
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmphiphilic carbosilane dendrons as a novel synthetic platform toward micelle formation†
Carlos E.
Gutierrez-Ulloa
ad,
Marina Yu.
Buyanova
b,
Evgeny K.
Apartsin
 *b,
Alya G.
Venyaminova
b,
F. Javier
de la Mata
ad,
Mercedes
Valiente
*c and
Rafael
Gómez
*ad
*b,
Alya G.
Venyaminova
b,
F. Javier
de la Mata
ad,
Mercedes
Valiente
*c and
Rafael
Gómez
*ad
aDepartamento de Química Inorgánica, Universidad de Alcalá, Campus Universitario, Alcalá de Henares, Madrid, Spain. E-mail: rafael.gomez@uah.es; Tel: (+34) 91 8854685
bInstitute of Chemical Biology and Fundamental Medicine SB RAS, Novosibirsk, Russia. E-mail: eka@niboch.nsc.ru; Tel: (+7) 383 3635129
cDepartamento de Química Analítica, Química Física e Ingeniería Química, Universidad de Alcalá, Campus Universitario, Alcalá de Henares, Madrid, Spain. E-mail: mercedes.valiente@uah.es; Tel: (+34) 91 8854670
dNetworking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Madrid, Spain
First published on 22nd August 2017
Abstract
A novel family of amphiphilic ionic carbosilane dendrons containing fatty acids at the focal point were synthesized and characterized. They spontaneously self-assembled in aqueous solution into micelles both in the absence and presence of salt, as confirmed by surface tension, conductivity, and DLS measurements. Dendron based micelles have spherical shapes and increase in size on decreasing dendron generation. These dendritic micelles have been demonstrated to be able to form complexes with therapeutic macromolecules such as siRNA and show a high loading capacity for drugs such as procaine, suggesting their potential use as nanocarriers for therapeutics.
Introduction
In the last few decades, there has been increased interest in the use of nanoparticles in nanomedicine and, in particular, their use for drug delivery applications.1,2 Among them, special attention was given to the nanosized supramolecular assemblies. These soft nanoparticles are colloidal particles composed of one layer (micelles) or one or several bilayers (liposomes and vesicles) formed by the aggregation of amphiphilic precursors in water media.3 The particles formed are capable of encapsulating a cargo (a drug, a reporter or a stimuli-sensitive molecule) in the hydrophobic part or inside a bilayer. The generation of such supramolecular structures by the self-assembly of molecular building blocks is an attractive option. Hence, the use of amphiphilic block copolymers to form different types of nanoparticles like micelles, polymersomes, nanospheres, nanocapsules or polyplexes has been extensively explored.4,5 The composition, molecular geometry and relative block length of the copolymers are essential parameters for the formation of these nanoparticles. Another strategy is the use of amphiphilic dendrons or dendritic amphiphiles as building blocks towards the formation of such supramolecular entities.6,7 Dendritic molecules provide a high degree of precision with well-defined structures compared to polymers, along with a high local density of multiple functionalities (multivalency). In this way, dendritic assemblies have shown higher stabilities and lower critical micellar concentrations (CMCs) than those formed from small surfactants.8,9 The multivalency, as a consequence of their conic shape, allows multiple interactions with biomolecules and other substrates, therefore increasing the binding capability.10 The main condition to form supramolecular structures is an adequate balance between hydrophobicity and hydrophilicity. According to the previous work of Percec et al.,11 self-assembly patterns can be predicted by controlling the four parameters: size, shape, dendron surface chemistry and flexibility. With this in mind, two main strategies have been used for this purpose: the binding at the focal point of a dendron of (i) hydrophilic chains like PEG or (ii) hydrophobic chains like fatty acids or alkyl chains. With respect to the first approach, several examples were described. Hydrophobic dendrons with phenyl acetamide as end groups sensitive to enzymatic response have been prepared. They were able to form micellar containers that can disassemble and release the encapsulated cargo upon enzymatic activation.12 Another example was the gallic acid dendron to originate polyion complex (PIC) micelles. These micelles displayed enhanced stability toward ionic strength compared to conventional PIC micelles from linear copolymers, a fact ascribed to the more rigid dendritic architecture.13 Concerning the second approach, hydrophilic poly(amidoamine)14,15 or polylysine16 dendrons featuring hydrophobic tails of different length have been synthesized. The ordered micellar aggregation depended on the optimal balance between hydrophilicity and lipophilicity and in the case of polylysine structures, only dendrons with alkyl chain lengths above C12 afforded micelles. Spermine-containing dendrons with a variety of lipophilic units such as cholesterol have been used as a tunable synthetic platform for controlling DNA binding and transfection ability.17 In all these latter cases, hydrophilicity was provided by the whole dendron structure.A different dendritic typology is that based on the carbosilane structure which is hydrophobic, constituting interesting building blocks to be developed from a biomedical point of view.18,19 Silicon strongly increases the lipophilicity of the systems and facilitates their membrane crossing and bioavailability.20 Kim et al. reported on the behavior of amphiphilic linear block copolymers based on PEG and allyl-terminated carbosilane dendrons.21 For them, the formation of micellar aggregates highly depended on the size of the hydrophobic part (i.e. dendron generation). In the last few years, our research groups have been interested in the synthesis and biomedical applications of the dendritic carbosilane molecules. Cationic carbosilane dendrimers have been used as antibacterial22,23 and antiparasite24 agents and as carriers for short nucleic acids like siRNA or antisense oligonucleotides for transfection purposes.25,26 In the case of anionic dendrimers, these were applied as antiviral agents mainly for HIV infection.27,28 Recently, we also reported a synthetic platform of carbosilane dendrons29,30 able to dendronize nanostructured materials such as poly(D,L-lactide-co-glycolide) acid,31 mesoporous silica32 or gold33 NPs, as well as decorating their focal point with molecules of interest like ferrocene,34 DO3A (1,4,7,10-tetraazacyclodocecane-N,N′,N′′,N′′′-triacetic acid) chelator35 or drugs,36 among others.
In this study, we report on the synthesis of new amphiphilic systems formed from ionic-terminated carbosilane dendrons containing a hydrophobic tail at the focal point with two different lengths. In this particular case, the total hydrophobic contribution will be the result of the addition of both the tail and the dendritic structure, leaving only the terminal ionic groups of the dendrons as a major part for hydrophilicity. This situation gives rise to a different scenario within the dendritic amphiphile engineering towards the formation of new supramolecular assemblies for biomedical applications.
Results and discussion
Synthesis of amphiphilic dendrons
Carbosilane dendrons with a fatty acid at the focal point were synthesized from previously described dendrons containing a Br atom at the focal point and allyl or vinyl functions at the periphery.37 The employed nomenclature to describe these dendrons was of the type XGn(Y)m, where Gn stands for the dendritic generation and (Y)m for the peripheral function and its number, while X refers to the nature of the focal point (n = 1, m = 2 (G1); n = 2, m = 4 (G2); n = 3, m = 8 (G3)), (see Fig. 1).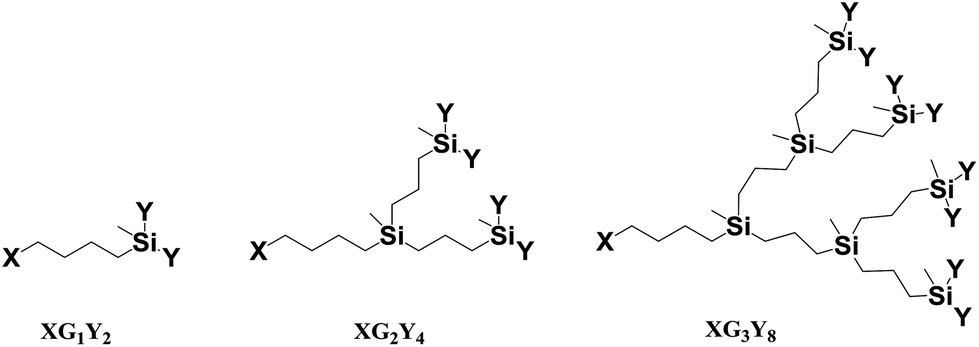 | ||
| Fig. 1 Structures of the dendrons described in this work. | ||
For the synthesis of ionic carbosilane dendrons, thiol–ene reactions were used. In the case of formation of sulfonate-terminated dendrons, allyl-terminated dendrons (BrGnAm) were employed as the starting materials for the reaction with the thiol derivatives containing sulfonate groups. However, in the case of ammonium-terminated dendrons, more reactive vinyl-terminated dendrons (BrGnVm) were necessary in order to proceed with the corresponding thiol derivative. In addition, two different fatty acids such as hexanoic (caproic) and hexadecanoic (palmitic) acids were used to study the implications of the chain length in the self-assembly processes. The coupling reaction of fatty acids with the carbosilane dendrons to replace the bromine atom by this new aliphatic chain was performed by ester bond formation. This procedure afforded new families of dendrons, with palmitic acid, C16GnAm (n = 1, m = 2 (1); n = 2, m = 4 (2); n = 3, m = 8 (3)) and C16GnVm (n = 1, m = 2 (4); n = 2, m = 4 (5); n = 3, m = 8 (6)), and with hexanoic acid, C6GnAm (n = 1, m = 2 (7); n = 2, m = 4 (8); n = 3, m = 8 (9)) and C6GnVm (n = 1, m = 2 (10); n = 2, m = 4 (11); n = 3, m = 8 (12)), all of them obtained as yellow oils in high yields (see Scheme 1).
