
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catching the chloride: searching for non-Hofmeister selectivity behavior in systematically varied polyamide macrocyclic receptors†
Kajetan
Dabrowa
 ,
Filip
Ulatowski
,
Dawid
Lichosyt
and
Janusz
Jurczak
*
,
Filip
Ulatowski
,
Dawid
Lichosyt
and
Janusz
Jurczak
*
Institute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: jurczak_group@icho.edu.pl
First published on 23rd June 2017
Abstract
The binding selectivity of structurally simple anion receptors is governed by the Hofmeister series (SO42− > HPO42− > carboxylates ∼ H2PO4− > HCO3− > Cl−), and exceptions to this rule are rare and require utilization of structurally sophisticated receptors. In this paper we examined a set of 48 structurally diverse anion receptors, barely one fourth of which exhibit selectivity for chloride over more basic dihydrogen phosphate (H2PO4−) or carboxylates (MeCO2− and PhCO2−). Searching for regularities in the properties of these mainly macrocyclic-derived receptors across quite systematic changes in structure, combined with analysis of multiple crystal structures, allowed us to identify the crucial structural features that are likely required for the occurrence of the phenomenon of selective chloride binding. Examination of a subset of other ‘case study’ receptors reported in the literature as being particularly chloride-selective served as a confirmation of our hypotheses. As such, our findings are valid for all artificial receptors with exceptional selectivity for chloride, as well as for natural chloride channel proteins (ClC).
1. Introduction
Nearly 130 years ago, the German scientist Wilhelm Hofmeister observed that various inorganic salts of common cations exhibit differing abilities to precipitate egg-white globulin, depending on the anion in the salt.1 In a number of further studies, the so-called Hofmeister series of anions was determined to arise from the overall impact of multiple parameters, such as ion size, charge density, enthalpy of solvation, hydration sphere, and polarizability.2 The experimentally determined and still not fully theoretically justified3 order of anions in Hofmeister series is as follows: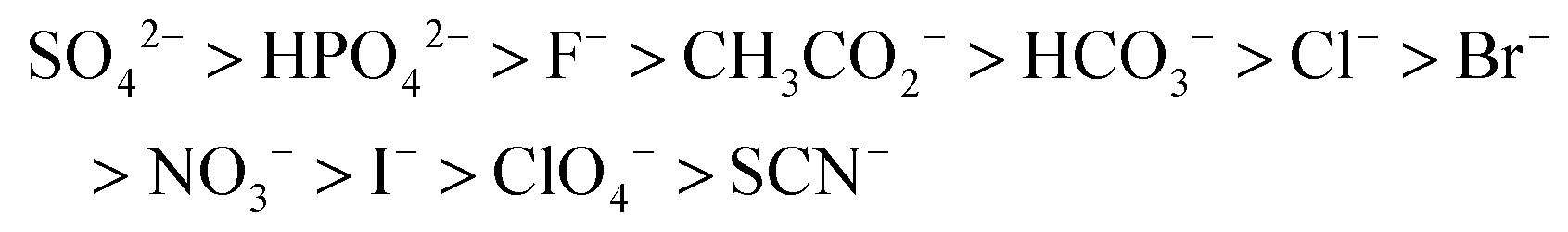
It begins with hard anions bearing high surface electrostatic potential regarded as kosmotropic (increasing viscosity of water) and ends with soft anions with low charge density and high polarizability – chaotropes (decreasing viscosity of water).3 Kosmotropes are strong H-bond acceptors and highly hydrophilic while chaotropes are much weaker hydrogen bond (H-bond) acceptors and exhibit lipophilic character.
Although, the Hofmeister bias was initially determined in buffered water solutions as a measure of the ability of anions to precipitate proteins, it was also found to reflect the natural order of anions’ affinities toward simple putative receptors equipped with H-bond donors even in organic solvents, as proved by direct measurments4a and analysis of multiple reports.4b–h Such correlation is not surprising since H-bond acceptor strength of anions relies on the aforementioned set of multiple parameters. In order to analyze the phenomena of selective anion binding in biological systems, one can restrict the Hofmeister series to several anions possessing the highest abundance in living organisms:5
| Phosphates ≈ carboxylates > HCO3− > Cl− |
Since concentrations of all other anions such as F−, Br−, sulfates, and nitrates are far lower one can easily recognize that chloride is the most difficult to be selectively bound in the presence of other anions and in general has a tendency to form weak supramolecular complexes. Despite these objective difficulties, chloride is the sole, ubiquitous inorganic anion which is used by cells in cooperation with cations in the formation of transmembrane gradients, which are substantial for various cell functions as they are used to produce electrical signals, regulate cell volume and facilitate electrolyte transport.
Apparently, therefore, Nature found its own way to overcome the Hofmeister bias and can bind chloride effectively and selectively to maintain these gradients through the utilization of various active and passive ion channels. Their common feature is high selectivity toward particular ionic species. Each channel permits only certain ionic species to flow through its selectivity filter, i.e. the pore in the protein responsible for the selective binding and passing the ions. The chloride channel proteins (ClCs) belong to several classes, with differing mechanisms of transport and its regulation.6 Their fundamental importance for the control of homeostasis is best expressed by the occurrence of various channelopathies, i.e. inherited diseases associated with mutations in genes coding channel proteins. Chloride channelopathies affect different organs and include diseases such as cystic fibrosis, myotonia, hyperekplexia, lysosomal storage disease, deafness, renal salt loss, kidney stones, and osteopetrosis.7
The structures and exact properties of most chloride channel (ClC) proteins remain unknown. To date, only a few structures of such proteins have been determined by single crystal X-ray analysis. The X-ray structure of ClC from Salmonella enterica revealed that in its selectivity filter, the chloride anion is bound by just four H-bond donors (Fig. 1).8
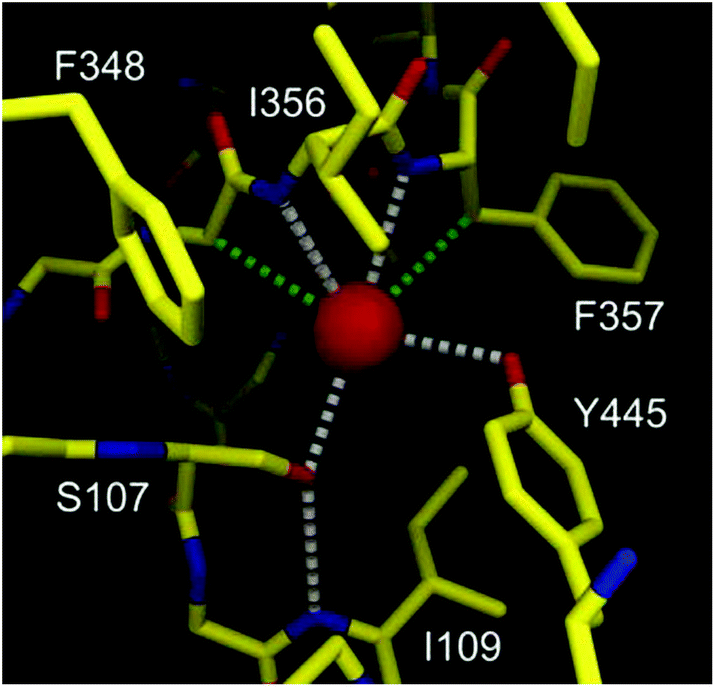 | ||
| Fig. 1 Graphical representation of binding of chloride (red sphere) by the selectivity filter of the ClC protein. Hydrogen and C–H⋯Cl− bonds are colored as gray and green dashed lines. Adapted by permission from Macmillan Publishers Ltd: Nature (ref. 8), copyright (2002). | ||
Two of them are NH groups from the peptide chain, the other two are OH groups of serine and tyrosine side chains. It is also noteworthy that apart from these four H-bond donors, the chloride anion is surrounded by hydrophobic amino acid side chains and contacts with hydrophobic CH2 groups, as indicated in Fig. 1. Notably, the chloride ion is not involved in any direct contact with the full positive charges from the neighboring lysine or arginine residues; instead the charge is balanced by the partial positive charges originating from the helix dipole interactions and from polarized residues adjacent to nitrogen and oxygen atoms. This pore, although equipped with a relatively low number of anchoring points, exhibits high selectivity towards chloride over other anions, such as carboxylates and phosphates, which means that it can overcome the natural Hofmeister series. Several studies have revealed that chloride anion binds to the selectivity filter of the ClC protein with association constant Ka = 0.3–1.4 M−1. The ClC protein consists of 473 amino acids and only four of them (less than 1%) are formally engaged in chloride binding in the selectivity filter. The vast majority (>99%) of the amino acids build different domains responsible for various functions: locating the protein in the cellular membrane, regulation of transport, directing anions to the pore, etc. Moreover, the majority of amino acids outside the binding pockets is in fact essential for spatially arranging those in the selectivity filter with very high precision and proper rigidity, which is necessary for obtaining the high selectivity and affinity.
Contrary to the described proteins, artificial anion receptors rarely exhibit selectivity for chloride. High selectivity for fluoride or phosphate has been highlighted in many studies,9 but this is not so much a result of good receptor design as a simple reflection of the intrinsic properties of these anions. In fact, developing a host molecule capable of binding chloride in the presence of anions situated at the top of Hofmeister series is a very challenging task and multiple research efforts are underway in this field, encouraged by several factors.
The first is the need to gain a better understanding of how natural chloride channels work and which structural parameters are crucial for their unique selectivity. Such knowledge may be obtained by analyzing multiple model receptors that systematically differ in structure. There is also curiosity about whether appropriately designed, simple receptors can mimic the action of these large biomolecules.
Secondly, artificial chloride transmembrane carriers are being intensively investigated in the search for therapeutics for different kinds of chloride channelopathies.10 Such vehicles must not only selectively transport chloride in the presence of highly abundant phosphate and carboxylates, but should also be resistant to inhibition caused by the formation of very stable complexes with these interfering anions. This means that the carriers should exhibit preferential chloride binding in solution.
Lastly, chloride receptors have been found to be useful in catalyzing nucleophilic substitution reactions, where they make the chloride a better leaving group and can control the stereochemical course of the reaction.11 Such a catalyst, if intended to work in aqueous buffers that resemble biological conditions, should, like transporters, exhibits low affinity for anions forming the buffer (e.g. carbonates, phosphates). This will ensure that catalyst is not deactivated in such a complex medium.
In the first part of the paper we present a large set of rationally selected and systematically varied anion receptors (mostly polyamide macrocycles), either designed by our group or found in the literature, which we analyzed for their selectivity in anion binding. Thermodynamic and available structural data of these hosts were then carefully analyzed in order to identify structural parameters which are crucial for obtaining both high affinity and selectivity for chloride. Such a generalization is needed to serve as a guideline for the further studies in the field and to explain the great selectivity of natural ClC proteins.
In the second part of this study, we additionally considered literature reports of receptors of other types which exhibit remarkable selectivity toward chloride, but cannot be organized in a series of analogues and are analyzed as individual cases.
2. Results and discussion
When a novel artificial anion receptor is announced in the literature, its affinity toward certain anions is examined, albeit there is no standard solvent nor standard set of anions which have to be arbitrarily employed in such studies. The anions most often used as guest include chloride (Cl−), dihydrogenphosphate (H2PO4−) and simple carboxylates (acetate MeCO2− or benzoate PhCO2−), whereas nitrate (NO3−), hydrogensulfate (HSO4−), sulfate (SO42−), and bromide (Br−) are far less frequently employed in such studies. In addition, except one report, we were unsuccessful in finding any suitable binding data for bicarbonate anion (HCO3−). Therefore, in this study we decided to focus on the following set of biologically relevant anions: chloride, dihydrogenphosphate and simple carboxylates (MeCO2− and PhCO2−). Selected properties of these anions are presented in Table 1.| Property | H2PO4− | CH3CO2− | Cl− |
|---|---|---|---|
| a pKb – 14 - pKa, where pKa is negative logarithm of dissociation constant of the conjugate acid; ΔGhyd. – experimental molar Gibbs energy of hydration in kJ mol−1 (taken from ref. 12). b V c – calculated anion volume in Å3; electron density surface calculated at 0.001 au; all calculations were carried out at DFT/M06-2X/6-311g(d) level of theory. c Calculated interaction energy of pyrrole with anion (corrected for ZPE): Eint. = Ecomplex − (Epyrrole + Eanion) in kcal mol−1. | |||
| Shapeb |
|
|

|
| pKb | 11.8 | 9.2 | 17 |
| ΔGhyd. | −465 | −365 | −340 |
V
c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
62.6 | 59.4 | 23.7 |
|
E
int.c |
−22.9 | −28.8 | −20.2 |
Since all selected anions are singly charged, they can be easily compared in terms of their ability to form supramolecular assemblies. Typically, H-bond energy is correlated with the basicity of the H-bond acceptor. In our set, carboxylates are the most basic, dihydrogenphosphate is situated in the middle, and chloride is only very weakly basic. Enthalpy of hydration also indicates that interaction of anion with H-bond donors of water molecules is weakest in the case of chloride. In order to provide further justification that anion binding affinities of simple receptors are correlated with their position in the Hofmeister series we performed quantum mechanical calculations of interactions of these anions with pyrrole which we have chosen as a model single H-bond donor lacking any preference on shape and size of the guest (see ESI† for details).13 Calculated interaction energies of its complexes with anions are consistent with the Hofmeister bias, i.e. more basic anions are bound more strongly by this putatively simple host (MeCO2− > H2PO4− > Cl−).
The fact that measurements of the determined association constants of different receptors were carried out in various solvents makes any meta-analysis of the data particularly difficult. Keeping in mind, however, that in this study we are focused on selective binding of chloride, we decided to use the relative binding affinities (Krel) defined in eqn (1) to facilitate such an analysis.
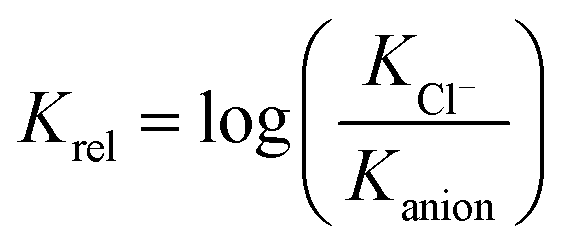 | (1) |
Such treatment of data allows us to compare receptors which were analyzed in various solvents, and directly provides the selectivity we are looking for. Negative values of Krel indicate that a given anion is bound more strongly than chloride, while positive values indicate that the receptor is selective for chloride – the property of our interest.
Although, to date, a great number of anion receptors of various architecture and properties have been reported, in the following analysis we decided to include only receptors which resemble natural ClC protein selectivity filter and, what is even more important, enable systematic studies. The inclusion criteria for hosts were as follows:
(1) the association constants (Ka) for chloride and at least for the following anions: H2PO4−, MeCO2− or PhCO2− are available;
(2) are neutral in charge;
(3) bind anions via multiple H-bond donors;
(4) belong to a family of receptors with systematic structural modifications.
