
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
13C-Carbamylation as a mechanistic probe for the inhibition of class D β-lactamases by avibactam and halide ions†
Christopher T.
Lohans
 a,
David Y.
Wang
a,
Christian
Jorgensen
b,
Samuel T.
Cahill
a,
Ian J.
Clifton
a,
Michael A.
McDonough
a,
Henry P.
Oswin
c,
James
Spencer
c,
Carmen
Domene
ab,
Timothy D. W.
Claridge
a,
Jürgen
Brem
*a and
Christopher J.
Schofield
*a
a,
David Y.
Wang
a,
Christian
Jorgensen
b,
Samuel T.
Cahill
a,
Ian J.
Clifton
a,
Michael A.
McDonough
a,
Henry P.
Oswin
c,
James
Spencer
c,
Carmen
Domene
ab,
Timothy D. W.
Claridge
a,
Jürgen
Brem
*a and
Christopher J.
Schofield
*a
aDepartment of Chemistry, University of Oxford, Oxford, OX1 3TA, UK. E-mail: christopher.schofield@chem.ox.ac.uk; jurgen.brem@chem.ox.ac.uk; Fax: +44 (0)1865 285002; Tel: +44 (0)1865 275625
bDepartment of Chemistry, King's College London, London, SE1 1DB, UK
cSchool of Cellular and Molecular Medicine, University of Bristol, Bristol, BS8 1TD, UK
First published on 26th June 2017
Abstract
The class D (OXA) serine β-lactamases are a major cause of resistance to β-lactam antibiotics. The class D enzymes are unique amongst β-lactamases because they have a carbamylated lysine that acts as a general acid/base in catalysis. Previous crystallographic studies led to the proposal that β-lactamase inhibitor avibactam targets OXA enzymes in part by promoting decarbamylation. Similarly, halide ions are proposed to inhibit OXA enzymes via decarbamylation. NMR analyses, in which the carbamylated lysines of OXA-10, -23 and -48 were 13C-labelled, indicate that reaction with avibactam does not ablate lysine carbamylation in solution. While halide ions did not decarbamylate the 13C-labelled OXA enzymes in the absence of substrate or inhibitor, avibactam-treated OXA enzymes were susceptible to decarbamylation mediated by halide ions, suggesting halide ions may inhibit OXA enzymes by promoting decarbamylation of acyl-enzyme complex. Crystal structures of the OXA-10 avibactam complex were obtained with bromide, iodide, and sodium ions bound between Trp-154 and Lys-70. Structures were also obtained wherein bromide and iodide ions occupy the position expected for the ‘hydrolytic water’ molecule. In contrast with some solution studies, Lys-70 was decarbamylated in these structures. These results reveal clear differences between crystallographic and solution studies on the interaction of class D β-lactamases with avibactam and halides, and demonstrate the utility of 13C-NMR for studying lysine carbamylation in solution.
Introduction
The development of antibiotic resistance in bacteria represents a major threat to human health. The β-lactamases enable resistance to β-lactam antibiotics (e.g., penicillins, cephalosporins, and carbapenems) by catalyzing β-lactam hydrolysis.1,2 β-Lactamases are organized into four classes based on sequence homology, structure, and enzyme mechanism.3 The serine β-lactamases (SBLs; classes A, C, and D) employ a mechanism involving a nucleophilic serine residue, whilst the metallo-β-lactamases (MBLs; class B) use one or two active site zinc ions to activate β-lactam substrates and water to enable hydrolysis.The class D serine β-lactamases have proliferated extensively, and represent the largest β-lactamase class with >400 variants described.4,5 Some OXA variants impart resistance to late generation β-lactam antibiotics, including carbapenems.6 OXA enzymes are associated with clinically relevant Gram-negative bacteria,6 and the production of OXA enzymes by Gram-positive bacteria has been recently described.5
The first crystal structure of OXA-10 revealed a similar overall fold to class A and C β-lactamases;7 a subsequent OXA-10 structure showed that Lys-70 was carbamylated.8,9 Lys-70 is located in a hydrophobic pocket, which is proposed to lower the pKa of the protonated lysine ε-amino group.10 OXA-10 lysine carbamylation is stabilised by hydrogen bonds between the carbamyl group and the nucleophilic serine (Ser-67), the side chain of Trp-154, and a water molecule.8 In addition to their use of a carbamate in catalysis, OXA enzymes are unusual in that they are inhibited by halide ions.11
Carbamylated Lys-70 acts as a general acid/base in the two-step mechanism by which OXA enzymes catalyze β-lactam hydrolysis (Fig. 1).10 In the acylation step, the nucleophilic serine reacts with the substrate β-lactam, leading to formation of an acyl-enzyme intermediate (Fig. 1A). Nucleophilic attack of water onto the acylated serine residue then produces the hydrolyzed β-lactam (Fig. 1B). The carbamylated lysine of the OXA enzymes is proposed to be involved in both steps, deprotonating the nucleophilic serine and the ‘hydrolytic’ water.12
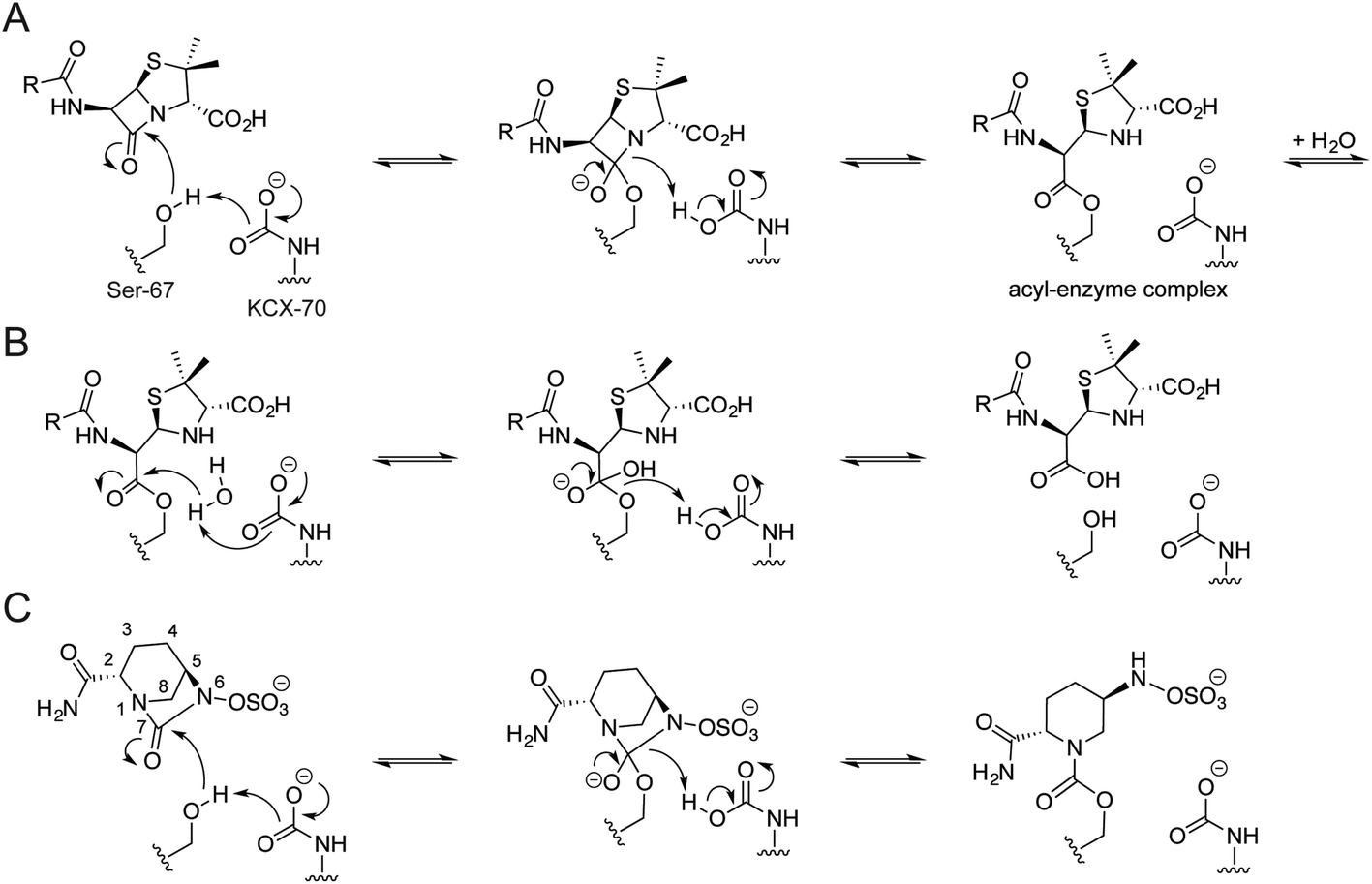 | ||
| Fig. 1 Outline catalytic mechanism for the class D OXA β-lactamases. Proposed mechanism for the (A) acylation and (B) hydrolysis steps of OXA catalysis with a penicillin substrate, and (C) the reversible acylation reaction of avibactam with an OXA enzyme. Residue numbering is based on OXA-10; the carbamylated lysine is indicated (KCX). | ||