 | ||
| Scheme 1 Synthesis of allyl- or vinyl-terminated dendrons with fatty acids at the focal point. | ||
Once allyl-terminated dendrons with adequate focal points were obtained, modification of their periphery via thiol–ene addition was accomplished. Thus, solutions of dendrons of the type C16GnAm (1–3) and C6GnAm (7–9) and sodium 3-mercapto-1-propanesulfonate in THF/MeOH/H2O were stirred for 1 h under UV light to afford the new dendritic wedges C16Gn(SO3Na)m (n = 1, m = 2 (13); n = 2, m = 4 (14); n = 3, m = 8 (15)) and C6Gn(SO3Na)m (n = 1, m = 2 (16); n = 2, m = 4 (17); n = 3, m = 8 (18)) (Scheme 2). All compounds were obtained as white solids in high yields.
 | ||
| Scheme 2 Synthesis of sulfonate-terminated dendrons with fatty acids at the focal point. | ||
Analogously, solutions of dendrons C16GnVm (4–6) or C6GnVm (10–12) and 2-dimethylethylenethiol hydrochloride, HS(CH2)2NMe2·HCl, in THF/MeOH were stirred under UV irradiation to afford the dendrons C16Gn(NMe2HCl)m (n = 1, m = 2 (19); n = 2, m = 4 (20); n = 3, m = 8 (21)), and C6Gn(NMe2HCl)m (n = 1, m = 2 (22); n = 2, m = 4 (23); n = 3, m = 8 (24)) as white solids in high yields (Scheme 3). These systems were neutralized with Na2CO3 to give the amine-terminated dendrons C16Gn(NMe2)m (n = 1, m = 2 (25); n = 2, m = 4 (26); n = 3, m = 8 (27)) and C6Gn(NMe2)m (n = 1, m = 2 (28); n = 2, m = 4 (29); n = 3, m = 8 (30)). After that, the latter systems were quaternized with MeI to obtain the dendritic wedges C16Gn(NMe3+I)m (n = 1, m = 2 (31); n = 2, m = 4 (32); n = 3, m = 8 (33)), and C6Gn(NMe3+I)m (n = 1, m = 2 (34); n = 2, m = 4 (35); n = 3, m = 8 (36)) (see Scheme 3). For all these reactions, the presence of 2,2′-dimethoxy-2-phenylacetophenone (DMPA) as a photoinitiator was necessary. The addition of the thiol to the allyl or vinyl groups was regioselective at the β-position (vide infra). All the anionic dendrons 13–18 and the cationic ones 19–24 and 31–36 were soluble in water.
 | ||
| Scheme 3 Synthesis of ammonium- or amine-terminated dendrons with fatty acids at the focal point. | ||
NMR spectroscopy confirmed the modification at the focal point from the –CH2Br group that showed one triplet at δ 3.40 in the 1H NMR spectra and one signal at δ 33.0 in the 13C NMR, to the new –CH2O– group that presented one triplet at δ 4.04 in the 1H NMR spectra and one signal at δ 63.8 in the 13C NMR (see the ESI† for spectra). One more signal at δ 173.9 in the 13C NMR corroborated the formation of the ester bond with respect to the –COOH group of the free fatty acids which appeared at δ 180.3. NMR spectroscopy, mass spectrometry (MS) and analytical data of all derivatives were consistent with their proposed structures. The presence of the new chain –Si(CH2)3S– in the anionic systems was confirmed by the presence of three multiplets at δ 0.53, 1.47 and 2.44 in the 1H NMR spectra. The outer chain –S(CH2)3SO3Na was observed as two multiplets at δ 2.52 and 2.85 for the protons of the –SCH2– and the –CH2SO3Na groups, respectively, and a multiplet at δ 1.89 for the inner methylene group of the chain (see Fig. S.4.3†). The –CH2– carbon atoms of the chain –Si(CH2)3S– were detected in the 13C NMR spectra at δ 12.8 for the –SiCH2– group, 23.9 for the middle –CH2– group, and δ 35.2 for the –CH2S– group. Those carbon atoms belonging to the chain –S(CH2)3SO3Na appeared at δ 30.3 for the –SCH2– group and δ 50.1 for the –CH2SO3Na group while the methylene group at the β position with respect to both S atoms was shown at δ 24.5 (see Fig. S.4.3 and S.4.4†). For the cationic compounds, the presence of the chain –Si(CH2)2S– was confirmed by 1H NMR, which showed two multiplets at δ 0.94 and 2.74, whereas the outer chain –S(CH2)2N– gave rise to two multiplets at δ 3.00 and 3.60 for the protons of the –SCH2– and –CH2N–, respectively. Furthermore, a singlet at δ 3.20 confirmed the presence of –NMe3+ (see Fig. S.4.9†). The carbon atoms of the –Si(CH2)2S– chain were observed in the 13C NMR spectra at δ 13.0 for the –SiCH2– group and at δ 29.4 for the –CH2S– group, whereas those belonging to the –S(CH2)2N– chain arose at δ 23.3 for the –SCH2– and δ 65.1 for the –CH2N– group. The –NMe3+ group was also detected at δ 53.6 by 13C NMR spectroscopy.
The structures of the new families of cationic and anionic carbosilane dendrons with fatty acids at the focal point are depicted in Fig. 2 and 3. To represent more clearly the structure–property relationship of these systems, the following notation was also used: CmGn+ or CmGn−, where Cm denotes the number of carbon atom of the aliphatic chain, Gn stands for the dendritic generation and + and − indicate the nature of the peripheral groups.
