
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of tetrafluoroethylene- and tetrafluoroethyl-containing azides and their 1,3-dipolar cycloaddition as synthetic application†
Svatava
Voltrová
a,
Mickaël
Muselli
a,
Josef
Filgas
a,
Václav
Matoušek
 b,
Blanka
Klepetářová
a and
Petr
Beier
*a
b,
Blanka
Klepetářová
a and
Petr
Beier
*a
aThe Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo náměstí 2, 166 10 Prague, Czech Republic. E-mail: beier@uochb.cas.cz
bCF Plus Chemicals s.r.o., Kamenice 771/34, 625 00 Brno, Czech Republic
First published on 18th May 2017
Abstract
Tetrafluoroethylene-containing azides are accessed in two steps (one pot) from tetrafluoroalkyl bromides by metalation and reaction with electrophilic azides. Subsequent copper(I)-catalyzed azide–alkyne cycloaddition afforded N-tetrafluoroethyl and N-tetrafluoroethylene 4-substituted 1,2,3-triazoles. In addition, the protocol for the synthesis of 4,5-disubstituted 1,2,3-triazoles is presented.
Tetrafluoroethyl- and tetrafluoroethylene-containing compounds have been recently intensively investigated in agrochemistry, medicinal chemistry, and materials science as alternatives to well established fluorinated functional groups such as trifluoromethyl, perfluoroalkyl, and difluoromethyl.1–9 For instance, tetrafluorinated sugar analogues were found to be significantly less hydrophilic than the parent carbohydrates, and thus exhibit enhanced protein–carbohydrate binding capabilities.10–14 The tetrafluoroethylene motif is found in insecticides15,16 and liquid crystals.17,18 The tetrafluoroethyl- and triazole-containing molecule tetraconazole displays fungicidal activity.19 Despite important recent advances in synthetic methods toward these compounds,20–26 protocols affording CF2CF2-containing molecules of the unprecedented structure are still in demand. Our contribution to the field has been in the design and application of nucleophilic,27–29 electrophilic30 and radical31 reagents for the CF2CF2 transfer which provided straightforward synthetic access to a variety of tetrafluoroethyl- and tetrafluoroethylene-containing compounds. In the latest report, tetrafluoroethyl bromides were reacted with i-PrMgCl·LiCl to afford metalated species stable at low temperature and displaying excellent reactivity with a range of electrophiles with one example of an electrophilic azide.32 Azides are highly valuable compounds in organic synthesis – some fluoroalkyl azides are known,33–40 including CF3N3
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 41,42 and perfluoroalkyl analogues for which we have recently reported a new and practical synthetic method.43 Perfluoroalkyl azides were investigated in copper(I)-catalyzed azide–alkyne cycloadditon (CuAAC)44–46 furnishing N-perfluoroalkyl-1,2,3-triazoles.43
41,42 and perfluoroalkyl analogues for which we have recently reported a new and practical synthetic method.43 Perfluoroalkyl azides were investigated in copper(I)-catalyzed azide–alkyne cycloadditon (CuAAC)44–46 furnishing N-perfluoroalkyl-1,2,3-triazoles.43
In this article, the synthesis of tetrafluoroethylene-containing azides is disclosed by metalation of tetrafluoroethyl bromides through organomagnesium or silicon species and their application in CuAAC is shown in the preparation of tetrafluoroethyl- and tetrafluoroethylene-containing triazoles.‡
Magnesiation of tetrafluoroethyl bromides (1) was performed with i-PrMgCl·LiCl (turbo Grignard reagent) in THF at −78 °C. We have earlier found that the metalated species are stable at this temperature and metalation times range from less than 5 min for R1 = PhS and alkyl to half an hour or slightly longer for R1 = imidazolyl, pyrazolyl and ArO.32 An electrophilic azide source (tosyl or nonaflyl azide working equally well) was then added to the generated Grignard reagent and the temperature was allowed to rise to ambient temperature over a period of 3 hours. In this way, good yields of tetrafluoroethyl azides (2) were obtained with electron-neutral, -withdrawing or -donating groups on the aryl ring of the aryloxy substituent (R1 = ArO) (Table 1). The 4-bromopyrazolyl derivative (1h) afforded the corresponding azide 2h in moderate yield; however, the reaction was highly regioselective (only the bromine atom on the fluorinated carbon reacted). In contrast, the phenylthio derivative 1i, under the same reaction conditions, provided selectively and in high conversion PhSCF2CF2SPh instead of the expected azide 2i. This outcome can be explained by high susceptibility of the Grignard intermediate to oxidation to radicals, partial fragmentation giving PhS radicals, which prefer to react with electrophilic PhSCF2CF2 radicals. Therefore, for the synthesis of 2i, an alternative method was used starting from PhSCF2CF2SiMe3.47 Nucleophilic fluoroalkyl transfer, after activation with CsF, to either tosyl or nonaflyl azide afforded high yields of 2i (Scheme 1).