The first three, formal requirements significantly narrowed the number of acceptable receptors. Within the remaining set we picked multiple acyclic, macrocyclic, and macrobicyclic receptors equipped with either amide or thioamide functionalities for anion via H-bond interactions. The largest subset of such receptors bearing common structural motif consists of hosts based on 1,3-disubstituted aromatic acids. The structures of receptors belonging to this family which satisfy the aforementioned criteria are presented in Fig. 2a and corresponding binding data are plotted in Fig. 2b. Whenever possible, the thermodynamic data from interactions in solution are accompanied with crystal structures, whose careful analysis provides deeper insight into the phenomenon of chloride binding.
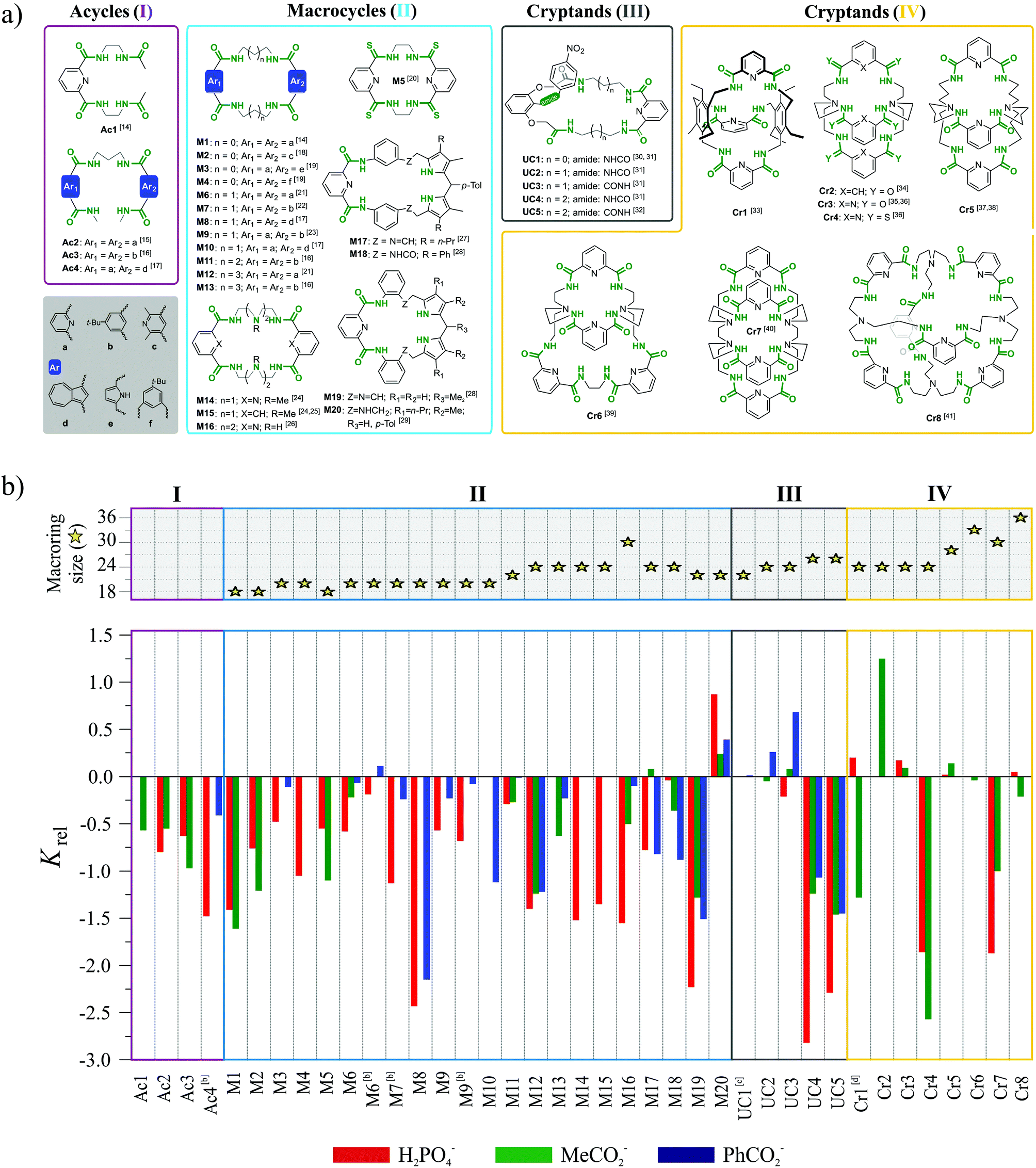 | ||
| Fig. 2 (a) Structures of polyamide receptors considered in this comparative study and (b) their relative binding affinities (Krel) for chloride; Krel = log(KCl−/Kanion) ≥ 0 indicates preference for chloride over other anions; the binding constants were derived from 1H NMR titrations conducted in DMSO-d6 (for UCs1–5 in DMSO-d6/0.5% water solution) at 298 K if not otherwise stated; [b] DMSO-d6 + 5% H2O; [c] Krel for benzoate and UC1 is close to 0; [d] CD2Cl2/CD3CN. | ||
In order to clarify the conclusions drawn from this meta-analysis, the discussion has been divided into four sections, depending on the assumed increase of the structural preorganization of a given receptor system in the following series: acycles (I) < macrocycles (II) < unclosed cryptands (III) < cryptands (IV).
3. Systematically varied polyamide receptors
Acycles (group I)
Acyclic receptors are characterized by relatively high structural flexibility, which is typically associated with the poor preorganization of the anion binding sites, hence the obtained receptor–anion complexes are rather weak (low Ka values). Within our series Ac1,14Ac2,15Ac3,16 and Ac417 none of the receptors displays anti-Hofmeister binding selectivity.It is most likely that, due to their flexibility, these hosts can adjust their geometries to effectively complex each of the analyzed anions. Although acyclic host Ac2 exhibits no preference for chloride, the crystal structure of its chloride complex indicates certain structural parameters which may be crucial for obtaining good selectivity in further designed receptors. Prior to anion binding, two strong H-bonds have to be broken and the acyclic receptor has to significantly change its structure, as concluded from the analysis of structures of free acyclic host Ac2vs. its chloride complex (Fig. 3).
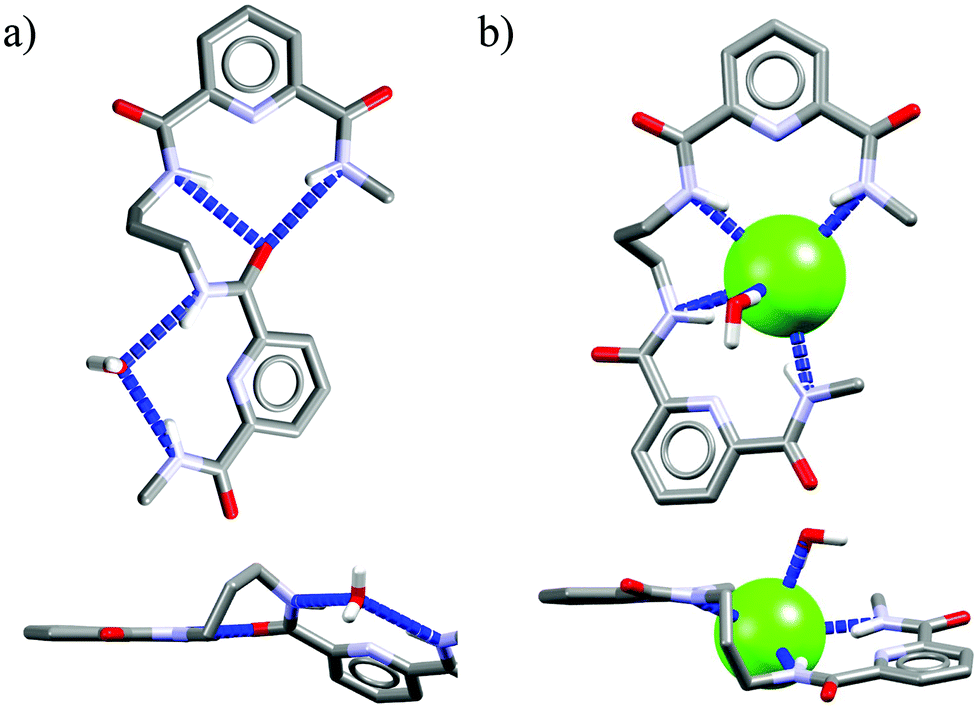 | ||
| Fig. 3 Crystal structures of the free acyclic receptor Ac2 (a) and its chloride complex (b). Non-acidic protons, counterions, and disorder have been omitted for clarity. | ||
The structure of the complex demonstrates that chloride is surrounded by a set of five H-bonds originating from four amide NH groups (d(N⋯Cl−) = 3.25–3.31 Å, ≤146–155°) and one water molecule (dO⋯Cl− = 3.18 Å, ≤170°), respectively. The water H-bond donor produces significantly stronger interaction than those from the amides. Although the energetic cost of this transformation is plausibly too high to achieve strong binding of chloride over more basic anions in solution, the structure of chloride complex provides some directions which may be crucial in the construction of future chloride-selective receptors.
Macrocycles (group II)
Appropriately designated macrocyclic receptors are generally considered to be more effective hosts as compared to their acyclic analogues, since the former benefit from the macrocyclic effect.15,42Macrocyclic structure imposes a higher degree of preorganization and guest binding requires fewer conformational changes, which means lower loss of entropy and results in higher association constants. The cost for this higher affinity is paid during synthesis, which is generally less efficient as compared to linear hosts for steric and entropic reasons.43 The rigidness of the scaffold in macrocycles is expected to assure not only higher binding strength, but also higher selectivity, since the receptor cannot easily adapt its geometry to match the size and shape of every guest.
The macrocyclic receptors M1–M2014,16–29 reviewed in this study are constructed from two binding pockets connected by various linkers. The binding pockets are based on aromatic diacid scaffolds which include benzene, pyridine, pyrrole and azulene.
The diversity of these building blocks results in differences in the geometries, preferential conformations, and acidity of H-bonds. Moreover, in macrocycles M17–M20 two pyrrole moieties provide additional H-bond donors. For macrocycles M1–M13 the aliphatic linkers vary from two to five methylene units, thus providing incremental increase of the binding cavity from 18- up to 26-membered rings. Only in the case of M14–M16 are the linkers constructed from oligo-(etyleneamine) chains, and in the case of M17–M20 the linker is constructed from benzene moiety.
Despite the aforementioned general advantages of a macrocyclic scaffold, most of the receptors in our subset M1–M20 do not exhibit substantially better selectivity as compared to the structurally related acyclic hosts. It is only for a few hosts, namely M3, M6, M9, and M11, that Krel reaches the value close to zero, which is already an indication of anti-Hofmeister selectivity. The only macrocycle clearly selective for chloride is the rather stiff dipyrromethane-based M20, equipped with six H-bond donors.
Receptors M3,19M6,21 and M923 are 20 membered macrocycles, with either ethylene (M3) or propylene linkers between amide groups (M6, M9).
In M3, both aromatic groups are based on dimethylenepyrrole, in M6 both are dipicolinic acids, while macrocycle M9 is non-symmetrical with dipicolinic and isophthalic moieties. Only these combinations of diacids were optimal for receptors with 20-membered rings, since hosts containing either two isophthalic groups or azulene moieties exhibit typical selectivity for phosphate and carboxylates.
Among the 22-membered macrocycles, M1116 with two isophthalic groups is moderately selective for chloride, while the non-symmetrical M2029 bearing dipicolinic and dipyrromethane moieties with aromatic linker, is very selective. Other macrocycles of this size possess highly negative Krel values, i.e. they more strongly bind dihydrogenphosphate and carboxylates.
Receptors of either smaller (18-membered) or larger macrocyclic size (24 and 32-membered) simply follow the normal Hofmeister series. Quite possibly, the rather small cavity of an 18-membered ring cannot accommodate chloride, whereas larger macrocycles are flexible enough to adjust their geometry for every anion, which results in a lack of selectivity.
In two cases (M6 and M9) we observed that increasing the water content in the solvent mixture has a positive influence on the selectivity for chloride. We attribute this effect to the fact that chloride possess the lowest enthalpy of hydration among the anions analyzed. In the process of supramolecular complex formation the solvent molecules in the anion solvation sphere are replaced by the receptor.
High water content in the solvent mixture strongly hampers binding of dihydrogen phosphate and carboxylates, while having a weaker impact on chloride. The net effect is an increase in Krel value.
Similar non-linear reduction of complex stability in more water-containing solvent mixtures has already been reported in the literature.44
Analysis of a quite large number of available crystal structures of complexes of macrocyclic receptors may explain some features of the observed dependence of selectivity on receptor structure.
The X-ray structure of chloride complex with M114 shows that the Cl− anion is complexed by all four amide NH donors. It is noteworthy that the anion does not occupy the center of the macrocyclic cavity of M1, but sits atop it, 1.9 Å above the mean plane of the four amidic nitrogen atoms. The binding of the anion by this tetralactam is not symmetrical; two of the four amide H-bonds are considerably shorter than the two others. What is more, the apical position of the chloride anion is occupied by a water molecule, acting as an additional H-bond donor.
The binding cavity of this 18 membered macrocycle is clearly too small to accommodate the chloride anion. A very similar structure was observed for the complex of the analogous tioamidic host M5.20 In this structure, however, short contacts (dC⋯Cl− = 3.46–3.48 Å) were found between Cl− and methylene groups from the linkers. Moreover, no additional interaction with a water molecule at apical position was observed. The oxygen to sulfur substitution resulted in a significant increase in the absolute association constants, but no improvement in selectivity was found.