 | ||
| Fig. 2 Representative structures of anionic dendrons with palmitic or hexanoic acid at the focal points 13–18. | ||
 | ||
| Fig. 3 Representative structures of cationic dendrons with palmitic or hexanoic acid at the focal points 31–36. | ||
Self-assembly of amphiphilic dendrons into dendritic micelles
The amphiphilic behavior of ionic dendrons containing fatty acids at the focal point, 13–18 and 31–36, was studied towards the formation of micelles in aqueous solution without and with ionic strength. For that, specific conductivity and surface tension measurements were used, respectively. A study of the thermodynamics for micelle formation was carried out by conductivity measurements in deionized water at different amphiphilic dendron concentrations. The addition of the ionic dendron to an aqueous solution caused an increase in the number of charge carriers and, consequently, an increase in the conductivity. Since a micelle is much larger than a monomer, it diffuses more slowly throughout the solution, being therefore a less efficient charge carrier. Thus, a plot of conductivity vs. amphiphilic dendron concentration showed a break point at the so-called critical micellar concentration (CMC), as expected. Above the CMC, further addition of dendrons increased the micelle concentration while the amphiphilic monomer concentration remained approximately constant and equal to the CMC (see Fig. 4). | ||
| Fig. 4 Conductivity measurements of selected dendrons containing palmitic acid (13–15 (A) and 31–33 (B)) and hexanoic acid (36 and 18) (C) at the focal point. | ||
Both cationic and anionic systems of all generations containing palmitic acid at the focal point were able to form micelles with a CMC of around 240–110 μM. There were not big differences between the CMC of cationic and anionic amphiphiles with the same hydrophobic part (Table 1), indicating that the nature of the charged groups on the periphery appeared not to affect drastically the CMC values. However, among the ones bearing hexanoic acid at the focal point, only the third generation dendrons formed micelles. It should be noted that as a rule for amphiphilic dendrons, the amphiphilicity arises from the difference in hydrophilicity between the functional dendritic scaffold and the dendron tail. In this kind of dendron, the head groups are composed both for hydrophobic and hydrophilic parts. In amphiphilic carbosilane dendrons from the first to the third generation, the hydrophilicity increases due to the presence of a greater number of charged groups, but the lipophilicity also increases on increasing the carbosilane scaffold. Therefore, the behavior of the amphiphilic carbosilane dendrons in water medium is defined by the hydrophilic–lipophilic balance. In particular, the peculiarities of such balance in the dendrons C6G3 allow them to form micelles in water despite the short hydrophobic tail in the focal point.
| Dendron | CMC (μM) | Ionization degree (α) | Gibbs energy of micellization (kJ mol−1) | |||
|---|---|---|---|---|---|---|
| CmGn− | CmGn+ | CmGn− | CmGn+ | CmGn− | CmGn+ | |
| C16G1 | 240 ± 26 | 120 ± 10 | 0.69 ± 0.02 | 0.37 ± 0.02 | −41 | −65 |
| C16G2 | 140 ± 8 | 110 ± 10 | 0.78 ± 0.01 | 0.20 ± 0.02 | −127 | −130 |
| C16G3 | 140 ± 10 | 190 ± 6 | 0.79 ± 0.01 | 0.49 ± 0.01 | −255 | −250 |
| C6G3 | 200 ± 20 | 120 ± 20 | 0.53 ± 0.02 | 0.33 ± 0.01 | −202 | −258 |
The ionization degrees (α) of the micelles obtained from the ratio of slopes method of specific conductivity measurements along with the Gibbs energy estimation of micelle formation for the pseudo-phase separation model as ΔmicG° ≈ iRT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnXCMC are presented in Table 1. Anionic micelles of palmitic dendrons were highly ionized in water media (α = 0.7–0.8). In contrast, the ionization degrees were considerably lower (α = 0.2–0.5) for analogous cationic micelles indicating that these cationic dendrons assembled into micelles with smaller charge density than the homologous micelles consisting of anionic dendrons. The smaller values for cationic micelles could be related to the nature of counterion. The counterions used in these cases were I− for cationic micelles and Na+ for anionic micelles. Larger ions (I−) are more polarizable tending to be less hydrated, and subsequently binding more effectively on the micellar surface. In addition, the values of micellization Gibbs energy were negative indicating spontaneous micelle formation and more favorable on increasing the dendron generation.
lnXCMC are presented in Table 1. Anionic micelles of palmitic dendrons were highly ionized in water media (α = 0.7–0.8). In contrast, the ionization degrees were considerably lower (α = 0.2–0.5) for analogous cationic micelles indicating that these cationic dendrons assembled into micelles with smaller charge density than the homologous micelles consisting of anionic dendrons. The smaller values for cationic micelles could be related to the nature of counterion. The counterions used in these cases were I− for cationic micelles and Na+ for anionic micelles. Larger ions (I−) are more polarizable tending to be less hydrated, and subsequently binding more effectively on the micellar surface. In addition, the values of micellization Gibbs energy were negative indicating spontaneous micelle formation and more favorable on increasing the dendron generation.
The strong electrostatic repulsion between head groups in both types of micelles can be reduced by increasing the ionic strength. One of the factors known to affect the CMC in aqueous solution is the presence of added electrolyte. It is a general rule that the addition of salt decreases the CMC and increases the micelle aggregation number in ionic micelles.38,39 In solutions of increasing ionic strength, the forces of electrostatic repulsion between the head groups in a micelle are considerably reduced, enabling micelles to form more easily, that is, at lower concentration. The electrostatic screening effect decreases the ionic head group repulsion to assist self-assembly formation with increasing aggregation number of micelles. Electrical conductivity measurements were not sensitive enough to evaluate CMC in the presence of salt; therefore surface tension curves of the dendrons with and without salts were generated. The ionic strength effect on the CMC and adsorption properties at the air/water interface are depicted in Fig. 5 and 6. NaCl addition facilitated the formation of micelle-like structures as was observed by decreasing the corresponding CMC values (see Table 2).
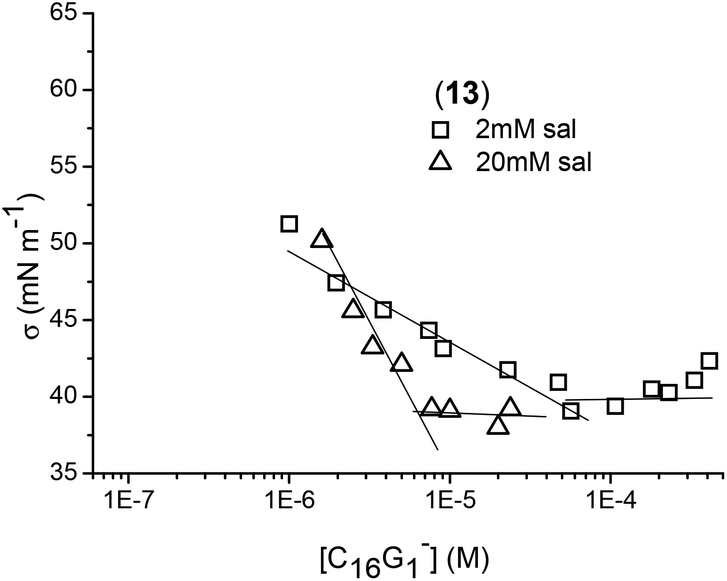 | ||
| Fig. 5 Example of surface tension measurements of dendron 13 at different salt concentrations. | ||
 | ||
| Fig. 6 Surface tension measurements of selected dendrons containing palmitic acid (13–15 (B) and 31–33 (C)) and hexanoic acid (17–18 (A)) at the focal point at a fixed salt concentration of 20 mM. | ||
| Dendron | CMC (μM) | 106Γ (mol m−2) | σ CMC (mN m−1) | A (nm2 per molecule) |
|---|---|---|---|---|
| a Salt concentration equal to 2 mM. | ||||
| Anionic dendrons (CmGn−) | ||||
| C16G1− (13)a | 50 ± 10 | 0.77 | 40 | 2.1 |
| C16G1− (13) | 6 ± 1 | 2.7 | 39 | 0.61 |
| C16G2− (14) | 6 ± 1 | 2.2 | 50 | 0.75 |
| C16G3− (15) | 8 ± 1 | 1.7 | 49 | 0.98 |
| C6G2− (17) | 50 ± 5 | 1.5 | 43 | 1.1 |
| C6G3− (18) | 1.5 ± 0.2 | 0.68 | 50 | 2.4 |
| Cationic dendrons (CmGn+) | ||||
| C16G1+ (31) | 25 ± 1 | 1.0 | 39 | 1.7 |
| C16G2+ (32) | 19 ± 1 | 0.77 | 45 | 2.1 |
| C16G3+ (33) | 17 ± 1 | 0.69 | 51 | 2.4 |
| C6G3+ (36) | 12 ± 1 | 0.83 | 50 | 2.0 |
Based on the surface tension data, the geometrical parameters of the dendron packaging in micelles were estimated. The required area per head group, A, at the interface can be calculated using the Gibbs equation
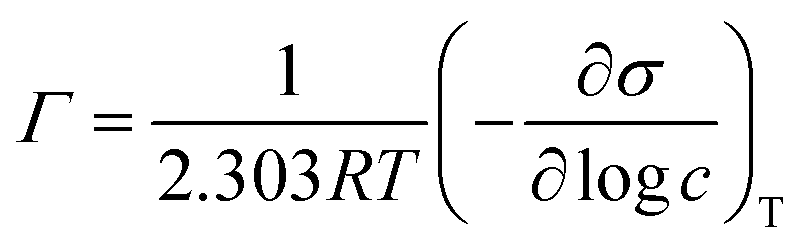 logc can be obtained from the slope of the plot surface tension versus the logarithm of the concentration at concentrations below the CMC. The reciprocal of Γ gives the area of surface occupied by a mole of adsorbed molecules. The values of CMC, Γ, A, and the surface tension at CMC (σCMC) for micelles formed from anionic and cationic dendrons are listed in Table 2.
logc can be obtained from the slope of the plot surface tension versus the logarithm of the concentration at concentrations below the CMC. The reciprocal of Γ gives the area of surface occupied by a mole of adsorbed molecules. The values of CMC, Γ, A, and the surface tension at CMC (σCMC) for micelles formed from anionic and cationic dendrons are listed in Table 2.
