
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of bioactive peptides, peptide conjugates, and peptide mimetic materials
Charlotte J. C.
Edwards-Gayle
and
Ian W.
Hamley
 *
*
Department of Chemistry, University of Reading, Whiteknights, Reading, RG6 6AD, UK. E-mail: I.W.Hamley@reading.ac.uk
First published on 26th June 2017
Abstract
Molecular self-assembly is a multi-disciplinary field of research, with potential chemical and biological applications. One of the main driving forces of self-assembly is molecular amphiphilicity, which can drive formation of complex and stable nanostructures. Self-assembling peptide and peptide conjugates have attracted great attention due to their biocompatibility, biodegradability and biofunctionality. Understanding assembly enables the better design of peptide amphiphiles which may form useful and functional nanostructures. This review covers self-assembly of amphiphilic peptides and peptide mimetic materials, as well as their potential applications.
Peptide self-assembly
Self-assembly is defined as the ability of a molecule, without guidance of external factors, to associate through non-covalent interactions to form highly ordered 3-dimensional structures.1 This occurs through a bottom-up approach. Most self-assembling molecules have both hydrophobic and hydrophilic components, and are termed amphiphilic.1 Lipids are perhaps the simplest molecule displaying amphiphilicity, with a hydrophilic head group, and a hydrophobic tail group. Peptides and proteins are more complex in their amphiphilicity. This is largely due to folding, giving rise to ‘faces’, different folded surfaces which are exposed to different environments. For example, a β-sheet peptide may contain alternating hydrophilic and hydrophobic residues, resulting in the side chains being exposed on opposite sides of the sheet.2Self-assembly is commonly found in nature. A natural example of a self-assembled structure is the phospholipid bilayer, which is the basis of cell membranes, vesicles and organelle membranes in cells and bacteria.3 Another example is microtubules, which are cytoskeletal components of eukaryotic and some prokaryotic cells. Microtubules are a main component of the mitotic spindle in cell division which contracts to pull apart chromosomes in eukaryotes, and in prokaryotes make up the internal structure of cilia and flagella in bacteria enabling movement.4,5 Other examples include protein folding in enzymes, DNA double helix formation and the formation of the virus protein capsid around a nucleic acid core.6
In aqueous environments, self-assembly is driven by non-covalent forces to form ordered structures ranging from the manometer to micron size.7 These non-covalent interactions include van der Waals forces, hydrophobic interactions, electrostatic interactions, hydrogen bonding and π–π stacking (aromatic) interactions.1,8 These interactions are weak, for example the backbone hydrogen bonding in peptides having an estimated energy of 4.2 kcal mol−1 in a gaseous environment, which decreases in solution.9 However, these interactions are enough to stabilise these robust structures. In turn, this means self-assembly can be influenced by temperature, pH and concentration. Some of the most common structures (Fig. 1) include micelles, vesicles and fibrillar structures (nanotubes, fibres).10
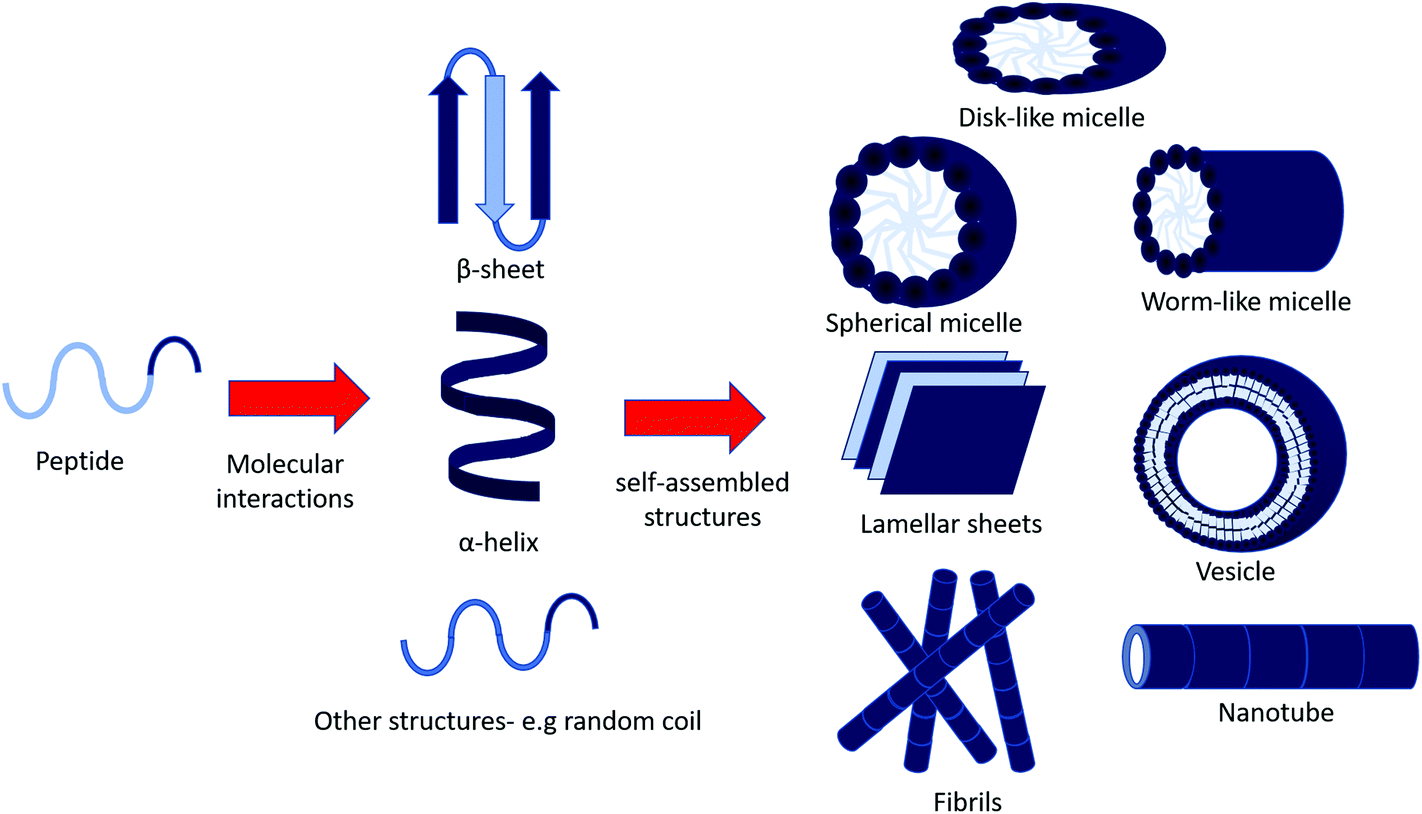 | ||
| Fig. 1 Possible self-assembled structures of peptide amphiphiles. Amphiphilic peptides may assemble into secondary structure through inter and intra molecular interactions (e.g. electrostatic interactions and hydrogen bonding), and continue to aggregate into larger self-assembled structures.8 | ||
Micelles can be spherical, worm-like or disk-shaped assemblies and form spontaneously above a critical micelle aggregation concentration and temperature.11,12 Vesicles are spherical, lamellar structures which are hollow, surrounding an aqueous core. Hydrophilic layers are exposed to the inner and outer aqueous environments, whilst hydrophobic residues pack together between hydrophilic interfaces.13
Hydrogen bonding between the backbone of peptide chains is an important factor in peptide self-assembly, as it drives longitudinal packing of peptide monomers into β-sheets. Inter β-sheet interactions among side chains of peptide molecules regulate the lateral packing of the β-sheet at a slow rate compared to the fast growth in the hydrogen-bonding direction. Packing of β-sheets, which are naturally twisted due to chirality, favours reduced twisting. Lateral interactions overcome the energy penalty due to untwisting.2 Thus, the final assembled morphologies are the result of these confinements, interactions and/or their interplay.2
Peptide amphiphiles (PAs)
A peptide amphiphile (PA) (Fig. 2) is defined by having a hydrophobic tail group, usually an alkyl chain, which leads to hydrophobic interactions, a peptide sequence that is able to form intermolecular hydrogen bonds, which determines the interfacial curvature of self-assembly, a section of charged amino acids to promote solubility and a functional peptide epitope that has bioactivity.14 The amphiphilicity of the molecule drives self-assembly, which draws the functional peptide group to the surface of the structure. Self-assembly occurs above a critical aggregation concentration (CAC). This can be measured using fluorescence methods, light scattering or proton NMR solubility measurements.1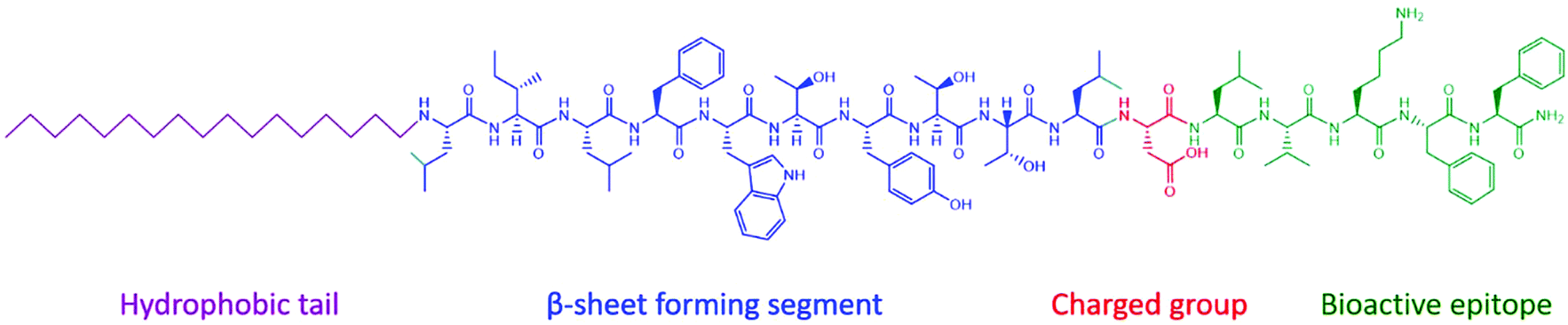 | ||
| Fig. 2 Typical structure of a peptide amphiphile. The four domains that incorporated into the Stupp group's design of bioactive β-sheet forming PA assemblies.14 | ||
Self-assembly of peptide amphiphiles has been widely studied due to their potential to assemble into a large range of novel nanostructures which are of interest commercially and biomedically. These structures can be monolayer-based (for example micelles), or bilayer-based (for example vesicles).15 The self-assembled structure of a peptide can largely be controlled by amino acid sequence, length of sequence and lipidation. Thus they are a malleable tool in order to make novel biomaterials. Some of their potential uses include drug delivery, tissue engineering and antimicrobial agents.1
The self-assembly of PAs and peptide-based molecules can be tuned by control of pH, temperature, concentration and other factors, as discussed in other reviews.1,8,16–18 This gives excellent scope to create biomaterials responsive to many or multiple environmental cues.