One might expect that elongation of the linkers and thus expansion of the macrocyclic cavities should make them large enough to accommodate chloride. However, structural analysis indicates that even the 20-membered ring macrocycles cannot complexate any guest in the center of their cavity. X-ray structures of solvates of receptors M6,21M7,22M8,17 and M10,17 which possess propylene linkers, are presented in Fig. 5.
 | ||
| Fig. 4 Crystal structures of the free (a, d) and anion complexes of (b–c, e) of 18-membered tetra-amide M1 and thioamide M5 macrocycles. | ||
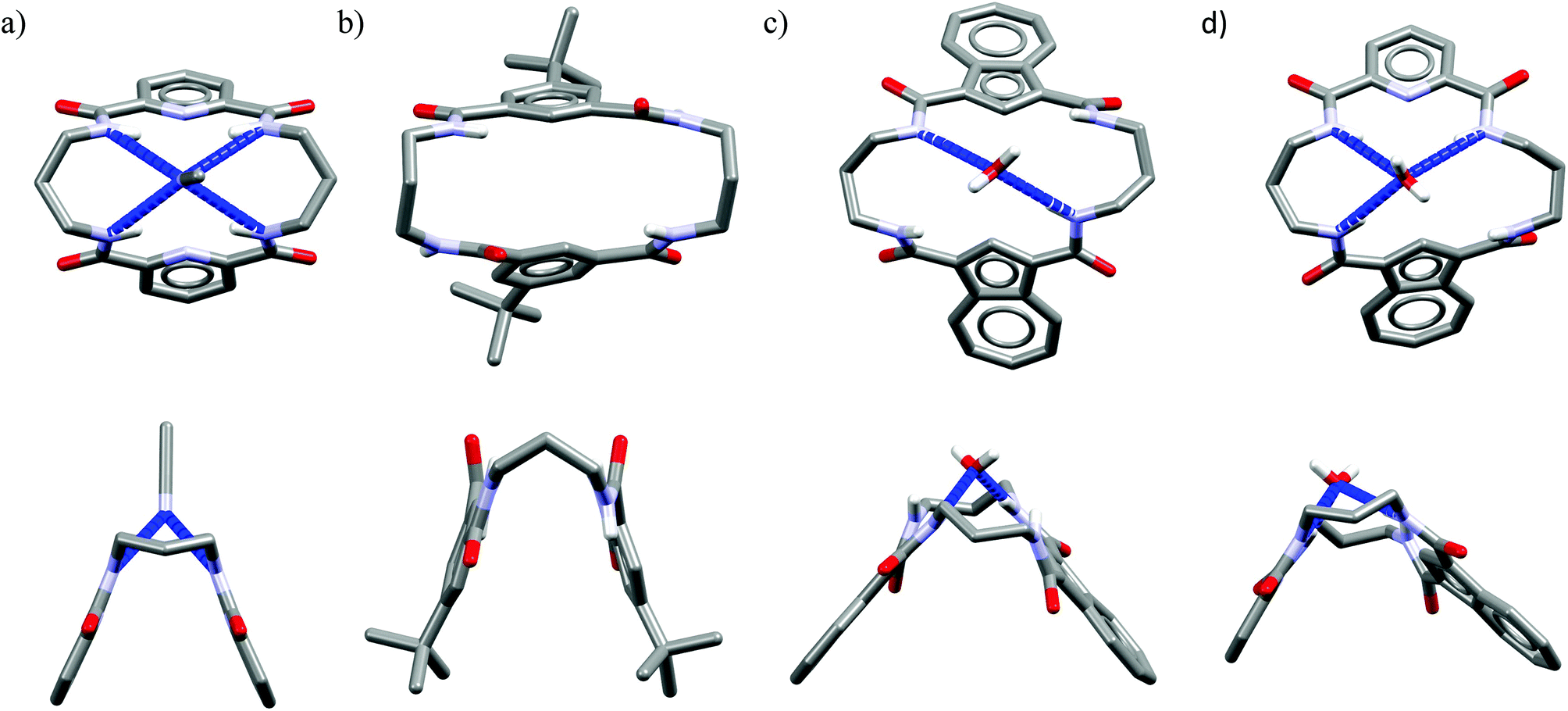 | ||
| Fig. 5 Crystal structures of the 20-membered macrocycles M6 (a), M7 (b), M8 (c), and M10 (d). | ||
In these structures the respective hosts form complexes with molecules of water or acetonitrile which act as H-bond acceptors. Although the guest molecules are very small, the hosts adopted non-planar V-shaped conformation with amide H-bond donors pointing to the guest molecule, located at the apex of pyramid. Since the small molecules of water or acetonitrile cannot fit into the cavity of 20-membered macrocycles, this should also be valid for larger anionic guests. Indeed, X-ray structures of chloride complexes of M6, M7, M9 macrocycles (Fig. 6) show that these receptors adopt the V-shape and the four H-bonds donors interact with the anion on its side, which is similar to the aforementioned structures of 18-membered ring macrocycles. Elongation of the linkers resulted merely in a minor expansion of the macrocyclic cavity. In these complexes, chloride is a bit closer to the plane of the amidic nitrogen atoms (1.81, 1.65, and 1.61 Å for M6, M7, M9, respectively) compared to the M1@Cl− complex, yet the complexation mode of chloride is still far from the optimal one, such as found in the crystal structure of ClC protein. In two cases (Fig. 6a and c) short contacts with the CH2 group of another receptor molecule are observed in apical position of the chloride anion. In addition, other anions considered in this paper (dihydrogen phosphate and benzoate) seem to fit the binding pocket of the V-shaped receptors very well (as depicted in Fig. 7). In each case both oxygen atoms of the guest are coordinated by H-bond donors, much like in the case of the weaker complexes with chloride.
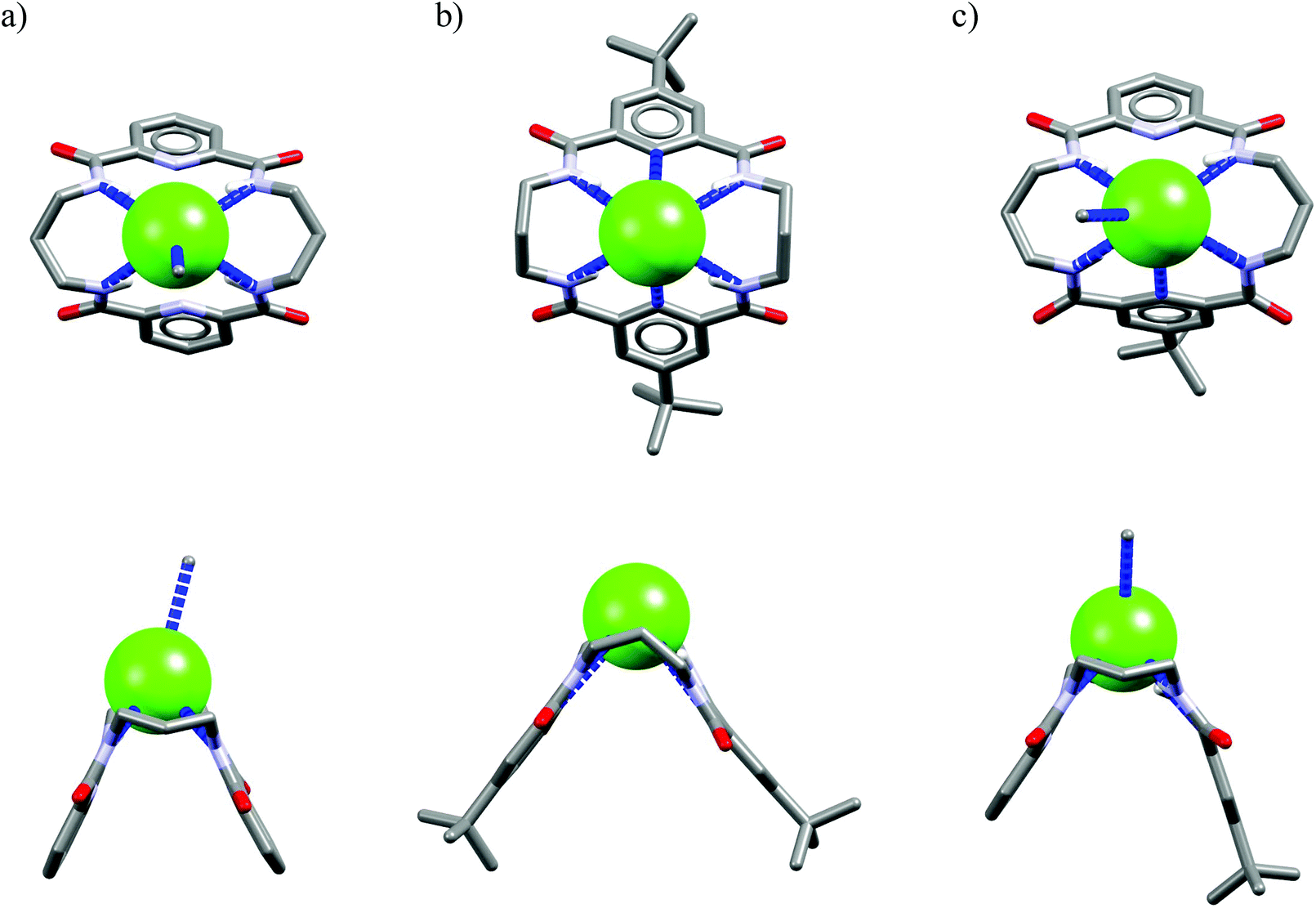 | ||
| Fig. 6 Top and side views of the crystal structures of the complexes of 20-membered macrocycles M6 (a), M7 (b), and M9 (c) with chloride. | ||
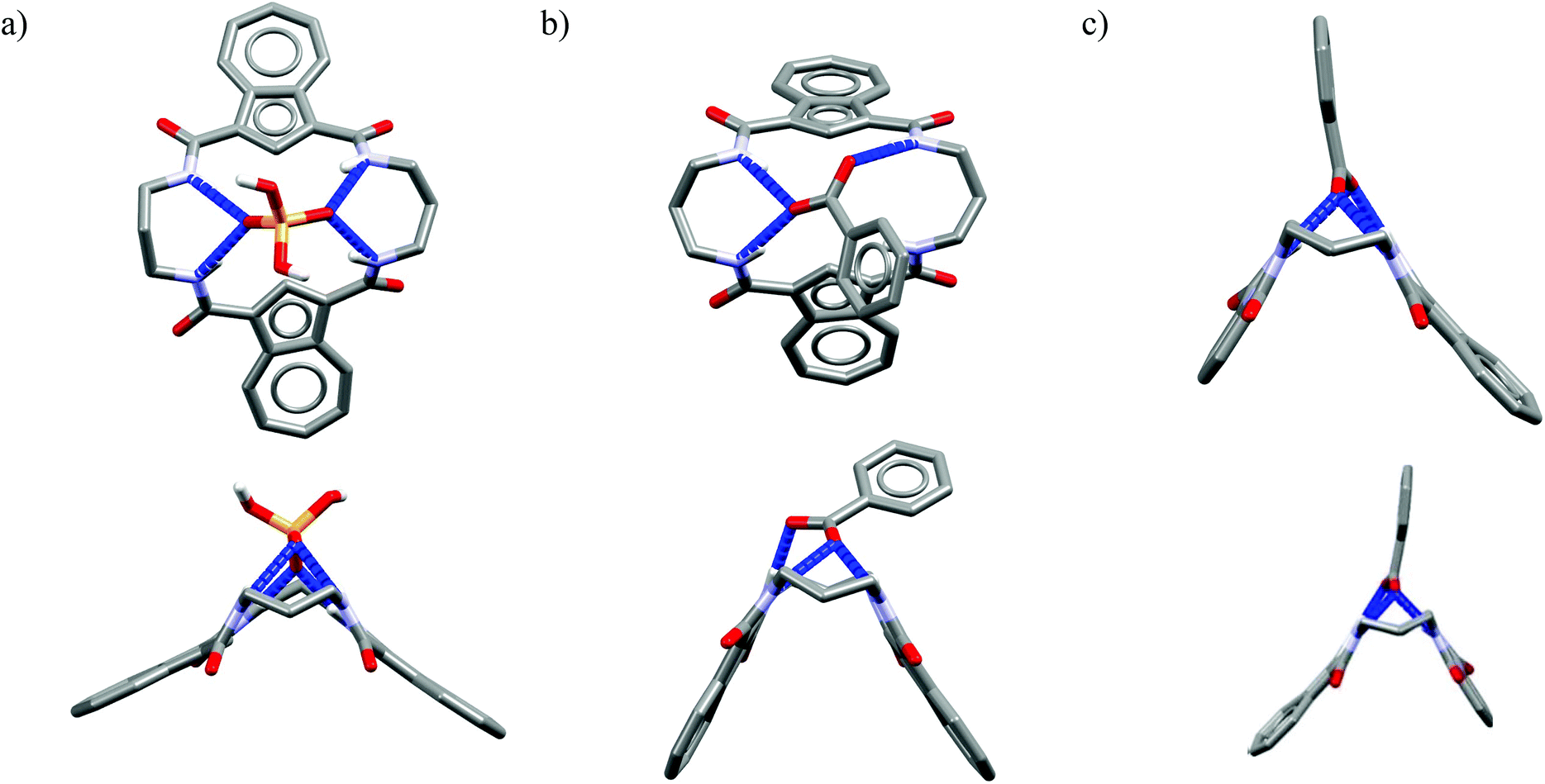 | ||
| Fig. 7 Top and side views of the crystal structures of the complexes of 20-membered macrocycles M8 with dihydrogenphosphate (a) and M9 (b) and M10 (c) with benzoate. | ||
From these data it seems that the type of aromatic diacid moiety has little impact on the adopted geometry. Likewise, comparison of the crystal structures cannot rationalize the observed distribution of Krel values.
In general, the scaffold with two meso diacid moieties cannot assure the optimal chloride encapsulation. Probably, the presence of a wider dipyrromethane fragment in the host M2029 resulted in a macrocyclic cavity large enough to accommodate chloride, which resulted in the highest Krel among this group. However, as indicated by crystal structure of Ac2, even a close to perfect geometric match between chloride and macrocyclic cavity does not saturate all the binding sites at the surface of chloride. Therefore, apical positions above and/or below the macrocyclic plane can be occupied by solvent molecules.
A general lesson from the analysis performed so far is that even a close to perfect geometric match between chloride and macrocyclic cavity does not guarantee selectivity for chloride, at least for tetraamide-derived receptors. In addition, one can assume that equipping the macrocyclic framework with substituent/s able to interact with the apical position of chloride, preferably via H-bond interactions, should positively influence selectivity for chloride.
Both indications are realized in the following subset of macrocyclic receptors with a flexible tether, i.e. unclosed cryptands.
Unclosed cryptands (group III)
Unclosed cryptands Uc1–Uc530–32 are structurally related to previously mentioned macrocycles M1–M13, however, only one side contains meso diacid moiety (dipicolinic acid), while the other side is constructed here from diacid derived from 2,6-dihydroxyaniline. The nitrogen atom of the latter group is a part of amide equipped with an additional p-nitrophenyl substituent (lariat arm) which is introduced at the intraannular position of the macroring. Presence of this flexible tether is expected to strongly influence the binding of the guest that is realized within the macrocyclic cavity.The smallest among the series, the 22-membered Uc1 has the ethylene linker and binds chloride as strongly as benzoate, which is against Hofmeister bias.30,31 The slightly larger, 24-membered Uc2 and Uc3, with the propylene spacers, exhibit association constants higher for chloride complexes as compared to complexes with carboxylates.31 Moreover, subtle structural variation in the position of heteroatoms between macrocyclic framework and lariat arm results in higher Krel values in the case of Uc3 over Uc2 (see Fig. 2a). Quite interestingly, selectivity is completely lost for the largest 26-membered UCs Uc431 and Uc5,32 having much more flexible 1,4-diaminobutane spacers. Comparative analysis of the crystal structures of monohydrate vs. chloride complex of Uc1 reveals some interesting aspects of these compounds (Fig. 8). Firstly, although macrocyclic scaffold is quite well preorganized for the binding of various guests via H-bond interactions,31,32,45,46 complexation of a guest which is solely an H-bond acceptor (such as chloride), requires a 180° degree rotation of the lariat arm accompanied with the breaking of the intramolecular H-bond. This in turn, enables very strong, albeit not highly directional interaction with an anion (dN⋯Cl− = 3.12 Å, ≤159°), which has no direct contact with the TBA+ counterion (Fig. 8b, inset) and is unsymmetrically bound in the macrocyclic cavity by just three NH donors from the macrocycle (dN⋯Cl− = 3.25–3.47 Å, ≤148–165°).