With respect to anionic micelles, the CMC for C16G1− (13) changed from 240 μM without salt to 50 μM with 2 mM of salt, and 6 μM with 20 mM of salt. Salt concentrations above 20 mM almost did not affect the CMC value, suggesting that the results obtained are applicable under physiological conditions. It seems that this amount of salt was high enough to reduce the electrostatic repulsions between the head groups. In this case, the CMC showed very poor sensitivity to the generation of palmitic dendrons. For hexanoic dendrons, the presence of salt permitted the formation of micelles even for the second generation above 50 μM, while the third generation formed micelles at a lower CMC value than those for palmitic dendrons. With respect to cationic micelles, the salt effect produced a decrease in CMC as well, ranging from 17 to 25 μM for all the generations of palmitic dendrons. Again, the third generation for hexanoic dendrons showed a lower CMC value than those observed for palmitic dendrons. Comparing the CMC data of the anionic and cationic systems, the latter afforded slightly higher values. It is worth noting that the lower CMC values obtained for the third generation dendrons containing hexanoic acids suggest the important role of the hydrophobic character of the dendritic scaffold in the amphiphilic behavior toward micelle formation.
With respect to the air/water interface, the hydrophilic charge of the head groups should be introduced in the water phase while the hydrophobic alkyl chain should be oriented to the air. From Table 2, the molecular area (A) occupied of C16Gn− (13–15) increased from n = 1 (0.61 nm2 per molecule) to n = 3 (0.98 nm2), suggesting that the volume of the head group was responsible for this increase. There was also observed an increase in σCMC with increasing generation.40 Regarding the salt effect, for the dendron C16G1− (13) with 2 and 20 mM of salt, the molecular area of the head group decreased as the salt concentration increased. This can be explained by a decrease in the strong electrostatic repulsion forces between the head groups due to the salt addition. Comparing the results for dendrons of the same generation with palmitic or hexanoic tails, the area occupied per molecule of dendron into the surface was clearly smaller for palmitic dendrons. The higher hydrophobic part of these systems can form an oriented monolayer in the interface allowing the molecule to be in closer packing than for hexanoic dendrons. For cationic micelles in the presence of salt, the amphiphilic dendrons were closer packing when generation decreased as it occurs with the anionic micelles; however, in this case, there was no difference between palmitic and hexanoic dendrons.
Hydrodynamic radii were determined by DLS for anionic and cationic micelles in the presence of salt (see Table 3) showing diameters between 3.8 and 6.4 nm for all of them, although no significant differences between anionic and cationic micelles were observed, taking the standard deviation into consideration. Hydrodynamic radii obtained were independent of dendron concentrations in the micellar regime at least for concentrations three fold higher than CMC. The increase in dendron concentrations appeared to increase the number of micelles and not their size, which showed the typical behavior of spherical ionic micelles. At low ionic dendron concentrations, we can assume that the dendritic micelles were spherical structures as is usual for conventional ionic surfactants.
| Anionic dendron | R H (nm) | Cationic dendron | R H (nm) |
|---|---|---|---|
| C16G1− (13) | 3.4 ± 0.1 | C16G1+ (31) | 3.2 ± 0.2 |
| C16G2− (14) | 2.9 ± 0.1 | C16G2+ (32) | 2.3 ± 0.1 |
| C16G3− (15) | 2.5 ± 0.1 | C16G3+ (33) | 2.3 ± 0.2 |
| C6G2− (17) | 2.3 ± 0.2 | ||
| C6G3− (18) | 2.2 ± 0.1 | C6G3+ (36) | 1.9 ± 0.1 |
A theory of micellar structure based on the geometry of various micellar shapes and the space occupied by the hydrophilic and hydrophobic groups of the surfactant molecules was developed by Israelachvili, Mitchell and Ninham.41 The volume occupied by the hydrophobic groups in the micellar core (V), the length of the hydrophobic groups in the core (L) and the cross-sectional area occupied by the hydrophilic groups in the micelle–solution interface (a0) have been used to calculate a “molecular packing parameter”, P = V/a0L, which determine the shape of micelles. P ≤ 1/3 accounts for spherical micelles, 1/3 < P < 1/2 for cylindrical ones, and 1/2 < P < 1 for bilayer structures such as vesicles. For ionic charged head groups at low concentrations, P must be lower than 1/3 due to the high value of a0. However, a0 decreases with salt addition due to a decrease in ionic repulsions between the head groups, which results in an increase of P and in the size of the micelles. With respect to dendron generation, although the RH values were close to each other, the tendency of the micelle size was detected to increase on decreasing the generation (see Fig. 7). The palmitic dendrons showed a systematic trend; the larger the head group the smaller the micelles. The change in generation affects a0 and also affects V and L values. The effect is more important in the cross-sectional area occupied by the hydrophilic groups in the micelle–solution interface. The salt addition permits us not to consider the increase in the number of charges with the generation. Basically, geometrical considerations can explain the decrease in hydrodynamic radius when the generation increases due to an increase in a0. The same effect was also observed in the cross-sectional area (A) occupied by the hydrophilic groups in the water–air interface (planar interface) (see Table 2).
 | ||
| Fig. 7 Schematic representation of dendritic micelles and their sizes depending on the generation. | ||
Nucleic acid binding by cationic dendritic micelles
One of the useful applications of micellar assemblies in biomedicine is to bind therapeutically relevant macromolecules and deliver them into target cells, tissues and organs. For example, they are successfully used to deliver therapeutic nucleic acids, e.g. siRNA, providing higher biological effect as compared with other methods. In particular, complexes of siRNA with micelles formed from amphiphilic PAMAM dendrons penetrated quickly across the cell membrane by macropinocytosis, and caused the inhibition of target protein expression up to 90%.14,15,42 These results evidenced that the development of soft dendritic nanoparticles as carriers for nucleic acid therapeutics is highly promising.As a preliminary work to demonstrate the capability of the family of carbosilane dendritic micelles to bind and carry therapeutic nucleic acids, complexes obtained by mixing nucleic acids with cationic micelles have been studied. The ability of cationic dendrons 31–36 to bind nucleic acids was tested using pro-apoptotic siRNA Mcl-143 as a model. The siRNA was mixed with cationic dendrons at different charge ratios. Such ratios correspond to dendron concentrations both below and above their CMC, and the amount of free siRNA in samples estimated from agarose gel electrophoregrams (Fig. 8A). The dendrons were found to bind siRNA in a different manner depending on the length of the fatty acid moiety at the focal point. Among the hexanoic acid-bearing dendrons 34–36, the siRNA binding efficiency increased with the dendron generation as G1 ≪ G2 < G3, indicating the positive dendritic effect on the complexation. Only the dendron C6G3+ (36) was able to form complexes with siRNA at a charge ratio of 1:1. In the case of dendrons with palmitic acid at the focal point C16Gn+ (31–33), no dendritic effect was observed, all of them efficiently binding the siRNA at the charge ratio of 1:2. These observations correlated with the dynamics of CMC values found and suggested that the siRNA binding by dendrons was the result of micelle formation. Hexanoic acid moieties for the first and second generations did not cause micelle formation as efficiently as the palmitic ones, thus affording an inefficient siRNA binding by dendrons 34 and 35 at charge ratio <5. Zeta potential profiles obtained by mixing siRNA with dendrons 31–36 at different charge ratios corroborated the effects observed (Fig. 8B).