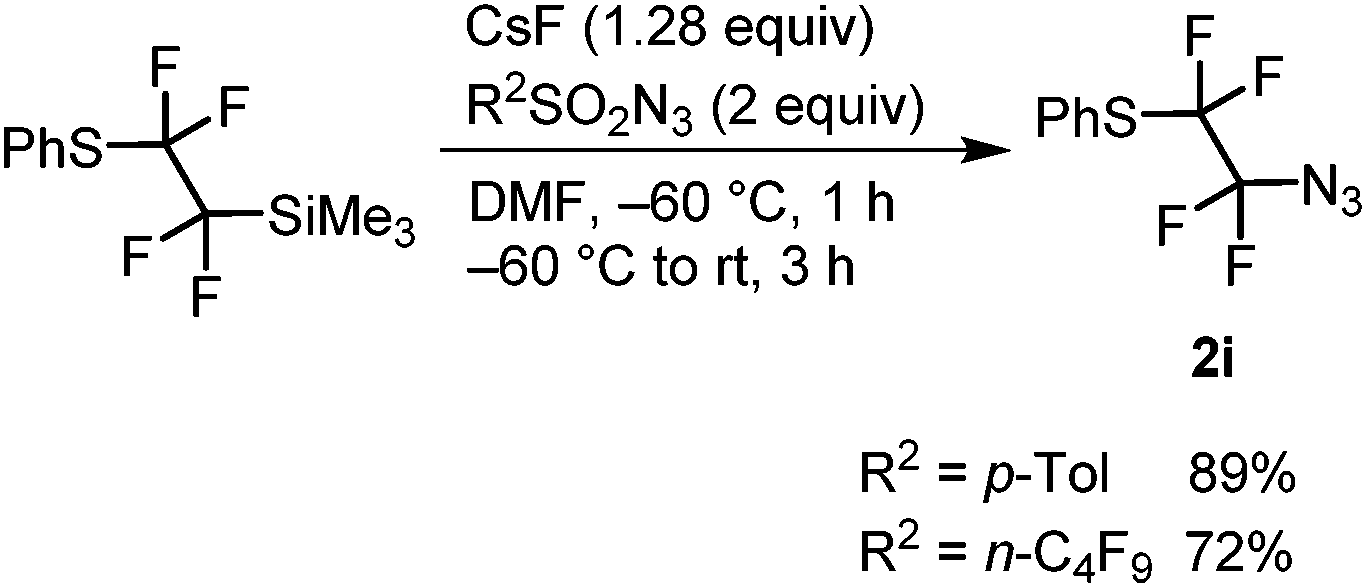 | ||
| Scheme 1 Synthesis of (2-azido-1,1,2,2-tetrafluoroethyl)(phenyl)sulfane (2i). | ||
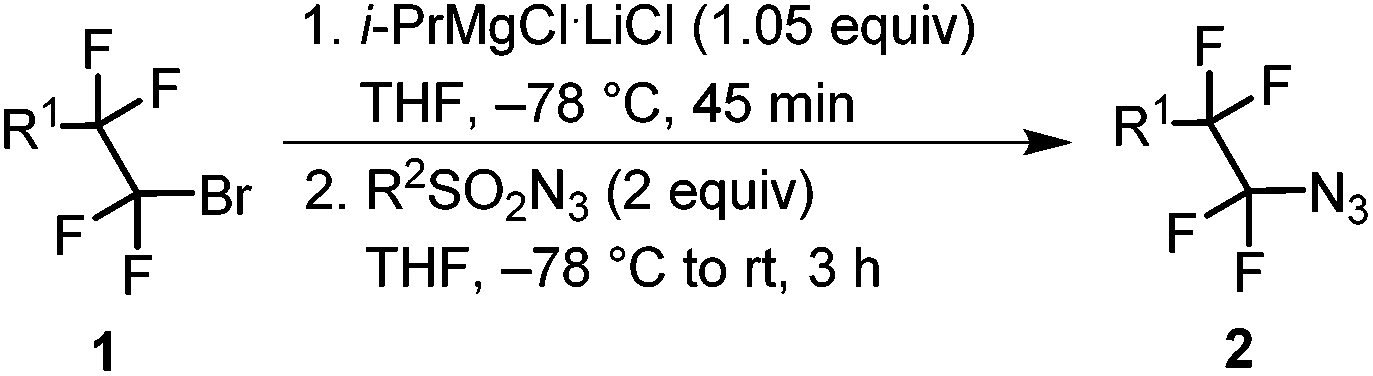
With azides 2, CuAAC was investigated. Our earlier work with azidotrifluoromethane and other azidoperfluoroalkanes43 showed that copper(I) 3-methylsalicylate (CuMeSal) was a preferred catalyst in terms of catalytic activity, good solubility in organic solvents and high stability. Therefore, it was favored over other Cu(I) sources. In the reaction, equimolar amounts of aryl, heteroaryl or alkyl acetylenes and 1 mol% of CuMeSal afforded good to high yields of N-tetrafluoroalkylated 4-substituted 1,2,3-triazoles 4 (Table 2). The examples showing moderate yields resulted from isolation difficulties rather than a lack of reactivity or side reactions. Exclusive formation of 1,4-disubstituted triazoles was observed in all cases. Compounds of the type 4 are unknown in the literature with the exception of a triazole formed by thermal cycloaddition of N3CF2CF2CO2Me.48 Reductive cleavage of the phenylsulfanyl group provided N-tetrafluoroethyl-containing triazole 5 in good yield (Scheme 2). However, attempts to prepare HCF2CF2N3 from 2i under the same reaction conditions met with limited success partially due to product volatility and also due to some unknown side reactions. Nevertheless, the in situ formed HCF2CF2N3 transformed to N-tetrafluoroethyl triazole 5 (Scheme 2).