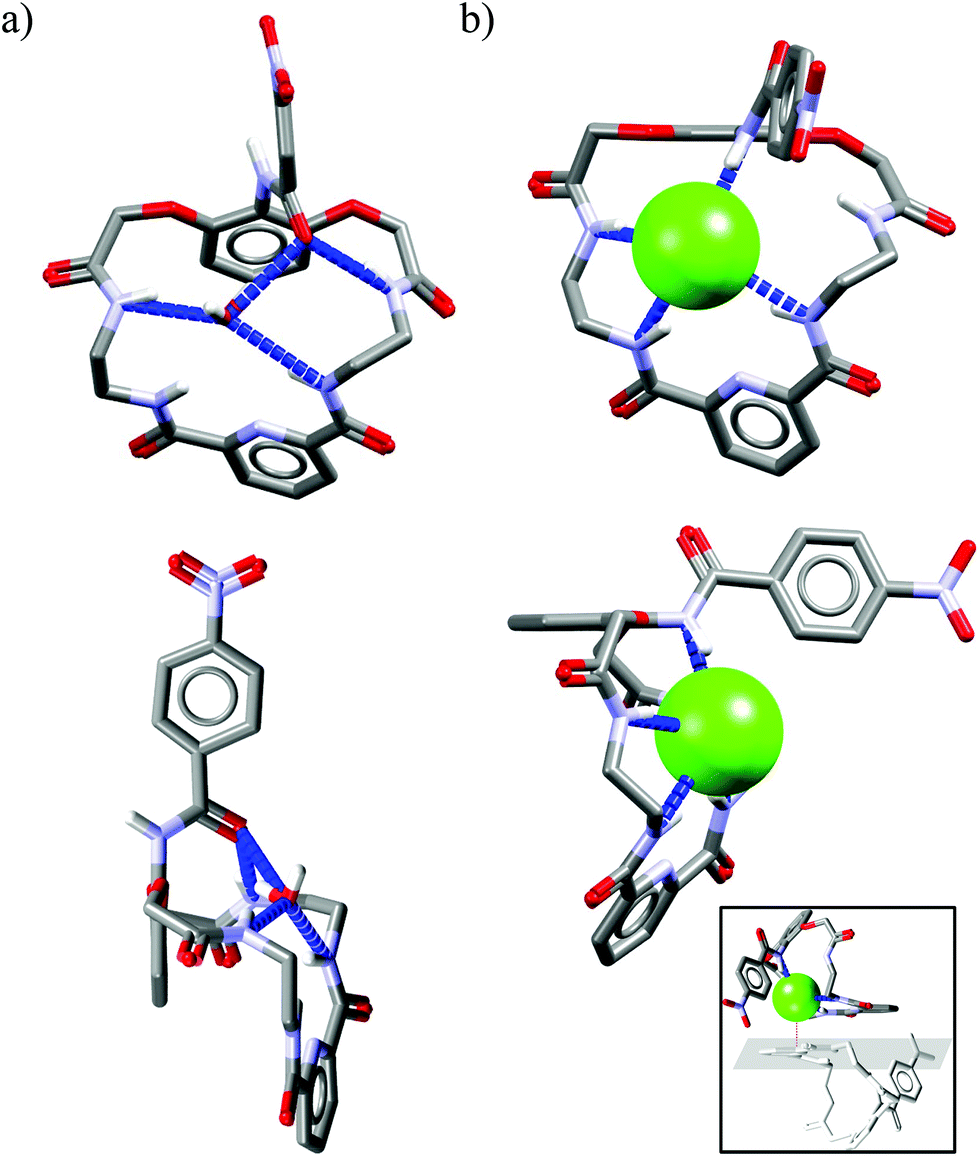 | ||
| Fig. 8 Crystal structures of the free (a) and chloride complex (b) of the Uc1. | ||
The remaining NH amide proton does not participate in any H-bond interaction, despite pointing toward the anion. Seemingly, similar binding behavior also occurs in solution, as deduced from titration data.31 Namely, virtually no shift of this proton is observed upon gradual addition of TBACl to the solution of Uc1 in DMSO:water mixture (99.5:0.5 v/v), whereas acetate and benzoate cause a pronounced downfield shift of this signal. In addition, for the larger Uc2 and Uc3 the same binding mode occurs, except that the NH amide proton shifted upfield (−0.45 ppm for Uc2 and −0.70 ppm for Uc3) upon titration with TBACl. These observations indicate that chloride is too small to establish effective H-bond interaction with these ‘unlocked’ NH amide protons, at least in solution. The experimental data, however, indicate that receptors built on this scaffold exhibit higher Krel values compared to previous groups, which is quite promising. In addition, recently developed efficient post-functionalization of the lariat arm after the macrocyclization step45 makes multiple analogues of these receptors synthetically available, which will eventually allow fine-tuning of this chloride-lariat arm interaction to obtain even higher selectivity.
Cryptands (group IV)
The last class of compounds in our set consists of structurally rigid cryptands Cr1–Cr833–41 constructed from either dipicolinic or isophthalic acids connected by either hexa-substituted benzenes or bis- or tris-amines, and equipped with six (Cr1,33Cr2,34Cr3,35,36Cr4,36 and Cr537,38), eight (Cr639 and Cr740), and twelve (Cr8)41 amide groups (see Fig. 2a for structures).Nearly all receptors of this set exhibit moderate selectivity for chloride expressed by Krel values being close to zero or slightly positive. From this set, the best selectivities were observed for smaller cryptands Cr1–Cr3, whereas expanded cryptands Cr5, Cr6, and Cr8 gave only moderate selectivities.
Two cryptands (Cr4 and Cr7) simply follow the Hofmeister series. The thioamidic host Cr4 is less efficient than the analogous amidic Cr3, which supports the assumption that substitution of oxygen with sulfur has either a small (compare M1vs.M5) or even a negative impact (compare Cr2vs.Cr3) on Krel.
The observation than host Cr2, constructed from isophthalic binding units, is more selective than its analog Cr3 with dipicolinic moieties can be explained by assuming that the three isophthalic units in Cr2 provide additional CH⋯Cl interactions for chloride binding. An alternative explanation assume that for the Cr2 no intramolecular H-bonds, that preorganize the 2,6-pyridinedicarboxamide units in Cr3, need to be cleaved prior to anion complex formation to occur.
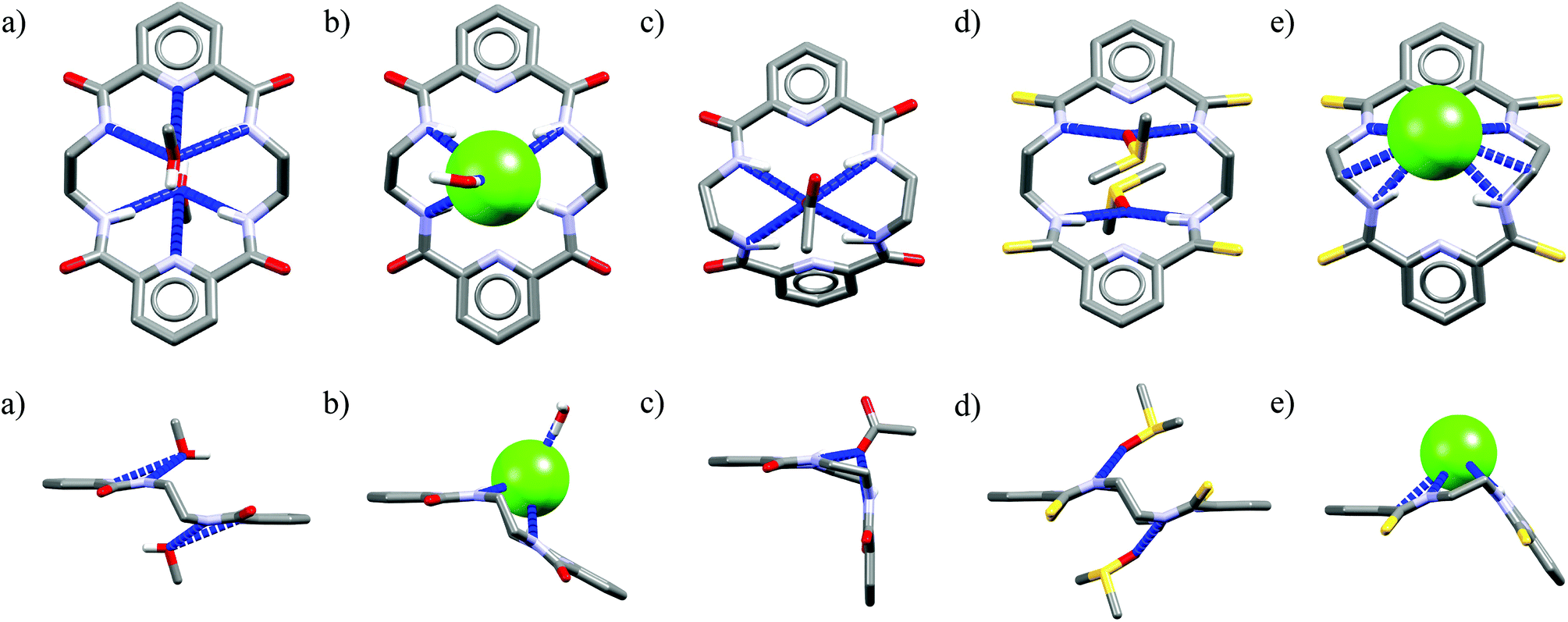
Crystal structure analysis for solvate and two anion complexes of Cr1 nicely demonstrate that the spherical Cl− cannot penetrate the cryptand cavity (Fig. 9b). In fact, two chloride anions sit at the cryptand's surface, quite similarly to the mode observed for macrocyclic hosts (Fig. 4 and 6).
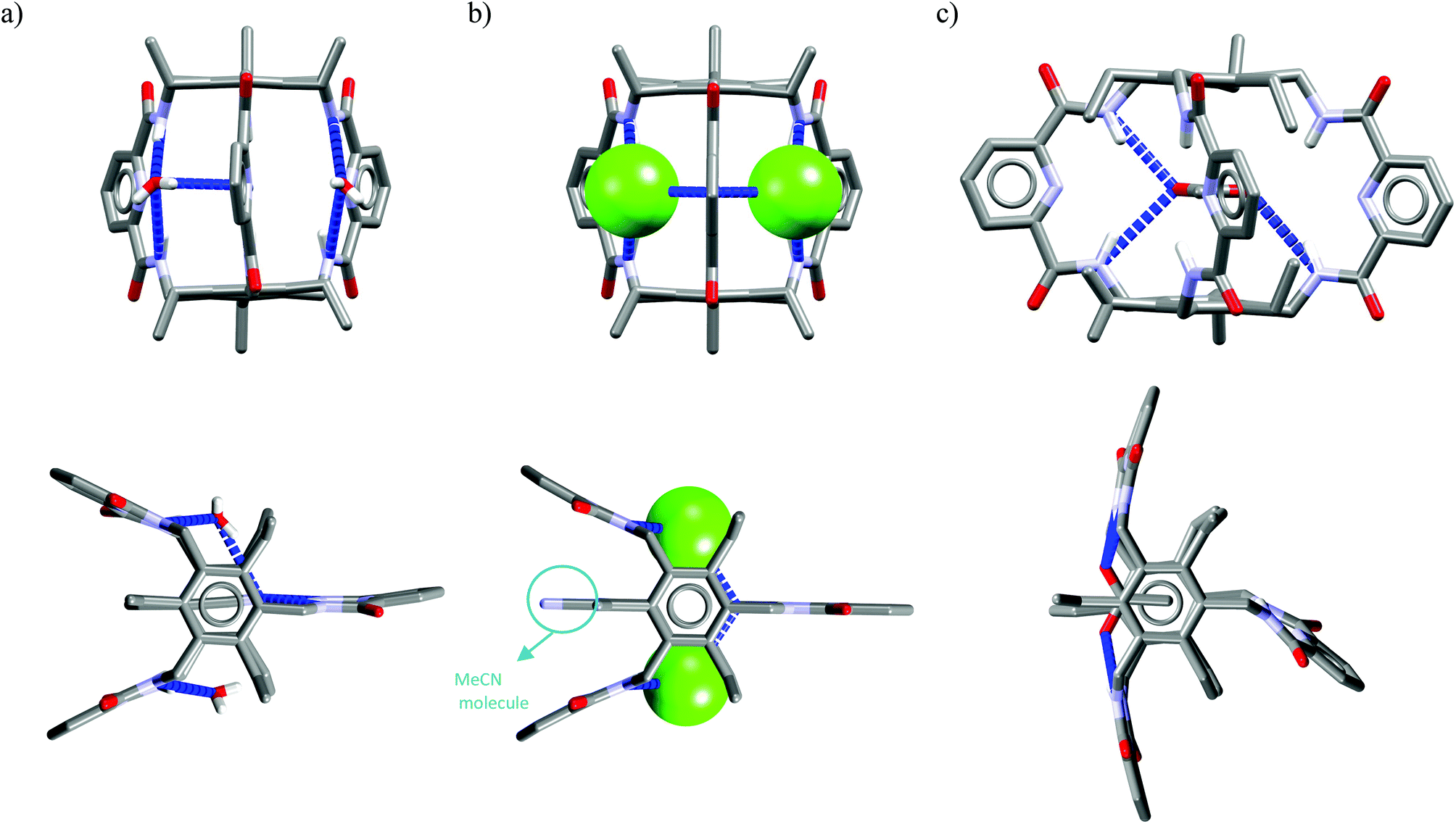 | ||
| Fig. 9 Crystal structures of the Cr1 solvate (a), and chloride (b) and acetate (c) complexes. | ||
In contrast, trigonal acetate satisfy the geometrical requirements of cavity of Cr1, which is able to accommodate either small neutral solvent molecule (Fig. 9a) or aliphatic moiety of anionic guest (Fig. 9c).