 | ||
| Fig. 8 (A) Agarose gel electrophoresis and (B) zeta potential profiles of complexes of Mcl-1 siRNA with cationic dendrons 31–36 at different charge ratios. An siRNA concentration of 4 μM in PBS. | ||
The results suggested an interesting regularity of siRNA binding. As it has been shown in several works devoted to the physico-chemical studies of the formation of dendriplexes prepared from nucleic acids and cationic dendrimers, the charge ratios corresponding to the full binding obtained using agarose gel electrophoresis or using other methods (ethidium bromide intercalation assay, fluorescence polarization measurements) were usually lower than those corresponding to the plateau in the zeta potential profiles.44–46 As we have observed, this phenomenon did not depend on the number of positive charges per molecule or the dendrimer architecture.46 One can assume that the interaction of nucleic acids with pre-organized supramolecular assemblies was more favourable than with individual macromolecules. Alternatively, siRNA can facilitate the formation of micelles by stabilizing them via electrostatic interactions. Earlier, Liu et al. have demonstrated14 that the amphiphilic PAMAM dendrons bound siRNA completely at an N:P ratio of 1:2.5, whereas its cationic part (8 cationic groups on the periphery) without a lipid tail was not able to bind siRNA even at 10-fold excess of cations. Recently, the formation of nano-sized dendrimer aggregates has been shown to increase the efficiency of nucleic acid delivery.47 This is in agreement with the data on the comparison of the behaviour of the hexanoic and palmitic acid-bearing carbosilane dendrons presented here and support the above mentioned assumption. Thus, the organization of dendritic molecules into soft nanoparticles was essential for efficient nucleic acid binding.
Drug encapsulation into dendritic micelles
Preliminary studies of the use of the new micelles as drug delivery systems and their loading capacity of drugs in water have been conducted via UV-Vis experiments. For that, the interaction of the water-soluble drug procaine hydrochloride with different concentrations of the anionic micelle formed from the first generation anionic dendrons containing palmitic acid at the focal point (13) was used as a working model. A small red shift was observed on increasing the dendron concentrations, i.e. on going from aqueous to micellar solutions (see Fig. 9). This is consistent with procaine transferred from a highly polar phase (H2O) to a less polar site in the micellar phase, therefore suggesting that procaine was solubilized in the micelles to some extent. The absorbance value has been used to estimate the partition coefficient (PMW) by using a pseudo-phase model and used in conventional micelles from surfactants.48 The partition coefficient obtained (PMW) of 922 was more than three times higher than that obtained with micelles formed from the SDS surfactant (PMW = 281) under the same conditions.49 From this comparison, one can conclude that micelles prepared with carbosilane dendrons are more adequate media to solubilize this drug than conventional micelles from surfactants. The high value obtained for the partition coefficient is due mainly to the attractive electrostatic forces between the negatively charged surface of micelles and the cationic charge of drugs. In this way, procaine is probably located between branches with the cationic charged groups pointing towards the anionic groups of dendrons. | ||
| Fig. 9 Absorption profiles in water of procaine hydrochloride in the presence of the dendron C16G1 (13) in the range of dendron concentrations of 1–80 μM. | ||
Conclusions
A new family of dendritic amphiphiles have been designed and synthesized with the hydrophobic part consisting of both the tail and the dendritic structure, and the terminal ionic groups as the main hydrophilic region. This new structural situation led to novel self-assembled constructions that need further study. Our investigations showed that not only the length of the aliphatic chain but also the carbosilane scaffold played important roles in the self-assembly process. It was not necessary to include large hydrophobic tails in the carbosilane dendritic structure to observe an ordered aggregation behavior. That is, small lipophilic fragments at the focal point and the use of a high dendritic generation was enough to observe aggregation. Therefore, an optimal balance between hydrophobicity and hydrophilicity was fundamental. The amphiphilic dendrons self-assembled into spherical micelles with defined sizes of ca. 4–6 nm. The aggregation dimensions diminished upon increasing the generation, as a matter of modification of the molecular packing parameter. Because of this behavior, amphiphilic dendrons can be used as carriers for therapeutically relevant macromolecules or drugs. As a proof of concept of the usefulness of these ionic dendritic micelles, cationic amphiphilic systems have been demonstrated to bind efficiently nucleic acids when micellar aggregation occurred. Dendritic building blocks with a small number of cationic groups were not able to bind siRNA unless their self-assembly capacity is increased. Therefore, amphiphilic carbosilane dendrons allowed fully controllable siRNA binding to be achieved. Moreover, there was no need to use a large excess of polycations to form siRNA-loaded dendritic micelles, thus being potentially able to decrease unwanted toxicity of the formulations. This feature can be highly useful for in vitro and in vivo experiments. Additionally, the controllable composition of the formulations obtained might facilitate their certification for further application as therapeutics. In addition, anionic micelles are able to encapsulate drug molecules like procaine in water with a payload capacity almost four times higher than that of conventional surfactants. These studies represent the starting point in the use of this dendritic amphiphile platform for the design of novel supramolecular aggregates, as drug and/or nucleic acid delivery systems in future biomedical applications.Experimental section
General information
All solvents were dried and freshly distilled under argon before use, unless otherwise stated. Reagents were obtained from commercial sources and used as received. Dendron precursors BrGnAm, BrGnVm (n = 1, m = 2 (G1); n = 2, m = 4 (G2); n = 3, m = 8 (G3)) were obtained as described elsewhere.37Analytical and spectroscopic techniques
Synthetic protocols
Only a selection of first generation dendrons containing the palmitic acid residue at the focal point will be described. For the rest, see the ESI.†![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 1.61 (m, 4H, OCH2CH2CH2CH2Si, CH3(CH2)12CH2CH2CO2), 2.26 (t, 2H, CH3(CH2)12CH2CH2CO2), 4.04 (t, 2H, OCH2CH2CH2CH2Si), 4.85 (m, 4H, SiCH2CHCH2), 5.75 (m, 2H, SiCH2CHCH2). 13C NMR (CDCl3) δ −5.29 (SiMe), 12.5 (OCH2CH2CH2CH2Si), 14.1 (CH3CH2CH2(CH2)10CH2CH2CO2), 20.7 (OCH2CH2CH2CH2Si), 21.2 (SiCH2CHCH2), 22.6 (CH3CH2CH2(CH2)10CH2CH2CO2), 24.6 (CH3CH2CH2(CH2)10CH2CH2CO2), 29.42 (CH3CH2CH2(CH2)10CH2CH2CO2), 31.9 (CH3CH2CH2(CH2)10CH2CH2CO2), 32.3 (OCH2CH2CH2CH2Si), 34.3 (CH3CH2CH2(CH2)10CH2CH2CO2), 63.8 (OCH2CH2CH2CH2Si), 113.2 (SiCH2CHCH2), 134.6 (SiCH2CHCH2), 173.9 (CH3CH2CH2(CH2)10CH2CH2CO2). MS: [M + H]+ = 437.43 Da. Anal. calculated for C27H52O2Si (436.78 g mol−1) %: C, 74.24, H, 12.00. Exp. %: C, 74.13, H, 11.86.
