
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reductive amination catalyzed by iridium complexes using carbon monoxide as a reducing agent†
Alexey P.
Moskovets
a,
Dmitry L.
Usanov
b,
Oleg I.
Afanasyev
a,
Vasilii A.
Fastovskiy
a,
Alexander P.
Molotkov
a,
Karim M.
Muratov
a,
Gleb L.
Denisov
a,
Semen S.
Zlotskii
c,
Alexander F.
Smol'yakov
ad,
Dmitry A.
Loginov
a and
Denis
Chusov
 *ad
*ad
aA. N. Nesmeyanov Institute of Organoelement Compounds of the Russian Academy of Sciences, 119991, Vavilova St. 28, Moscow, Russian Federation. E-mail: Chusov@ineos.ac.ru; Denis.chusov@gmail.com
bDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA 02138, USA
cUfa Oil Institute, Kosmonavtov Str., 1, Ufa, 450062, Russian Federation
dFaculty of Science, RUDN University, 6 Miklukho-Maklaya St., Moscow 117198, Russian Federation
First published on 11th July 2017
Abstract
Development of novel, sustainable catalytic methodologies to provide access to amines represents a goal of fundamental importance. Herein we describe a systematic study for the construction of a variety of amines catalyzed by a well-defined homogeneous iridium complex using carbon monoxide as a reducing agent. The methodology was shown to be compatible with functional groups prone to reduction by hydrogen or complex hydrides.
Among the transformations which are conventionally classified as reductions, reductive amination is notable for particularly high preparative significance, being one of the most convenient and versatile methods of C–N bond formation.1,2 Notably, a representative screening of publications in the field of medicinal chemistry demonstrated that reductive amination is used more frequently than the eight most popular reductive processes combined,3 which is not surprising in light of the numerous advantages of this type of transformation, such as accessibility of starting materials, modular nature and the possibility of rapid build-up of molecular complexity.
