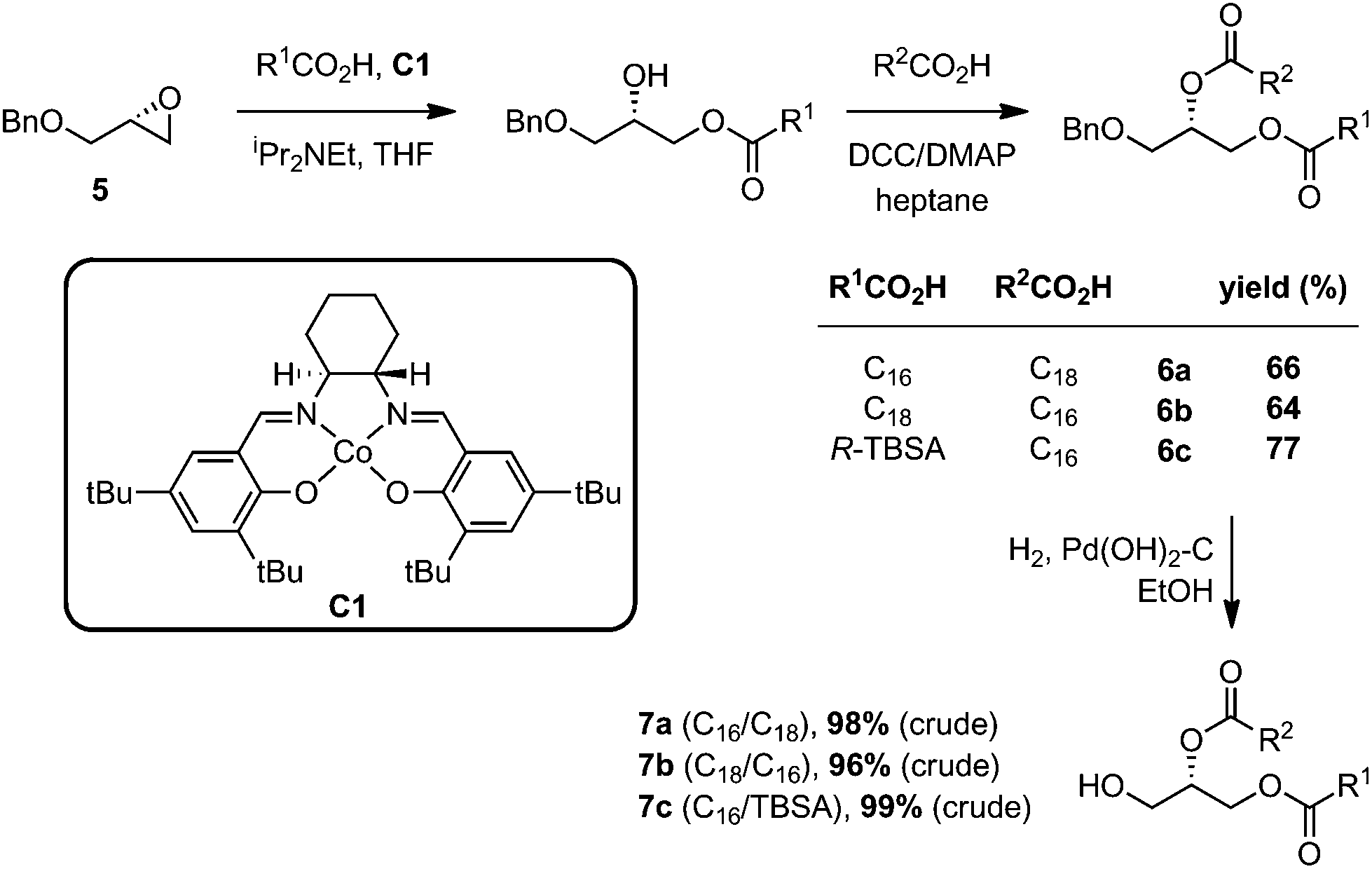 | ||
| Scheme 2 Preparation of diacylglycerols. | ||
Assembly of the PG structure relied upon the use of a phosphordiamidite linchpin, benzyl bis(N,N-diisopropyl)phosphordiamidite (Scheme 3).17,29 Reaction of the diacylglycerols 7a–c with this phosphordiamidite, catalyzed by diisopropyl ammonium tetrazolide (DIPAT), a weak acid that does not activate the product,25 afforded the unstable phosphoramidites 8a–c. Compounds 8a–c were purified by a quick passage over alumina to remove any residual reactant or reagents, and then immediately subjected to a second phosphitylation reaction with dibenzylglycerol 9, catalyzed by 1H-tetrazole. Upon consumption of the phosphoramidate, t-BuOOH was added to oxidize the intermediate phosphite to the protected PGs 10a–c. Finally, hydrogenolysis of the benzyl groups provided Mtb PG 1, and analogues 11a and 11b.