β-Lactamase inhibitors are used clinically to preserve and extend the utility of β-lactam antibiotics. The recently approved β-lactamase inhibitor avibactam has a diazabicyclooctane core, unlike previous generations of inhibitors which contain a β-lactam ring (Fig. S1†).13–15 Avibactam prevents substrate hydrolysis by acylating the nucleophilic serine (Fig. 1C), and is released from the enzyme either by irreversible hydrolysis or reversible reformation of avibactam (Fig. S2†).13,16 Importantly, the hydrolysis of avibactam by OXA enzymes is disfavoured.17 Crystallographic analyses have led to the proposal that OXA-bound avibactam disfavours carbamylation, rationalising its disfavoured hydrolysis.18
Protein carbamylation may be characterised in solution using NMR spectroscopy. As lysine carbamylation is reversible, the carbamylated residue can be site-specifically labelled using 13C-labelled bicarbonate.10,19,20 Efficient 13C incorporation can be achieved by dialysing the OXA enzyme against a low pH buffer (to effect decarbamylation), then dialysing against a higher pH buffer containing NaH13CO3.10,21 Despite the potential utility of a selectively introduced NMR active nucleus in the OXA active site, this approach has been little used in mechanistic and inhibition studies.9
During ongoing investigations into the interactions of avibactam with the different classes of β-lactamases,15,22 we developed an assay for screening OXA enzyme inhibitors.23 While previous work suggests that carbamylation promotes deacylation of the OXA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :avibactam complex,17 we found that the inhibition of OXA-23 and OXA-48 by avibactam is largely independent of the concentration of added sodium bicarbonate. However, as indicated above, crystallographic studies have led to the proposal that reaction with avibactam causes decarbamylation of OXA enzymes and so contributes to the extent of inhibition.18,24
:avibactam complex,17 we found that the inhibition of OXA-23 and OXA-48 by avibactam is largely independent of the concentration of added sodium bicarbonate. However, as indicated above, crystallographic studies have led to the proposal that reaction with avibactam causes decarbamylation of OXA enzymes and so contributes to the extent of inhibition.18,24
To clarify the role of lysine carbamylation in avibactam inhibition, we 13C-labelled clinically relevant OXA enzymes and used 13C-NMR spectroscopy to investigate the impact of avibactam. In combination with X-ray crystallographic and kinetic analyses, as well as modelling studies, the NMR results contribute to a detailed understanding of the mechanism of avibactam inhibition. The results also inform on the interplay between halides and OXA lysine carbamylation, and clarify the mechanism by which halides inhibit the OXA enzymes.
Experimental
13C labelling of OXA enzymes
OXA-10, OXA-23, and OXA-48 were produced and purified as described,25,26 and 13C labelled according to the reported protocol.10 Purified enzyme (Fig. S3†) was dialyzed (10000 MWCO) overnight against degassed 25 mM sodium acetate (pH 4.5), then overnight against 50 mM sodium phosphate (pH 7.4), 1 mM NaH13CO3 (Sigma-Aldrich). Enzymes were then dialyzed overnight against 50 mM sodium phosphate (pH 7.4), 10 mM NaH13CO3, aliquoted, and frozen in liquid N2. The buffers used for OXA-10 dialysis were supplemented with 0.1 mM ethylenediaminetetraacetic acid (EDTA).
Fluorometric kinetic assays
Kinetic and inhibition analyses and UV-vis studies were performed using a fluorogenic assay monitoring FC5 hydrolysis (5 μM) by OXA-10 (1 nM), OXA-23 (12.5 nM), and OXA-48 (12.5 nM), adapted from a previous protocol for OXA-10.23,27 Inhibitors and enzymes were pre-incubated for 0, 10, 30, 60 and 360 minutes in the appropriate buffer (either 50 mM MES, pH 6.0, 100 mM sodium phosphate, pH 7.4, or 50 mM Tris phosphate, pH 8.5, supplemented with 0.01% Triton X-100, with and without 50 mM sodium bicarbonate). The inhibitory activity of iodide was studied using OXA-23 (5 nM), FC5 (from 781 nM to 100 μM) and NaI (from 500 μM to 250 mM), in 100 mM sodium phosphate, pH 7.4, 50 mM sodium bicarbonate and 0.01% Triton X-100.NMR spectroscopy
13C-NMR spectra were acquired using a Bruker AVIII HD 600 spectrometer equipped with a BB-F/1H Prodigy N2 cryoprobe. 13C spectra consisted of 2048 scans, using a 2.0 s relaxation delay, and were processed with a 10 Hz line broadening. Unless stated otherwise, samples consisted of OXA enzyme (0.6 to 0.8 mM) prepared in 50 mM sodium phosphate, pH 7.4, 10 mM NaH13CO3 with 10% D2O; OXA-10 samples were supplemented with 0.1 mM EDTA. Avibactam was tested at a concentration of 15 mM. 1H NMR datasets were acquired on a Bruker Avance III 700 MHz spectrometer with a TCI inverse cryoprobe. The 1H spectra used 16 scans and a 2.0 s relaxation delay. All experiments were measured at 298 K.The impact of OXA-10, OXA-23 and OXA-48 on avibactam was monitored by incubating 400 μM avibactam and 150 nM enzyme, with or without 10 mM NaHCO3, in 50 mM Tris-d11, pH 7.5, 10% D2O at room temperature for 4 h. The impact of avibactam on the activity of OXA-10, OXA-23 and OXA-48 was monitored using 75 nM enzyme, 100 μM or 10 mM avibactam and 4 mM ampicillin in 50 mM Tris-d11 pH 7.5, 10% D2O. Ampicillin was either added directly to the enzyme avibactam mixture, or following 1 h pre-incubation. The impact of fluoride, chloride, bromide and iodide on ampicillin hydrolysis was measured similarly. Reactions consisted of 75 nM enzyme, 50 mM or 500 mM of sodium halide, and 10 mM ampicillin in 50 mM Tris-d11 pH 7.5, 10% D2O.