Our group has recently discovered a catalytic reductive methodology4 which takes advantage of the deoxygenative potential of carbon monoxide and does not require an external hydrogen source unlike the conventional approaches to e.g. reductive amination.
So far only rhodium and ruthenium complexes were found to be capable of catalyzing CO-assisted5,6 reductive reactions.7 In this work, we tried to expand our knowledge about this type of processes and tried both iridium(I) and iridium(III) complexes with weakly and strongly bonded ligands with η-1, η-2, η-3, η-5 and η-6 coordination types (Scheme 1) as potential catalysts. As a result, herein we report the first example of iridium-catalyzed CO-assisted reductive amination.
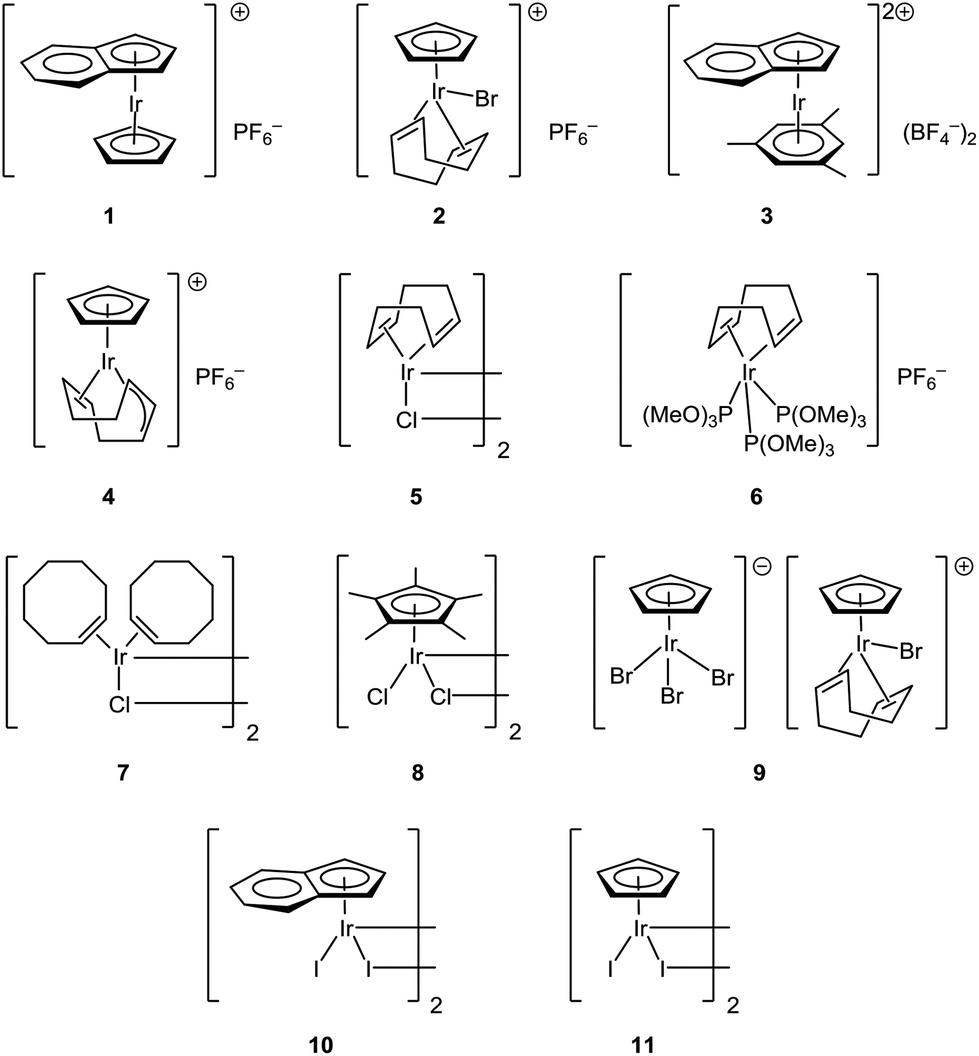 | ||
| Scheme 1 Iridium complexes tested as pre-catalysts for the reductive amination reaction. | ||
Complexes [CpIrI2]2, [Cp*IrCl2]2, [CpIr(cod)Br]PF6, [CpIr(cod)Br][CpIrBr3], [(η5-indenyl)IrI2]2, [(η5-indenyl)IrCp]PF6, [(η5-indenyl)Ir(η6-C6H3Me3)](BF4)2 were synthesized by known procedures.8–11 Cyclooctadienyl complex [CpIr(η3,η2-C8H11)]PF6 was prepared by the reaction of CpIr(cod) with silver trifluoroacetate (Scheme 2; anions are omitted in the schemes for clarity). Probably, this reaction proceeds via intermediate formation of dication [CpIr(cod)]2+ which undergoes spontaneous deprotonation to give [CpIr(η3,η2-C8H11)]+. Earlier, Maitlis and coworkers synthesized the pentamethyl analog [Cp*Ir(η3,η2-C8H11)]PF6 by interaction of [Cp*Ir(Me2CO)3]2+ with cyclooctadiene.12