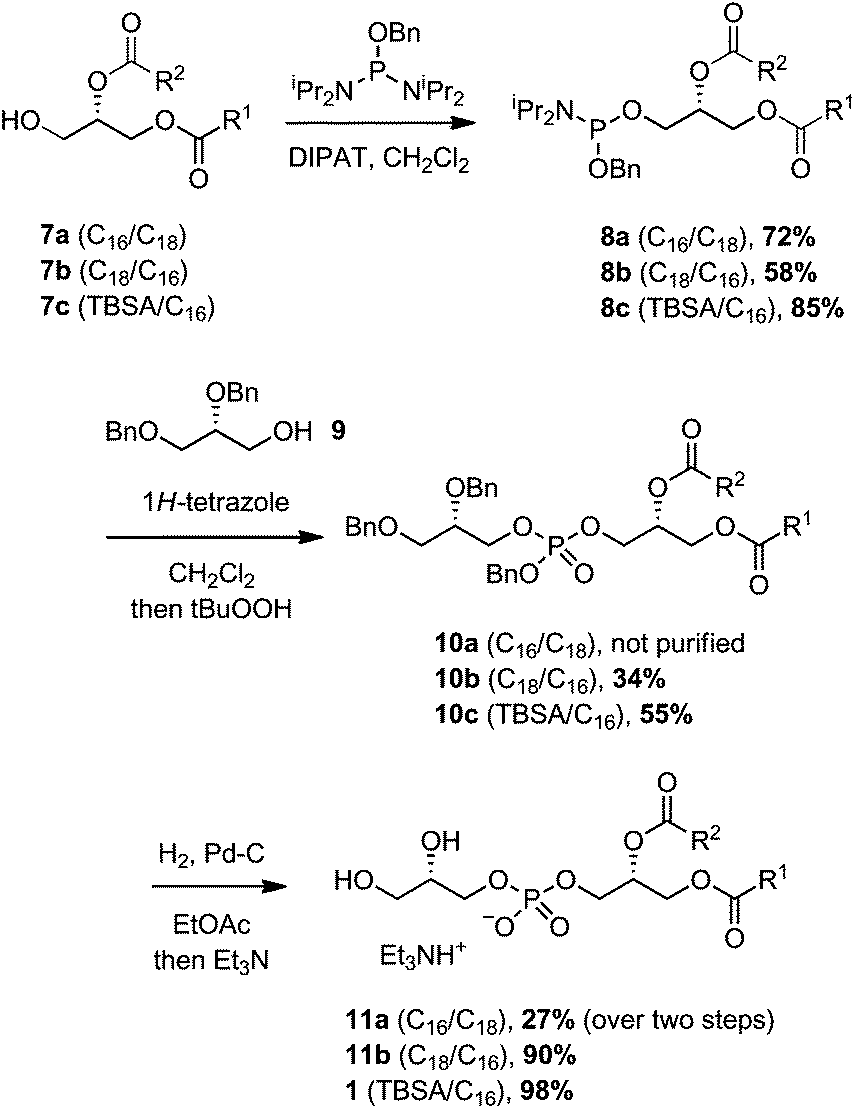 | ||
| Scheme 3 Preparation of Mtb PG 1 and analogues. | ||
Collision-induced dissociation mass spectrometry of phosphatidylglycerols
Early studies reported that negative-ion fast-atom bombardment or electrospray-ionization with collision-induced dissociation/tandem mass spectrometry of phosphatidylethanolamine (PE) and PG results in fragmentation to provide high abundance fatty acid carboxylate (RCO2−) fragment ions.30,31 These studies revealed similar fragmentation pathways for PE and PG, and showed that regioisomeric PEs fragment in a characteristic way to produce RCO2− ions for which those derived from the sn-2 position are of greater intensity, thereby allowing structural characterization of fatty acyl regiochemistry.10,31 Subsequently, it was shown that CID/MS-MS of PG occurs with similar results, producing RCO2− ions in which those from the sn-2 position are more abundant, in what has been described as a charge-driven process.32,33 In order to confirm these fragmentation patterns we studied the chemically synthesized regioisomers 11a and 11b in negative ion CID/MS2 experiments using electro-spray ionization. Fragmentation of the parent anions [M − H]− (m/z 749.5) formed from the regioisomers 11a (C16/C18) and 11b (C18/C16), yielded characteristic fatty acid carboxylate (RCO2−) fragment ions at m/z 255.2 and 283.3 (Fig. 2a and b). In each case the more abundant ion is that derived from the fatty acid located in the sn-2 position. This work confirms that CID-induced fragmentation patterns can be used to identify the structures of asymmetrical PG molecules. Fig. 2c shows the CID-MS2 spectrum of 1, revealing that fragmentation of the parent ion [M − H]− (m/z 763.5) affords daughter ions at m/z 255.2 and 297.3. Here, the most abundant fragment (m/z 255.2) is that derived from the palmitic acid located at the sn-2 position. We note that these and earlier observations for CID-MS of PGs differ from those for the structurally-related phosphatidylinositols32 and phosphatidylinositolmannosides,11,17 which under similar conditions fragment by neutral loss of fatty acids and fatty acid ketenes in a diagnostic way. While these systems also yield RCO2− fragment ions, their relative intensities do not reflect the regiochemistry of their location on the phosphatidyl group because other pathways also yield these ions in abundances that are sensitive to the applied collision energy.32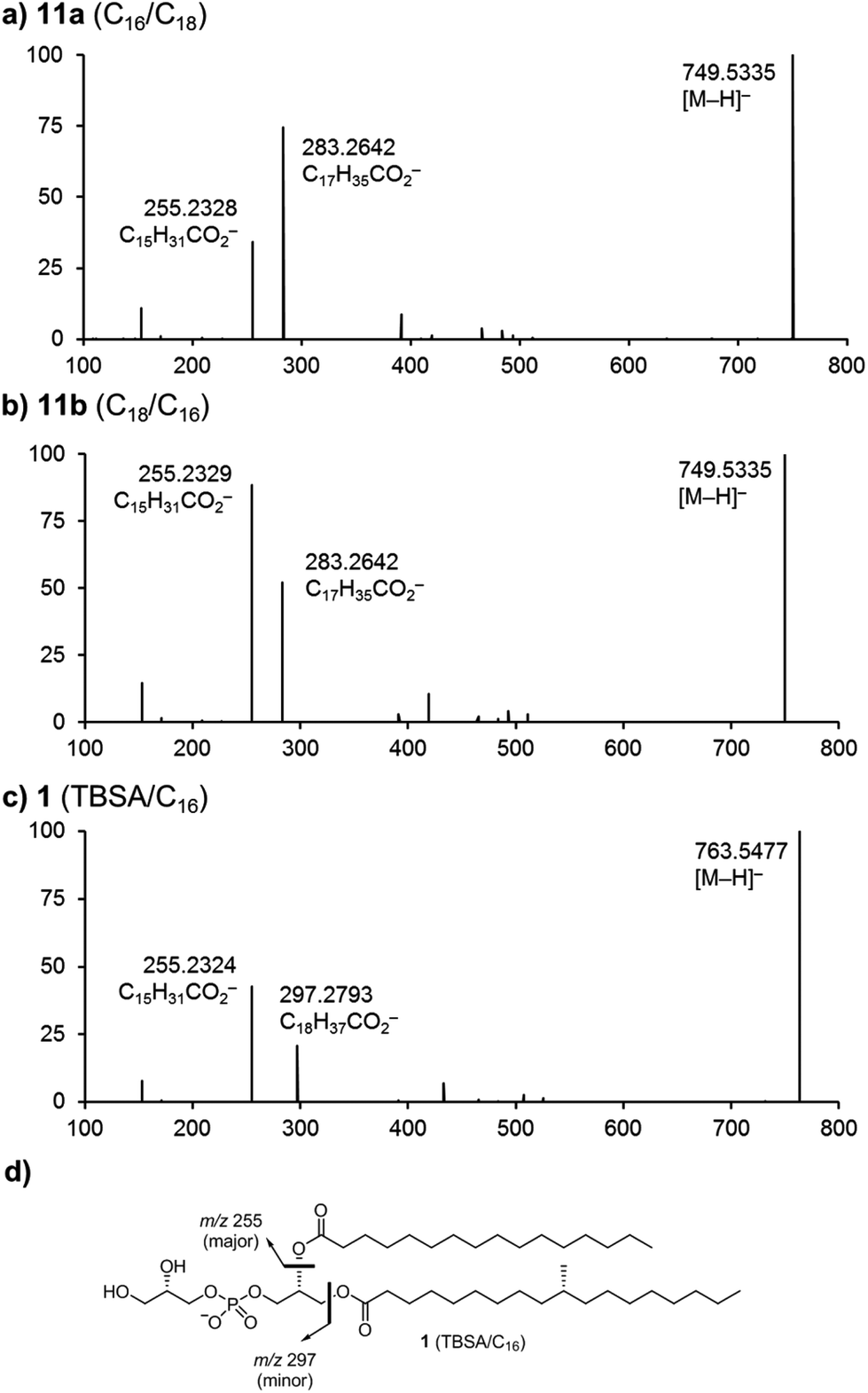 | ||