X-ray crystallography
OXA-10 was buffer exchanged into 50 mM sodium phosphate, pH 7.4 (with or without 50 mM NaHCO3), and sitting drops were set up using the PACT premier broad screen (Molecular Dimensions). The structures published herein used the following conditions for crystallisation: PDB 5MMY – 0.1 M HEPES, pH 7.0, 20% PEG 6000; PDB 5MOX – 0.2 M sodium citrate tribasic dihydrate, 0.1 M Bis–Tris propane, pH 7.5, 20% w/v PEG 3350; PDB 5MNU – 0.2 M NaBr, 0.1 M Bis–Tris propane, pH 6.5, 20% w/v PEG 3350; PDB 5MOZ – 0.2 M NaI, 20% w/v PEG 3350. Crystals were cryoprotected using well solution diluted to 25% (v/v) glycerol before being flash cooled in liquid nitrogen. Data for OXA-10 were collected at Diamond Light Source synchrotron beamline I04 at 100 K. Data were indexed, integrated and scaled using Mosflm and Scala, respectively.28 The OXA-10 structures were solved by molecular replacement using Phaser29 using PDB 5FQ927 as a search model. Iterative cycles of fitting and refinement continued using COOT30 and PHENIX28 until Rwork and Rfree no longer converged. Data collection and refinement statistics are provided in Table S2.†Results and discussion
Kinetic characterization
To study the inhibition of OXA enzymes by avibactam, we developed assays employing nitrocefin and FC5 as substrates (Table S1†).23,27 We chose to work with OXA-10, a well-characterised enzyme first identified in Pseudomonas isolates,6 as well as OXA-23 and OXA-48 that have broader substrate scopes and which are of clinical importance.6 In the absence of added sodium bicarbonate, OXA-23 and OXA-48 were strongly inhibited by avibactam, while relatively poor OXA-10 inhibition was observed (Table 1, Fig. S4†). While the addition of sodium bicarbonate (50 mM) did not substantially impact on avibactam-mediated inhibition of OXA-23 and OXA-48 (Table 1), the extent of OXA-10 inhibition was increased by >100 fold. Furthermore, the extent of inhibition for the three OXA enzymes was enhanced according to the extent of pre-incubation with avibactam (Table 1).| Avibactam IC50 (μM) | ||||
|---|---|---|---|---|
| 0 min | 10 min | 30 min | 60 min | |
| OXA-10 (no NaHCO3) | >100 | 19.7 | 11.18 | 10.01 |
| OXA-10 (50 mM NaHCO3) | 0.4452 | 0.1735 | 0.0424 | 0.0217 |
| OXA-23 (no NaHCO3) | 0.544 | 0.0535 | 0.0175 | 0.0362 |
| OXA-23 (50 mM NaHCO3) | 1.781 | 0.0870 | 0.170 | 0.0870 |
| OXA-48 (no NaHCO3) | 1.624 | 0.550 | 0.123 | 0.0601 |
| OXA-48 (50 mM NaHCO3) | 0.593 | 0.496 | 0.141 | 0.0566 |
The kinetics governing the interaction of OXA-10 and OXA-48 with avibactam have been previously examined.17 The poor inhibition of OXA-10 by avibactam may reflect a relatively slow reaction rate, as compared to OXA-48 (k2/Ki values of 1.1 ± 0.1 × 101 M−1 s−1 and 1.4 ± 0.1 × 103 M−1 s−1, respectively).17 Given that OXA-48 is likely fully acylated within 5 min in these conditions,17 the continuing decrease in IC50 after 30 min pre-incubation was unexpected. Potential enzymatic reaction of the avibactam complex to a more stable inhibitory complex is possible, but as yet there is no evidence for this. No OXA-catalysed degradation of avibactam was observed by 1H-NMR spectroscopy within limits of detection (Fig. S5†).17
The potential impact of lysine carbamylation on the time dependent effect was next considered. While addition of bicarbonate did not change the extent of OXA-23 and OXA-48 inhibition by avibactam, it did increase the extent of OXA-10 inhibition (Table 1). Sodium bicarbonate has previously been observed to promote deacylation of the avibactam complex with OXA-10 by recyclisation.17 Taken together, these results suggest that the reaction rate of avibactam with OXA-10 is more sensitive to the concentration of carbon dioxide, and, thus to lysine carbamylation, than are either OXA-23 or OXA-48.
Impact of avibactam on OXA carbamylation
To monitor the interplay between carbamylation and the interaction with avibactam, we 13C-labelled the carbamylated lysine residue of the three OXA variants.10 By 13C-NMR, all three showed signals consistent with lysine 13C-carbamylation (Fig. 2A).10 Labelled OXA-23 was buffer exchanged to remove most of the labelled bicarbonate/carbon dioxide, resulting in loss of signal; the 13C-signal was restored upon adding more 13C-bicarbonate (Fig. 2B). Titration of OXA-48 with 13C-bicarbonate showed labelling was saturated with only a moderate excess (e.g., 10:1) of 13C-bicarbonate (Fig. 2C). The impact of pH on the extent of carbamylation of OXA-23 was then examined (Fig. 2B). In the presence of an excess of 13C-bicarbonate (10 mM), the 13C-carbamylated lysine peak intensity was greater at higher pH. When the majority of the 13C-bicarbonate was removed by buffer exchange, the trend was reversed (Fig. 2B), i.e., carbamylation was enhanced in a lower pH buffer, possibly due to the pH-dependence of the equilibrium between bicarbonate and carbon dioxide.31
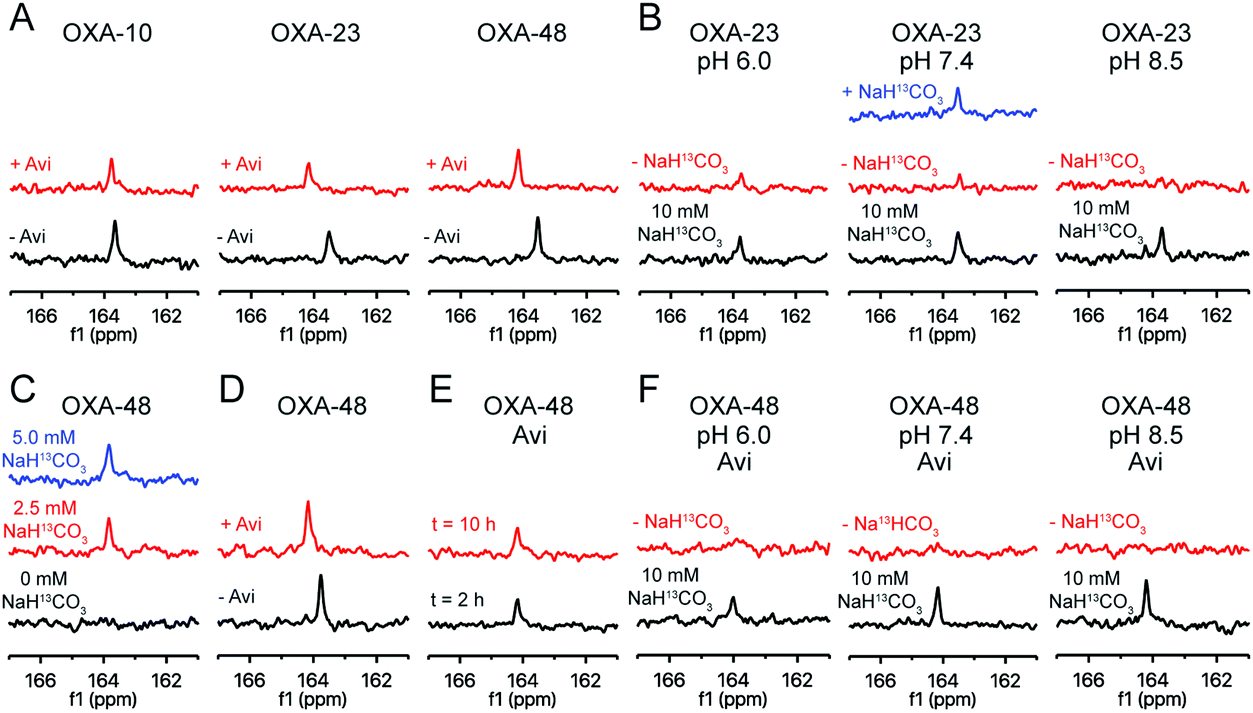 | ||