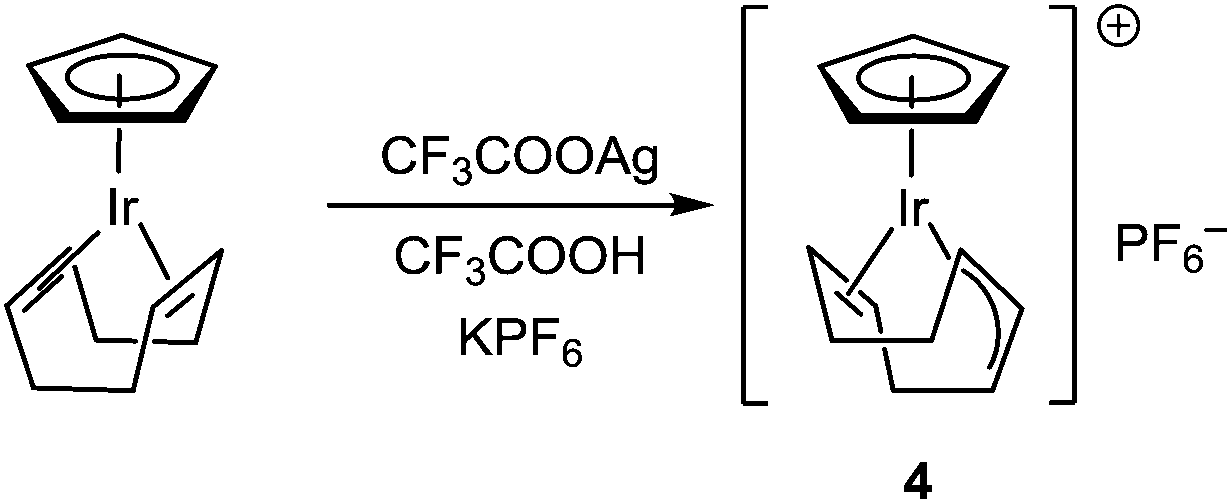 | ||
| Scheme 2 Synthesis of [CpIr(η3,η2-C8H11)]+. | ||
Phosphite complexes [(cod)Ir{P(OR)3}3]PF6 (R = Me, Et) were unexpectedly formed in the reaction of [CpIr(cod)Br]PF6 with phosphites (Scheme 3). Notably, phosphites play a dual role in this reaction: they serve both as ligands and reducing agents. Indeed, there are signals for five-valence phosphorus species (2.5–30 ppm) along with those for coordinated and free phosphite (89 and 140 ppm, respectively) in the 31P NMR spectrum of the reaction mixture. Usually, binding of cyclopentadienyl ligand to transition metals is considerably stronger than that of cyclooctadiene. Probably, reduction of Ir(III) to Ir(I) facilitates substitution of Cp in this case. In contrast, a similar reaction of [CpIr(cod)Br]PF6 with 2,2′-bipyridyl, which does not possess reducing ability, leads to substitution of cyclooctadiene with the formation of [CpIr(2,2′-bipy)Br]PF6 (Scheme 3).
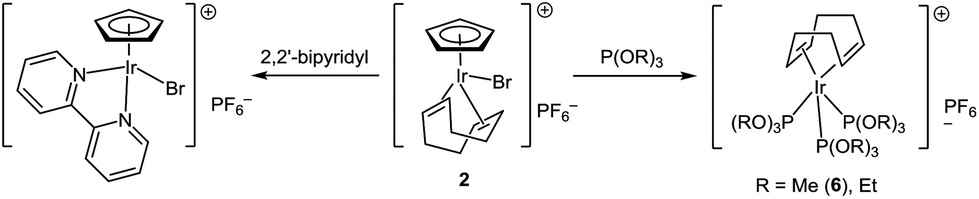 | ||
| Scheme 3 Reactivity of [CpIr(cod)Br]+. | ||
The structure of [(cod)Ir{P(OMe)3}3]PF6 was determined by X-ray diffraction (Fig. 1). The Ir–P bonds (2.248–2.317, av. 2.278 Å) in cation [(cod)Ir{P(OMe)3}3]+ are considerably shorter than those in the previously reported phosphide complex [(cod)Ir(PMe3)3]+ (2.327–2.379, av. 2.348 Å).13 The strong Ir–P bonds correlate well with low catalytic activity of [(cod)Ir{P(OMe)3}3]PF6 in reductive amination reactions (see below). In accordance with the trans-effect, the Ir–cod bonds in the phosphite complex (2.182–2.312, av. 2.248 Å) are longer than the corresponding bonds in the phosphide analog (2.178–2.237, av. 2.207 Å).