Different classes of peptide-based molecules which will be reviewed include here peptides with polar and nonpolar residues giving rise to hydrophobic and hydrophilic properties, named surfactant-like peptides (SLPs), and hydrophilic peptides attached to a hydrophobic lipid alkyl chains, named PAs.
Amphiphilic lipopeptides
Self-assembling amphiphilic lipopeptides are a class of molecules defined as having a one or more lipid chains attached to a peptide head group. The self-assembly of this class is thought to depend upon the hydrophile/lipophile balance (HLB).19 They offer advantages compared to peptides, as they offer increased amphiphilicity and are compatible with the phospholipid bilayer (which makes up cell membranes), enabling them to deliver actives into cells via endocytosis. Additionally, self-assembly of lipopeptides facilitates the presentation of peptide functionalities at high density at the surface of nanostructures (micelles, vesicles or fibrils).The length of lipid chain on the self-assembly of PAs has been considered in several studies which are discussed in a recent paper.20 Essentially, studies so far21 indicate that a minimal chain length (typically C6–C10) is required for the conjugate to exhibit self-assembly. Shorter chains attached to hydrophilic peptides lead to PAs that are just “too soluble”. Of course the sequence and length of the peptide will also influence amphiphilicity of the PA conjugate as well as many other factors such as electrostatic interactions, hydrogen bonding, van der Waals interactions π–π stacking interactions etc. There are no universal rules for this, however PAs with less than a couple of charged residues will not be water soluble and at least 2 or more heptad repeats is required to observe α-helix formation.
Many naturally expressed bioactive lipopeptides contain one lipid chain (C14–C18) with a cyclic head group. Cyclisation is thought to have evolved to reduce proteolysis, thus enhancing in vivo stability. However, these conformational peptide constraints may be relevant to bioactivity. In bacteria, many naturally occurring amphiphilic lipopeptides are produced as part of a host defence response against other organisms.22 This has many interesting uses clinically. An example is daptomycin, produced by the Gram positive bacterium, Streptomyces roseoporous, which is used to treat MRSA and self-assembles into micelle structures.23,24
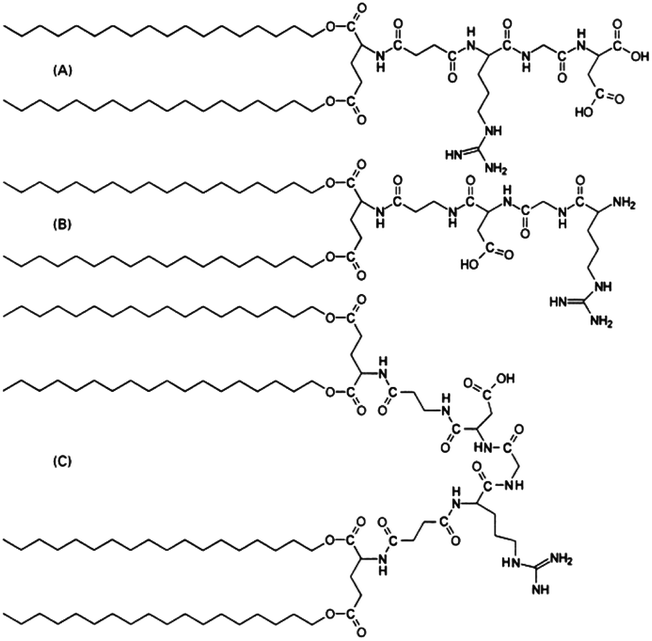
Synthetic amphiphilic lipopeptides tend to be based on a bio-derived sequence, often containing a linear peptide head group with 1–3 (usually palmitoyl) lipid chains attached. An example is Toll-like receptor agonists. Toll-like receptors (TLRs) are transmembrane proteins which are part of the innate immune response, making them an important therapeutic target. Agonists of TLRs were designed based on the Pam3Cys peptide (where Pam denotes a C16 chain). This synthetic peptide has been shown to stimulate cytotoxic T lymphocyte (CTL) responses against cells infected with influenza virus.25 Another related lipopeptide to this is Pam3CysSer2 which has been shown to stimulate antibodies against foot and mouth disease.26 Pam1CKS4 and Pam2CSK4 (Fig. 3) have recently been shown to self-assemble into spherical micelles in contrast to Pam3CSK4 which forms bilayer-based structures.27
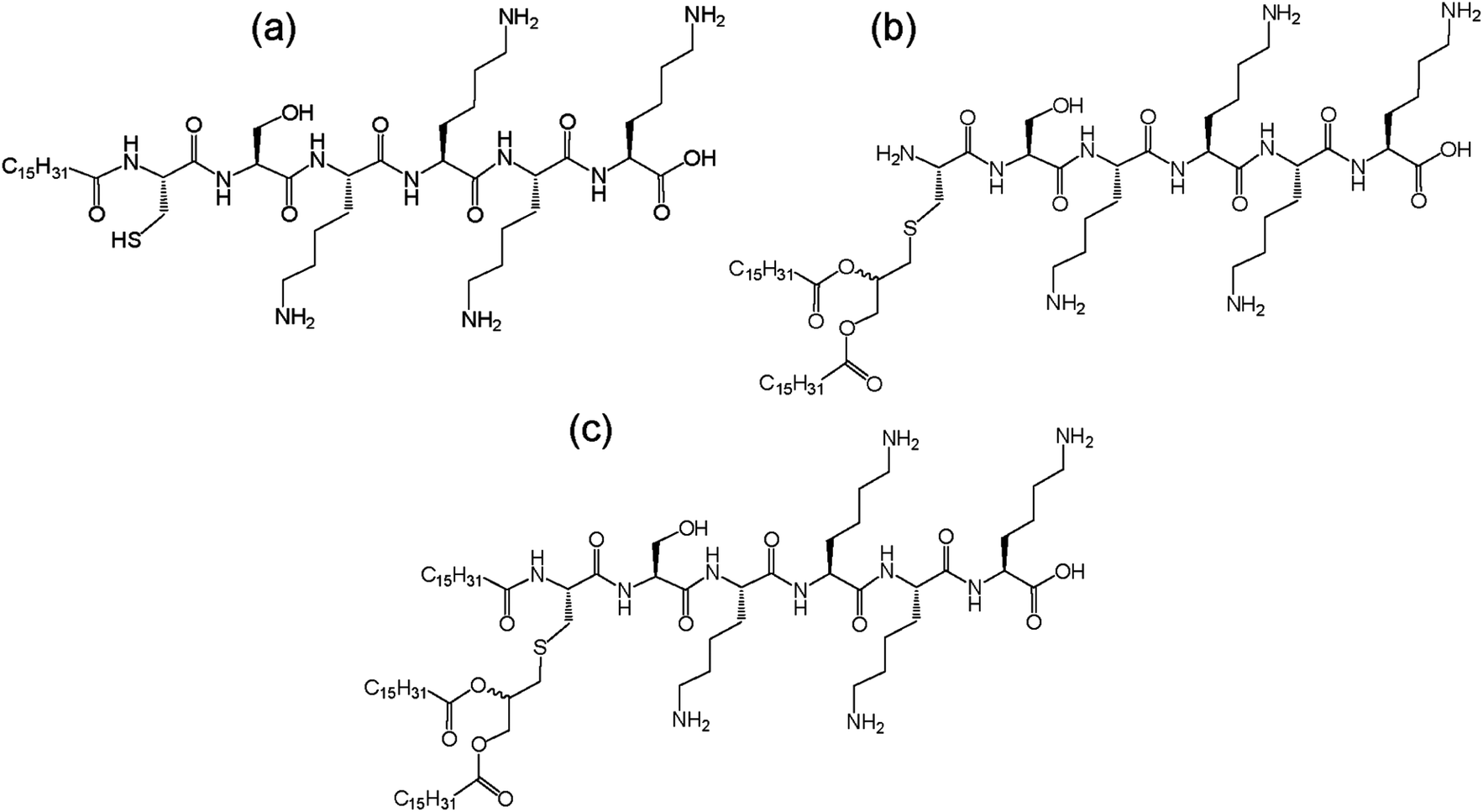 | ||
| Fig. 3 Structure of Toll-like receptor agonists (a) Pam1CSK4, (b) Pam2CSK4 and (c) Pam3CSK4.27 | ||
Surfactant-like peptides
Surfactant-like peptides (SLPs) are peptides containing sequences of both hydrophobic and hydrophilic amino acids, which self-assemble in a similar way to PAs. Popular examples of this include A6D and V6D.28 Alanine-containing SLPs have found to self-assemble into more stable structures due to stronger hydrophobic interactions, in contrast to other hydrophobic amino acid residues.SLPs tend to feature 1–2 charged amino acids that form a hydrophilic head group, and 4 or more consecutively hydrophobic amino acids.29 Early studies of self-assembly of this group showed that in water they undergo self-assembly to from bilayered nanovesicles and nanotubes, with an average diameter of 30–50 nm.30 Nanotubes are thought to form from bilayers of peptide molecules, which resemble sheets. These sheets then roll up to form tubes, which have a defined diameter, and continue to grow from the edges. More recently, SLPs have been prepared that form micelles through packing of hydrophobic tails, or nanofibers. Two variants of A6K were shown to have differences in assembly due to deprotection of the C-terminis or change in the pH of the solution. Peptide A6K with the sequence Ac-AAAAAAK-CONH2 was observed to form nanofibers at low pH and pH 6, and amorphous aggregates at high pH. When the protecting NH2 group at the C-terminus was removed, leaving the sequence Ac-AAAAAAK-COOH (named A6K±), it was shown to form short nanofibers at low pH, longer fibres at pH 5, and nanospheres at high pH.31 This was studied using atomic force microscopy, dynamic light scattering and transition electron microscopy (TEM). The difference at high pH was thought to be due to the ability of the carboxyl group in the second variant to disassociate. A6K has also been shown to form single layer nanotubes using cryo-TEM and SAXS (small angle-X-ray scattering), and NMR (Fig. 4).32,33