The spherical chloride, in contrast to larger H2PO4−, could potentially accommodate between benzene rings of cryptand cavity, however, in such an arrangement formation of all potential six H-bonds provided by the three neighboring 2,6-pyridinedicarboxamide units is simply impossible (dN⋯Cl− would be 4.15 Å as calculated for Cl− sitting in the centroid of cryptand from Cr1·TBACl X-ray structure). For this reason, it locates between two pyridines on the peripherally side of the cryptand via four H-bonds (dN⋯Cl− = 3.20 Å, ≤154°).
Interestingly, much like in the case of the structure of Cr1 solvate (Fig. 9a), the third dipicolinic unit binds to a water molecule which is also hydrogen-bonded to chloride (dO⋯Cl = 3.24 Å), thus enabling the binding of second chloride (dCl⋯Cl = 5.42 Å), at least in the solid state. In contrast to Cr1 solvate, the MeCN molecule is bound in the cleft between carbamoyl pyridine units in a reverse manner that facilitates the weak non-covalent interactions between negatively charged chlorides and positively charged methyl group of MeCN (d = 3.70 Å). Moreover, the small distance between hexasubstituted benzenes and MeCN molecule (d = 3.47 Å) possibly indicates the occurrence of hydrophobic interactions. On the other hand, in contrast to chloride complex, the acetate is fully encapsulated via four H-bonds and weak CH interactions within the cryptand cavity of Cr1 in a similar way as MeCN molecule, except the methyl group of acetate locates almost perfectly between centroids of hexasubstituted benzenes (d = 3.42 Å). To diminish repulsion between negative charge of anion and benzene rings, the carboxylate fragment is faced outwards cryptand cavity. These size- and geometry-sensitive hydrophobic interactions might explain the favorable binding of acetate over chloride in solution.
Interestingly, as depicted in Fig. 10a, the cryptand Cr5 exhibits similar binding behavior with chloride as Cr1, i.e. it utilizes a water molecule to mediate strong and unsymmetrical binding of two chlorides (dN⋯Cl− = 3.15–3.25 Å, ≤157–170°) on the peripheral domains of the cryptand via just three H-bonds from the 2,6-pyridinedicarboxamide units (dN⋯Cl = 3.23–3.36 Å, ≤140–146°).
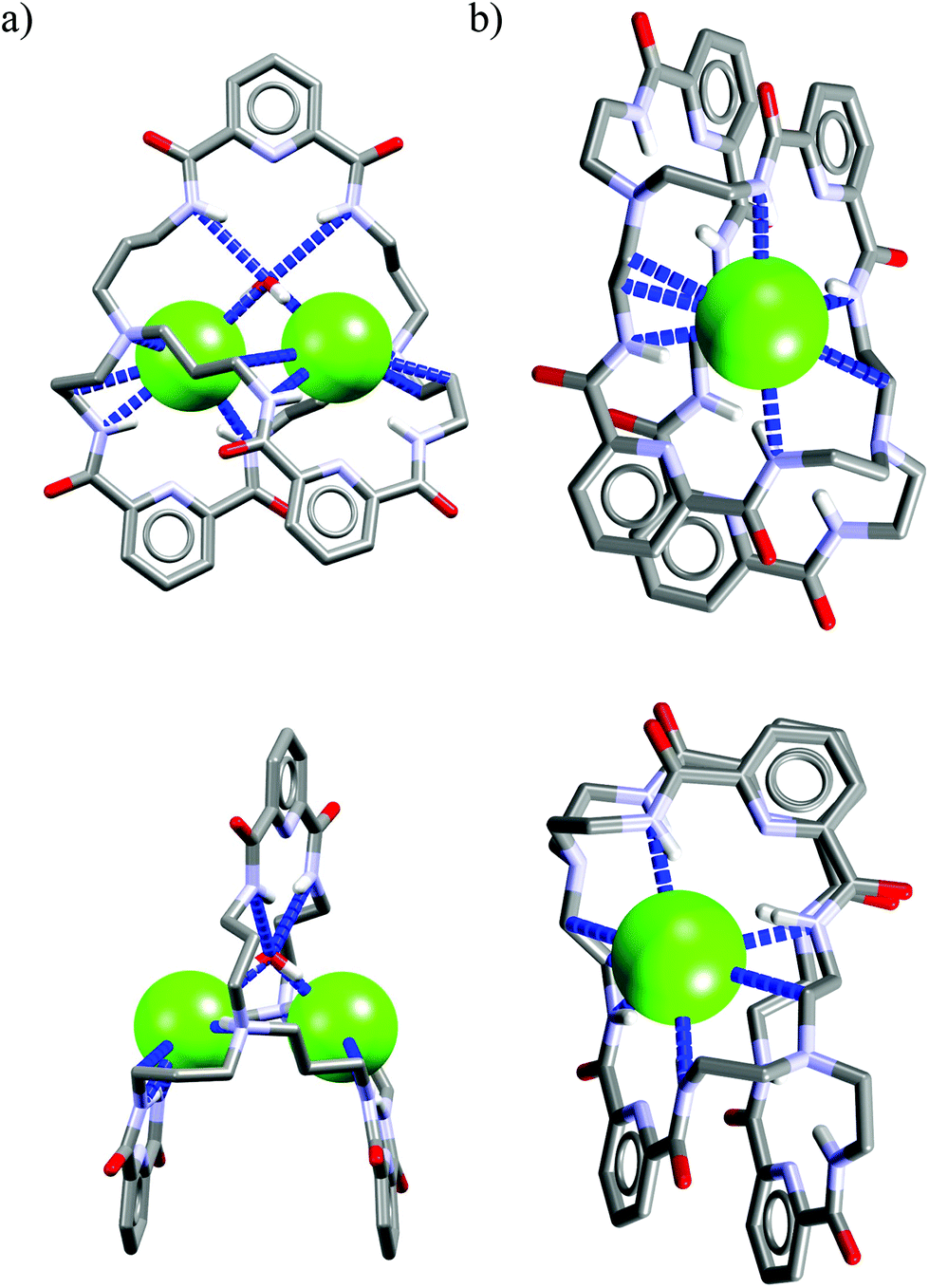 | ||
| Fig. 10 Crystal structures of the complexes of Cr5 (a) and Cr7 (c) with chloride. | ||
In addition, binding of chlorides is strengthened and interatomic distance is shortened (dCl⋯Cl = 4.95 Å) by two strong electrostatic interactions originating from protonated nitrogen atoms of tris(2-aminoethyl)amine linkers (dN⋯Cl = 3.16–3.19 Å, ≤156–168°).
The reasons why cryptand Cr7 is non selective for Cl− in solution cannot be easily rationalized on the basis of solid-state structure analysis of its chloride complex (Fig. 10b).
More specifically, cryptand Cr7, in contrast to Cr1 and Cr5, can bind only one chloride by the array of four strong H-bonds (dN⋯Cl− = 3.26–3.28 Å, ≤143–148°), and interestingly, water does not participate in this interaction, despite the fact that five water molecules are present in the elemental cell. Moreover, the apical positions of chloride are not saturated, although they are limited by the second macroring of the same cryptand and two neighboring cryptands, hence the chloride is not in direct contact with tetraethylammonium cation. Nevertheless, solution and crystal data for cryptands Cr1–Cr8, suggest that these bimacrocyclic structures are inappropriate for achieving high chloride selectivity, since in neither case is there evidence of chloride encapsulation within the cryptand cavity, and this suggests that complexation occurs primary in the cleft-binding sites.
Although these hosts are equipped with multiple H-bond donors, the rigid cryptand scaffold prevents these donors from creating a sphere-like cavity that is complementary to the chloride ionic radius. Therefore, it is conceptually similar to the binding behavior of simple macrocyclic receptors described in the preceding section. Moreover, synthesis of cryptands is rather troublesome and structures of these hosts do not easily allow for further modification required for the tuning of chloride binding selectivity.
4. Literature case-reports
In this section, we consider the cases reported in the literature of electrically neutral chloride-selective receptors which are not based on meta aromatic diamides. This set shown in Fig. 11 includes four diverse acycles (Ac5–Ac8),47–50 five macrocycles (M21–M25),51–53 and five structurally similar cryptands (Cr9–Cr13).54,55 Furthermore, at the end of section we briefly discuss bambus[6]uril (M26)56 and biotin[6]uril (M27),57 the new members of cucurbituril family able to efficiently encapsulate weakly basic chaotropic anions.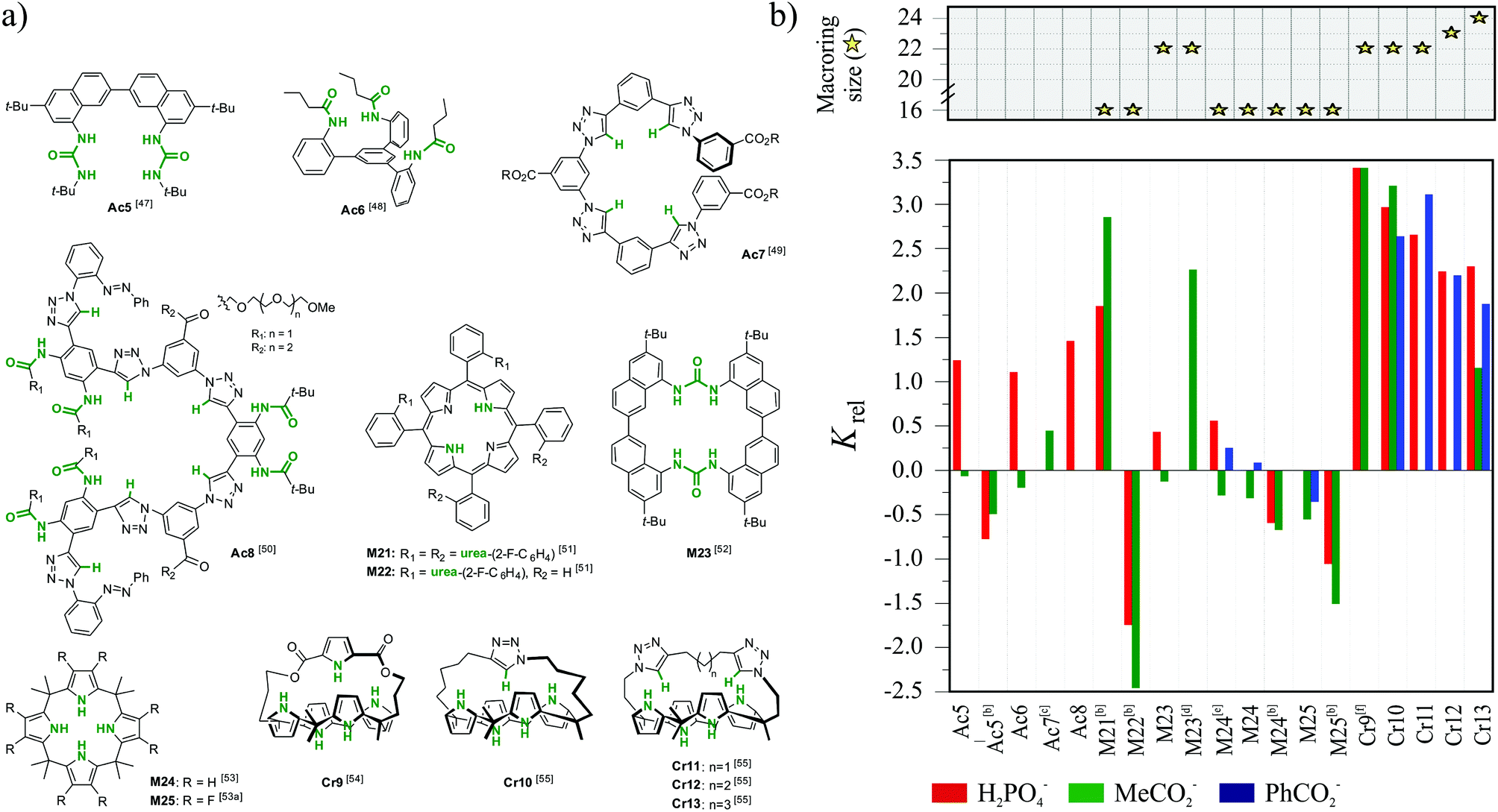 | ||
| Fig. 11 (a) Structures for the literature case-reports exhibiting selectivity for chloride and (b) their relative binding affinities (Krel) for chloride in MeCN; Krel = log(Kanion/KCl−) ≥ 0 indicates preference for chloride over other anions; for details of the titration data see ESI;† [b] DMSO; [c] Acetone; [d] MeCN + 5% H2O; [e] CD2Cl2; [f] DMSO + 20% H2O. | ||
Acycle Ac547 can be considered a monomer equivalent of macrocycle M23,52 therefore we decided to analyze them together. The very simple Ac5 equipped with just two urea moieties already exhibits anti-Hofmeister selectivity. From the comparison of Krel for Ac5vs.M23 it is clearly evident that the latter benefits from the macrocyclic effect. In addition, the urea groups seem to be too close to each other to enable cooperative binding with larger anions such as MeCO2− and H2PO4−. Quite interestingly, an increase in the solvent polarity renders distinct changes in selectivity for chloride over H2PO4− for these two receptors. Namely, the acyclic Ac5 completely lost selectivity for chloride when MeCN was changed to the more competitive DMSO, whereas in case of macrocyclic M23 improved selectivity for chloride is observed when going from MeCN to a MeCN/H2O (95:5 v/v) solvent mixture (similarly as for M6 and M9).
Analysis of the crystal structure of chloride complex of M23 reveals that anionic guest is bound in a rigid and structurally preorganized cavity by four short H-bonds (dN⋯Cl− = 3.24–3.30 Å, ≤156–164°) delivered from the two urea groups. The negative charge is balanced via CH⋯Cl− interactions from the two TBA cations which are placed above, and below the macrocyclic ring. The two urea groups are not coplanar but twisted by 45°, which assures good encapsulation of the guest and restricts its interaction with solvent molecules. This twist is attributed to conformational restrictions of this macrocyclic scaffold (compare the structures of uncomplexed vs. complexed receptor shown in Fig. 12). These results also demonstrate that urea groups, when constrained in an appropriate rigid structure, might alter their natural preference for Y-shaped carboxylates.