| Fig. 2 Multistage mass spectrometry (MS2) experiments involving the collision-induced dissociation of negative ions formed by electro-spray ionization.† (a) MS2 spectrum of deprotonated 11a (C16/C18). (b) MS2 spectrum of deprotonated 11b (C18/C16). (c) MS2 spectrum of deprotonated 1. (d) Fragmentation of compound 1. See Experimental for details. | ||
Conclusions
In this work we disclose the first total synthesis of Mtb PG 1, and analogues 11a and 11b. Our approach includes the most concise preparation of R-TBSA reported to date. We note that while Mtb PG has only been studied in the context of a CD1d-presented lipid antigen, other closely related PGs from mammals, Escherichia coli, Corynebacterium glutamicum and Listeria monocytogenes have been identified as CD1b-presented lipid antigens.3 Access to synthetic Mtb PG 1 and analogues 11a and 11b should support studies to understand its immunological properties, and investigation of the effect of the branched-methyl group in the fatty acyl chain. Collision-induced dissociation mass spectrometry analysis confirms the desired acylation patterns in our synthetically acquired materials, and provides a foundation for structural analyses of mycobacterial PGs.Experimental
General methods
Proton (1H NMR, 400 or 500 MHz), proton-decoupled carbon-13 (13C NMR, 100 or 125 MHz), and phosphorus-31 (31P NMR, 202 MHz) nuclear magnetic resonance spectra were obtained in deuterochloroform or methanol-d4 (CD3OD) with residual protonated solvent or solvent carbon signals as internal standards. Abbreviations for multiplicity are s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Flash chromatography was carried out on silica gel 60 according to the procedure of Still et al.34 Chromatography on alumina was performed on a column using neutral, activated aluminium oxide (50–200 micron, Acros Organics). Analytical thin layer chromatography (t.l.c.) was conducted on aluminium-backed 2 mm thick silica gel 60 F254 and chromatograms were visualized with ceric ammonium molybdate (Hanessian's stain), potassium permanganate or 5% H2SO4/MeOH, with charring as necessary. High resolution mass spectra (HRMS) were obtained using an ESI-TOF-MS; all samples were run using 0.1% formic acid. Dry CH2Cl2, THF, and Et2O were obtained from a dry solvent apparatus (Glass Contour of SG Water, Nashua, USA) as per the procedure of Pangborn et al.35 Dry DMF was dried over 4 Å molecular sieves. Pet. spirits refers to petroleum ether, boiling range 40–60 °C.Synthesis of (R)-tuberculostearic acid (2)