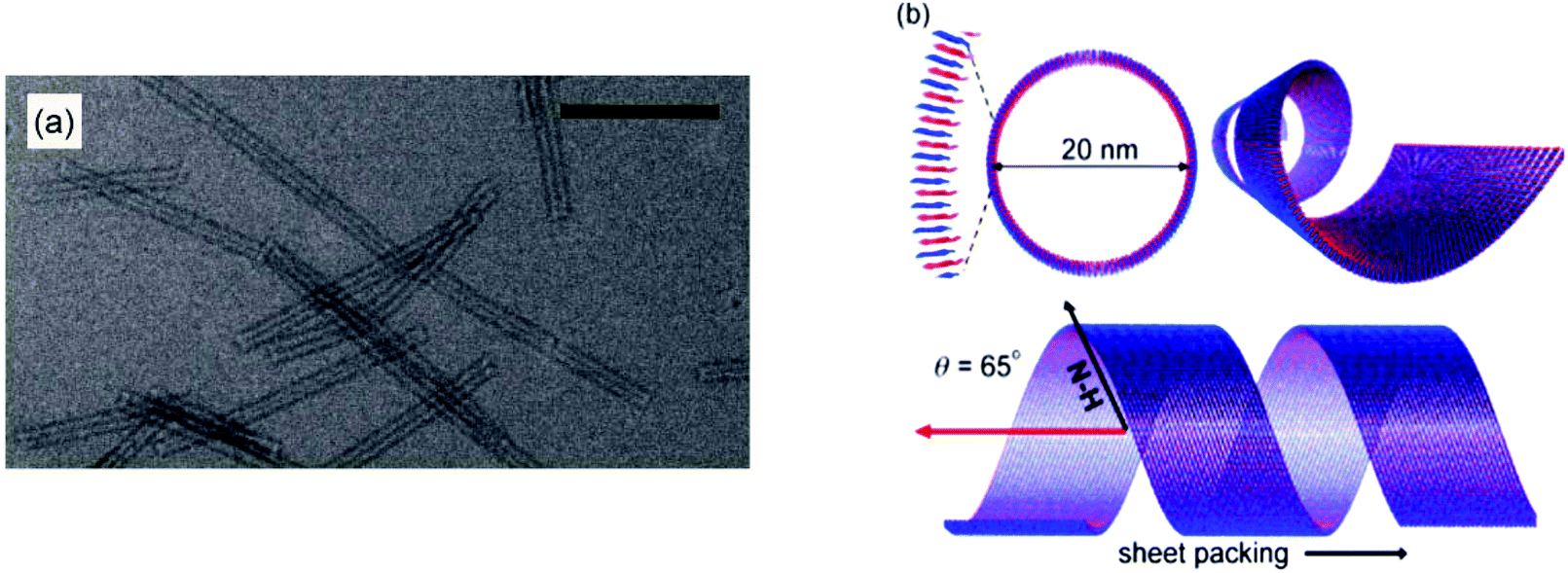 | ||
| Fig. 4 Structure of A6K self-assemblies in water. (a) TEM image of A6K nanotubes in water, scale bar = 200 μM (b) schematic diagram of three perspectives of the nanotubes formed by A6K as shown through TEM, NMR and FTIR.33 | ||
SLPs have promising potential to be used in the study of membrane proteins because they can form layered structures like lipids. Membrane protein purification is a complex process due to the size and interactions membrane proteins have with the lipid bilayer. Surfactant-like peptides could be used to bind the hydrophobic section of the membrane protein and sequester it from water, thus preventing denaturation. For example, surfactant-like peptides have been shown to enhance the stability of bovine rhodopsin, a G-protein coupled receptor.30
Applications
Peptide amphiphiles have many applications. Some applications, including biomineralization,34–36 membrane stability enhancement37–40 and antibody production41,42 are not covered here but are described in detail elsewhere.17,43Tissue scaffolds
Tissue scaffolds are engineered materials that mimic the extra-cellular matrix (ECM), and are designed to allow cell attachment/migration, enable diffusion of desired molecules, exert mechanical and biological influences to modify the behaviour of the cells, or to deliver and retain cells and biochemical factors. Peptide amphiphiles have been shown to have many potential applications in this field. As this has been extensively reviewed elsewhere,44,45 it is not discussed in detail again here. Furthermore, excellent overviews of the material requirements, including the incorporation of peptide motifs, in the development of biomaterials for tissue engineering applications are also available.46–51 Peptide motifs that have an important role in such biomaterials include cell adhesion sequences.One of the most commonly studied cell adhesion motifs is the integrin recognition motif RGDS from fibronectin.52–56 Tirrell's group attached RGD units either through amino or carboxyl units to dialky lipid chains (Fig. 5). They also prepared a conjugate which contained loops connected by linkers to dialkyl chains on both sides.57 It was found that lipid chain attachment to the RGD peptides effected melanoma spreading. The amino conjugated PA was found to hinder spreading, whilst the looped PA was found to promoyr concentration-dependent spreading, and indiscriminate spreading was observed for the carboxyl coupled PA. Further studies based on liposomes revealed that spontaneous metastasis could be inhibited by RGD based PAs.58
 | ||
| Fig. 5 Structure of RGD based peptides. (A) Amino-coupled RGD, (C18)2–Glu–C2–RGD, (B) carboxyl-coupled RGD, RGD–C2–Glu–(C18)2, and (C) looped RGD, (C18)2–Glu–C2–RGD–C2–Glu–(C18)2.57 | ||
PAs have also been shown to have promise in regenerative medicine. PA nanofibril gels containing peptides incorporating a cell adhesion motif, IKVAV have been observed to encapsulate cells and rapidly differentiate them into neurones.59 This same PA has been shown to effective in a mouse model for treating spinal cord injury, the self-assembling nanofibers inhibiting glial scar formation and promotion of axon elongation. Other PAs have been shown to be effective in cartilage regeneration using bone marrow derived stem and progenitor cells.60
The lipopeptide amphiphile Matrixyl (C16-KTTKS), is a component of an antiwrinkle cream which has shown to stimulate collagen production in fibroblasts,61 undergoes self-assembly in aqueous solution.17 At present, it is not known whether the self-assembly directly influences its bioactivity and nor is it established whether the prior studies showing increased collagen production in fibroblasts at concentrations where the PA begins to aggregate61 are relevant in vivo. The skin contains a significant barrier of keratin and lipid membranes in the stratum corneum and the transport of the PA across this barrier has not been carefully examined, although some stripping studies of the stratum corneum with radiolabelled peptide have been performed to probe penetration of PAs.62 The discovery that C16-KTTKS self-assembles into highly extended nanotape structures63 may be relevant to one aspect of anti-wrinkle treatment in that when the applied skincare cream is applied, dried micron scale fibrillar structures could act as filler. The PA C16-KTTKS has been shown to have β-sheet secondary structure and form bilayer-based nanotapes32 which are stable between the pH range of 3–7 at room temperature. However, pH reduction/increase64 or increased temperature65 favours random coil secondary structure, and spherical micelle self-assembly.