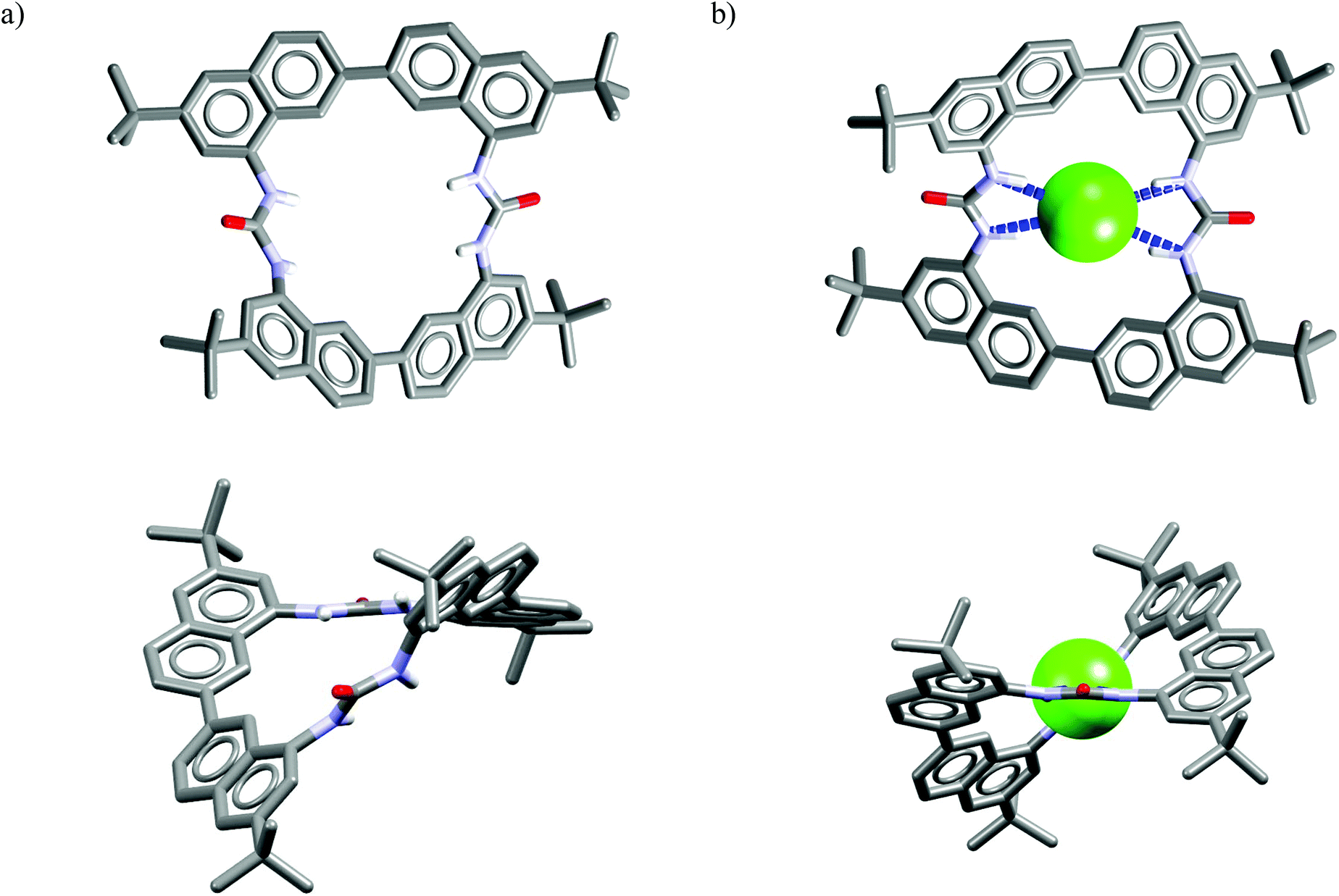 | ||
| Fig. 12 Crystal structures of the free (a) and chloride complex (b) of the M23. | ||
The C3-symmetric host Ac6 binds chloride more strongly than H2PO4−, whereas affinity for MeCO2− is slightly higher than for Cl−.48 The anion binding is likely realized by three cooperative H-bonds from ortho-phenyl-butanamide tethers, and the bottom apical position of chloride is limited by hydrophobic 1,3,5-trisubstituted benzene.
The following acycles Ac749 and Ac850 exploit the diaryl-1,2,3-triazole units which provide moderately polarized C–H bonds for the binding of anions. The ionic radius of the anion is postulated to be the primary factor governing the complex formation, when multiple triazole binding sites are incorporated within the receptor framework.
In both Ac7 and Ac8 the chloride binding triggers a pro-helical conformation of the oligomer that creates a deep electropositive cavity for this particular anion. The crystal structure of sodium chloride complex with acyclic receptor Ac8 depicted in Fig. 13 is particularly interesting since it reveals that chloride is totally encapsulated within electropositive hydrophobic cavity of the helical foldameric structure of Ac8. It is postulated that initial recognition of chloride by Ac8 triggers the folding of its structure into a highly concaved foldamer to maximize the interaction with chloride.
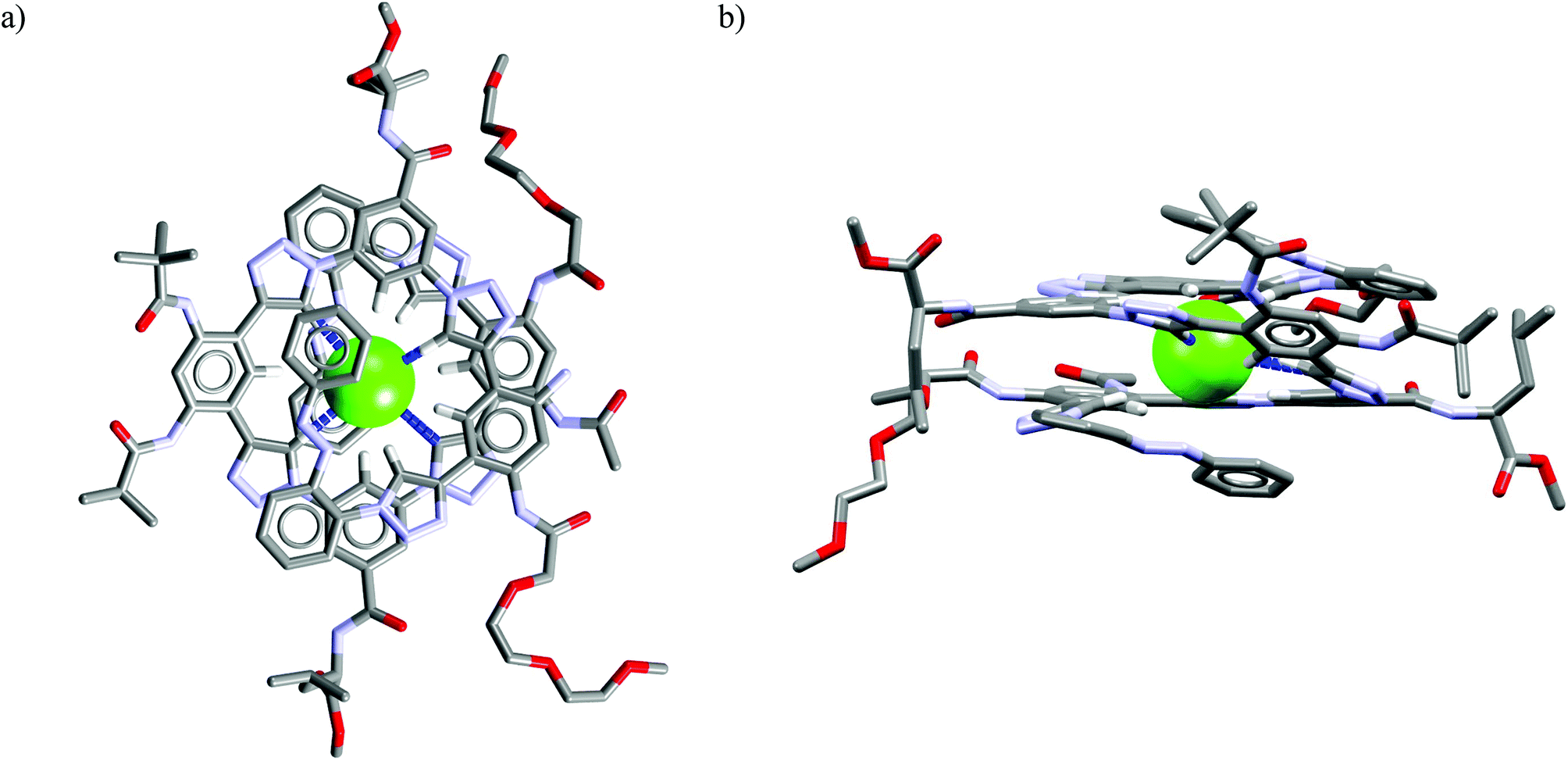 | ||
| Fig. 13 Front (a) and side (b) views of the crystal structure of the chloride complex of Ac8. | ||
Although the 1,2,3-triazole moieties form considerably longer interaction with chloride than receptors employing amide or urea groups (typical dN⋯Cl− are 3.4–3.5 Å and 3.2–3.3 Å, respectively), it is clearly evident that they exhibit an inherent preference for halogens, which might be exploited for the construction of successful chloride-selective receptors.
The macrocycles M21 and M22 are based on a rigid porphyrin core decorated with four (M21) and two (M22) urea-2-fluorophenyl tethers.51M21 exhibits pronounced selectivity for chloride over H2PO4− and MeCO2−, whereas M22, lacking two urea binding sites, exhibits natural Hofmeister binding selectivity. The crystal structure for M21/TBACl complex depicted in Fig. 14 indicates that only two urea-aromatic tethers are employed for the binding of chloride via four H-bond interactions (two of them are particularly strong: dN⋯Cl− = 3.14–3.15 Å, ≤155–171° and two are considerably weaker: dN⋯Cl− = 3.45–3.46 Å, ≤151–153°), whereas the two remaining ones are engaged in binding of two DMSO molecules (dN⋯O = 2.82–2.92 Å, ≤141–160°). What more, the presence of DMSO molecules is crucial for the observed anti-Hofmeister selectivity, since in a pure CD2Cl2 the receptor M21 exhibits usual binding selectivity: H2PO4−, MeCO2− ≫ Cl−. Moreover, crystallization of M21 without the presence of DMSO produces a very different complex in which two chloride ions are bound by two adjacent urea binding sites. Although the apical position of chloride is blocked by the porphyrin ring and two phenyl-substituted residues, access to the chloride is free from the side parallel to the porphyrin core. The negative charge is thus balanced by a TBA cation via close CH⋯Cl− contacts.
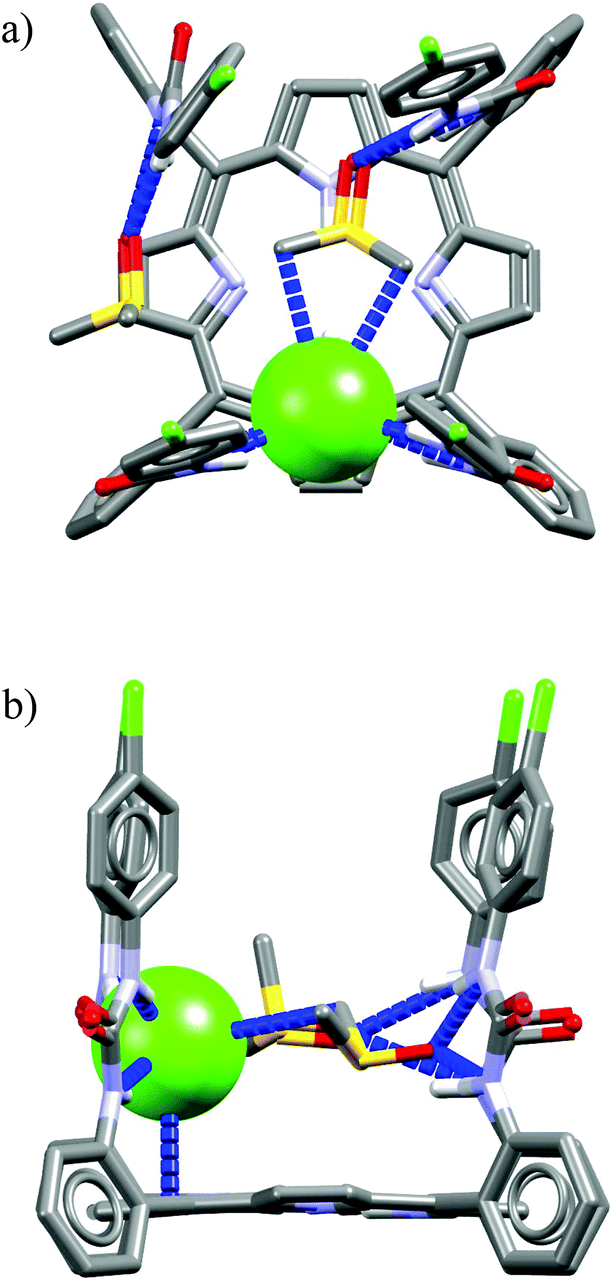 | ||
| Fig. 14 Front (a) and side (b) views of the crystal structure of the chloride complex of M21. | ||
The pivotal role of DMSO binding may be rationalized by the presence of hydrophobic interactions between highly polarized methyl groups of DMSO58 and electron cloud of chloride, which is potentially slightly deformed from an ideal sphere by the unsymmetrical interactions with urea NH protons.
The next macrocycles in this set are calix[4]pyrrole M2453 and its octafluoro-substituted analog M25.53a The M24 exhibits selectivity for chloride over the dihydrogenphosphate and benzoate, but not acetate, in CH2Cl2.53c,d The selectivity is lower in MeCN53b and is completely lost in a more competitive DMSO.53a According to the literature this may be attributed to presence of residual water and stronger solvation of salts in the latter solvents.59 On the other hand, although fluorinated M25 bind anions with increased affinity than M24, it is completely unselective for chloride, neither in MeCN nor in DMSO.53a The reason of lack of selectivity of M25 is not fully understood on the basis of their crystal structures, since the corresponding hydrogen bond parameters (dN⋯Cl− = 3.26–3.33 Å vs. 3.28–3.34 Å for chloride complexes of M24 and M25, respectively) and crystal structures are near identical as can be deduced from Fig. 15.