:petroleum ether) of the residue afforded 4 as a colorless oil (240 mg, 95%); [α]22D +0.75 (c 0.5 in CHCl3) (lit.22 [α]25D +1.5 in CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.88–0.83 (6 H, m, 2 × CH3), 1.15–1.06 (2 H, m, CH2), 1.31–1.22 (20 H, m, 10 × CH2), 1.60 (3 H, s, CH3), 1.68 (3 H, s, CH3), 1.95 (2 H, dd, J = 6.8, 13.7 Hz, CHCH2), 5.10 (1 H, t, J = 7.0 Hz, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C); 13C NMR (101 MHz, CDCl3) δ 14.2, 17.7, 19.2, 22.8, 25.7, 27.1, 29.5, 29.8, 30.1, 32.0, 32.5, 37.1, 37.3, 125.2, 131.0; HRMS calcd for C16H32 [M + H]+ 225.2538, found 225.2534.
OOCH2); 13C NMR (101 MHz, CDCl3) δ 14.2, 14.4, 19.8, 22.8, 25.1, 27.1, 27.2, 29.3, 29.4, 29.5, 29.6, 29.8, 30.0, 30.1, 32.0, 32.8, 32.8, 34.5, 37.2, 60.2, 174.0 (CO); HRMS calcd for C21H42O2 [M + H]+ 327.3218, found 327.3220.
C); 13C NMR (101 MHz, CDCl3) δ 14.2, 17.7, 19.2, 22.8, 25.7, 27.1, 29.5, 29.8, 30.1, 32.0, 32.5, 37.1, 37.3, 125.2, 131.0; HRMS calcd for C16H32 [M + H]+ 225.2538, found 225.2534.
OOCH2); 13C NMR (101 MHz, CDCl3) δ 14.2, 14.4, 19.8, 22.8, 25.1, 27.1, 27.2, 29.3, 29.4, 29.5, 29.6, 29.8, 30.0, 30.1, 32.0, 32.8, 32.8, 34.5, 37.2, 60.2, 174.0 (CO); HRMS calcd for C21H42O2 [M + H]+ 327.3218, found 327.3220.
From ethyl (R)-10-methyloctadecanoate. 2 M KOH (0.76 ml, 1.5 mmol) was added to a solution of the ethyl ester (50 mg, 0.153 mmol) in MeOH/THF (1
:1, 0.8 ml) and stirred under reflux overnight. The solution was acidified with aq. citric acid (1 M), extracted with EtOAc, and the organic phase washed with brine, dried (MgSO4) and concentrated under reduced pressure to afford 2 as a colorless oil (44 mg, 97%); [α]25D −0.9 (c 1.0 in CHCl3) (lit.22 [α]25D −0.34 in CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.83 (3 H, d, J = 6.5 Hz, CHCH3), 0.88 (3 H, t, J = 6.8 Hz, CH2CH3), 1.12–1.04 (2 H, m, CH2), 1.36–1.21 (25 H, m, 12 × CH2, 1 × CH), 1.67–1.59 (2 H, m, OCOCH2CH2), 2.35 (2 H, t, J = 7.5 Hz, OCOCH2); 13C NMR (101 MHz, CDCl3) δ 14.2, 19.8, 22.8, 24.8, 27.1, 27.2, 29.2, 29.4, 29.5, 29.6, 29.8, 30.0, 30.1, 32.0, 32.8, 34.0, 37.2, 179.5; HRMS calcd for C18H36O2 [M + H]+ 285.2749, found 285.2747.