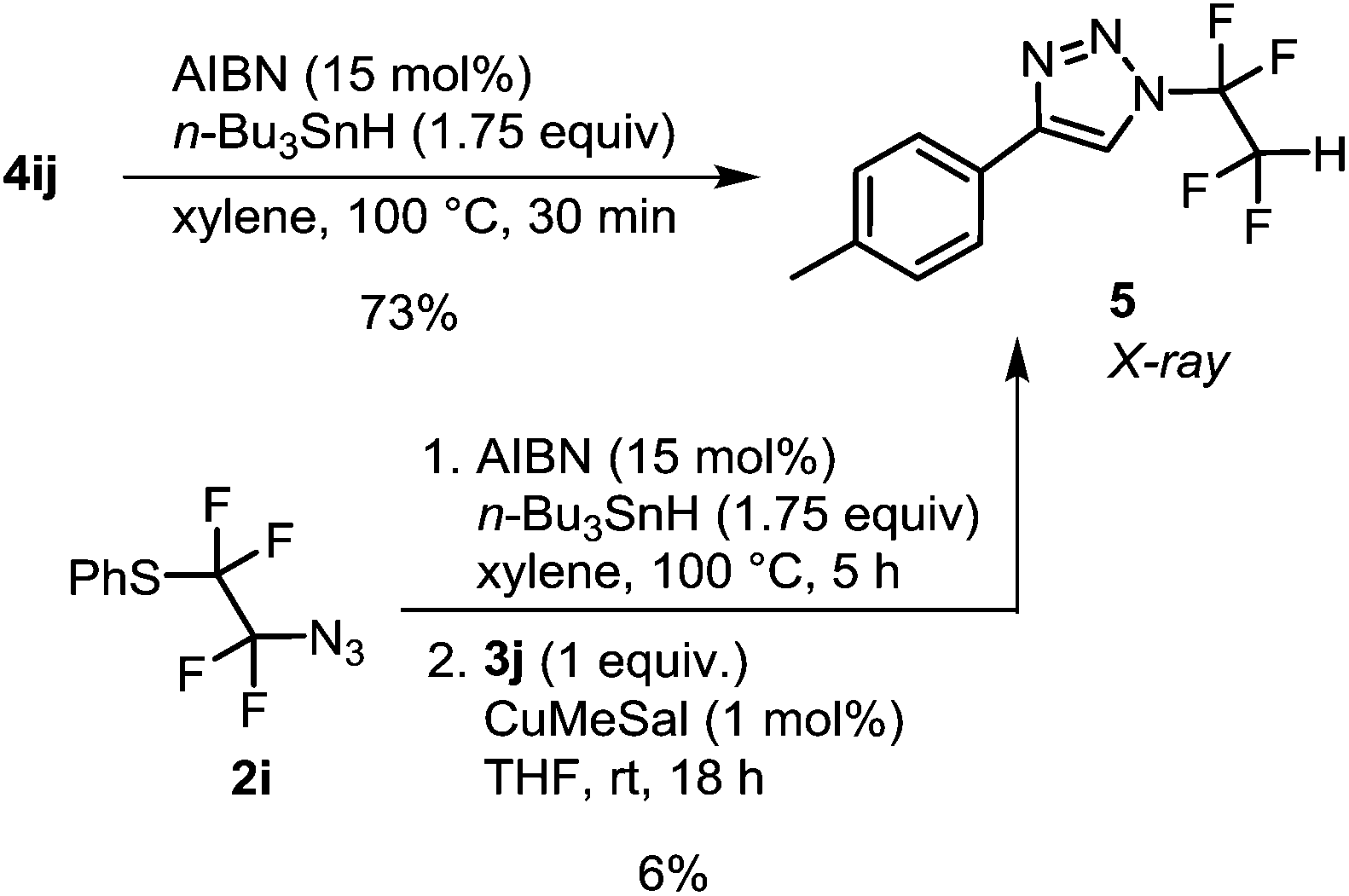 | ||
| Scheme 2 Preparation of N-tetrafluoroethyl 4-substituted 1,2,3-triazole 5. | ||
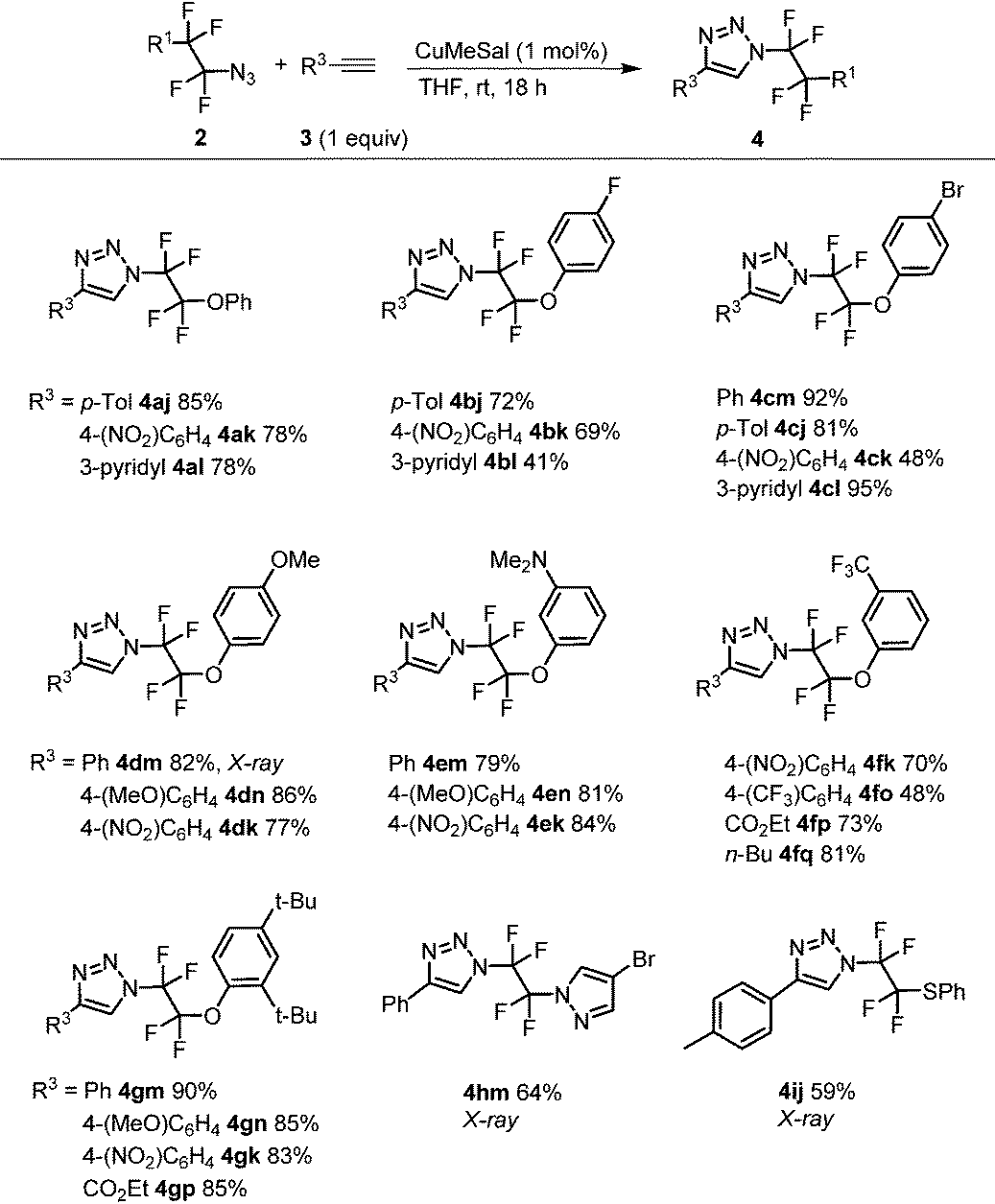
|
Finally, N-tetrafluoroethyl 4,5-disubstituted 1,2,3-triazoles were accessed in two steps from azides 2a and 2d and copper acetylides 6j and 6m, respectively. The reaction with iodine and a base afforded iodotriazoles 7aj and 7dm, respectively, in good to high yields which were amenable to Sonogashira or Suzuki–Miyaura cross-coupling reactions giving 4,5-disubstituted triazoles 8 and 9, respectively (Scheme 3).