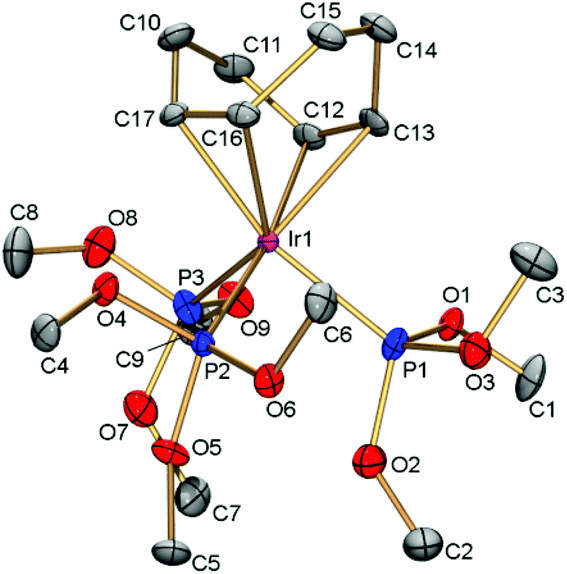 | ||
| Fig. 1 Cation [(cod)Ir{P(OMe)3}3]+6 with atoms shown as thermal ellipsoids at 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Ir1–C12 2.289(3), Ir1–C13 2.312(3), Ir1–C16 2.209(3), Ir1–C17 2.182(3), Ir1–P1 2.2691(8), Ir1–P2 2.2480(7), Ir1–P3 2.3177(8), C12–C13 1.395(5), C16–C17 1.433(4). | ||
After all the catalysts had been synthesized, we decided to compare them using p-methylbenzaldehyde and p-anisidine as model substrates (Table 1). It was found that dimeric iridium(III) complexes with bridging halide ligands are considerably more catalytically active than cationic monomeric species (Table 1, entries 8–11 vs. 1–4). Iridium(I) complexes give moderate yields (entries 5–7). Superior performance was observed for complexes of iridium(III) with Cp and halogens (Table 1, entries 8–10). Cyclopentadienyl iridium(III) diiodide was found to provide the best results among the tested metal complexes (entry 11) and was therefore used for the further optimization studies. The effect of iodine counterion seems to be general for the iridium complexes. When sodium iodide was added to iridium trichloride the yield increases from 33 to 47% (Table 1, entries 11 and 12).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Catalyst loading [mol%] | Yield [%] |
a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the amine and the aldehyde was employed. 0.2 mmol scale. :1 ratio of the amine and the aldehyde was employed. 0.2 mmol scale.
|
|||
| 1 | 1 | 1.0 | 5 |
| 2 | 2 | 1.0 | 8 |
| 3 | 3 | 1.0 | 8 |
| 4 | 4 | 1.0 | 12 |
| 5 | 5 | 0.5 | 14 |
| 6 | 6 | 1.0 | 25 |
| 7 | 7 | 0.5 | 29 |
| 8 | 8 | 0.5 | 28 |
| 9 | 9 | 1.0 | 21 |
| 10 | 10 | 0.5 | 39 |
| 11 | 11 | 0.5 | 57 |
| 12 | IrCl3 | 1.0 | 33 |
| 13 | IrCl3 + 3NaI | 1.0 | 47 |
Solvent screening revealed ethereal solvents and alcohols as the optimum media for the reaction (see ESI†). To avoid any possibility of hydrogen transfer reaction we chose tetrahydrofuran as a solvent instead of isopropanol. The temperature studies showed no significant trend at temperatures higher than 140 °C (Table 2, entries 1–3). The pressure had almost no influence on the reaction outcome above 30 bar, which can be explained by the supercritical nature of carbon monoxide (Table 2, entries 4–7). When the pressure was decreased from 30 to 20 bar the reaction rate was significantly lower (Table 2, entry 6 vs. 7). Nonetheless, the reaction still proceeded to some extent even at the pressure of as low as 5 bar (Table 2, entry 9). The yield becomes significantly higher at amine to aldehyde ratio of 2:1 (Table 2, entries 10–12). Comparison iridium catalysis with our previous data clearly shows that for the most nucleophilic amines rhodium still represents the best choice. For ruthenium catalyzed reactions with nucleophilic aliphatic amines, N-formamide was detected as a side product; no such byproduct was found in case of iridium catalysis.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solventb | T [°C] | P [bar] | Catalyst loading [mol%] | Yieldc [%] |
|
a 1.5:1 ratio of the amine and the aldehyde was employed. 0.1 mmol scale.