From alkene 4. HG-II (2.80 mg, 4 mol%) was added to a solution of 4 (25 mg, 0.114 mmol) and heptenoic acid (59 mg, 0.334 mmol) in dry CH2Cl2 (1.14 ml) and stirred for 24 h under reflux. The reaction mixture was concentrated under reduced pressure. EtOAc (5.0 ml) was added to the residue and the solution was subjected to hydrogenation under high pressure (37.5 bar) in a Buchi® hydrogenator overnight at room temperature after which solvent was removed under reduced pressure. Flash chromatography (2% Et2O/petroleum spirits) of the crude mixture afforded 2 as a colorless oil (25 mg, 73%).
Synthesis of diacylglycerols
:pentane) to afford the title compound as a colorless thick liquid (178 mg, 77%); [α]25D +6.2 (c = 0.5, CHCl3), [α]23D lit.19 +5.45 (c = 3.9, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.83 (3 H, d, J = 6.5 Hz, CCH3 (TBSA)), 0.88 (6 H, t, J = 6.8 Hz, 2 × CH3), 1.07 (2 H, m, CH2 (TBSA)), 1.25 (48 H, m, 24 × CH2), 1.60 (4 H, m, 2 × COCH2CH2), 2.25–2.34 (4 H, m, 2 × COCH2), 3.59 (2 H, d, J = 5.2 Hz, H3a,3b), 4.19 (1 H, dd, J = 11.9, 6.4 Hz, H1a), 4.34 (1 H, dd, J = 11.9, 3.8 Hz, H1b), 4.50–4.58 (2 H, m, CH2Ph), 5.24 (1 H, m, H2), 7.27–7.37 (5 H, m, Ph); 13C NMR (125 MHz, CDCl3) δ 14.2, 19.8, 22.8, 25.0, 25.1, 27.2, 29.2, 29.2, 29.4, 29.5, 29.6, 29.6, 29.8, 30.1, 30.1, 32.0, 32.9, 34.2, 34.4, 37.2, 62.8, 68.4, 70.1, 73.4, 127.7, 127.9, 128.5, 137.8 (Ph), 173.2, 173.5 (CO); HRMS: calcd for C45H80O5 [M + H]+ 701.6039, found 701.6038.
O); HRMS: calcd for C44H78O5 [M + H]+ 687.5883, found 687.5884.
Synthesis of phosphatidylglycerols
O); 31P NMR (202 MHz, CDCl3) δ 148.8; HRMS-ESI: [M + H]+ calcd for C51H95O6NP 848.6897; found 848.6885.
38 (10.7 mg, 0.0392 mmol) in dry CH2Cl2 (2 ml) at 0 °C under nitrogen. The reaction mixture was stirred for 15 min at 0 °C and 2 hours at room temperature. The reaction mixture was cooled to 0 °C and t-BuOOH (5 M in decane) (5 μl, 0.0582 mmol) was added and stirred for 2 h warming to room temperature. Excess oxidant was quenched with sodium metabisulfite (1 M, 0.1 ml), the reaction mixture was diluted with CH2Cl2, extracted, dried (MgSO4), filtered and concentrated. The residue was purified by column chromatography on silica gel (35% EtOAc/petroleum ether, 0.5% Et3N) to afford the title compound as an oil (25 mg, 55%); [α]24D 6.3 (c 0.25, CHCl3); 1H NMR (600 MHz, CDCl3) δ 0.83 (3 H, d, J = 6.6 Hz, CCH3 (TBSA)), 0.88 (6 H, t, J = 6.9 Hz, 2 × CH3 (TBSA, palmitoyl)), 1.06–1.07 (2 H, m, CH2), 1.26 (49 H, m, 24 × CH2, 1 × CH), 1.57 (4 H, m, 2 × COCH2CH2), 2.26 (4 H, m, 2 × COCH2), 3.56 (2 H, t, J = 5.6 Hz, H3′a,3′b), 3.77 (1 H, m, H2′), 4.03–4.16 (4 H, m, H3a,3b,1′a,1′b), 4.23 (2 H, m, H1a,1b), 4.51 (2 H, d, J = 4.8 Hz, CH2Ph), 4.60–4.67 (2 H, m, CH2Ph), 5.02–5.05 (2 H, m, CH2Ph), 5.16 (1 H, m, H2′), 7.32 (15 H, m, Ph); 13C NMR (126 MHz, CDCl3) δ 14.2, 19.8, 22.8, 24.9, 27.2, 29.2, 29.2, 29.4, 29.5, 29.6, 29.7, 29.8, 30.1, 30.2, 32.0, 32.9, 34.1, 34.2, 37.2, 61.8, 65.5, 67.3, 67.3, 69.1, 69.4, 69.4, 69.5, 69.6, 72.3, 73.6, 127.7, 127.8, 127.9, 128.0, 128.5, 128.5, 128.7 (Ph), 172.9, 173.3 (CO); 31P NMR (202 MHz, CDCl3) δ −1.01, −0.98; HRMS: [M + H]+ calcd for C62H99O10P, 1035.7009; found 1035.7006.