Delivery and cell internalization
Transportation of hydrophobic drugs and other active molecules into cells is an important and ongoing biomedical challenge. PA-based nanocarrier systems have shown potential and are usually designed based upon natural sequences. An example is a PA comprising of a tandem dimer, containing binding sites for LDL (low-density lipoprotein) receptor and cell-surface heparin sulphate proteoglycans. Internalisation of the PA into brain capillary endothelial cells was imaged using fluorescent techniques, showing potential drug transport applications.66 Another example of a delivery system is a PA containing cell adhesive MMP-2 sensitive peptide domain, which was shown to form fibrillar hydrogels. This PA can delivery anti-cancer agent cisplatin through enzymatic degradation.67Cationic PAs have also been shown to be useful in gene therapy. The PAs H5R10 and H10R10 were conjugated to cholesterol.68 They were shown to assemble into cationic micelles, which lead to increased localization of charge and increased DNA binding. H10R10 was shown to stimulate higher gene expression, probed through use of a reporter gene in HEK293 and Hep62 cells.68 Moreover, A12H5K10 and A12H5K15 were also investigated.69 Through the same reporter assay, these PAs were shown to have improved gene expression compared to non-amphiphilic control peptide, and a more favourable cytotoxicity profile compared to polyethyleneimine (PEI), a commonly used synthetic DNA-condensing polymer. Simultaneous delivery of genes (either p53 tumour suppressor or luciferase reporter gene) and doxorubicin was examined using Ac-(AF)6H5K15-NH2.70 This PA self-assembled into micelles with the ability to encapsulated condensed DNA and doxorubicin, with delivery efficiency and in vitro expression examined in HePG2 cells.
It has also been possible to use PAs to study endocytosis. One such PA, based on p53 tumour suppressor, comprised this pro-apoptotic peptide attached to C16 lipid chain (Fig. 6). Internalization was investigated using fluorescent methods. This PA was shown to self-assemble into rod-shaped micelles. FRET (fluorescence resonance energy transfer) showed internalization of monomers into SJSA-1 human osteosarcoma cells as opposed to micelles, and uptake was enhanced by lipidation.71
 | ||
| Fig. 6 Schematic diagram of PA based on p53 tumour suppressor protein internalisation into SJSA-1 human osteosarcoma cell line.71 | ||
Antimicrobial peptides
Short cationic peptides can exhibit antimicrobial properties. Lipidation is thought to enhance antimicrobial properties of cationic peptides.72 Lipidation causes changes in secondary structure as the peptide interacts with the membranes. The exact mechanism by which these peptides act upon bacteria and fungi is unknown. Reports on di- and tri-lysine PAs showed that antimicrobial activity against Gram-positive and Gram-negative bacterial occurred through leakage, caused by cell membrane disruption.73Surfactins, iturins and lichenysin are lipopeptides with molecular structures shown in Fig. 7, which have antifungal properties.73 Surfactin, produced by B. subtilis, has the ability to reduce the surface tension of water to 27 mN m−1 at concentrations as low as 20 μM. It was found through neutron scattering to self-assemble into spherical micelles in bulk aqueous solution, and to adopt a more globular conformation at the interface (for example air/water).19 The self-assembly of surfactin and two other lipopeptides produced by B. subtilis, plipastatin and mycosubtilin were also examined using biophysical techniques. Small-angle X-ray scattering (SAXS) and Cryo-TEM confirmed that surfactin and plipastatin self-assembled into spherical micelles, whereas mycosubtilin formed nanotapes based on bilayer stacking.74 It remains an open question as to whether the self-assembly influences bioactivity, changing the interaction of the peptide with receptor directly or via shifts in the molecule-aggregate equilibrium.
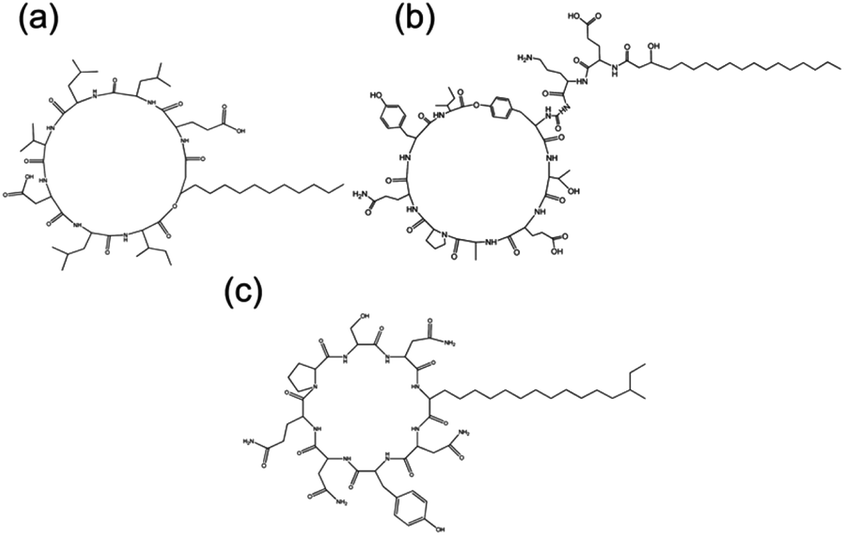 | ||
| Fig. 7 Chemical structure of three bacterially derived lipopeptides. (A) Surfactin, (b) plipastatin and (c) mycosubtilin.74 | ||
Arginine rich SLPs have been shown to have some antimicrobial properties. An example is the TAT (YGRKKRRQRRR) peptide from HIV retrovirus which has been shown to have antimicrobial properties, and contains 6 arginine and 2 lysine residues. It was found that substitution of any basic residues lead to a decrease in antimicrobial activity. TAT peptide is commonly used to transport peptides and proteins across cell membranes.75 Liposomes decorated with TAT peptide have also been investigated with potential as a drug delivery vehicle for the anticancer drug paclitaxel.76 Conjugation of TAT to polyarginine and cholesterol shows improvements in membrane translocation, as well as in antimicrobial strength. This PA assembles into micelles, and has shown to be effective in rabbit and mouse models against S. aureus.77 PAs have also demonstrated promise in sepsis treatment. Sepsis (blood poisoning) is induced by bacterial endotoxins, namely lipopolysacharrides. A PA was designed based on natural immune response antibodies against sepsis. It was found that increased effective neutralisation of lipopolysacharrides by the PAs correlated with increased lipid chain length. Interestingly, two other designed PAs with lipopolysacharride binding sequences, attached to C16 alkyl lipid chain were shown to self-assemble into fibrils.78
Peptide hydrogels
A hydrogel is defined as a water swollen, cross-linked or entangled polymeric network. Hydrogels may form through non-covalent processes such as the formation of a sample-spanning fibrillar network or they may be created through covalent cross-linking within pre-existing or produced network structures. Some hydrogels are able to absorb large amounts of water and they may also be designed to have mechanical properties similar to natural tissue. Hydrogel formation can be very dependent upon temperature, pH, concentration of polymer or salts.79Hydrogels can be made of natural or synthetic materials, and can be prepared through use of homopolymers, copolymers or multi-polymer networks. Homopolymeric hydrogels contain one polymer, copolymer networks contain two or more polymers interacting with each other to form the fibrillar network, and co-networks contain two independent fibril forming monomers. They can be non-crystalline, semi-crystalline or crystalline.79 Formation can occur through covalent interactions between monomers, or through electrostatic interactions or hydrogen bonding, van der Waals or ionic interactions.