| Fig. 2 13C-NMR reveals the impact of avibactam on OXA carbamylation status. (A) 13C-NMR (150 MHz) spectra for OXA-10, OXA-23 and OXA-48 labelled with 10 mM NaH13CO3 (black), and the impact of avibactam addition (15 mM; red) at pH 7.4. (B) Impact of pH on 13C-labelling of OXA-23 in 10 mM NaH13CO3 before (black) and after buffer exchange which removes most of the NaH13CO3 (red). The impact of adding further NaH13CO3 (final concentration 9.3 mM) following buffer exchange was tested at pH 7.4 (blue). (C) Impact of NaH13CO3 concentration on OXA-48 carbamylation, starting with unlabelled enzyme (black), 2.5 mM NaH13CO3 (red), and 5 mM NaH13CO3 (blue) at pH 7.4. (D) Spectra resulting from incubation of OXA-48 with avibactam (15 mM; red) or buffer (black) prior to adding NaH13CO3 (10 mM). (E) Impact of avibactam (15 mM) on carbamylation after 2 h (black) and 10 h incubation (red). (F) Impact of pH on 13C-labelling of the avibactam OXA-48 complex (15 mM avibactam) in 10 mM NaH13CO3 (black), or after buffer exchange which removes most of the NaH13CO3 (red). | ||
Addition of avibactam to the 13C-labelled OXA enzymes in the presence of 10 mM 13C-bicarbonate resulted in a decrease (5–20%) in the 13C signal, which was shifted by 0.2 to 0.6 ppm (Fig. 2A). As all three enzymes are likely fully reacted with avibactam under the conditions used,17 this suggests that reaction with avibactam decreases, but does not ablate the extent of lysine carbamylation. 13C-Bicarbonate (10 mM) was added to decarbamylated OXA-48 which had been pre-incubated with avibactam (Fig. 2D); the resulting 13C-carbamylated lysine peak was of a similar intensity to that for OXA-48 without avibactam, demonstrating lysine carbamylation is reversible following reaction with avibactam. The impact of avibactam on the carbamylation status of OXA-48 was stable, with no change observed over 10 hours (Fig. 2E).
The impact of avibactam on OXA-48 carbamylation was explored at different pH values (Fig. 2F). In the presence of 10 mM 13C-bicarbonate, the 13C signal decreased at lower pH values. When excess 13C-bicarbonate was removed via buffer exchange, no signal was observed for avibactam-treated OXA at any pH tested. Collectively, these results suggest that avibactam destabilizes OXA enzyme carbamylation. This may arise through the loss of a hydrogen bond donor upon carbamoylation of Ser-67 with avibactam, by steric effects, or by an increase in the lysine side chain pKa due to dipolar interactions with avibactam.24
These results do not explain the impact of sodium bicarbonate on the inhibition of OXA-10 by avibactam when compared to OXA-23 and OXA-48 (see above). The 13C-NMR data suggest that OXA-10 is carbamylated with only a mild (10:1) excess of bicarbonate, and is likely fully carbamylated in the absence of added bicarbonate at the enzyme concentrations (<20 nM) used for the inhibition assays.
Impact of halide ions on OXA carbamylation
Like avibactam,18 halide ions are proposed to hinder carbamylation of OXA enzymes.11,32 To examine the impact of halide ions on lysine carbamylation, 13C-labelled OXA-23 was treated with NaF, NaCl, NaBr, and NaI (Fig. 3). No decarbamylation was observed at 50 mM halide, while only 500 mM fluoride shifted and broadened the signal, potentially due to a pH effect. Unexpectedly, the extent of carbamylation was enhanced in the presence of 50 mM halide (Fig. 3).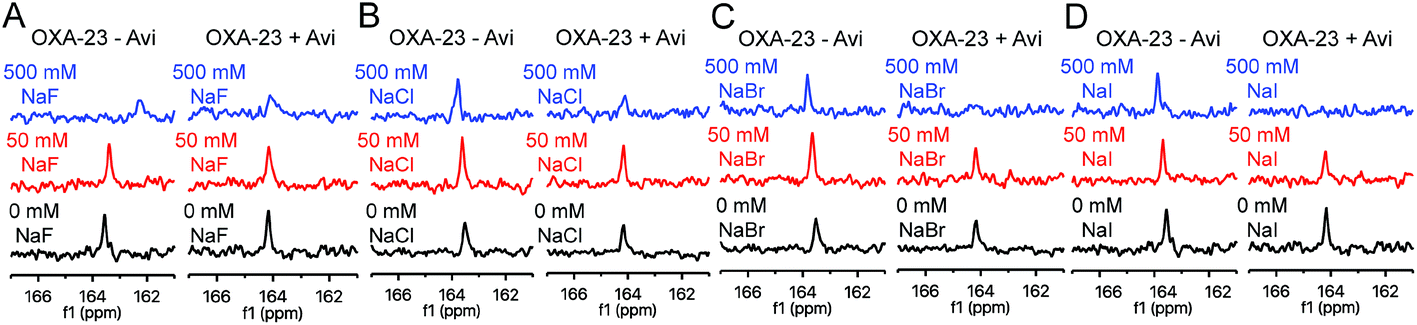 | ||
| Fig. 3 13C-NMR solution studies on the impact of halide ions on OXA lysine carbamylation status. 13C-NMR (150 MHz) spectra showing the effect of increasing (A) sodium fluoride, (B) sodium chloride, (C) sodium bromide, and (D) sodium iodide concentrations (black, 0 mM; red, 50 mM; blue, 500 mM) on lysine carbamylation of OXA-23 in the absence and in the presence of avibactam (15 mM). | ||
To investigate whether reaction of the nucleophilic serine changes the impact of halides on carbamylation, OXA-23 was treated with avibactam and 50 mM or 500 mM of the sodium halides (Fig. 3). At 50 mM, sodium fluoride and sodium chloride slightly enhanced carbamylation, sodium bromide had no effect, and sodium iodide decreased the extent of carbamylation. At 500 mM, a decrease in signal was observed, with the extent of decarbamylation increasing with the halide size. A similar trend was observed for the inhibitory activity of halide ions on OXA-10 (Fig. S12†). Analysis of the kinetics of inhibition of OXA-23 by iodide using FC5 as a reporter substrate suggests a mixed mode of inhibition (IC50 = 75.2 mM; Ki = 34 mM; Fig. S13†). This is consistent with the (at least) dual effects of halides promoting carbamylation in the absence of Ser-70 modification, but decreasing carbamylation in the avibactam-modified complex. The sensitivity of OXA-40 to halide ions is likely influenced by a phenylalanine residue located on an active site loop; replacement of this residue with tyrosine results in increased susceptibility to inhibition by halide ions.33 As OXA-23 has a phenylalanine in this position (i.e., Phe-152), it may be expected to be relatively insensitive to halide ions. Our 13C-NMR results suggest that OXA-10, which has a tyrosine residue at this position, is more easily decarbamylated by sodium bromide than OXA-23 (Fig. S14†). Preliminary experiments suggest that alkali metal ions (e.g., sodium, potassium) do not impact on OXA carbamylation (Fig. S15†).