:methanol (5 ml, 5:1) was stirred under hydrogen (20 bar) for 4 h at room temperature. The reaction mixture is filtered through a short plug of Celite to remove the catalyst. Triethylamine (5 μl) was added to the filtrate and solvent removed under reduced pressure. The resulting residue was purified by flash chromatography (20% methanol:chloroform) to afford 1 as a colorless oil (9 mg, 98%); [α]24D +5.8 (c 0.25, CHCl3); 1H NMR (500 MHz, CD3OD:CDCl3 (1:2)) δ 0.80 (3 H, d, J = 6.6 Hz, CCH3 (TBSA)), 0.85 (6 H, t, J = 6.8 Hz, 2 × CH3 (TBSA, palmitoyl)), 1.03–1.06 (2 H, m, CH2), 1.17–1.36 (62 H, m, 26 × CH2, 1 × CH, HN(CH2CH3)3), 1.57 (4 H, m, 2 × COCH2CH2), 2.29 (4 H, dd, J = 15.6, 8.0 Hz, 2 × COCH2), 3.11 (4 H, q, J = 7.3 Hz, NH(CH2CH3)3) 3.58 (2 H, s, H3′a,3′b), 3.76 (1 H, s, H2′), 3.90 (2 H, s, H1′a,1′b), 3.98 (2 H, s, H3a,3b), 4.15 (1 H, dd, J = 11.8, 6.5 Hz, H1b), 4.38 (1 H, d, J = 12.0 Hz, H1a), 5.20 (1 H, s, H2); 13C NMR (126 MHz, CD3OD:CDCl3 (1:2)) δ 14.2, 19.9, 23.0, 25.2, 25.2, 27.4, 29.5, 29.5, 29.7, 29.9, 29.9, 30.0, 30.3, 30.4, 32.3, 33.1, 34.4, 34.6, 37.4, 46.8, 62.9, 64.2, 67.1, 70.7, 71.4 (glycerol C), 174.0, 174.3 (CO); 31P NMR (202 MHz, CD3OD:CDCl3 (1:2)) δ 4.78; HRMS: [M + H]+ calcd for C47H96NO10P 866.6805; found 866.6806.
O); 31P NMR (202 MHz, CDCl3) δ 148.8; HRMS-ESI: [M + H]+ calcd for C50H92NO6P 834.6696; found 834.6696.
O); 31P NMR (202 MHz, CDCl3) δ 148.0; HRMS-ESI: [M + H]+ calcd for C50H92NO6P 834.6696; found 834.6696.
38 (9.4 mg, 0.034 mmol) in dry CH2Cl2 (2 ml) at 0 °C under nitrogen. The reaction mixture was stirred for 15 min at 0 °C and 2 h at room temperature. The reaction mixture was cooled to 0 °C and t-BuOOH (5 M in decane) (4.1 μl, 0.042 mmol) was added to the reaction mixture and stirred for 2 h with warming to room temperature. Excess oxidant was quenched with sodium metabisulfite (1 M, 0.1 ml), the reaction mixture was diluted with CH2Cl2, extracted, dried (MgSO4), filtered and concentrated. The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel (35% EtOAc/petroleum ether, 0.5% Et3N) which resulted in a 1:1 mixture the product and unreacted dibenzyl glyercol, which was directly subjected to hydrogenolysis.
O); 31P NMR (202 MHz, CDCl3) δ −1.01, −0.98; HRMS: [M + H]+ calcd for C61H97O10P 1021.6853; found 1021.6852.
:CDCl3 (1:2)) δ 0.84 (6 H, t, J = 6.4 Hz, 2 × CH3), 1.10–1.14 (9 H, m, HN(CH2CH3)3), 1.17–1.36 (56 H, m, 28 × CH2), 1.52 (4 H, s, 2 × COCH2CH2), 2.23 (4 H, dd, J = 14.5, 7.2 Hz, 2 × COCH2), 3.46–3.61 (6 H, s, H3′a,3′b, HN(CH2CH3)3), 3.65–3.73 (1 H, m, H2′), 3.83–3.90 (4 H, m, H1′a,1′b,3a,3b), 3.95 (2 H, m, H3a,3b), 4.11 (1 H, dd, J = 12.0, 6.6 Hz, H1b), 5.16 (1 H, s, H2); 13C NMR (126 MHz, CD3OD:CDCl3 (1:2)) δ 14.2, 23.0, 25.2, 25.2, 29.5, 29.5, 29.7, 29.7, 29.9, 30.0, 30.0, 32.3, 34.4, 34.6, 46.5, 62.8, 63.0, 63.7, 66.8, 70.8, 70.9, 71.5, 72.7, 174.0, 174.4; 31P NMR (202 MHz, CDCl3) δ 4.78; HRMS: [M + H]+ calcd for C40H79O10P 751.5444; found 751.5444.