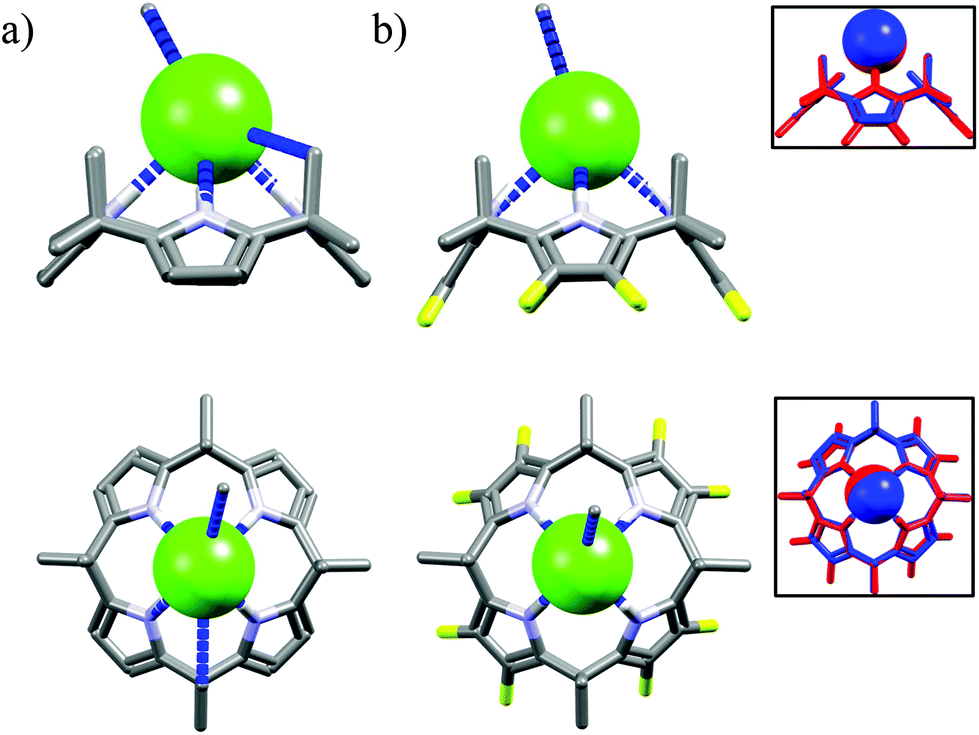 | ||
| Fig. 15 Crystal structures of the chloride complexes of the M24 (a) and M25 (b), inset: superposition of chloride complexes of M24 (blue) and M25 (red). | ||
Further functionalization of the calix[4]pyrrole core by strapping by either pyrrole (Cr9),54 or with one (Cr10) or two (Cr11–Cr13)55 1,2,3-triazole moieties allows preparation of the very effective cryptand-like compounds. Among all the receptors analyzed, they exhibit the best selectivities for chloride over both dihydrogenphosphate and carboxylates. Of this group, the 22-membered cryptands Cr9–Cr11 are slightly better than the larger Cr12 and Cr13. The chloride selectivity of Cr9–Cr13 likely originates from the binding properties of their components, since as was mentioned above, the parent calix[4]pyrrole53b–d as well as oligomers of 1,2,3-triazoles29,60,61 are known to form complexes with enhanced stabilities with chloride and fluoride. A combination of these binding sites raises the chloride selectivity to a considerably higher level, as is nicely exemplified in the structures of solvates and chloride complexes of Cr12 and Cr13 (Fig. 16). The structures of solvates of these cryptands demonstrate that they are quite well preorganized, since they exhibit rather small conformational deviations upon encapsulation of chloride. The calix[4]pyrrole macroring, however, adopts a cone conformation in which all four NH pyrrole protons are faced inside the cavity, just above the macroring plane. Likewise, 1,2,3-triazole moieties also slightly reorganize to direct their CH proton toward the cavity (Fig. 16b and d). As a result, the chloride anion is tightly bound by eight interactions (very similar for both Cr12 and Cr13, hence only values for Cr12 are provided), half of which are strong and highly directional H-bonds (dN⋯Cl− = 3.27–3.32 Å, ≤168–178°), the other half are weaker and more diffused C–H bonds originating from 1,2,3-triazoles (dCH⋯Cl− = 3.63–3.67 Å) and aliphatic linkers (dCH⋯Cl− = 3.66 Å).
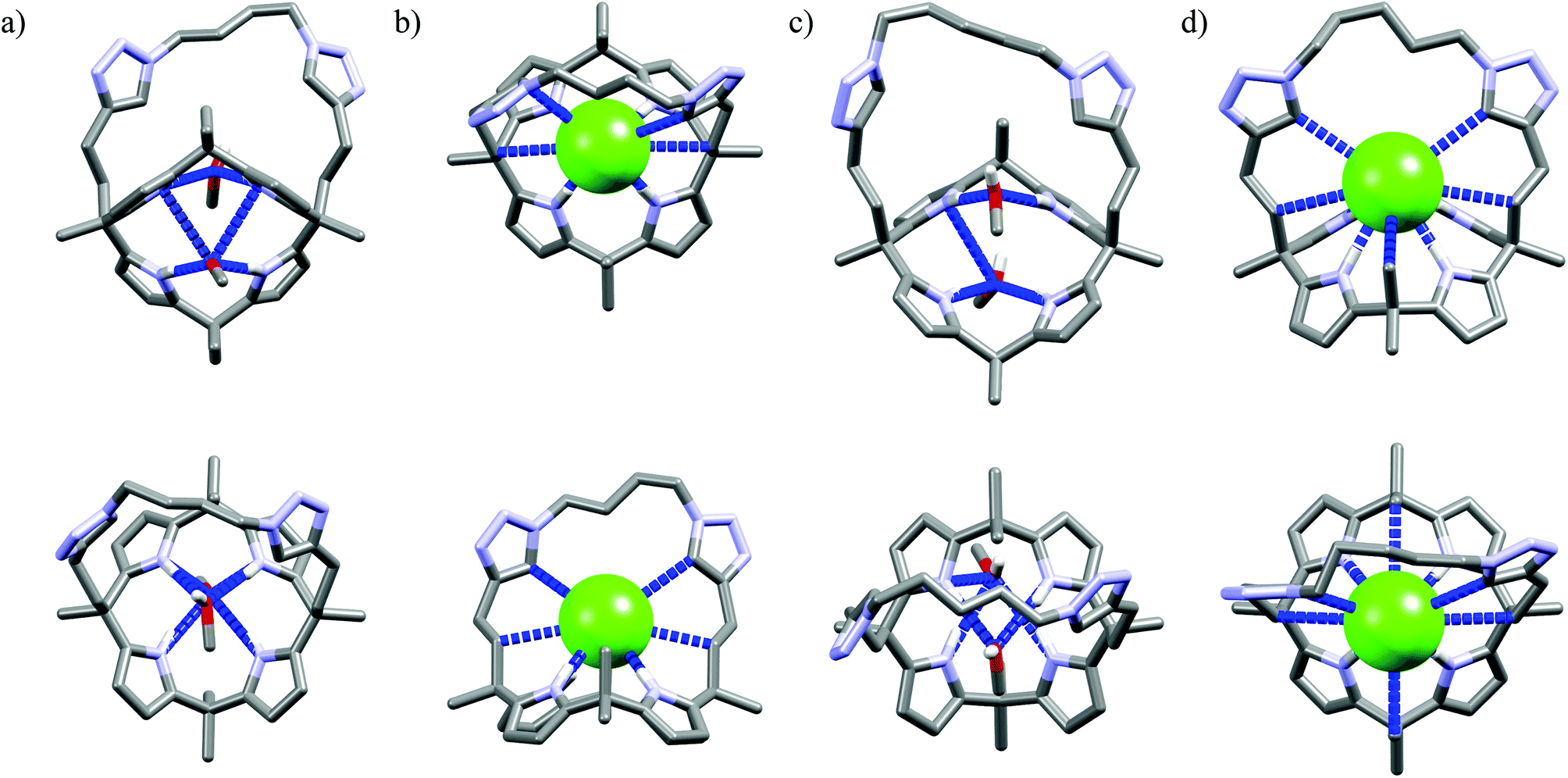 | ||
| Fig. 16 Crystal structures of the free (a, c) and chloride complexes (b, d) of the cryptand Cr12 and Cr13, respectively. Non-acidic protons, counterions, and disorder have been omitted for clarity. | ||
The last analyzed compounds are cucurbituril-like macrocycles bambus[6]uril (M26)56 and biotin[6]uril (M27) (Fig. 17).57
 | ||
| Fig. 17 Structures of the cucurbituril analogues. | ||
Unusual binding properties of these high-molecular weight compounds which rely exclusively on polarized C(sp3)–H close-contacts certainly diminish any interaction with more basic and strongly solvated anions, especially in aqueous environments. Although direct binding data for either carboxylates or dihydrogenphosphate are not available, one should note that affinities of M26 and M27 were measured in phosphate buffer, therefore with a high excess of competing anion. For example, the M26 is able to selectively recognize chloride (Ka = 910 M−1) over significantly more basic fluoride (Ka = 110 M−1) in D2O, buffered with K2DPO4 at pH 7.1.
However, more lipophilic anions such as Br−, NO3−, and I− are bound with even higher affinities (Ka = 1.4 × 105, 4.8 × 105, and 1.0 × 107 M−1, respectively). Nevertheless, experimental evidences, in particular negative entropy of binding determined in ITC measurements, suggest that expulsion of the weakly bound D2PO4− from the host cavity is required prior to binding of these chaotropic anions.57a
The analysis of the crystal structure of chloride complex with analog of M26 depicted in Fig. 18 suggest that high binding affinity for lipophilic anionic guests results from the full isolation of the bound anion from the water molecules and counteraction by its deep inclusion in the receptor's hydrophobic cavity having electropositive character.56b The anion is stabilized by multiple weak C(sp3)H⋯Cl− hydrogen bonding interactions between the methine hydrogen atoms of the macrocycle and the anion (dC⋯Cl− = 3.41–3.69 Å). The macrocycle M27 bind soft and weakly-coordinating anions in a similar fashion as M26, albeit with considerably lower affinity.56b,c,57a Nevertheless, it is able to recognize chloride with moderate affinity (Ka = 32–63 M−1 for NaCl) over the phosphates (H2PO4−, HPO42−), carbonate (CO32−), and sulphate (SO42−) both in phosphate and carbonate buffered-water solutions. Similarly as M26, the negative entropy of binding was determined from ITC experiments, however, for this macrocycle it was attributed to the release of two water molecules from the macrocyclic cavity of M27 during complexation.
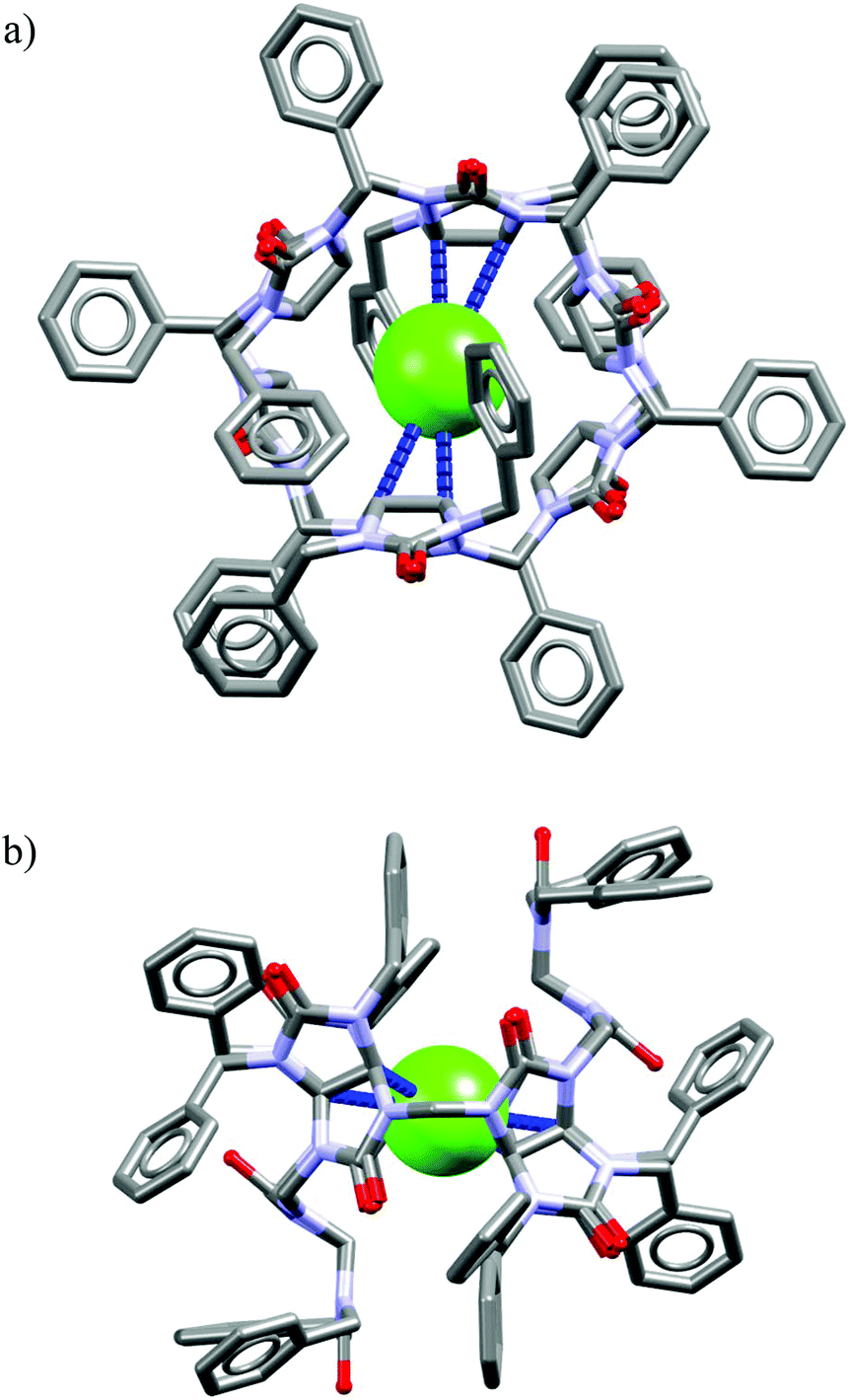 | ||
| Fig. 18 Front (a) and side (b) views of the crystal structure of the chloride complex of analog of M26 lacking carboxylic acid functions. | ||
5. Conclusions and future remarks
Chloride anion ranks far below H2PO4− and carboxylates in the Hofmeister series and its interactions with H-bond donors are relatively weak. Yet there are some features unique to chloride which can be utilized to achieve its selective complexation. Firstly, chloride is significantly smaller compared to H2PO4− and carboxylates. Moreover, it possesses a spherical shape and is capable of accepting multiple H-bond donors from different sites as already proved in the solid state.62 The remaining anions are significantly larger and are likely to accept H-bonding only at their specified side. Secondly, among this series chloride is characterized by the lowest enthalpy of hydration, i.e. it interacts fairly weakly with the solvent molecules, in particular in polar, protic solvents. In other words, its desolvation is relatively easy, which can be used as an advantage in the development of chloride-selective receptors.These considerations are in perfect agreement with the aforementioned principles concluded from the analysis of large set of receptors presented in this paper. We found that to achieve good selectivity for chloride, the following five criteria should be met:
1. The host possesses a rigid scaffold – preferentially a macrocyclic one,
2. The size of the binding pocket match the radius of chloride but is too small for competing anions,
3. Chloride is surrounded by the receptor's binding sites from all spatial directions, not just from one side or in one plane,
4. Chloride in the supramolecular complex is separated from all the solvent molecules and countercation,
5. Hydrophobic groups are close to the binding pocket and can aid chloride binding with hydrophobic effect (in water or polar solvents).