b 2:1 ratio of the amine and the aldehyde was employed. 22 h.
c Yields were determined by GC.
|
|||||
| 1 | THF | 140 | 50 | 1.0 | 49 |
| 2 | THF | 150 | 50 | 1.0 | 54 |
| 3 | THF | 160 | 50 | 1.0 | 57 |
| 4 | THF | 150 | 60 | 0.5 | 39 |
| 5 | THF | 150 | 50 | 0.5 | 37 |
| 6 | THF | 150 | 30 | 0.5 | 34 |
| 7 | THF | 150 | 20 | 0.5 | 11 |
| 8 | THF | 150 | 10 | 0.5 | 9 |
| 9 | THF | 150 | 5 | 0.5 | 2 |
| 10b | THF | 130 | 50 | 1.0 | 67 |
| 11b | THF | 140 | 50 | 1.0 | 67 |
| 12b | THF | 150 | 50 | 1.0 | 72 |
With the optimized conditions in hand, we turned to investigation of the scope of the developed methodology (Scheme 4). We found that chlorinated cyclopropanes could react with amines without any erosion of either cyclopropane rings or halogen substitution (12f, 12n). The stability of the dioxalane moiety was also successfully validated (12e). N-Benzyl moiety, which is usually fragile under various reductive conditions, remained fully intact in our system. Both aromatic and aliphatic amines are suitable for the reaction, and both primary and secondary amines can be used.
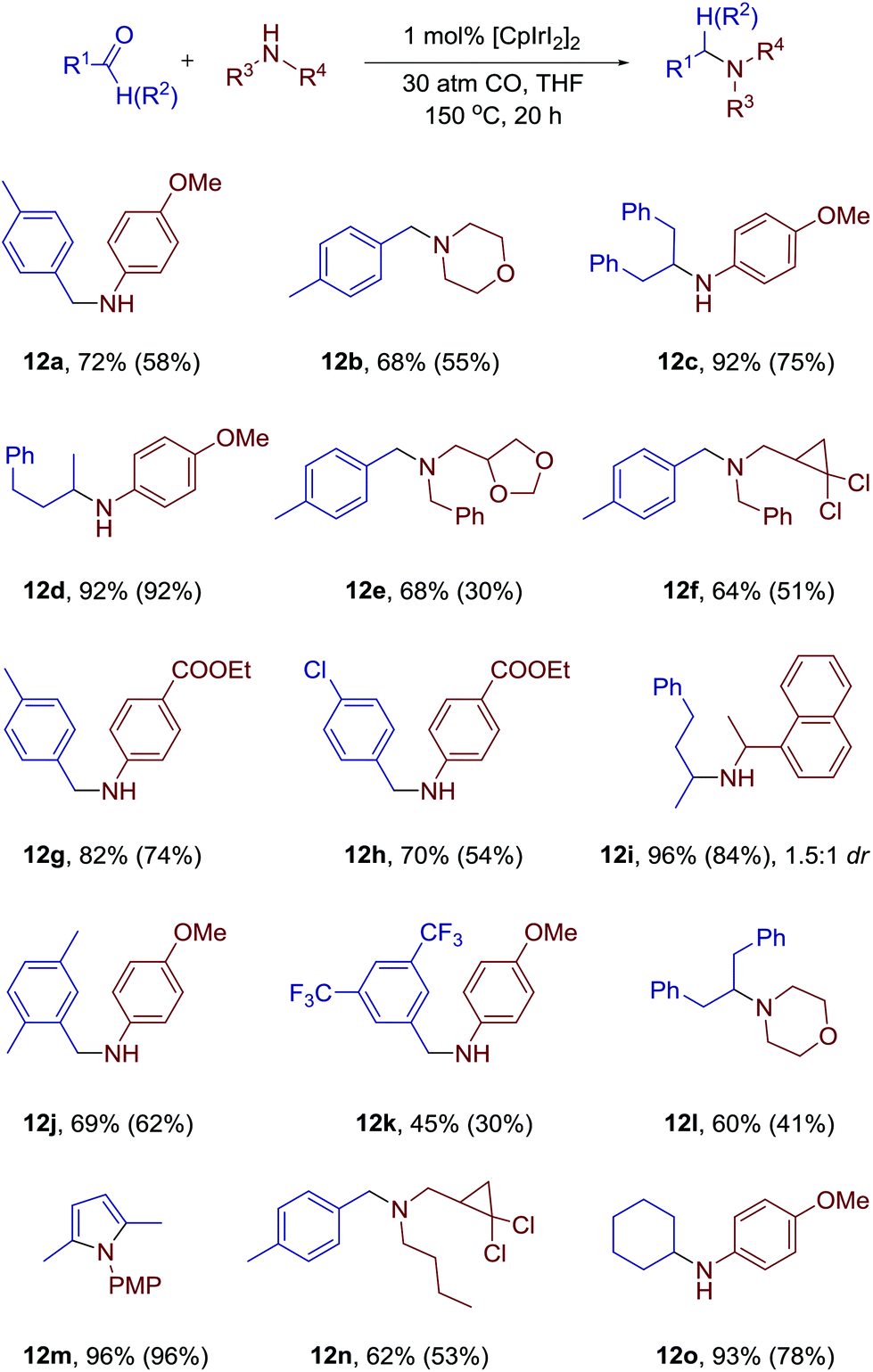 | ||
| Scheme 4 Substrate scope of reductive amination catalyzed by CpIrI2 in the presence of carbon monoxide. Yields were determined by NMR with an internal standard. Isolated yields are given in parentheses. | ||