Previous crystallographic studies show chloride and iodide ions bound between decarbamylated Lys-70 and Trp-154 of OXA-10 V117T (PDB 2WGV)32 and OXA-163 (PDB 4S2M)11 (Fig. S10 and S11†). This observation led to the suggestion that halide ions inhibit OXA enzymes via promoting decarbamylation.11,32 However, our NMR data only showed halide ion induced decarbamylation when OXA-23 was treated with avibactam. It is possible that reaction of the nucleophilic serine may destabilize carbamylation such that halide ions cause decarbamylation. Thus, halide ions may inhibit OXA enzymes by selectively promoting decarbamylation of the acyl-enzyme complex during substrate turnover. However, given that crystallography indicates multiple halides bind to the OXA enzymes, other factors may be involved (Fig. S10†).
Crystallography and modelling
To investigate the structural basis for the impact of avibactam and halides on OXA carbamylation, we carried out crystallographic analyses using OXA-10.27 Datasets for crystals obtained from precipitants covering a broad range of pH values and halide ion content were screened. Four datasets were selected for refinement based on the presence of different ions and small molecules bound in the active site. All crystals were of the space group P212121, and contain two protein molecules in the asymmetric unit (chains A and B). Small structural differences exist between chains A and B in all four structures, notably in the conformation of the active site residue Leu-155.The refined OXA-10 structure obtained in the absence of avibactam (1.88 Å, PDB 5MMY; Fig. 4B) indicated full carbamylation of Lys-70, and a molecule of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) present at the active site of chain A, but not chain B. The other three crystal structures, showing complexed avibactam in both chains, did not show lysine carbamylation. Indeed, no lysine carbamylation was observed in any of the electron density maps derived from initial crystals obtained in the presence of avibactam (data not shown).
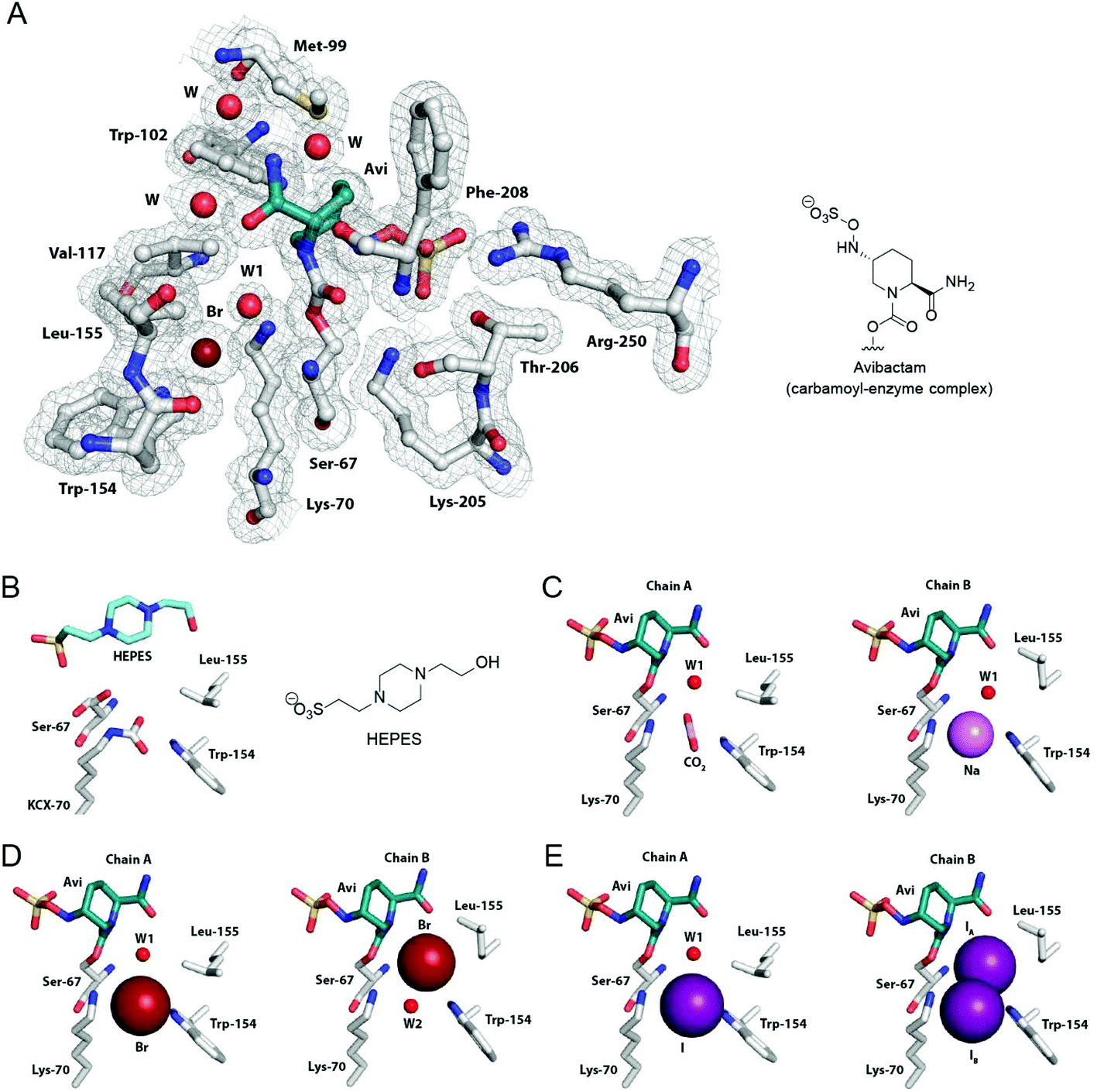 | ||
| Fig. 4 Crystallographic analyses of OXA-10:avibactam complexes. (A) View of the active site derived from a crystal structure of the OXA-10:avibactam complex with a bromide ion bound between Lys-70 and Trp-154 (PDB 5MNU, 1.56 Å, 2mFo-DFc map contoured to 1.0 σ). (B) View of the active site from a crystal structure of OXA-10 in complex with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; PDB 5MMY, 1.88 Å, chain A). The carbamylated Lys-70 is labeled as KCX. Active site views from crystal structures of the OXA-10:avibactam complex with assigned bound (C) carbon dioxide (PDB 5MOX, 1.41 Å, chain A), a sodium ion (PDB 5MOX, 1.41 Å, chain B), (D) bromide ions (PDB 5MNU, 1.56 Å), and (E) iodide ions (PDB 5MOZ, 1.34 Å; occupancies of 70% for IA and 30% for IB). Note we cannot rule out relatively low partial occupancy of other ions at the assigned binding positions. See Fig. S8† for views from other avibactam OXA complexes, Fig. S9† for views of bound carbon dioxide in avibactam OXA complexes, and Fig. S10 and S11† for views of bound halide ions to OXA enzymes. | ||
In chain A of the OXA-10 HEPES complex (Fig. 4B), Ser-67 is observed in two conformations, while in chain B (which does not contain HEPES) Ser-67 is observed in a single conformation, positioned to form a hydrogen bond to Lys-70 (Fig. S7†). Unexpectedly, the positioning and conformation of the bound HEPES molecule is similar to that of the avibactam enzyme complex (Fig. 4 and S6†), with the conformation of the HEPES piperazine ring resembling that of the avibactam piperidine core. While the HEPES sulfonate group is slightly off-set relative to the avibactam sulfate group (sulfur atoms differ by 1 Å), both are positioned to interact with Arg-250 and Thr-206.