Peptides and peptide conjugates have the ability to form hydrogels. Hydrogelation can result from non-covalent self-assembly processes under appropriate conditions.80 For example, for fibre-based hydrogels, self-assembly might occur involving the formation of nanofibers from β-sheet peptides. These fibres may then elongate in three dimensions, leading to increased fibre thickness and length, ultimately leading to fibrillar network formation. These complex networks of peptides may then entrap water, thus providing a self-supporting hydrogel. Peptides and peptide conjugates with other secondary structures, for example α-helical, have also been shown to be able to form hydrogels.81
For biomedical applications, it is essential that the gels are biocompatible. Synthetic hydrogels may induce an immune inflammatory response, which could have potential cytotoxicity. For example, although PEG is often considered an “inert” polymer, high molar mass PEG is not degraded in vivo and can cause toxicity, as can high doses of PEG polymers of low molar mass.82–85 Immunogenic response to PEG has been reported.84,86 Furthermore, synthetic PEG may contain impurities or unreacted monomer which is highly toxic. PEG-peptide and PEG-protein hydrogels and alternatives based on other synthetic polymers are the subject of recent reviews.84,87,88 Natural hydrogel polymers, for example peptide functionalised polymer hydrogels,89–95 can be designed to be highly biocompatible. A number of other physical parameters, for example degradation and performance (for example cell adhesion) must also be considered. The mechanism by which the hydrogel forms is important. Gels formed by ionic cross-linking may be prematurely degraded in vivo due to electrostatic interactions in body fluids, making covalent cross-linking preferred in this way. Some examples of other naturally occurring polymers include fibrin, collagen and gelatin25 and hyaluronate. The most popular FDA approved synthetic polymers are poly(acrylic acid) derivatives and poly(ethylene oxide) (PEO).96
Hydrogels have also been shown to have applications in cell culturing. Peptide hydrogels for cell culturing must mimic the extracellular matrix and incorporate peptide adhesion motifs. RGD, from is one of the most widely used peptide sequences for this purpose. In one example, an Fmoc-RGD peptide was shown to form β-sheet secondary structure, and self-assemble into amyloid fibrils. The hydrogel formed by Fmoc-RGD was shown to be capable of sustaining cells and to support fibroblasts compared to a control scrambled sequence.97 Other applications for peptide and peptide conjugate hydrogels include biosensors,98 regenerative medicine and tissue engineering,99–109 and slow release drug delivery systems.110–115
Peptide-mimetic polymers and self-assembly
The abundant uses of peptides biomedically, biologically and commercially has led to significant interest in polymers designed to have similar characteristics. One class of peptide-mimetic materials are peptoids or N-substituted glycines (Fig. 8). They are structural isomers of peptides, with the side chain attached to the amide nitrogen as opposed to the α-carbon,116 resulting in novel morphologies. The positioning of the side chain interrupts intra- and inter-back bone hydrogen bonding which holds together peptide secondary structure. Helices, ribbons and sheets may be formed, through careful molecular design including selection of the sidechains. Their nature also makes them resistant to proteolysis and biodegradation, making them suitable for some applications including long-term biointerfaces and biomaterials, as well as antimicrobial peptidomimics.116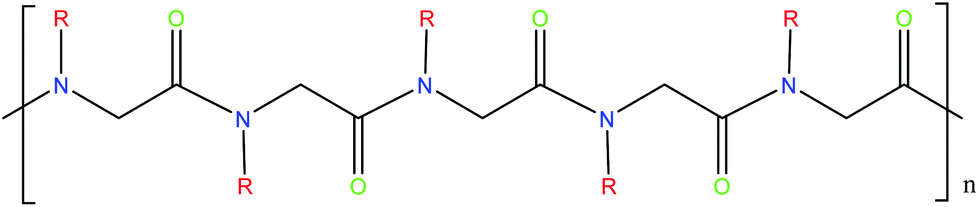 | ||
| Fig. 8 Structure of peptoid polymer. Peptoids are structural isomers of peptides, with the R group attached to the amide group as opposed to the α-carbon. | ||
There is a defined method for synthesising sequence-specific peptoids, and bioactive sequences can be developed through use of combinatorial library searches.116 One example of a clinically used peptide is poly(N-methyl glycine, polysarcosine) which is prescribed as a non-immunogenic. Biomedical studies also show promise, including a study cell viability assays showing that they can be tolerated at significant concentrations before they are cytotoxic. This makes them an interesting material for drug delivery. Other studies have included development of protein-mimetics and membrane spanning helices.116
Self-assembly of peptoids is currently attracting much attention. They have been shown to assemble into nanosheets dependent on sequence (Fig. 9), with a mixture of hydrophobic and ionic monomer units, which may extend to micron size laterally.117–119 Hollow nanotubes assemblies have been observed in water, without a central hydrophobic core, chirality, electrostatic interactions, hydrogen bond network or π-stacking.120 The nanotube is thought to be stabilised by van der Waals interactions between side-chains as a pose to hydrogen bonds which stabilise peptide structure.120 Worm-like vesicles and micelles structures have been observed.121 Lipidation of peptoids, inspired by lipopeptide assembly has been shown to enhance designability of assembly. Ultra-small spherical micelles of 5 nm diameter have been designed based on this principle, with subtle variations in size due to sequence changes.122
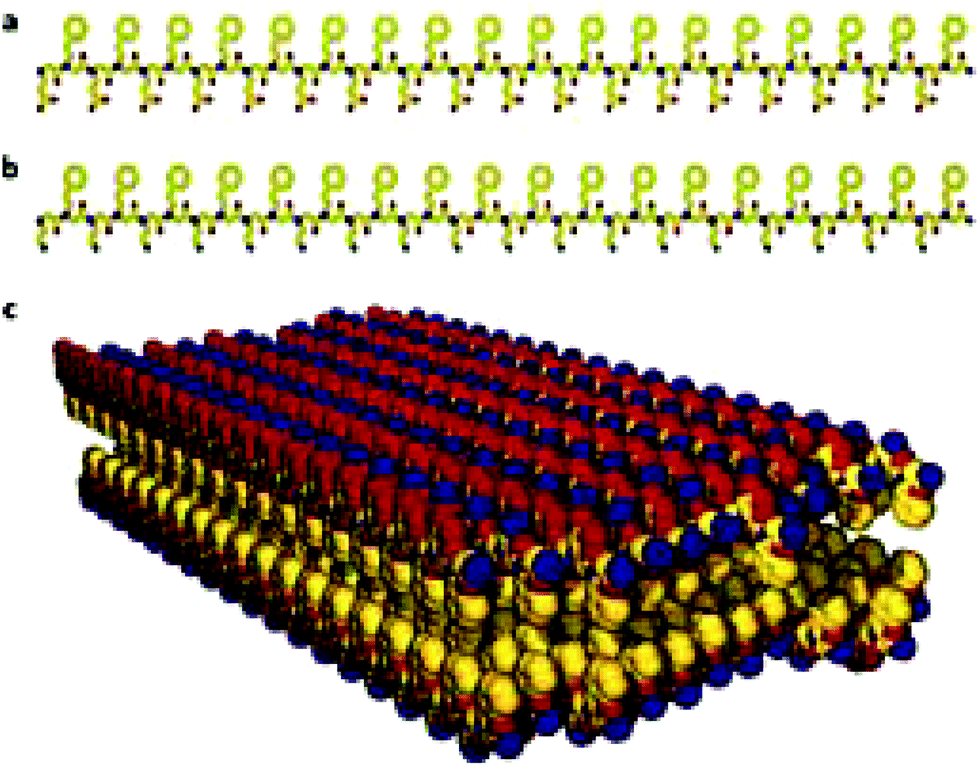 | ||
| Fig. 9 The structure of nanosheets formed from oppositely charged peptoid polymers. (a) Chemical structure of a negatively charged periodic amphiphilic peptoid (Nce–Npe)18. (b) Chemical structure of a positively charged periodic amphiphilic peptoid (Nae–Npe)18. (c) Molecular model of the sheets assembled from (Nae–Npe)18 and (Nce–Npe)18. The modelled conformation shows that hydrophobic groups face each other in the interior of the sheet and oppositely charged hydrophilic groups are alternating and surface-expose. Atom colours: carbon, yellow; nitrogen, blue; oxygen, red.119 Nce denotes N-2(carboxyethyl)glycine, Npe denotes N-2(phenylethyl)glycine and Nae denotes N-2(aminoethyl)glycine. | ||