:CDCl3 (1:2)) δ 0.84 (6 H, t, J = 6.4 Hz, 2 × CH3), 1.17–1.36 (56 H, m, 28 × CH2), 1.57 (4 H, s, 2 × COCH2CH2), 2.29 (4 H, dd, J = 14.5, 7.2 Hz, 2 × COCH2), 3.57 (2 H, s, H3′a,3′b), 3.70–3.78 (1 H, m, H2′), 3.84–3.90 (2 H, s, H1′a, H1′b), 3.95 (2 H, m, H3a,3b), 4.15 (1 H, dd, J = 11.9, 6.7 Hz, H1b), 4.37 (1 H, m, H1a), 5.20 (1 H, s, H2); 13C NMR (126 MHz, CD3OD:CDCl3 (1:2)) δ 14.2, 23.0, 25.2, 25.2, 29.5, 29.5, 29.7, 29.7, 29.9, 30.0, 30.0, 32.3, 34.4, 34.6, 46.5, 62.8, 63.0, 63.7, 66.8, 70.8, 70.9, 71.5, 72.7, 174.0, 174.4; 31P NMR (202 MHz, CDCl3) δ 5.25; HRMS: [M + H]+ calcd for C40H79O10P 751.5444; found 751.5444.
Collision-induced dissociation mass spectrometry of phosphatidylglycerols
Characterization of lipid ions, introduced to a Q Exactive Plus Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) via nanoESI (nESI) using an Advion Triversa Nanomate (Advion, Ithaca, NY), was performed in negative ionization mode by higher-energy collision induced dissociation (HCD)-tandem mass spectrometry (MS/MS). Deprotonated precursor ions were mono-isotopically isolated using a window of ±0.5 m/z. Product ion spectra were acquired using a mass resolving power of 70000. Spectra shown are the average of 100 scans.
Conflicts of interest
There are no conflicts to declareAcknowledgements
We acknowledge the Australian Research Council for financial support (DP160100597). Prof. Gavin Reid and Mengxuan Fang are thanked for assistance with the mass spectrometric analysis of PGs.References
- S. Furse, J. Chem. Biol., 2017, 10, 1–9 CrossRef PubMed.
- S. E. Horvath and G. Daum, Prog. Lipid Res., 2013, 52, 590–614 CrossRef CAS PubMed.
- I. Van Rhijn, T. van Berlo, T. Hilmenyuk, T. Y. Cheng, B. J. Wolf, R. V. Tatituri, A. P. Uldrich, G. Napolitani, V. Cerundolo, J. D. Altman, P. Willemsen, S. Huang, J. Rossjohn, G. S. Besra, M. B. Brenner, D. I. Godfrey and D. B. Moody, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 380–385 CrossRef CAS PubMed.
- R. V. Tatituri, G. F. Watts, V. Bhowruth, N. Barton, A. Rothchild, F. F. Hsu, C. F. Almeida, L. R. Cox, L. Eggeling, S. Cardell, J. Rossjohn, D. I. Godfrey, S. M. Behar, G. S. Besra, M. B. Brenner and M. Brigl, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1827–1832 CrossRef CAS PubMed.
- B. J. Wolf, R. V. Tatituri, C. F. Almeida, J. Le Nours, V. Bhowruth, D. Johnson, A. P. Uldrich, F. F. Hsu, M. Brigl, G. S. Besra, J. Rossjohn, D. I. Godfrey and M. B. Brenner, J. Immunol., 2015, 195, 2540–2551 CrossRef CAS PubMed.
- E. Maloney, D. Stankowska, J. Zhang, M. Fol, Q. J. Cheng, S. Lun, W. R. Bishai, M. Rajagopalan, D. Chatterjee and M. V. Madiraju, PLoS Pathog., 2009, 5, e1000534 Search PubMed.
- M. Jackson, D. C. Crick and P. J. Brennan, J. Biol. Chem., 2000, 275, 30092–30099 CrossRef CAS PubMed.
- P. V. Sarma, L. Srikanth, K. Venkatesh, P. S. Murthy and P. U. Sarma, Bioinformation, 2013, 9, 690–695 CrossRef PubMed.
- H. Okuyama, T. Kankura and S. Nojima, J. Biochem., 1967, 61, 732–737 CrossRef CAS PubMed.
- B. ter Horst, C. Seshadri, L. Sweet, D. C. Young, B. L. Feringa, D. B. Moody and A. J. Minnaard, J. Lipid Res., 2010, 51, 1017–1022 CrossRef CAS PubMed.
- F. F. Hsu, J. Turk, R. M. Owens, E. R. Rhoades and D. G. Russell, J. Am. Soc. Mass Spectrom., 2007, 18, 479–492 CrossRef CAS PubMed.
- J. Rossjohn, S. Gras, J. J. Miles, S. J. Turner, D. I. Godfrey and J. McCluskey, Annu. Rev. Immunol., 2015, 33, 169–200 CrossRef CAS PubMed.
- M. V. Dhodapkar and V. Kumar, J. Immunol., 2017, 198, 1015–1021 CrossRef CAS PubMed.
- F. S. Prout, J. Cason and A. W. Ingersoll, J. Am. Chem. Soc., 1948, 70, 298–305 CrossRef CAS.
- S. Mekala and R. C. Hahn, J. Org. Chem., 2015, 80, 1610–1617 CrossRef CAS PubMed.
- R. P. Linstead, J. C. Lunt and B. C. L. Weedon, J. Chem. Soc., 1951, 1130–1132 RSC.