A structure of the OXA-10:avibactam complex, crystallised in the absence of added halide ions or sodium bicarbonate, indicated the presence of either a sodium ion or carbon dioxide bound between Lys-70 Nε and Trp-154 Nε1 in the two chains (1.41 Å, PDB 5MOX; Fig. 4C). Two additional structures were obtained by crystallising the OXA-10:avibactam complex with 50 mM sodium bicarbonate, and 0.2 M sodium bromide or iodide. The resulting structures had either bromide (1.56 Å, PDB 5MNU) or iodide (1.34 Å, PDB 5MOZ) ions bound in the active site and elsewhere (Fig. 4D, E and S10, S11†). The positions of the bromide ion in the active site differed between the two chains, either positioned between Lys-70 Nε and Trp-154 Nε1 (chain A; Fig. 4D) or in a position proposed for the ‘hydrolytic’ water molecule (chain B; Fig. 4D). In the OXA-10 structure obtained in the presence of sodium iodide, an iodide ion was observed in chain A positioned between Lys-70 Nε and Trp-154 Nε1. However, chain B has iodide ions bound in two different positions in the active site, with one ion positioned in the same position as chain A (30% occupancy) and the other in the proposed position of the ‘hydrolytic’ water molecule (70% occupancy; Fig. 4E).
The conformations of avibactam in the three different OXA-10:avibactam complex structures resemble those previously reported (Fig. 4).18 In particular, the avibactam sulfate group is positioned to form a ‘bidentate’ salt bridge interaction with Arg-250, and to hydrogen bond with the side chain of Thr-206. The rate of recyclisation of the avibactam-derived OXA complex likely depends on the conformation of avibactam (Fig. 5A). The orientations of the sulfate and amide groups (on N6 and C2, respectively; numbering from Fig. 1C) seen in our structures differ from a previously reported OXA-10 avibactam complex structure (PDB 4S2O; Fig. 5B and S8†).18 The nucleophilic nitrogen in the structures described herein is positioned such that nucleophilic attack on the carbonyl of carbamoylated Ser-67 is stereoelectronically favourable (i.e., it is close to the ideal Bürgi–Dunitz trajectory),16,34,35 whereas this nitrogen adopts an unfavourable orientation for recyclisation in the previously reported structure.18
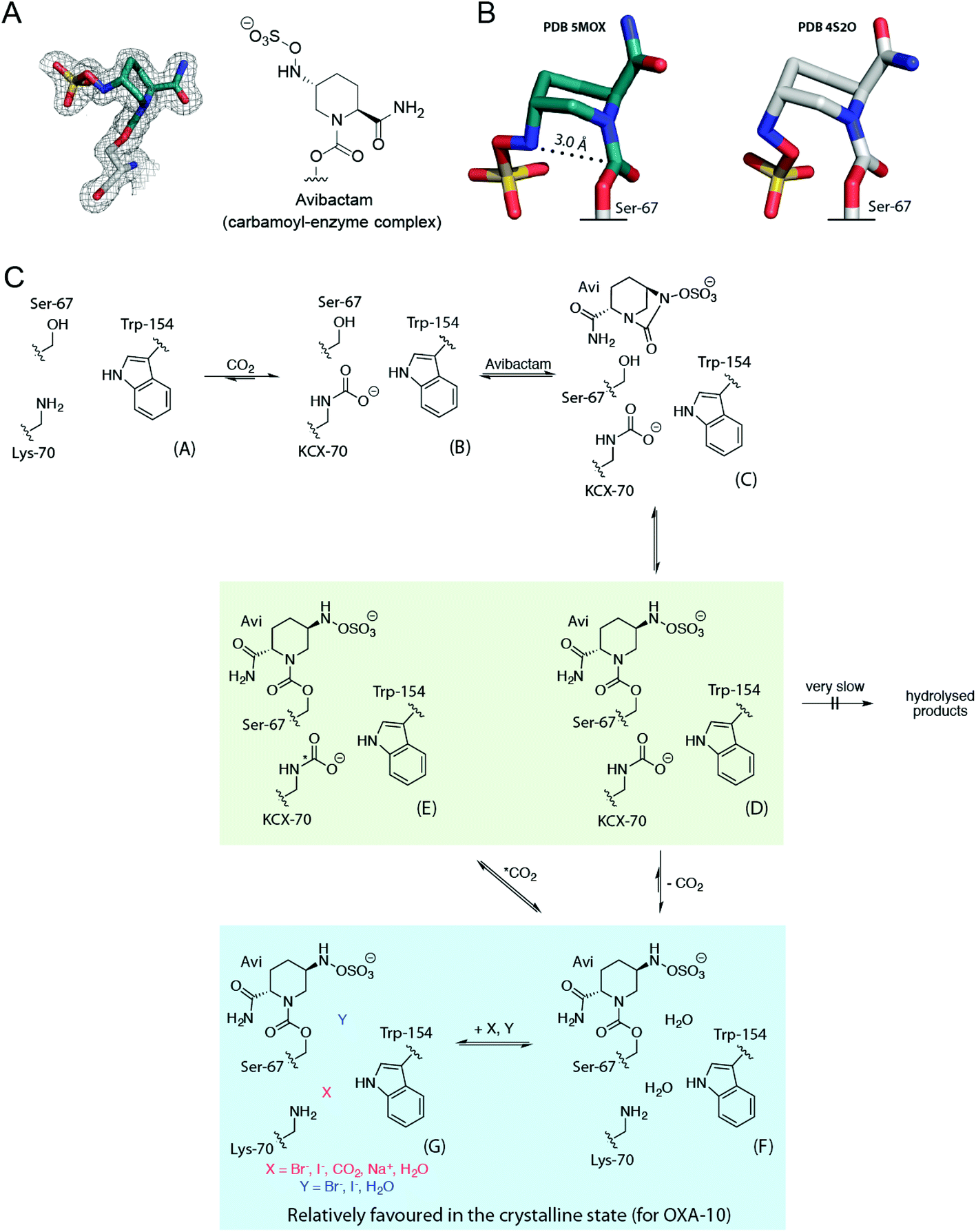 | ||
| Fig. 5 Summary of the interactions of OXA β-lactamases with avibactam in solution and in crystals. (A) Conformation of the covalently bound avibactam in the OXA-10:carbon dioxide crystal structure (PDB 5MOX, 1.41 Å; 2mFo-DFc map contoured to 1.0 σ), and (B) comparison of this avibactam conformation (teal carbons) with that from a reported structure of the OXA-10:avibactam complex (PDB 4S2O; white carbons).44 The approximate Bürgi–Dunitz trajectory required for recyclisation is indicated with a dashed line.34 (C) Scheme showing the behaviour of the OXA-10:avibactam complex in the solution and crystalline states. Unreacted OXA-10 A undergoes reversible carbamylation of Lys-70 giving B, which reacts with avibactam to form non-covalent complex C (which has not been observed). C reacts to give the carbamoyl-enzyme complex D. In solution, D can undergo exchange of its carbamyl group, to E via F. However, in the crystalline state, D may decarbamylate to give F, which may then bind carbon dioxide, halides, or sodium ions as in G. In solution, reaction with avibactam decreases lysine carbamylation, relative to that in its absence. The equilibrium between D and F/G relatively favours the decarbamylated forms (F/G), compared to that between B and A, in which B is relatively favoured. A green background indicates structures observed by NMR spectroscopy and a blue background corresponds to crystallographically observed complexes. | ||
Considered with the solution data, the lack of carbamylation seen in these structures suggests that carbamylation of the OXA:avibactam complex may be disfavoured by the crystalline state, consistent with previous crystallographic studies of avibactam with OXA enzymes.18,24 At pH 6.5 and 7.5, crystal structures of the OXA-48:avibactam complex (PDB 4S2J, 4S2K) were not carbamylated, while only partial carbamylation was seen at pH 8.5 (PDB 4S2N).18 Another reported structure suggests partial carbamylation of the OXA-48:avibactam complex at pH 7.5 in two out of eight molecules in the asymmetric unit (PDB 4WMC).24 Only bound carbon dioxide was observed adjacent to the decarbamylated lysine residue for the OXA-24:avibactam complex (PDB 4WM9),24 while the avibactam complex structure with OXA-10 (PDB 4S2O) did not show carbamylation or bound carbon dioxide.18 The carbon dioxide in these prior structures is apparently positioned so as to ‘π-stack’ with the adjacent Trp-154 side chain, whilst in our OXA-10 structure, the carbon dioxide binds in a position apparently poised for reaction with (non-protonated) Lys-70 (Fig. S9†).