:25) and a 0.5 mL aqueous solution containing sodium 3-mercapto-1-propanesulfonate (0.45 g, 2.51 mmol) was prepared. Over the dendron solution, one fourth of the aqueous solution and of the photoinitiator 2,2 dimethoxy-2-phenylactenophenone (DMPA) (0.06 g, 0.25 mmol) were added. The mixture was deoxygenized and stirred under UV light for 1 h. The aqueous solution was added stepwise each 1 h with the photoinitiator. The total irradiation time was 4 h. Afterwards, solvents were removed under vacuum and the products were dissolved in distilled water and purified by nanofiltration with cellulose membranes with a cutoff limit MWCO = 500–1000 Da. Finally, water was removed to obtain 13 as a white powder with a high yield (0.82 g, 91%). Data for 13 are as follows. NMR (D2O): 1H NMR δ −0.13 (m, 3H, SiMe), 0.53 (m, 6H, OCH2CH2CH2CH2Si, SiCH2CH2CH2S), 0.75 (m, 3H, CH3(CH2)14CO2), 1.15 (m, 26H, CH3(CH2)12(CH2)2CO2, OCH2CH2CH2CH2Si), 1.47 (m, 8H, CH3(CH2)12CH2CH2CO2, OCH2CH2CH2CH2Si, SiCH2CH2CH2S), 1.89 (m, 4H, SCH2CH2CH2SO3,), 2.14 (m, 2H, CH3(CH2)12CH2CH2CO2), 2.44 (m, 4H, SiCH2CH2CH2S), 2.52 (m, 4H, SCH2CH2CH2SO3), 2.85 (m, 4H, SCH2CH2CH2SO3), 3.92 (m, 2H, OCH2CH2CH2CH2Si). 13C NMR (D2O) δ −5.35 (SiMe), 12.8 (SiCH2CH2CH2S), 13.2 (OCH2CH2CH2CH2Si), 13.92 (CH3 (CH2)12CH2CH2CO2), 20.1 (OCH2CH2CH2CH2Si), 23.9 (SiCH2CH2CH2S), 24.5 (SCH2CH2CH2SO3), 24.9 (CH3(CH2)10CH2CH2CO2), 29.3–29.9 (CH3(CH2)12CH2CH2CO2), 30.29 (SCH2CH2CH2SO3), 32.3 (OCH2CH2CH2CH2Si), 34.1 (CH3(CH2)12CH2CH2CO2), 35.2 (SiCH2CH2CH2S), 50.1 (SCH2CH2CH2SO3), 63.9 (OCH2CH2CH2CH2Si), 173.3 (CH3CH2CH2(CH2)10CH2CH2CO2). Anal. calculated for C33H66Na2O8S4Si (793.20 g mol−1) %: C, 49.97, H, 8.39. Exp. %: C, 49.95, H, 8.35.
:25) solution (5 ml) were mixed. The reaction mixture was deoxygenated and irradiated for 2 h. Another 5% mol of DMPA was added, and the reaction mixture was irradiated for 2 h again and monitored by 1H NMR. The products were dissolved in distilled water and purified by nanofiltration with cellulose membranes with a cutoff limit MWCO = 500–1000 Da. Finally, water was removed to obtain 19 as a white powder with a high yield (1.5 g, 88%). Data for 19 are as follows. NMR (D2O): 1H NMR δ 0.00 (s, 3H, SiMe), 0.54 (t, 2H, OCH2CH2CH2CH2Si), 0.84 (t, 3H, CH3(CH2)14CO2), 0.90, (t, 4H, SiCH2CH2S), 1.25 (s, 24H, CH3(CH2)12(CH2)2CO2), 1.30 (m, 2H, OCH2CH2CH2CH2Si), 1.58 (m, 4H, OCH2CH2CH2CH2Si, CH3(CH2)12CH2CH2CO2), 2.24 (t, 2H, CH3(CH2)12CH2CH2CO2), 2.62 (t, 4H, SiCH2CH2S), 2.87 (s, 12H, SCH2CH2NMe2HCl), 3.0 (m, 4H, SCH2CH2NMe2HCl), 3.27 (m, 4H, SCH2CH2NMe2HCl), 4.00 (t, 2H, OCH2CH2CH2CH2Si). 13C NMR (D2O) δ −5.4 (SiMe), 12.5 (OCH2CH2CH2CH2Si), 13.4 (SiCH2CH2S), 14.0 (CH3CH2CH2(CH2)10CH2CH2CO2), 20.0 (OCH2CH2CH2CH2Si), 22.6 (CH3CH2CH2(CH2)10CH2CH2CO2), 24.3 (SCH2CH2NMe2HCl), 24.9 (CH3CH2CH2(CH2)10CH2CH2CO2), 26.0 (SiCH2CH2S), 29.0–29.6 (CH3CH2CH2(CH2)10CH2CH2CO2), 31.8 (CH3CH2CH2(CH2)10CH2CH2CO2), 32.2 (OCH2CH2CH2CH2Si), 34.2 (CH3CH2CH2 (CH2)10CH2CH2CO2), 41.4 (SCH2CH2NMe2HCl), 55.3 (SCH2CH2NMe2HCl), 63.7 (OCH2CH2CH2CH2Si), 173.7 (CH3CH2CH2(CH2)10CH2CH2CO2). Anal. calculated for C33H72Cl2N2O2S2Si (692.05 g mol−1) %: C, 57.27, H, 10.49, N, 4.51. Exp. %: C, 57.15, H, 10.35, N, 4.39.
CH2), 1.61 (m, 4H, OCH2CH2CH2CH2Si, CH3(CH2)12CH2CH2CO2), 2.26 (t, 2H, CH3(CH2)12CH2CH2CO2), 4.04 (t, 2H, OCH2CH2CH2CH2Si), 4.85 (m, 4H, SiCH2CHCH2), 5.75 (m, 2H, SiCH2CHCH2). 13C NMR (CDCl3) δ −5.29 (SiMe), 12.5 (OCH2CH2CH2CH2Si), 14.1 (CH3CH2CH2(CH2)10CH2CH2CO2), 20.7 (OCH2CH2CH2CH2Si), 21.2 (SiCH2CHCH2), 22.6 (CH3CH2CH2(CH2)10CH2CH2CO2), 24.6 (CH3CH2CH2(CH2)10CH2CH2CO2), 29.42 (CH3CH2CH2(CH2)10CH2CH2CO2), 31.9 (CH3CH2CH2(CH2)10CH2CH2CO2), 32.3 (OCH2CH2CH2CH2Si), 34.3 (CH3CH2CH2(CH2)10CH2CH2CO2), 63.8 (OCH2CH2CH2CH2Si), 113.2 (SiCH2CHCH2), 134.6 (SiCH2CHCH2), 173.9 (CH3CH2CH2(CH2)10CH2CH2CO2). MS: [M + H]+ = 437.43 Da. Anal. calculated for C27H52O2Si (436.78 g mol−1) %: C, 74.24, H, 12.00. Exp. %: C, 74.13, H, 11.86.
:25) and a 0.5 mL aqueous solution containing sodium 3-mercapto-1-propanesulfonate (0.45 g, 2.51 mmol) was prepared. Over the dendron solution, one fourth of the aqueous solution and of the photoinitiator 2,2 dimethoxy-2-phenylactenophenone (DMPA) (0.06 g, 0.25 mmol) were added. The mixture was deoxygenized and stirred under UV light for 1 h. The aqueous solution was added stepwise each 1 h with the photoinitiator. The total irradiation time was 4 h. Afterwards, solvents were removed under vacuum and the products were dissolved in distilled water and purified by nanofiltration with cellulose membranes with a cutoff limit MWCO = 500–1000 Da. Finally, water was removed to obtain 13 as a white powder with a high yield (0.82 g, 91%). Data for 13 are as follows. NMR (D2O): 1H NMR δ −0.13 (m, 3H, SiMe), 0.53 (m, 6H, OCH2CH2CH2CH2Si, SiCH2CH2CH2S), 0.75 (m, 3H, CH3(CH2)14CO2), 1.15 (m, 26H, CH3(CH2)12(CH2)2CO2, OCH2CH2CH2CH2Si), 1.47 (m, 8H, CH3(CH2)12CH2CH2CO2, OCH2CH2CH2CH2Si, SiCH2CH2CH2S), 1.89 (m, 4H, SCH2CH2CH2SO3,), 2.14 (m, 2H, CH3(CH2)12CH2CH2CO2), 2.44 (m, 4H, SiCH2CH2CH2S), 2.52 (m, 4H, SCH2CH2CH2SO3), 2.85 (m, 4H, SCH2CH2CH2SO3), 3.92 (m, 2H, OCH2CH2CH2CH2Si). 13C NMR (D2O) δ −5.35 (SiMe), 12.8 (SiCH2CH2CH2S), 13.2 (OCH2CH2CH2CH2Si), 13.92 (CH3 (CH2)12CH2CH2CO2), 20.1 (OCH2CH2CH2CH2Si), 23.9 (SiCH2CH2CH2S), 24.5 (SCH2CH2CH2SO3), 24.9 (CH3(CH2)10CH2CH2CO2), 29.3–29.9 (CH3(CH2)12CH2CH2CO2), 30.29 (SCH2CH2CH2SO3), 32.3 (OCH2CH2CH2CH2Si), 34.1 (CH3(CH2)12CH2CH2CO2), 35.2 (SiCH2CH2CH2S), 50.1 (SCH2CH2CH2SO3), 63.9 (OCH2CH2CH2CH2Si), 173.3 (CH3CH2CH2(CH2)10CH2CH2CO2). Anal. calculated for C33H66Na2O8S4Si (793.20 g mol−1) %: C, 49.97, H, 8.39. Exp. %: C, 49.95, H, 8.35.