Hybrid oligo(peptoid-peptides) are a combination of peptides and peptoids with potential biomedical applications and interesting self-assembly behaviour. Few studies have been done on this type of molecule, however there are a few examples such as, cyclosporin123 isolated from Hypocladium inflatum gams fungus, which is a naturally occurring and clinically used immunosuppressant.124 Self-assembly of this class of conjugate has not been explored extensively. It has been shown that these nanosheets may curl to form nanotubes or microtubes.125 Core–shell micelle structures have recently been observed from synthetic oligo(peptoid-peptide) hybrid molecules.126
Conclusions
Self-assembly is a vital process in nature, driving aggregation in a manner that depends on the conditions (concentration, pH, temperature, presence of salts etc.). Peptide amphiphiles and designed peptide-mimetic molecules are able to self-assemble into complex stable nanostructures. The examples presented above include peptides and conjugates with bioactive functions which can be exploited, for clinical use, commercial use, or to further understand the self-assembly process.Surfactant-like peptides are simple peptide systems which can potentially be produced inexpensively and at large scale (by genetic engineering for instance) for applications such as antimicrobials or membrane protein support. Their self-assembly behaviour is remarkably rich and is the subject of ongoing investigation, exploring the role of charge, sequences and the nature of the constituent amino acids. There is also great scope to prepare novel peptide-mimetics based on peptoids and their conjugates for example. These will have new applications and are likely to show unanticipated self-assembly behaviour. This will be a rich field for future research.
New antimicrobial peptides are being discovered in nature, a process likely to continue given the need to create new agents to overcome antimicrobial resistant infections. Newly discovered natural antimicrobial peptides and their derivatives can be complemented with novel designed peptides. Cyclisation is an important tool to stabilize these molecules in vivo, for applications as antimicrobials and for other therapeutic peptide-based compounds. The influence of cyclisation on self-assembly is an interesting avenue for further research. Certainly one can expect the packing of the molecules to be influenced due to the large steric bulk of cyclic peptides which can form the “headgroup” in peptide amphiphiles.
A key question that remains unresolved is whether there is a relationship between bioactivity and self-assembly of peptide amphiphiles. The reason the answer remains elusive is due to the complexity of the question. It is very difficult to decouple the effect of self-assembly from the concentration effect on bioactivity. One method suggested would be to cross-link self-assembled structures, trapping them in a specific thermodynamic state.19 However, this may interfere with the self-assembled structure and potentially impact the resultant recorded bioactivity. It is also not straightforward to observe substrate and molecule/aggregate interactions without perturbing the molecule-aggregate equilibrium. It is likely that there will be some cases where self-assembled structures themselves impart bioactivity, and others where there is no direct relationship between self-assembly and bioactivity, particularly in the circumstances where bioactivity is observed below the critical aggregation concentration. It is possible that there may be an indirect relationship, where amphiphilicity leads to greater bioactivity, through for example increased membrane compatibility, this amphiphilicity, in turn, leading to self-assembly at high concentrations.
Acknowledgements
This work was supported by EPSRC grant EP/L020599/1 to IWH. The PhD studentship of CJCEG was supported by the University of Reading and Diamond Light Source.References
- A. Dehsorkhi, V. Castelletto and I. W. Hamley, J. Pept. Sci., 2014, 20, 453–467 CrossRef CAS PubMed.
- P. Zhou, L. Deng, Y. Wang, J. R. Lu and H. Xu, J. Colloid Interface Sci., 2016, 464, 219–228 CrossRef CAS PubMed.
- D. Marsh, Biophys. J., 2012, 102, 1079–1087 CrossRef CAS PubMed.
- K. A. Johnson and G. G. Borisy, J. Mol. Biol., 1979, 133, 199–216 CrossRef CAS PubMed.
- M. K. Gardner, B. D. Charlebois, I. M. Janosi, J. Howard, A. J. Hunt and D. J. Odde, Cell, 2011, 146, 582–592 CrossRef CAS PubMed.
- D. Mandal, A. Nasrolahi Shirazi and K. Parang, Org. Biomol. Chem., 2014, 12, 3544–3561 CAS.
- M. C. Branco and J. P. Schneider, Acta Biomater., 2009, 5, 817–831 CrossRef CAS PubMed.
- J. A. Hutchinson, S. Burholt and I. W. Hamley, J. Pept. Sci., 2017, 23, 82–94 CrossRef CAS PubMed.
- S.-Y. Sheu, D.-Y. Yang, H. L. Selzle and E. W. Schlag, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12683–12687 CrossRef CAS PubMed.
- J. C. Stendahl, M. S. Rao, M. O. Guler and S. I. Stupp, Adv. Funct. Mater., 2006, 16, 499–508 CrossRef CAS.
- V. P. Torchilin, Pharm. Res., 2007, 24, 1–16 CAS.
- R. S. Tu and M. Tirrell, Adv. Drug Delivery Rev., 2004, 56, 1537–1563 CrossRef CAS PubMed.
- K. Kita-Tokarczyk, J. Grumelard, T. Haefele and W. Meier, Polymer, 2005, 46, 3540–3563 CrossRef CAS.
- H. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS PubMed.
- D. W. P. M. Löwik and J. C. M. van Hest, Chem. Soc. Rev., 2004, 33, 234–245 RSC.
- D. W. P. M. Löwik, E. H. P. Leunissen, M. van den Heuvel, M. B. Hansen and J. C. M. van Hest, Chem. Soc. Rev., 2010, 39, 3394 RSC.
- I. W. Hamley, Soft Matter, 2011, 7, 4122 RSC.
- J. B. Matson, R. H. Zha and S. I. Stupp, Curr. Opin. Solid State Mater. Sci., 2011, 15, 225–235 CrossRef CAS PubMed.
- I. W. Hamley, Chem. Commun., 2015, 51, 8574–8583 RSC.
- V. Castelletto, A. Kaur, R. M. Kowalczyk, I. W. Hamley, M. Reza and J. Ruokolainen, Biomacromolecules DOI:10.1021/acs.biomac.7b00057.
- J. T. Meijer, M. Roeters, V. Viola, D. W. P. M. Löwik, G. Vriend and J. C. M. Van Hest, Langmuir, 2007, 23, 2058–2063 CrossRef CAS PubMed.
- T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020–13041 CrossRef CAS PubMed.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef CAS PubMed.
- S. Kirkham, V. Castelletto, I. W. Hamley, K. Inoue, R. Rambo, M. Reza and J. Ruokolainen, ChemPhysChem, 2016, 2118–2122 CrossRef CAS PubMed.
- K. Deres and H.-G. Rammensee, Nature, 1989, 342, 189–192 CrossRef PubMed.
- M. Krug, G. Folkers, B. Haas, G. Hess, K. H. Wiesmuller, S. Freund and G. Jung, Biopolymers, 1989, 28, 499–512 CrossRef CAS PubMed.
- I. W. Hamley, S. Kirkham, A. Dehsorkhi, V. Castelletto, M. Reza and J. Ruokolainen, Chem. Commun., 2014, 50, 15948–15951 RSC.