The differences in carbamylation observed between the crystalline and solution states for the OXA enzymes are similar to differences observed in the carbamylation status of the BlaR1 β-lactam sensor proteins. Like the OXA enzymes, BlaR1 uses a carbamylated lysine residue to catalyse nucleophilic attack of a serine residue onto a β-lactam substrate; however, BlaR1 then undergoes decarbamylation, preventing deacylation and stabilising the acyl-enzyme complex.36 BlaR1 carbamylation was initially observed by spectroscopy (including NMR),37 but early crystal structures of BlaR1 (without substrate) did not show carbamylation.38,39 However, in a more recent crystal structure, carbamylation of BlaR1 was observed.36
The carbamylation status of lysine has ramifications for the mechanism of avibactam inhibition. Recyclisation of avibactam from OXA-10 is accelerated in the presence of bicarbonate,17 likely dependent on lysine carbamylation. Additionally, decarbamylation was proposed to limit avibactam hydrolysis.18 However, our results indicate that the carbamylation status is largely maintained despite the presence of avibactam, thus, reduced carbamylation may not account for the lack of avibactam-catalysed hydrolysis. Instead, the lack of avibactam hydrolysis has been proposed to arise from non-productive positioning of a potential ‘hydrolytic’ water molecule,24 consistent with our solution data.
Our crystal structures of the OXA-10:avibactam complex reveal two different active site binding modes for halide ions. Structures with bromide or iodide ions bound between Lys-70 and Trp-154 (e.g., chain A in Fig. 4D and E) resemble those described previously (Fig. S9,†).7,11,32 QM/MM calculations were performed to investigate the protonation state of the Lys-70 ε-amino group in these OXA-10:avibactam complexes (Fig. S16, Tables S3–S5†). The calculations for all six systems examined (i.e., with bound carbon dioxide, sodium ions, the two different bromide ion configurations, and the two different iodide ion configurations) suggest that the lysine ε-amino group is likely neutral.
Reaction of the OXA nucleophilic serine with avibactam prevents the serine from acting as a hydrogen bond donor to the carbamylated lysine residue. This may allow halide ions to effect decarbamylation, likely by occupying a position as seen in chain A of Fig. 4D. The OXA-avibactam complex is expected to resemble the acyl-enzyme complex formed during substrate hydrolysis (Fig. 1). These results suggest that halides inhibit the OXA enzymes in part by promoting the decarbamylation of the acyl-enzyme complex. This mechanism is in agreement with the mixed inhibition mode observed for iodide and OXA-23 (Fig. S13†).
The observation of bromide and iodide ions adjacent to the carbonyl of the modified nucleophilic serine (e.g., chain B of Fig. 4D and E) is unprecedented. This site is expected to be occupied by the ‘hydrolytic’ water, and so binding of halide ions at this site may inhibit hydrolysis. Our analyses suggest that OXA inhibition by halide ions may involve two distinct mechanisms, i.e., displacement of a hydrolytic water molecule, and decarbamylation of the acyl-enzyme complex. Halide ions do not cause decarbamylation of OXA enzymes in solution when the nucleophilic serine is not reacted, suggesting that the observation of halide ions between Lys-70 and Trp-154 may arise primarily during crystallographic analysis. A summary of our proposals for the behaviour of OXA enzymes with avibactam in solution and crystalline states is given in Fig. 5C.
Conclusions
The combined biophysical analyses on the OXA enzymes, both in solution and in the crystalline state, inform on the relationship between avibactam-mediated inhibition, lysine carbamylation status, and halide ion binding. Given that lysine carbamylation is sensitive to salts, small molecules and structural perturbations, 13C-labelling of the Lys-70 carbamyl group is an efficient solution method to monitor behaviour of this catalytically important residue. 13C-NMR could be a valuable tool not only for further mechanistic studies on the OXA enzymes, but also for the development of novel OXA inhibitors. In this regard, developing successors to avibactam that react with OXA enzymes and then efficiently displace CO2 is of interest, perhaps in a manner analogous to that of β-lactam sensing by BlaR.36The general importance of lysine carbamylation is beginning to be more fully appreciated, and the number of proteins bearing this post-translational modification is growing. Given the historical reliance on crystallography to identify carbamylation, the number of proteins with carbamylated lysines is likely underestimated. Computational studies suggest that >1% of proteins in the Protein Data Bank (PDB) have a carbamylated lysine.40 However, care must be taken when monitoring protein carbamylation using 13C-NMR, as carbamylation of residues other than lysine (e.g., the N-terminus) can occur.41 Many carbamylated proteins are potential antibiotic targets, including enzymes involved in bacterial cell wall biosynthesis (e.g., alanine racemase, MurD, MurF).42,43 Given that studying carbamylation by 13C-NMR is inexpensive, requiring only 13C-labelled sodium bicarbonate and access to an NMR spectrometer capable of measuring 13C spectra, the use of NMR to study carbamylation is likely to grow.
Acknowledgements
C. T. L. thanks the Canadian Institutes of Health Research for a postdoctoral fellowship. C. J. thanks King's College London for a Graduate Teaching Assistant studentship. C. D. acknowledges use of ARCHER, the UK National Supercomputing Service (http://www.archer.ac.uk) and the National Service for Computational Chemistry Software (NSCCS). This work was supported by Medical Research Council (MRC) grant MR/L007665/1, an MRC Confidence in Concept award, MRC/Canadian Grant G1100135, AstraZeneca, and the MRC SWON alliance. We are grateful to the Wellcome Trust for funding the 600 MHz NMR spectrometer.References
- K. Bush and P. A. Bradford, Cold Spring Harbor Perspect. Med., 2016, 6, a025247 CrossRef PubMed.
- L. I. Llarrull, S. A. Testero, J. F. Fisher and S. Mobashery, Curr. Opin. Microbiol., 2010, 13, 551–557 CrossRef CAS PubMed.
- S. M. Drawz and R. A. Bonomo, Clin. Microbiol. Rev., 2010, 23, 160–201 CrossRef CAS PubMed.
- K. Bush, Ann. N. Y. Acad. Sci., 2013, 1277, 84–90 CrossRef CAS PubMed.
- M. Toth, N. T. Antunes, N. K. Stewart, H. Frase, M. Bhattacharya, C. A. Smith and S. B. Vakulenko, Nat. Chem. Biol., 2016, 12, 9–14 CrossRef CAS PubMed.
- B. A. Evans and S. G. Amyes, Clin. Microbiol. Rev., 2014, 27, 241–263 CrossRef PubMed.
- M. Paetzel, F. Danel, L. de Castro, S. C. Mosimann, M. G. Page and N. C. Strynadka, Nat. Struct. Biol., 2000, 7, 918–925 CrossRef CAS PubMed.
- L. Maveyraud, D. Golemi, L. P. Kotra, S. Tranier, S. Vakulenko, S. Mobashery and J. P. Samama, Structure, 2000, 8, 1289–1298 CrossRef CAS PubMed.