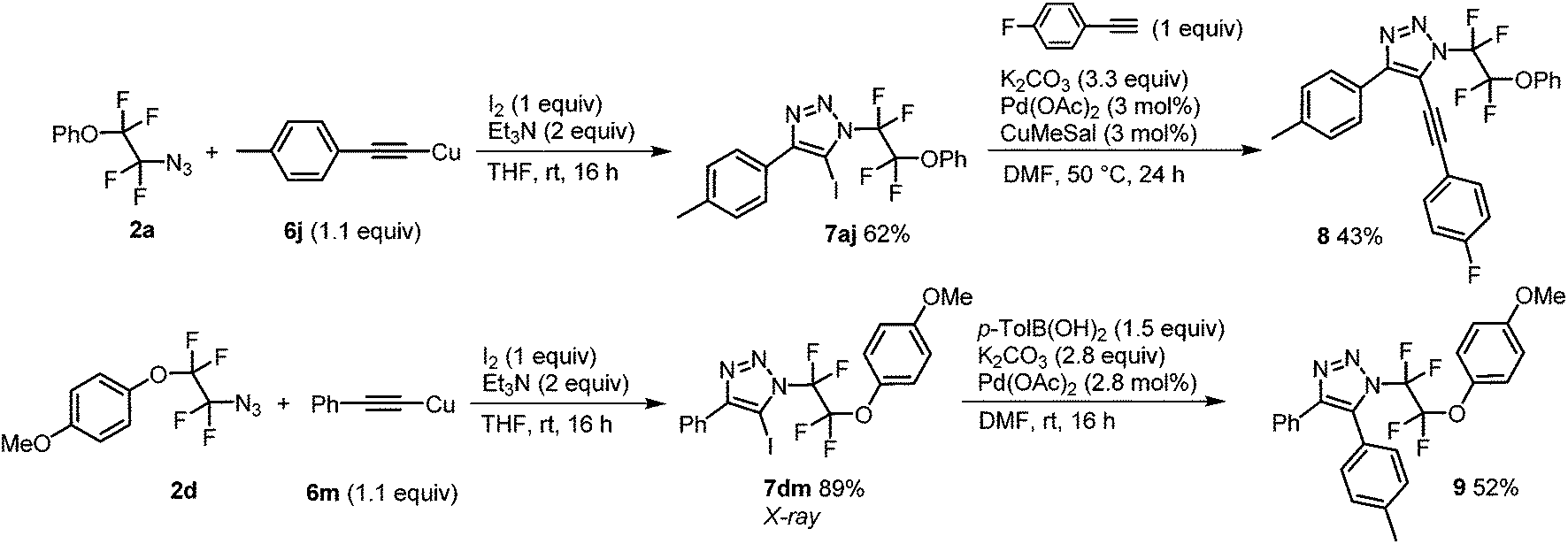 | ||
| Scheme 3 Synthesis of 4,5-disubstituted 1,2,3-triazoles 8 and 9. | ||
Crystallographic analysis of triazoles 4dm, 4hm, 4ij, 5 and 7dm confirmed their molecular structures (see the ESI†) and also showed an interesting difference in conformation at the N-CF2CF2-X unit. While in compounds 4hm (X = N) and 4ij (X = S) the N and X atoms are approximately anti-periplanar, in compounds 4dm, 7dm (X = O) and 5 (X = H) the N and X atoms are gauche as illustrated in Fig. 1. The reasons for this difference is unclear; however, an interplay between the steric repulsion of N and X atoms favoring anti-periplanar conformation and favorable vicinal fluorine–fluorine gauche conformation (three vs. two F, F gauche arrangements) should be taken into consideration.