- S. Vauthey, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5355–5360 CrossRef CAS PubMed.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- X. Zhao, Curr. Opin. Colloid Interface Sci., 2009, 14, 340–348 CrossRef CAS.
- F. Qiu, Y. Chen and X. Zhao, J. Colloid Interface Sci., 2009, 336, 477–484 CrossRef CAS PubMed.
- V. Castelletto, D. R. Nutt, I. W. Hamley, S. Bucak, C. Cenker and U. Olsson, Chem. Commun., 2010, 46, 6270–6272 RSC.
- D. A. Middleton, J. Madine, V. Castelletto and I. W. Hamley, Angew. Chem., Int. Ed., 2013, 52, 10537–10540 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- E. D. Spoerke, S. G. Anthony and S. I. Stupp, Adv. Mater., 2009, 21, 425–430 CrossRef CAS PubMed.
- S. Cavalli, D. C. Popescu, E. E. Tellers, M. R. J. Vos, B. P. Pichon, M. Overhand, H. Rapaport, N. A. J. M. Sommerdijk and A. Kros, Angew. Chem., Int. Ed., 2006, 45, 739–744 CrossRef CAS PubMed.
- X. Zhao, Y. Nagai, P. J. Reeves, P. Kiley, H. G. Khorana, S. Zhang and J. M. Buchanan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17707–17712 CrossRef CAS PubMed.
- K. Matsumoto, M. Vaughn, B. D. Bruce, S. Koutsopoulos and S. Zhang, J. Phys. Chem. B, 2009, 113, 75–83 CrossRef CAS PubMed.
- R. Das, P. J. Kiley, M. Segal, J. Norville, A. A. Yu, L. Wang, S. A. Trammell, L. E. Reddick, R. Kumar, F. Stellacci, N. Lebedev, J. Schnur, B. D. Bruce, S. Zhang and M. Baldo, Nano Lett., 2004, 4, 1079–1083 CrossRef CAS.
- P. Kiley, X. Zhao, M. Vaughn, M. A. Baldo, B. D. Bruce and S. Zhang, PLoS Biol., 2005, 3, 1180–1186 CAS.
- F. Boato, R. M. Thomas, A. Ghasparian, A. Freund-Renard, K. Moehle and J. A. Robinson, Angew. Chem., Int. Ed., 2007, 46, 9015–9018 CrossRef CAS PubMed.
- C.-L. McGregor, L. Chen, N. C. Pomroy, P. Hwang, S. Go, A. Chakrabartty and G. G. Privé, Nat. Biotechnol., 2003, 21, 171–176 CrossRef CAS PubMed.
- S. I. Stupp, Nano Lett., 2010, 10, 4783–4786 CrossRef CAS PubMed.
- J. B. Matson and S. I. Stupp, Chem. Commun., 2012, 48, 26–33 RSC.
- E. Arslan, I. C. Garip, G. Gulseren, A. B. Tekinay and M. O. Guler, Adv. Healthcare Mater., 2014, 3, 1357–1376 CrossRef CAS PubMed.
- M. P. Lutolf and H. M. Blau, Adv. Mater., 2009, 21, 3255–3268 CrossRef CAS PubMed.
- E. S. Place, N. D. Evans and M. M. Stevens, Nat. Mater., 2009, 8, 457–470 CrossRef CAS PubMed.
- E. S. Place, J. H. George, C. K. Williams and M. M. Stevens, Chem. Soc. Rev., 2009, 38, 1139 RSC.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- J. J. Rice, M. M. Martino, L. De Laporte, F. Tortelli, P. S. Briquez and J. A. Hubbell, Adv. Healthcare Mater., 2013, 2, 57–71 CrossRef CAS PubMed.
- A. J. Keung, S. Kumar and D. V. Schaffer, Annu. Rev. Cell Dev. Biol., 2010, 26, 533–556 CrossRef CAS PubMed.
- E. Ruoslahti and M. D. Pierschbacher, Cell, 1986, 44, 517–518 CrossRef CAS PubMed.
- E. Ruoslahti and M. Pierschbacher, Science, 1987, 238, 491–497 CAS.
- R. O. Hynes, Cell, 1992, 69, 11–25 CrossRef CAS PubMed.
- J. A. Hubbell, Biotechnology, 1995, 13, 565–576 CAS.
- M. Tirrell, E. Kokkoli and M. Biesalski, Surf. Sci., 2002, 500, 61–83 CrossRef CAS.
- T. Pakalns, K. L. Haverstick, G. B. Fields, J. B. McCarthy, D. L. Mooradian and M. Tirrell, Biomaterials, 1999, 20, 2265–2279 CrossRef CAS PubMed.
- N. Oku, C. Koike, Y. Tokudome, S. Okada, N. Nishikawa, H. Tsukada, M. Kiso, A. Hasegawa, H. Fujii, J. Murata and I. Saiki, Adv. Drug Delivery Rev., 1997, 24, 215–223 CrossRef.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS PubMed.
- V. M. Tysseling-Mattiace, V. Sahni, K. L. Niece, D. Birch, C. Czeisler, M. G. Fehlings, S. I. Stupp and J. A. Kessler, J. Neurosci., 2008, 28, 3814–3823 CrossRef CAS PubMed.
- R. R. Jones, V. Castelletto, C. J. Connon and I. W. Hamley, Mol. Pharm., 2013, 10, 1063–1069 CrossRef CAS PubMed.
- K. Lintner and O. Peschard, Int. J. Cosmet. Sci., 2000, 22, 207–218 CrossRef CAS PubMed.
- V. Castelletto, I. Hamley, J. Perez, L. Abezgauz and D. Danino, Chem. Commun., 2010, 46, 9185–9187 RSC.
- A. Dehsorkhi, V. Castelletto, I. W. Hamley, J. Adamcik and R. Mezzenga, Soft Matter, 2013, 9, 6033 RSC.
- J. F. Miravet, B. Escuder, M. D. Segarra-Maset, M. Tena-Solsona, I. W. Hamley, A. Dehsorkhi and V. Castelletto, Soft Matter, 2013, 9, 3558–3564 RSC.
- E. Leupold, H. Nikolenko, M. Beyermann and M. Dathe, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 2781–2789 CrossRef CAS PubMed.
- J. K. Kim, J. Anderson, H. W. Jun, M. A. Repka and S. Jo, Mol. Pharm., 2009, 6, 978–985 CrossRef CAS PubMed.
- X. D. Guo, F. Tandiono, N. Wiradharma, D. Khor, C. G. Tan, M. Khan, Y. Qian and Y. Y. Yang, Biomaterials, 2008, 29, 4838–4846 CrossRef CAS PubMed.
- N. Wiradharma, M. Khan, Y. W. Tong, S. Wang and Y. Y. Yang, Adv. Funct. Mater., 2008, 18, 943–951 CrossRef CAS.
- N. Wiradharma, Y. W. Tong and Y. Y. Yang, Biomaterials, 2009, 30, 3100–3109 CrossRef CAS PubMed.
- D. Missirlis, H. Khant and M. Tirrell, Biochemistry, 2009, 48, 3304–3314 CrossRef CAS PubMed.
- A. F. Chu-Kung, K. N. Bozzelli, N. A. Lockwood, J. R. Haseman, K. H. Mayo and M. V. Tirrell, Bioconjugate Chem., 2004, 15, 530–535 CrossRef CAS PubMed.
- A. Makovitzki, A. Viterbo, Y. Brotman, I. Chet and Y. Shai, Appl. Environ. Microbiol., 2007, 73, 6629–6636 CrossRef CAS PubMed.
- I. W. Hamley, A. Dehsorkhi, P. Jauregi, J. Seitsonen, J. Ruokolainen, F. Coutte, G. Chataigné and P. Jacques, Soft Matter, 2013, 9, 9572 RSC.
- J. S. Wadia, R. V. Stan and S. F. Dowdy, Nat. Med., 2004, 10, 310–315 CrossRef CAS PubMed.
- H. Fu, K. Shi, G. Hu, Y. Yang, Q. Kuang, L. Lu, L. Zhang, W. Chen, M. Dong, Y. Chen and Q. He, J. Pharm. Sci., 2015, 104, 1160–1173 CrossRef CAS PubMed.
- L. Liu, K. Xu, H. Wang, P. K. J. Tan, W. Fan, S. S. Venkatraman, L. Li and Y.-Y. Yang, Nat. Nanotechnol., 2009, 4, 457–463 CrossRef CAS PubMed.