- L. Maveyraud, D. Golemi-Kotra, A. Ishiwata, O. Meroueh, S. Mobashery and J. P. Samama, J. Am. Chem. Soc., 2002, 124, 2461–2465 CrossRef CAS PubMed.
- D. Golemi, L. Maveyraud, S. Vakulenko, J. P. Samama and S. Mobashery, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14280–14285 CrossRef CAS PubMed.
- V. Stojanoski, D. C. Chow, B. Fryszczyn, L. Hu, P. Nordmann, L. Poirel, B. Sankaran, B. V. Prasad and T. Palzkill, Biochemistry, 2015, 54, 3370–3380 CrossRef CAS PubMed.
- J. Li, J. B. Cross, T. Vreven, S. O. Meroueh, S. Mobashery and H. B. Schlegel, Proteins, 2005, 61, 246–257 CrossRef CAS PubMed.
- D. E. Ehmann, H. Jahić, P. L. Ross, R. F. Gu, J. Hu, G. Kern, G. K. Walkup and S. L. Fisher, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11663–11668 CrossRef CAS PubMed.
- K. Coleman, Curr. Opin. Microbiol., 2011, 14, 550–555 CrossRef CAS PubMed.
- D. Y. Wang, M. I. Abboud, M. S. Markoulides, J. Brem and C. J. Schofield, Future Med. Chem., 2016, 8, 1063–1084 CrossRef CAS PubMed.
- H. Choi, R. S. Paton, H. Park and C. J. Schofield, Org. Biomol. Chem., 2016, 14, 4116–4128 CAS.
- D. E. Ehmann, H. Jahic, P. L. Ross, R. F. Gu, J. Hu, T. F. Durand-Réville, S. Lahiri, J. Thresher, S. Livchak, N. Gao, T. Palmer, G. K. Walkup and S. L. Fisher, J. Biol. Chem., 2013, 288, 27960–27971 CrossRef CAS PubMed.
- D. T. King, A. M. King, S. M. Lal, G. D. Wright and N. C. J. Strynadka, ACS Infect. Dis., 2015, 1, 175–184 CrossRef CAS PubMed.
- M. H. O'Leary, R. J. Jaworski and F. C. Hartman, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 673–675 CrossRef.
- Y. Li, X. Yu, J. Ho, D. Fushman, N. M. Allewell, M. Tuchman and D. Shi, Biochemistry, 2010, 49, 6887–6895 CrossRef CAS PubMed.
- V. Verma, S. A. Testero, K. Amini, W. Wei, J. Liu, N. Balachandran, T. Monoharan, S. Stynes, L. P. Kotra and D. Golemi-Kotra, J. Biol. Chem., 2011, 286, 37292–37303 CrossRef CAS PubMed.
- M. I. Abboud, C. Damblon, J. Brem, N. Smargiasso, P. Mercuri, B. Gilbert, A. M. Rydzik, T. D. Claridge, C. J. Schofield and J. M. Frère, Antimicrob. Agents Chemother., 2016, 60, 5655–5662 CrossRef CAS PubMed.
- S. S. van Berkel, J. Brem, A. M. Rydzik, R. Salimraj, R. Cain, A. Verma, R. J. Owens, C. W. Fishwick, J. Spencer and C. J. Schofield, J. Med. Chem., 2013, 56, 6945–6953 CrossRef CAS PubMed.
- S. D. Lahiri, S. Mangani, H. Jahić, M. Benvenuti, T. F. Durand-Reville, F. De Luca, D. E. Ehmann, G. M. Rossolini, R. A. Alm and J. D. Docquier, ACS Chem. Biol., 2015, 10, 591–600 CrossRef CAS PubMed.
- S. Baurin, L. Vercheval, F. Bouillenne, C. Falzone, A. Brans, L. Jacquamet, J. L. Ferrer, E. Sauvage, D. Dehareng, J. M. Frère, P. Charlier, M. Galleni and F. Kerff, Biochemistry, 2009, 48, 11252–11263 CrossRef CAS PubMed.
- S. T. Cahill, R. Cain, D. Y. Wang, C. T. Lohans, D. W. Wareham, H. P. Oswin, J. Mohammed, J. Spencer, C. W. Fishwick, M. A. McDonough, C. J. Schofield and J. Brem, Antimicrob. Agents Chemother., 2017, 61 DOI:10.1128/AAC.02260-16.
- J. Brem, R. Cain, S. Cahill, M. A. McDonough, I. J. Clifton, J. C. Jiménez-Castellanos, M. B. Avison, J. Spencer, C. W. Fishwick and C. J. Schofield, Nat. Commun., 2016, 7, 12406 CrossRef CAS PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkóczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L. W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- J. N. Butler, Carbon dioxide equilibria and their applications, CRC Press, Boca Raton, FL, 1991 Search PubMed.
- L. Vercheval, C. Bauvois, A. di Paolo, F. Borel, J. L. Ferrer, E. Sauvage, A. Matagne, J. M. Frère, P. Charlier, M. Galleni and F. Kerff, Biochem. J., 2010, 432, 495–504 CrossRef CAS PubMed.
- C. Héritier, L. Poirel, D. Aubert and P. Nordmann, Antimicrob. Agents Chemother., 2003, 47, 268–273 CrossRef.
- H. B. Burgi, J. D. Dunitz and E. Shefter, J. Am. Chem. Soc., 1973, 95, 5065–5067 CrossRef CAS.
- R. C. Wilmouth, S. Kassamally, N. J. Westwood, R. J. Sheppard, T. D. Claridge, R. T. Aplin, P. A. Wright, G. J. Pritchard and C. J. Schofield, Biochemistry, 1999, 38, 7989–7998 CrossRef CAS PubMed.
- O. Borbulevych, M. Kumarasiri, B. Wilson, L. I. Llarrull, M. Lee, D. Hesek, Q. Shi, J. Peng, B. M. Baker and S. Mobashery, J. Biol. Chem., 2011, 286, 31466–31472 CrossRef CAS PubMed.
- D. Golemi-Kotra, J. Y. Cha, S. O. Meroueh, S. B. Vakulenko and S. Mobashery, J. Biol. Chem., 2003, 278, 18419–18425 CrossRef CAS PubMed.
- F. Kerff, P. Charlier, M. L. Colombo, E. Sauvage, A. Brans, J. M. Frère, B. Joris and E. Fonzé, Biochemistry, 2003, 42, 12835–12843 CrossRef CAS PubMed.
- M. S. Wilke, T. L. Hills, H. Z. Zhang, H. F. Chambers and N. C. Strynadka, J. Biol. Chem., 2004, 279, 47278–47287 CrossRef CAS PubMed.
- D. Jimenez-Morales, L. Adamian, D. Shi and J. Liang, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 48–57 CAS.
- J. S. Morrow, P. Keim and F. R. Gurd, J. Biol. Chem., 1974, 249, 7484–7494 CAS.
- A. A. Morollo, G. A. Petsko and D. Ringe, Biochemistry, 1999, 38, 3293–3301 CrossRef CAS PubMed.
- S. Dementin, A. Bouhss, G. Auger, C. Parquet, D. Mengin-Lecreulx, O. Dideberg, J. van Heijenoort and D. Blanot, Eur. J. Biochem., 2001, 268, 5800–5807 CrossRef CAS PubMed.
- A. M. King, D. T. King, S. French, E. Brouillette, A. Asli, J. A. Alexander, M. Vuckovic, S. N. Maiti, T. R. Parr, E. D. Brown, F. Malouin, N. C. Strynadka and G. D. Wright, ACS Chem. Biol., 2016, 11, 864–868 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob01514c |
| This journal is © The Royal Society of Chemistry 2017 |