- B. S. Dyer, J. D. Jones, G. D. Ainge, M. Denis, D. S. Larsen and G. F. Painter, J. Org. Chem., 2007, 72, 3282–3288 CrossRef CAS PubMed.
- P. Fodran and A. J. Minnaard, Org. Biomol. Chem., 2013, 11, 6919–6928 CAS.
- X. Liu, B. L. Stocker and P. H. Seeberger, J. Am. Chem. Soc., 2006, 128, 3638–3648 CrossRef CAS PubMed.
- P. S. Patil, T.-J. R. Cheng, M. M. L. Zulueta, S.-T. Yang, L. S. Lico and S.-C. Hung, Nat. Commun., 2015, 6, 7239 CrossRef CAS PubMed.
- B. Cao, X. Chen, Y. Yamaryo-Botte, M. B. Richardson, K. L. Martin, G. N. Khairallah, T. W. Rupasinghe, R. M. O'Flaherty, R. A. O'Hair, J. E. Ralton, P. K. Crellin, R. L. Coppel, M. J. McConville and S. J. Williams, J. Org. Chem., 2013, 78, 2175–2190 CrossRef CAS PubMed.
- I. O. Roberts and M. S. Baird, Chem. Phys. Lipids, 2006, 142, 111–117 CrossRef CAS PubMed.
- P. D'Arrigo, L. de Ferra, G. Pedrocchi-Fantoni, D. Scarcelli, S. Servi and A. Strini, J. Chem. Soc., Perkin Trans. 1, 1996, 2657–2660 RSC; R. Sato, Y. Itabashi, H. Fujishima, H. Okuyama and A. Kuksis, Lipids, 2004, 39, 1025–1030 CrossRef CAS PubMed.
- P. Woolley and H. Eibl, Chem. Phys. Lipids, 1988, 47, 55–62 CrossRef CAS; R. Wohlgemuth, N. Waespe-Sarcevic and J. Seelig, Biochemistry, 1980, 19, 3315–3321 CrossRef PubMed; E. Baer and D. Buchnea, J. Biol. Chem., 1958, 232, 895–902 Search PubMed; R. M. Saunders and H. P. Schwarz, J. Am. Chem. Soc., 1966, 88, 3844–3847 CrossRef PubMed.
- M. H. Caruthers, A. D. Barone, S. L. Beaucage, D. R. Dodds, E. F. Fisher, L. J. McBride, M. Matteucci, Z. Stabinsky and J.-Y. Tang, Methods Enzymol., 1987, 154, 287–313 CAS.
- G. D. Prestwich, Acc. Chem. Res., 1996, 29, 503–513 CrossRef CAS.
- S. M. Ali, M. U. Ahmad, P. Koslosky, K. Kasireddy, U. Murali Krishna and I. Ahmad, Tetrahedron, 2006, 62, 6990–6997 CrossRef CAS.
- E. N. Jacobsen, F. Kakiuchi, R. G. Konsler, J. F. Larrow and M. Tokunaga, Tetrahedron Lett., 1997, 38, 773–776 CrossRef CAS.
- C. E. Dreef, C. J. J. Elie, P. Hoogerhout, G. A. van der Marel and J. H. van Boom, Tetrahedron Lett., 1988, 29, 6513–6515 CrossRef CAS.
- N. J. Jensen, K. B. Tomer and M. L. Gross, Lipids, 1987, 22, 480–489 CrossRef CAS PubMed.
- P. B. Smith, A. P. Snyder and C. S. Harden, Anal. Chem., 1995, 67, 1824–1830 CrossRef CAS PubMed.
- F.-F. Hsu and J. Turk, J. Am. Soc. Mass Spectrom., 2000, 11, 986–999 CrossRef CAS PubMed.
- R. V. Tatituri, B. J. Wolf, M. B. Brenner, J. Turk and F. F. Hsu, Anal. Bioanal. Chem., 2015, 407, 2519–2528 CrossRef CAS PubMed.
- W. C. Still, M. Kahn and A. M. Mitra, J. Org. Chem., 1978, 43, 2923–2925 CrossRef CAS.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- S. Front, N. Court, M.-L. Bourigault, S. Rose, B. Ryffel, F. Erard, V. F. J. Quesniaux and O. R. Martin, ChemMedChem, 2011, 6, 2081–2093 CrossRef CAS PubMed.
- G. M. Rankin, B. J. Compton, K. A. Johnston, C. M. Hayman, G. F. Painter and D. S. Larsen, J. Org. Chem., 2012, 77, 6743–6759 CrossRef CAS PubMed.
- N. Chen and J. Xie, J. Org. Chem., 2014, 79, 10716–10721 CrossRef CAS PubMed; C. A. A. van Boeckel, G. M. Visser and J. H. van Boom, Tetrahedron, 1985, 41, 4557–4565 CrossRef.
- Z. J. Wang, N. D. Spiccia, W. R. Jackson and A. J. Robinson, J. Pept. Sci., 2013, 19, 470 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob01786c |
| This journal is © The Royal Society of Chemistry 2017 |