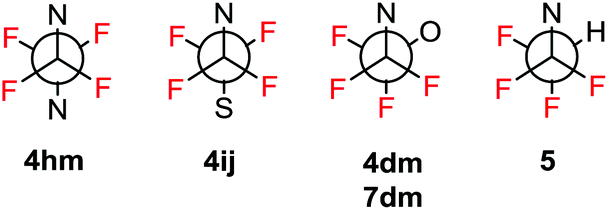 | ||
| Fig. 1 Schematic representation of the N–C–C–X conformation in the crystal structures of N-tetrafluoroethyl 1,2,3-triazoles. | ||
In conclusion, magnesiation of tetrafluoroethyl bromides with i-PrMgCl·LiCl and subsequent reaction with tosyl or nonaflyl azides afforded tetrafluoroethylated azides. (2-Azido-1,1,2,2-tetrafluoroethyl)(phenyl)sulfane was prepared by fluoride-initiated nucleophilic fluoroalkyl transfer from the corresponding silane. The azides proved to be competent partners in copper(I)-catalyzed azide alkyne cycloadditions with terminal alkynes to generate N-tetrafluoroalkylated 4-substituted 1,2,3-triazoles. Reductive cleavage of the PhS group in the tetrafluoroethyl unit afforded N-tetrafluoroethyl azide. 4,5-Disubstituted triazoles were accessed by CuAAC of the triazoles with copper acetylides in the presence of iodine followed by transition metal-catalyzed cross-coupling reactions.
Acknowledgements
This work was supported by the Czech Academy of Sciences (Research Plan RVO: 61388963) and the Initial Training Network, FLUOR21, funded by the FP7 Marie Curie Actions of the European Commission (FP7-PEOPLE-2013-ITN-607787).References
- P. Kirsch, Modern Fluoroorganic Chemistry, Wiley-VCH, Weinheim, 2nd edn, 2013 Search PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826–870 CrossRef CAS PubMed.
- C. Alonso, E. Martinez de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847–1935 CrossRef CAS PubMed.
- J. Wang, M. Sanchez-Rosello, J. L. Acena, C. Del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, New York, 2009 Search PubMed.
- J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465–7478 RSC.
- J.-A. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1–PR43 CrossRef CAS PubMed.
- A. J. Boydell, V. Vinader and B. Linclau, Angew. Chem., Int. Ed., 2004, 43, 5677–5679 CrossRef CAS PubMed.
- B. Linclau, A. J. Boydell, R. S. Timofte, K. J. Brown, V. Vinader and A. C. Weymouth-Wilson, Org. Biomol. Chem., 2009, 7, 803–814 CAS.
- T. Konno, T. Hoshino, T. Kida, S. Takano and T. Ishihara, J. Fluorine Chem., 2013, 152, 106–113 CrossRef CAS.
- L. N'Go, S. Golten, A. Arda, J. Canada, J. Jimenez-Barbero, B. Linclau and S. P. Vincent, Chem. – Eur. J., 2014, 20, 106–112 CrossRef PubMed.
- K. E. van Straaten, J. R. Kuttiyatveetil, C. M. Sevrain, S. A. Villaume, J. Jimenez-Barbero, B. Linclau, S. P. Vincent and D. A. Sanders, J. Am. Chem. Soc., 2015, 137, 1230–1244 CrossRef CAS PubMed.
- E. L. Plummer, US4737509, 1988 Search PubMed.
- E. L. Plummer, US4636523, 1987 Search PubMed.
- S. Yamada, S. Hashishita, T. Asai, T. Ishihara and T. Konno, Org. Biomol. Chem., 2017, 15, 1495–1509 CAS.
- P. Kirsch, M. Bremer, F. Huber, H. Lannert, A. Ruhl, M. Lieb and T. Wallmichrath, J. Am. Chem. Soc., 2001, 123, 5414–5417 CrossRef CAS PubMed.
- D. Bianchi, P. Cesti, S. Spezia, C. Garavaglia and L. Mirenna, J. Agric. Food Chem., 1991, 39, 197–201 CrossRef CAS.
- Y. Sakaguchi, S. Yamada, T. Konno, T. Agou and T. Kubota, J. Org. Chem., 2017, 82, 1618–1631 CrossRef CAS PubMed.
- T. Konno, S. Takano, Y. Takahashi, H. Konishi, Y. Tanaka and T. Ishihara, Synthesis, 2011, 33–44 CrossRef CAS.
- Y. Watanabe and T. Konno, J. Fluorine Chem., 2015, 174, 102–107 CrossRef CAS.
- J. Zhu, C. Ni, B. Gao and J. Hu, J. Fluorine Chem., 2015, 171, 139–147 CrossRef CAS.