:25) solution (5 ml) were mixed. The reaction mixture was deoxygenated and irradiated for 2 h. Another 5% mol of DMPA was added, and the reaction mixture was irradiated for 2 h again and monitored by 1H NMR. The products were dissolved in distilled water and purified by nanofiltration with cellulose membranes with a cutoff limit MWCO = 500–1000 Da. Finally, water was removed to obtain 19 as a white powder with a high yield (1.5 g, 88%). Data for 19 are as follows. NMR (D2O): 1H NMR δ 0.00 (s, 3H, SiMe), 0.54 (t, 2H, OCH2CH2CH2CH2Si), 0.84 (t, 3H, CH3(CH2)14CO2), 0.90, (t, 4H, SiCH2CH2S), 1.25 (s, 24H, CH3(CH2)12(CH2)2CO2), 1.30 (m, 2H, OCH2CH2CH2CH2Si), 1.58 (m, 4H, OCH2CH2CH2CH2Si, CH3(CH2)12CH2CH2CO2), 2.24 (t, 2H, CH3(CH2)12CH2CH2CO2), 2.62 (t, 4H, SiCH2CH2S), 2.87 (s, 12H, SCH2CH2NMe2HCl), 3.0 (m, 4H, SCH2CH2NMe2HCl), 3.27 (m, 4H, SCH2CH2NMe2HCl), 4.00 (t, 2H, OCH2CH2CH2CH2Si). 13C NMR (D2O) δ −5.4 (SiMe), 12.5 (OCH2CH2CH2CH2Si), 13.4 (SiCH2CH2S), 14.0 (CH3CH2CH2(CH2)10CH2CH2CO2), 20.0 (OCH2CH2CH2CH2Si), 22.6 (CH3CH2CH2(CH2)10CH2CH2CO2), 24.3 (SCH2CH2NMe2HCl), 24.9 (CH3CH2CH2(CH2)10CH2CH2CO2), 26.0 (SiCH2CH2S), 29.0–29.6 (CH3CH2CH2(CH2)10CH2CH2CO2), 31.8 (CH3CH2CH2(CH2)10CH2CH2CO2), 32.2 (OCH2CH2CH2CH2Si), 34.2 (CH3CH2CH2 (CH2)10CH2CH2CO2), 41.4 (SCH2CH2NMe2HCl), 55.3 (SCH2CH2NMe2HCl), 63.7 (OCH2CH2CH2CH2Si), 173.7 (CH3CH2CH2(CH2)10CH2CH2CO2). Anal. calculated for C33H72Cl2N2O2S2Si (692.05 g mol−1) %: C, 57.27, H, 10.49, N, 4.51. Exp. %: C, 57.15, H, 10.35, N, 4.39.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank Olga Krasheninina (ICBFM SB RAS) for the synthesis of oligonucleotides. This work has been supported by grants from the MINECO CTQ-2014-54004-P, RFBR 16-33-60152_mol_a_dk, by a Marie Curie International Research Staff Exchange Scheme Fellowship within the 7th European Community Framework Program, project no. PIRSES-GA-2012-316730 NANOGENE, Russian State funded budget project (VI.62.1.4, 0309-2016-0004), and by the Scholarship of the President of the Russian Federation (grant 882.2016.4). The CIBER-BBN is an initiative funded by the VI National R&D&i Plan 2008–2011, Iniciativa Ingenio 2010, Consolider Program, CIBER Actions and financed by the Instituto de Salud Carlos III with assistance from the European Regional Development Fund.References
- G. Chen, I. Roy, C. Yang and P. N. Prasad, Chem. Rev., 2016, 116, 2826–2885 CrossRef CAS PubMed.
- X. Yang, M. Yang, B. Pang, M. Varaand and Y. Xia, Chem. Rev., 2015, 115, 10410–10488 CrossRef CAS PubMed.
- Soft Nanoparticles for Biomedical Applications, ed. J. Callejas-Fernández, J. Estelrich, M. Quesada-Pérez and J. Forcada, RSC Nanoscience and Nanotechnology series, 2014 Search PubMed.
- K. Miyata, R. J. Christie and K. Kataoka, React. Funct. Polym., 2011, 71, 227–234 CrossRef CAS.
- N. Noshiyama and K. Kataoka, Pharmacol. Ther., 2006, 112, 630–648 CrossRef PubMed.
- C. Park, J. Lee and C. Kim, Chem. Commun., 2011, 47, 12042–12056 RSC.
- B. N. S. Thota, L. H. Urner and R. Haag, Chem. Rev., 2016, 116, 2079–2102 CrossRef CAS PubMed.
- V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim, F. S. Bates, K. P. Davis, T. P. Lodge, M. L. Klein, R. H. DeVane, E. Aqad, B. M. Rosen, A. O. Argintaru, M. J. Sienkoswka, K. Rissanen, S. Nummelin and J. Ropponen, Science, 2010, 328, 1009–1014 CrossRef CAS PubMed.
- C. Park, K. S. Choi, Y. Song, H.-J. Jeon, H. H. Song, J. Y. Chang and C. Kim, Langmuir, 2006, 22, 3812–3817 CrossRef CAS PubMed.
- M. A. Kostianen, O. Kasyutick, J. J. L. M. Cornelissen and R. J. M. Nolte, Nat. Chem., 2010, 2, 394–399 CrossRef PubMed.
- B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275–6540 CrossRef CAS PubMed.
- A. J. Harnoy, I. Rosenbaum, E. Tirosh, Y. Ebenstein, R. Shaharabani, R. Beck and R. J. Amir, J. Am. Chem. Soc., 2014, 136, 7531–7534 CrossRef CAS PubMed.
- A. Sousa-Herves, R. Novoa-Carballal, R. Riguera and E. Fernandez-Megia, APPS J., 2014, 16, 949–961 Search PubMed.
- X. Liu, J. Zhou, T. Yu, C. Chen, Q. Cheng, K. Sengupta, Y. Huang, H. Li, C. Liu, Y. Wang, P. Posocco, M. Wang, Q. Cui, S. Giorgio, M. Fermeglia, F. Qu, S. Pricl, Y. Shi, Z. Liang, P. Rocchi, J. J. Rossi and L. Peng, Angew. Chem., Int. Ed., 2014, 53, 11822–11827 CrossRef CAS PubMed.
- C. Chen, P. Posocco, X. Liu, Q. Cheng, E. Laurini, J. Zhou, C. Liu, Y. Wang, J. Tang, V. Dal Col, T. Yu, S. Giorgio, M. Fermeglia, F. Qu, Z. Liang, J. J. Rossi, M. Liu, P. Rocchi, S. Pricl and L. Peng, Small, 2016, 12, 3667–3676 CrossRef CAS PubMed.
- S. Mirsharghi, K. D. Knudsen, S. Bagherifam, B. Nyström and U. Boas, New J. Chem., 2016, 40, 3597–3611 RSC.
- S. P. Jones, N. P. Gabrielson, C.-H. Wong, H.-F. Chow, D. W. Pack, P. Posocco, M. Fermeglia, S. Pricl and D. K. Smith, Mol. Pharm., 2011, 8, 416–429 CrossRef CAS PubMed.