Surprisingly, ketones are even more suitable for this reaction. Only when hexane-2,5-dione was used as a starting material exclusive formation of N-PMP-2,5-dimethylpyrrole 12m was detected. When naphtylethylamine was used together with benzylacetone the product of 1.5:1 dr was obtained (12i). Based on our previous studies we suggest a plausible mechanism of the process (Scheme 5). The reaction between an amine and a carbonyl compound leads to the formation of a hemiaminal. The oxidative addition of iridium complex to C–O bond of the heminal leads to a new complex. After an attack of hydroxyl group on the coordinated CO and elimination of CO2, the iridium hydride complex should be formed. Reductive elimination then leads to the formation of the product and regeneration of the catalytic species.
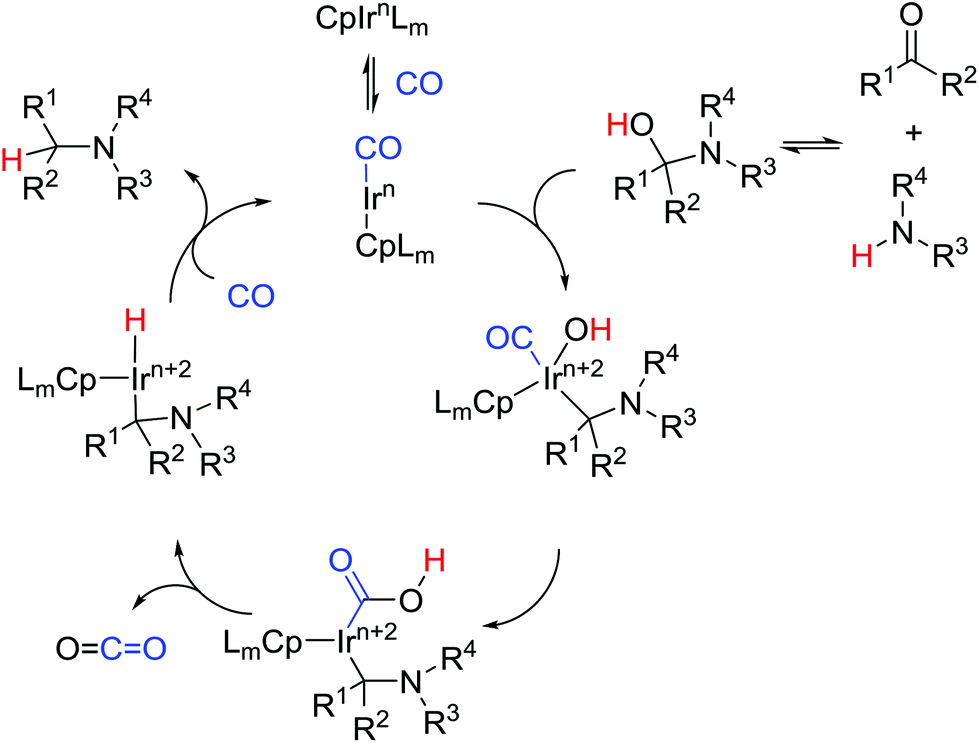 | ||
| Scheme 5 Plausible mechanism. | ||
Conclusions
In summary, we have found a new type of catalyst which can provide atom-economical reductive amination of aldehydes and ketones. The methodology takes advantage of the unique deoxygenative potential of carbon monooxide and does not require an external hydrogen source, which enables full compatibility with a range of functional groups prone to reduction (e.g. N-benzyl, dioxalane, halo-, cyclopropanes).Acknowledgements
We thank the Russian Academy of Sciences (P-8 grant), the Russian Foundation for Basic Research (grant no. 15-03-02548 A) and the Council of the President of the Russian Federation (Grant for Young Scientists No. MK-520.2017.3) for the financial support. D. A. L. gratefully acknowledges support of the Russian Foundation for Basic Research (project 16-33-60140 mol_a_dk). D. C. and A. F. S. thanks the Ministry of Education and Science of the Russian Federation (Agreement number 02.a03.21.0008). The contribution of Center for molecule composition studies of INEOS RAS is gratefully acknowledged.Notes and references