- M. O'Duill, E. Dubost, L. Pfeifer and V. Gouverneur, Org. Lett., 2015, 17, 3466–3469 CrossRef PubMed.
- H. Saijo, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2014, 136, 15158–15161 CrossRef CAS PubMed.
- M. Ohashi, Y. Ueda and S. Ogoshi, Angew. Chem., Int. Ed., 2017, 56, 2435–2439 CrossRef CAS PubMed.
- Y. Chernykh, K. Hlat-Glembová, B. Klepetářová and P. Beier, Eur. J. Org. Chem., 2011, 4528–4531 CrossRef CAS.
- J. Václavík, Y. Chernykh, B. Jurásek and P. Beier, J. Fluorine Chem., 2015, 169, 24–31 CrossRef.
- Y. Chernykh, B. Jurásek and P. Beier, J. Fluorine Chem., 2015, 171, 162–168 CrossRef CAS.
- V. Matoušek, J. Václavík, P. Hájek, J. Charpentier, Z. E. Blastik, E. Pietrasiak, A. Budinská, A. Togni and P. Beier, Chem. – Eur. J., 2016, 22, 417–424 CrossRef PubMed.
- Y. Chernykh and P. Beier, J. Fluorine Chem., 2013, 156, 307–313 CrossRef CAS.
- A. Budinská, J. Václavík, V. Matoušek and P. Beier, Org. Lett., 2016, 18, 5844–5847 CrossRef PubMed.
- C. G. Krespan and B. E. Smart, J. Org. Chem., 1986, 51, 320–326 CrossRef CAS.
- C. G. Krespan, J. Org. Chem., 1986, 51, 332–337 CrossRef CAS.
- J. Dai, Z. Li, T. Wang and R. Bai, Org. Biomol. Chem., 2016, 14, 4382–4386 CAS.
- S. A. Postovoi, Y. V. Zeifman and I. L. Knunyants, Russ. Chem. Bull., 1986, 35, 1183–1186 CrossRef.
- T. G. Archibald and K. Baum, J. Org. Chem., 1990, 55, 3562–3565 CrossRef CAS.
- T. Hiramatsu, Y. Guo and T. Hosoya, Org. Biomol. Chem., 2007, 5, 2916–2919 CAS.
- S. A. Lermontov, A. G. Polivanova and S. B. Shkavrov, Russ. J. Gen. Chem., 2010, 80, 1646–1651 CrossRef CAS.
- J. Dai, Z. Li, T. Wang, W. Bai and R. Bai, Org. Lett., 2017, 19, 1418–1421 CrossRef CAS PubMed.
- S. P. Makarov, A. Y. Yakubovich, A. S. Filatov, M. A. Enlin and T. Y. Nikiforova, Zh. Obshch. Khim., 1968, 38, 709–715 CAS.
- K. O. Christe and C. J. Schack, Inorg. Chem., 1981, 20, 2566–2570 CrossRef CAS.
- Z. E. Blastik, S. Voltrová, V. Matoušek, B. Jurásek, D. W. Manley, B. Klepetářová and P. Beier, Angew. Chem., Int. Ed., 2017, 56, 346–349 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- E. Haldon, M. C. Nicasio and P. J. Perez, Org. Biomol. Chem., 2015, 13, 9528–9550 CAS.
- F. Toulgoat, B. R. Langlois, M. Medebielle and J. Y. Sanchez, J. Org. Chem., 2007, 72, 9046–9052 CrossRef CAS PubMed.
- S. A. Lermontov, S. V. Shkavrov and A. N. Pushin, J. Fluorine Chem., 2000, 105, 141–147 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1545038–1545042. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ob01151b |
| ‡ The CF Plus Chemicals s.r.o. (http://www.cfplus.cz) company, an ETH Zurich spin-off, commercializes the compounds used in this publication. |
| This journal is © The Royal Society of Chemistry 2017 |