- C. Schlenk and H. Frey, Carbosilane dendrimers-synthesis, functionalization, application, in Silicon Chemistry, ed. U. Schubert, 1999, Springer-Verlag, pp. 3–14 Search PubMed.
- T. Gonzalo, M. I. Clemente, L. Chonco, N. D. Weber, L. Díaz, M. J. Serramía, R. Gras, P. Ortega, F. J. de la Mata, R. Gómez, L. A. López-Fernández, M. A. Muñoz-Fernández and J. L. Jiménez, ChemMedChem, 2010, 5, 921–929 CrossRef CAS PubMed.
- E. Remond, C. Martin, J. Martinez and F. Cavelier, Chem. Rev., 2016, 116, 11654–11684 CrossRef CAS PubMed.
- Y. Chang, Y. C. Kwon, S. C. Lee and C. Kim, Macromolecules, 2000, 33, 4496–4500 CrossRef CAS.
- E. Fuentes-Paniagua, J. M. Hernández-Ros, M. Sánchez-Milla, M. A. Camero, M. Maly, J. Pérez-Serrano, J. L. Copa-Patiño, J. Sánchez-Nieves, J. Soliveri, R. Gómez and F. J. de la Mata, RSC Adv., 2014, 4, 1256–1265 RSC.
- B. Rasines, J. M. Hernández-Ros, N. de las Cuevas, J. L. Copa-Patiño, J. Soliveri, M. A. Muñoz-Fernández, R. Gómez and F. J. de la Mata, Dalton Trans., 2009, 40, 8704–8713 RSC.
- I. Heredero-Bermejo, J. L. Copa-Patiño, J. Soliveri, E. Fuentes-Paniagua, R. Gómez, F. J. de la Mata and J. Pérez-Serrano, Parasitol. Res., 2015, 114, 473–486 CrossRef PubMed.
- M. J. Serramía, S. Álvarez, E. Fuentes-Paniagua, M. I. Clemente, J. Sánchez-Nieves, R. Gómez, J. de la Mata and M. A. Muñoz-Fernández, J. Controlled Release, 2015, 200, 60–70 CrossRef PubMed.
- N. Weber, P. Ortega, M. I. Clemente, D. Shcharbin, M. Bryszewska, F. J. de la Mata, R. Gómez and M. A. Muñoz-Fernández, J. Controlled Release, 2008, 132, 55–64 CrossRef CAS PubMed.
- B. Rasines, J. Sánchez-Nieves, M. Maiolo, M. Maly, L. Chonco, J. L. Jiménez, M. A. Muñoz-Fernández, F. J. de la Mata and R. Gómez, Dalton Trans., 2012, 41, 12733–12748 RSC.
- D. Sepúlveda-Crespo, M. J. Serramía, A. M. Tager, V. Vrbanac, R. Gómez, F. J. de la Mata, J. L. Jiménez and M. A. Muñoz-Fernández, Nanomedicine, 2015, 11, 1299–1308 CrossRef PubMed.
- M. Galán, E. Fuentes-Paniagua, R. Gómez and F. J. de la Mata, Organometallics, 2014, 33, 3977–3989 CrossRef.
- E. Fuentes-Paniagua, C. E. Peña-González, R. Gómez, F. J. de la Mata and J. Sánchez-Nieves, Organometallics, 2013, 32, 1789–1796 CrossRef CAS.
- C. Fornaguera, S. Grijalvo, M. Galán, E. Fuentes-Paniagua, F. J. de la Mata, R. Gómez, R. Eritja, G. Caldero and C. Solans, Int. J. Pharm., 2015, 478, 113–123 CrossRef CAS PubMed.
- A. Martínez, E. Fuentes-Paniagua, A. Baeza, J. Sánchez-Nieves, R. Gómez, F. J. de la Mata, B. González and M. Vallet-Regí, Chem. – Eur. J., 2015, 21, 15651–15666 CrossRef PubMed.
- C. E. Peña-González, P. García-Broncano, M. F. Ottaviani, M. Cangiotti, A. Fattori, M. Hierro-Oliva, M. L. González-Martín, J. Pérez-Serrano, R. Gómez, M. A. Muñoz-Fernández, J. Sánchez-Nieves and F. J. de la Mata, Chem. – Eur. J., 2016, 22, 2987–2999 CrossRef PubMed.
- T. Lozano-Cruz, P. Ortega, B. Batanero, J. L. Copa-Patiño, J. Soliveri, F. J. de la Mata and R. Gómez, Dalton Trans., 2015, 44, 19294–19304 RSC.
- S. Moreno, P. Ortega, F. J. de la Mata, M. F. Ottaviani, M. Cangiotti, A. Fattori, M. A. Muñoz-Fernández and R. Gómez, Inorg. Chem., 2015, 54, 8943–8956 CrossRef CAS PubMed.
- A. J. Perisé-Barrios, E. Fuentes-Paniagua, J. Sánchez-Nieves, M. J. Serramía, E. Alonso, R. M. Reguera, R. Gómez, F. J. de la Mata and M. A. Muñoz-Fernández, Mol. Pharm., 2016, 13, 3427–3438 CrossRef PubMed.
- J. Sánchez-Nieves, P. Ortega, M. A. Muñoz-Fernández, R. Gómez and F. J. de la Mata, Tetrahedron, 2010, 66, 9203–9213 CrossRef.
- P. Palladino and R. Ragone, Langmuir, 2011, 27, 14065–14070 CrossRef CAS PubMed.
- G. C. Kresheck, in Surfactants, Water: A Comprehensive Treatise, ed. F. Franks, Plenum Press, New York, 1975, vol. 4, pp. 95–167 Search PubMed.
- K. Torigoe, A. Tasaki, T. Yoshimura, K. Sakai, K. Esumi, Y. Takamatsue, S. C. Sharmad, H. Sakaia and M. Abe, Colloids Surf., A, 2008, 326, 184–190 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- X. Liu, C. Liu, J. Zhou, C. Chen, F. Qu, J. J. Rossi, P. Rocchif and L. Peng, Nanoscale, 2015, 7, 3867–3875 RSC.
- N. Chetoui, K. Sylla, J.-V. Gagnon-Houde, C. Alcaide-Loridan, D. Charron, R. Al-Daccak and F. Aoudjit, Mol. Cancer Res., 2008, 6, 42–52 CrossRef CAS PubMed.
- D. Shcharbin, E. Pedziwiatr, O. Nowacka, M. Kumar, M. Zaborski, P. Ortega, F. J. de la Mata, R. Gómez, M. A. Munoz-Fernandez and M. Bryszewska, Colloids Surf., B, 2011, 83, 388–391 CrossRef CAS PubMed.
- D. Shcharbin, V. Dzmitruk, A. Shakhbazau, N. Goncharova, I. Seviaryn, S. Kosmacheva, M. Potapnev, E. Pedziwiatr-Werbicka, M. Bryszewska, M. Talabaev, A. Chernov, V. Kulchitsky, A.-M. Caminade and J.-P. Majoral, Pharmaceutics, 2011, 3, 458–473 CrossRef CAS PubMed.
- M. Ionov, J. Lazniewska, V. Dzmitruk, I. Halets, S. Loznikova, D. Novopashina, E. Apartsin, O. Krasheninina, A. Venyaminova, K. Milowska, O. Nowacka, R. Gómez, F. J. de la Mata, J.-P. Majoral, D. Shcharbin and M. Bryszewska, Int. J. Pharm., 2015, 485, 261–269 CrossRef CAS PubMed.
- C. Liu, N. Shao, Y. Wang and Y. Cheng, Adv. Healthcare Mater., 2016, 5, 584–592 CrossRef CAS PubMed.
- R. Hosseinzadeh, M. Gheshlagi, R. Tahmasebi and F. Hojjati, Cent. Eur. J. Chem., 2009, 7, 90–95 CAS.
- L. J. Waters and B. Kasprzyk-Hordern, J. Therm. Anal. Calorim., 2010, 102, 343–347 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob01331k |
| This journal is © The Royal Society of Chemistry 2017 |