Among all the receptors which exhibit moderate to high Krel values at least three of these five postulates are fulfilled. For example Uc3 possess a rigid macrocyclic cavity of appropriate size with H-bond donors from multiple directions (postulates 1–3), while in the case of biotinurils or bambusurils the high selectivity is achieved by a perfect size of the rigid hydrophobic interior (postulates 1–2 and 4–5). Notably, fulfilling of all the five criteria is observed in the biological system of ClC protein, which is a strong indication of universal character of these criteria.
In this paper we examined 51 receptors in terms of selectivity for chloride versus either dihydrogen phosphate or carboxylates. Only about one-quarter these receptors are highly selective for chloride, but the unselective ones were likewise included in the analysis on solid grounds: only analysis of this full set, consisting largely of unselective hosts, could guide us to the final conclusions. Searching for regularities in receptor properties across quite systematic changes in structure, combined with analysis of multiple crystal structures, allowed us to hone in on the crucial structural features. A subset of ‘case study’ receptors, reported in the literature as being particularly selective, served as a confirmation of our hypotheses. This demonstrates the viability of the approach we took and indicates the strong need for large scale systematic research.
Large sets of experimental data can provide very useful guidelines, such as those found in this paper. We believe that they will be very useful in designing novel hosts, catalysts or transporters, and aide in understanding the biological systems. However, ‘Chem-is-try!’ and all novel systems, even built according to the criteria presented herein, will always need to be evaluated.
Acknowledgements
We would like to acknowledge financial support from Poland's National Science Centre (Project 2014/15/B/ST5/05038). FU acknowledge Foundation for Polish Science for financial support.References
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 24, 247–260 Search PubMed.
- R. Custelcean and B. A. Moyer, Eur. J. Inorg. Chem., 2007, 2007, 1321–1340 CrossRef.
- A. Salis and B. W. Ninham, Chem. Soc. Rev., 2014, 43, 7358–7377 RSC.
- Recent paper on direct measurement of properties of anions as H-bond donors, see: (a) S. J. Pike, J. J. Hutchinson and C. A. Hunter, J. Am. Chem. Soc., 2017, 139, 6700–6706 CrossRef CAS PubMed; This dependence is collaborated with multiple literature reports as well as our observations. For selected examples of relatively simple anion receptors exhibiting Hofmeister selectivity pattern, see: (b) D. E. Gross, V. Mikkilineni, V. M. Lynch and J. L. Sessler, Supramol. Chem., 2010, 22, 135–141 CrossRef CAS PubMed; (c) T. Zieliński, P. Dydio and J. Jurczak, Tetrahedron, 2008, 64, 568–574 CrossRef; (d) S. J. Brooks, P. A. Gale and M. E. Light, Chem. Commun., 2005, 4696–4698 RSC; (e) T. Zieliński and J. Jurczak, Tetrahedron, 2005, 61, 4081–4089 CrossRef; (f) M. J. Chmielewski, M. Charon and J. Jurczak, Org. Lett., 2004, 6, 3501–3504 CrossRef CAS PubMed; (g) S. Camiolo, P. A. Gale, M. B. Hursthouse and M. E. Light, Tetrahedron Lett., 2002, 43, 6995–6996 CrossRef CAS; (h) P. A. Gale, S. Camiolo, C. P. Chapman, M. E. Light and M. B. Hursthouse, Tetrahedron Lett., 2001, 42, 5095–5097 CrossRef CAS.
- Depending on the pH, either H2PO4− or HPO42− is the major component of this conjugate acid-base pair.
- A. Accardi and A. Picollo, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 1457–1464 CrossRef CAS PubMed.
- R. Planells-Cases and T. J. Jentsch, Biochim. Biophys. Acta, Mol. Basis Dis., 2009, 1792, 173–189 CrossRef CAS PubMed.
- R. Dutzler, E. B. Campbell, M. Cadene, B. T. Chait and R. MacKinnon, Nature, 2002, 415, 287–294 CrossRef CAS PubMed.
- For illustrative example, see: A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603–6782 CrossRef CAS PubMed.
- For reviews, see. (a) G. W. Gokel and S. Negin, Acc. Chem. Res., 2013, 46, 2824–2833 CrossRef CAS PubMed; (b) A. Vargas Jentzsch, A. Hennig, J. Mareda and S. Matile, Acc. Chem. Res., 2013, 46, 2791–2800 CrossRef CAS PubMed.
- For recent examples, see: (a) Y. Park, K. C. Harper, N. Kuhl, E. E. Kwan, R. Y. Liu and E. N. Jacobsen, Science, 2017, 355, 162–166 CrossRef CAS PubMed; (b) A. E. Metz, K. Ramalingam and M. C. Kozlowski, Tetrahedron Lett., 2015, 56, 5180–5184 CrossRef CAS PubMed.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995–2999 RSC.
- J. Jurczak, M. J. Chmielewski, P. Dydio, D. Lichosyt, F. Ulatowski and T. Zieliński, Pure Appl. Chem., 2011, 83, 1543–1554 CrossRef CAS.
- A. Szumna and J. Jurczak, Eur. J. Org. Chem., 2001, 4031–4039 CrossRef CAS.
- M. J. Chmielewski and J. Jurczak, Chem. – Eur. J., 2005, 11, 6080–6094 CrossRef CAS PubMed.
- M. J. Chmielewski and J. Jurczak, Chem. – Eur. J., 2006, 12, 7652–7667 CrossRef CAS PubMed.
- D. Lichosyt, P. Dydio and J. Jurczak, Chem. – Eur. J., 2016, 22, 17673–17680 CrossRef CAS PubMed.
- M. Chmielewski, PhD Thesis, Institute of Organic Chemistry PAS, Warsaw, 2003.
- J. Jurczak, P. Dydio, P. Stepniak and T. Zielinski, RSC Adv., 2016, 6, 41568–41571 RSC.
- Y. Inoue, T. Kanbara and T. Yamamoto, Tetrahedron Lett., 2003, 44, 5167–5169 CrossRef CAS.
- M. Chmielewski and J. Jurczak, Tetrahedron Lett., 2004, 45, 6007–6010 CrossRef CAS.
- M. J. Chmielewski, A. Szumna and J. Jurczak, Tetrahedron Lett., 2004, 45, 8699–8703 CrossRef CAS.
- M. J. Chmielewski and J. Jurczak, Tetrahedron Lett., 2005, 46, 3085–3088 CrossRef CAS.
- M. A. Hossain, S. O. Kang, D. Powell and K. Bowman-James, Inorg. Chem., 2003, 42, 1397–1399 CrossRef CAS PubMed.
- M. A. Hossain, J. M. Llinares, D. Powell and K. Bowman-James, Inorg. Chem., 2001, 40, 2936–2937 CrossRef CAS PubMed.
- I. V. Korendovych, M. Cho, O. V. Makhlynets, P. L. Butler, R. J. Staples and E. V. Rybak-Akimova, J. Org. Chem., 2008, 73, 4771–4782 CrossRef CAS PubMed.
- G. V. Kolesnikov, K. E. German, G. Kirakosyan, I. G. Tananaev, Y. A. Ustynyuk, V. N. Khrustalev and E. A. Katayev, Org. Biomol. Chem., 2011, 9, 7358–7364 CAS.
- J. L. Sessler, E. Katayev, G. D. Pantos and Y. A. Ustynyuk, Chem. Commun., 2004, 1276–1277 RSC.
- J. L. Sessler, E. Katayev, G. D. Pantos, P. Scherbakov, M. D. Reshetova, V. N. Khrustalev, V. M. Lynch and Y. A. Ustynyuk, J. Am. Chem. Soc., 2005, 127, 11442–11446 CrossRef CAS PubMed.
- M. Pawlaki, PhD Thesis, Institute of Organic Chemistry PAS, Warsaw, 2007.
- K. Dąbrowa, M. Pawlak, P. Duszewski and J. Jurczak, Org. Lett., 2012, 14, 6298–6301 CrossRef PubMed.
- K. Dąbrowa, M. Ceborska and J. Jurczak, Cryst. Growth Des., 2014, 14, 4906–4910 Search PubMed.
- A. P. Bisson, V. M. Lynch, M.-K. C. Monahan and E. V. Anslyn, Angew. Chem., Int. Ed. Engl., 1997, 36, 2340–2342 CrossRef CAS.
- S. O. Kang, M. A. Hossain and K. Bowman-James, Coord. Chem. Rev., 2006, 250, 3038–3052 CrossRef CAS.
- S. O. Kang, J. M. Llinares, D. Powell, D. VanderVelde and K. Bowman-James, J. Am. Chem. Soc., 2003, 125, 10152–10153 CrossRef CAS PubMed.
- M. A. Hossain, S. O. Kang, J. M. Llinares, D. Powell and K. Bowman-James, Inorg. Chem., 2003, 42, 5043–5045 CrossRef CAS PubMed.
- S. O. Kang, D. Powell and K. Bowman-James, J. Am. Chem. Soc., 2005, 127, 13478–13479 CrossRef CAS PubMed.
- S. O. Kang, J. M. Llinares, V. W. Day and K. Bowman-James, Chem. Soc. Rev., 2010, 39, 3980–4003 RSC.
- Q.-Q. Wang, R. A. Begum, V. W. Day and K. Bowman-James, Polyhedron, 2013, 52, 515–523 CrossRef CAS.
- S. O. Kang, D. Powell, V. W. Day and K. Bowman-James, Angew. Chem., Int. Ed., 2006, 45, 1921–1925 CrossRef CAS PubMed.
- Q.-Q. Wang, V. W. Day and K. Bowman-James, Angew. Chem., Int. Ed., 2012, 51, 2119–2123 CrossRef CAS PubMed.
- A. H. Flood, Beilstein J. Org. Chem., 2016, 12, 611–627 CrossRef CAS PubMed.
- (a) V. Martí-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Chem. Rev., 2015, 115(16), 8736–8834 CrossRef PubMed; (b) F. Diederich, P. J. Stang and R. R. Tykwinski, Modern supramolecular chemistry: strategies for macrocycle synthesis, John Wiley & Sons, 2008 Search PubMed.
- F. Sommer and S. Kubik, Org. Biomol. Chem., 2014, 12, 8851–8860 CAS.
- K. Dabrowa, P. Niedbala, M. Majdecki, P. Duszewski and J. Jurczak, Org. Lett., 2015, 17, 4774–4777 CrossRef CAS PubMed.
- K. Dąbrowa, M. Ceborska, M. Pawlak and J. Jurczak, Cryst. Growth Des., 2017, 17, 701–710 Search PubMed.
- S.-I. Kondo, M. Nagamine, S. Karasawa, M. Ishihara, M. Unno and Y. Yano, Tetrahedron, 2011, 67, 943–950 CrossRef CAS.
- P. Piątek, Tetrahedron Lett., 2007, 48, 4427–4430 CrossRef.
- H. Juwarker, J. M. Lenhardt, J. C. Castillo, E. Zhao, S. Krishnamurthy, R. M. Jamiolkowski, K.-H. Kim and S. L. Craig, J. Org. Chem., 2009, 74, 8924–8934 CrossRef CAS PubMed.
- Y. Hua, Y. Liu, C.-H. Chen and A. H. Flood, J. Am. Chem. Soc., 2013, 135, 14401–14412 CrossRef CAS PubMed.
- K. Calderon-Kawasaki, S. Kularatne, Y. H. Li, B. C. Noll, W. R. Scheidt and D. H. Burns, J. Org. Chem., 2007, 72, 9081–9087 CrossRef CAS PubMed.
- A. Satake, Y. Ishizawa, H. Katagiri and S.-I. Kondo, J. Org. Chem., 2016, 81, 9848–9857 CrossRef CAS PubMed.
- (a) J. L. Sessler, W.-S. Cho, D. E. Gross, J. A. Shriver, V. M. Lynch and M. Marquez, J. Org. Chem., 2005, 70, 5982–5986 CrossRef CAS PubMed; (b) J. L. Sessler, D. An, W.-S. Cho and V. Lynch, J. Am. Chem. Soc., 2003, 125, 13646–13647 CrossRef CAS PubMed; (c) C. N. Warriner, P. A. Gale, M. E. Light and M. B. Hursthouse, Chem. Commun., 2003, 1810–1811 RSC; (d) P. A. Gale, J. L. Sessler, V. Král and V. Lynch, J. Am. Chem. Soc., 1996, 118, 5140–5141 CrossRef CAS.
- D.-W. Yoon, D. E. Gross, V. M. Lynch, C.-H. Lee, P. C. Bennett and J. L. Sessler, Chem. Commun., 2009, 1109–1111 RSC.
- M. Yano, C. C. Tong, M. E. Light, F. P. Schmidtchen and P. A. Gale, Org. Biomol. Chem., 2010, 8, 4356–4363 CAS.
- (a) M. Lisbjerg, H. Valkenier, B. M. Jessen, H. Al-Kerdi, A. P. Davis and M. Pittelkow, J. Am. Chem. Soc., 2015, 137, 4948–4951 CrossRef CAS PubMed; (b) M. Lisbjerg, B. E. Nielsen, B. O. Milhøj, S. P. Sauer and M. Pittelkow, Org. Biomol. Chem., 2015, 13, 369–373 RSC; (c) M. Lisbjerg, B. M. Jessen, B. Rasmussen, B. E. Nielsen, A. Ø. Madsen and M. Pittelkow, Chem. Sci., 2014, 5, 2647–2650 RSC.
- (a) M. A. Yawer, V. Havel and V. Sindelar, Angew. Chem., Int. Ed., 2015, 54, 276–279 CrossRef CAS PubMed; (b) V. Havel, M. A. Yawer and V. Sindelar, Chem. Commun., 2015, 51, 4666–4669 RSC; (c) V. Havel, J. Svec, M. Wimmerova, M. Dusek, M. Pojarova and V. Sindelar, Org. Lett., 2011, 13, 4000–4003 CrossRef CAS PubMed.
- T. Clark, J. Murray, P. Lane and P. Politzer, J. Mol. Model., 2008, 14, 689–697 CrossRef CAS PubMed.
- (a) J. R. Blas, M. Márquez, J. L. Sessler, F. J. Luque and M. Orozco, J. Am. Chem. Soc., 2002, 124, 12796–12805 CrossRef CAS PubMed; (b) F. P. Schmidtchen, Org. Lett., 2002, 4, 431–434 CrossRef CAS PubMed.
- P. A. Gale, J. L. Sessler, V. Král and V. Lynch, J. Am. Chem. Soc., 1996, 118, 5140–5141 CrossRef CAS.
- C.-H. Lee, J.-S. Lee, H.-K. Na, D.-W. Yoon, H. Miyaji, W.-S. Cho and J. L. Sessler, J. Org. Chem., 2005, 70, 2067–2074 CrossRef CAS PubMed.
- C. A. Ilioudis, K. S. Hancock, D. G. Georganopoulou and J. W. Steed, New J. Chem., 2000, 24, 787–798 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1534255 and 1534256. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ob01385j |
| This journal is © The Royal Society of Chemistry 2017 |