- C. Mas-Moruno, L. Cascales, P. Mora, L. J. Cruz, E. Pérez-Payá and F. Albericio, Biopolymers, 2009, 92, 508–517 CrossRef CAS PubMed.
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS PubMed.
- N. Singh, M. Kumar, J. F. Miravet, R. V. Ulijn and B. Escuder, Chem. – Eur. J., 2017, 23, 981–993 CrossRef CAS PubMed.
- A. Dasgupta, J. H. Mondal and D. Das, RSC Adv., 2013, 3, 9117 RSC.
- B. Li, X. Dong, S. Fang, J. Gao, G. Yang and H. Zhao, Drug Chem. Toxicol., 2011, 34, 208–212 CrossRef CAS PubMed.
- R. Webster, V. Elliott, B. K. Park, D. Walker, M. Hankin and P. Taupin, in PEGylated Protein Drugs: Basic Science and Clinical Applications, 2009, pp. 127–146 Search PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- G. Pasut, Polymer, 2014, 6, 160–178 Search PubMed.
- A. C. Engler, X. Ke, S. Gao, J. M. W. Chan, D. J. Coady, R. J. Ono, R. Lubbers, A. Nelson, Y. Y. Yang and J. L. Hedrick, Macromolecules, 2015, 48, 1673–1678 CrossRef CAS.
- E. M. Pelegri-Oday, E. W. Lin and H. D. Maynard, J. Am. Chem. Soc., 2014, 136, 14323–14332 CrossRef CAS PubMed.
- I. W. Hamley, Biomacromolecules, 2014, 15, 1543–1559 CrossRef CAS PubMed.
- W. A. Petka, J. L. Harden, K. P. Mcgrath, D. Wirtz and D. A. Tirrell, Science, 1998, 281, 389–392 CrossRef CAS PubMed.
- W. Shen, K. Zhang, J. A. Kornfield and D. A. Tirrell, Nat. Mater., 2006, 5, 153–158 CrossRef CAS PubMed.
- I. W. Hamley, G. Cheng and V. Castelletto, Macromol. Biosci., 2011, 11, 1068–1078 CrossRef CAS PubMed.
- P. J. Stahl, N. H. Romano, D. Wirtz and S. M. Yu, Biomacromolecules, 2010, 11, 2336–2344 CrossRef CAS PubMed.
- P. Jing, J. S. Rudra, A. B. Herr and J. H. Collier, Biomacromolecules, 2008, 9, 2438–2446 CrossRef CAS PubMed.
- N. Tzokova, C. M. Fernyhough, M. F. Butler, S. P. Armes, A. J. Ryan, P. D. Topham and D. J. Adams, Langmuir, 2009, 25, 11082–11089 CrossRef CAS PubMed.
- N. Tzokova, C. M. Fernyhough, P. D. Topham, N. Sandon, D. J. Adams, M. F. Butler, S. P. Armes and A. J. Ryan, Langmuir, 2009, 25, 2479–2485 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS PubMed.
- G. Cheng, V. Castelletto, R. R. Jones, C. J. Connon and I. W. Hamley, Soft Matter, 2011, 7, 1326–1333 RSC.
- N. A. Peppas and D. S. Van Blarcom, J. Controlled Release, 2016, 240, 142–150 CrossRef CAS PubMed.
- S. Van Vlierberghe, P. Dubruel and E. Schacht, Biomacromolecules, 2011, 12, 1387–1408 CrossRef CAS PubMed.
- C. A. DeForest and D. A. Tirrell, Nat. Mater., 2015, 14, 523–531 CrossRef CAS PubMed.
- T. T. Yu and M. S. Shoichet, Biomaterials, 2005, 26, 1507–1514 CrossRef CAS PubMed.
- W. L. Murphy, T. C. McDevitt and A. J. Engler, Nat. Mater., 2014, 13, 547–557 CrossRef CAS PubMed.
- J. Thiele, Y. Ma, S. M. C. Bruekers, S. Ma and W. T. S. Huck, Adv. Mater., 2014, 26, 125–148 CrossRef CAS PubMed.
- J. S. Miller, C. J. Shen, W. R. Legant, J. D. Baranski, B. L. Blakely and C. S. Chen, Biomaterials, 2010, 31, 3736–3743 CrossRef CAS PubMed.
- M. J. Wilson, S. J. Liliensiek, C. J. Murphy, W. L. Murphy and P. F. Nealey, Soft Matter, 2012, 8, 390–398 RSC.
- S. Q. Liu, R. Tay, M. Khan, P. L. Rachel Ee, J. L. Hedrick and Y. Y. Yang, Soft Matter, 2010, 6, 67 RSC.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS PubMed.
- M. J. Cooke, K. Vulic and M. S. Shoichet, Soft Matter, 2010, 6, 4988 RSC.
- B. V. Slaughter, S. S. Khurshid and O. Z. Fisher, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed.
- Y. Bae and K. Kataoka, Adv. Drug Delivery Rev., 2009, 61, 768–784 CrossRef CAS PubMed.
- N. Nishiyama, S. Okazaki, H. Cabral, M. Miyamoto, Y. Kato, Y. Sugiyama, K. Nishio, Y. Matsumura and K. Kataoka, Cancer Res., 2003, 63, 8977–8983 CAS.
- F. Q. Hu, Y. Y. Zhang, J. You, H. Yuan and Y. Z. Du, Mol. Pharm., 2012, 9, 2469–2478 CrossRef CAS PubMed.
- A. S. Karakoti, S. Das, S. Thevuthasan and S. Seal, Angew. Chem., Int. Ed., 2011, 50, 1980–1994 CrossRef CAS PubMed.
- S. D. Brown, P. Nativo, J. A. Smith, D. Stirling, P. R. Edwards, B. Venugopal, D. J. Flint, J. A. Plumb, D. Graham and N. J. Wheate, J. Am. Chem. Soc., 2010, 132, 4678–4684 CrossRef CAS PubMed.
- A. Kolate, D. Baradia, S. Patil, I. Vhora, G. Kore and A. Misra, J. Controlled Release, 2014, 192, 67–81 CrossRef CAS PubMed.
- A. M. Rosales, R. A. Segalman and R. N. Zuckermann, Soft Matter, 2013, 8400–8414 RSC.
- E. J. Robertson, A. Battigelli, C. Proulx, R. V. Mannige, T. K. Haxton, L. Yun, S. Whitelam and R. N. Zuckermann, Acc. Chem. Res., 2016, 49, 379–389 CrossRef CAS PubMed.
- B. Sanii, T. K. Haxton, G. K. Olivier, A. Cho, B. Barton, C. Proulx, S. Whitelam and R. N. Zuckermann, ACS Nano, 2014, 8, 11674–11684 CrossRef CAS PubMed.
- K. T. Nam, S. A. Shelby, P. H. Choi, A. B. Marciel, R. Chen, L. Tan, T. K. Chu, R. A. Mesch, B.-C. Lee, M. D. Connolly, C. Kisielowski and R. N. Zuckermann, Nat. Mater., 2010, 9, 454–460 CrossRef CAS PubMed.
- J. Sun, X. Jiang, R. Lund, K. H. Downing, N. P. Balsara and R. N. Zuckermann, Proc. Natl. Acad. Sci. U. S. A., 2016, 15, 3954–3959 CrossRef PubMed.
- C. Fetsch, J. Gaitzsch, L. Messager, G. Battaglia and R. Luxenhofer, Sci. Rep., 2016, 6, 33491 CrossRef CAS PubMed.
- K. A. L. Lau, V. Castelletto, T. Kendall, J. Sefcik, I. W. Hamley, M. Reza and J. Ruokolainen, Chem. Commun., 2017, 53, 2178–2181 RSC.
- G. L. Butterfoss, K. Drew, P. D. Renfrew, K. Kirshenbaum and R. Bonneau, Biopolymers, 2014, 102, 369–378 CrossRef CAS PubMed.
- S. Matsuda and S. Koyasu, Immunopharmacology, 2000, 47, 119–125 CrossRef CAS PubMed.
- H. K. Murnen, A. M. Rosales, J. N. Jaworski, R. A. Segalman and R. N. Zuckermann, J. Am. Chem. Soc., 2010, 132, 16112–16119 CrossRef CAS PubMed.
- M. Hartweg, C. J. C. Edwards-Gayle, E. Radavar, D. Collis, M. Reza, J. Ruokolainen, C. Barner-Kowoliki, I. W. Hamley, H. S. Azevedo and R. Becer, submitt. publ.
| This journal is © The Royal Society of Chemistry 2017 |