- (a) E. W. Baxter and A. B. Reitz, in Organic Reactions, ed. L. E. Overman and A. Overman, Wiley-VCH, Weinheim, 2002, vol. 59 Search PubMed; (b) A. F. Abdel-Magid and S. J. Mehrman, Org. Process Res. Dev., 2006, 10, 971 CrossRef CAS; (c) R. P. Tripathi, S. S. Verma, J. Pandey and V. K. Tiwari, Curr. Org. Chem., 2008, 12, 1093 CrossRef CAS; (d) P. Margaretha, in Science of Synthesis Knowledge Updates 2010/4, Thieme, Stuttgart, 2011, p. 405 Search PubMed.
- For selected recent papers on reductive amination see: (a) H. Alinezhad, H. Yavari and F. Salehian, Curr. Org. Chem., 2015, 19, 1021 CrossRef CAS; (b) S. Pisiewicz, T. Stemmler, A.-E. Surkus, K. Junge and M. Beller, ChemCatChem, 2015, 7, 62 CrossRef CAS; (c) M. Zhu, Catal. Lett., 2014, 144, 1568 CrossRef CAS; (d) X. Yan, E. Sokol, X. Li, G. Li, S. Xu and R. Graham Cooks, Angew. Chem., Int. Ed., 2014, 53, 5931 CrossRef CAS PubMed; (e) E. E. Drinkel, R. R. Campedelli, A. M. Manfredi, H. D. Fiedler and F. Nome, J. Org. Chem., 2014, 79, 2574 CrossRef CAS PubMed; (f) D. Talwar, N. Poyatos Salguero, C. M. Robertson and J. Xiao, Chem. – Eur. J., 2014, 20, 245 CrossRef CAS PubMed; (g) S. Werkmeister, K. Junge and M. Beller, Green Chem., 2012, 14, 2371 RSC; (h) N. K. Pahadi, M. Paley, R. Jana, S. R. Waetzig and J. A. Tunge, J. Am. Chem. Soc., 2009, 131(46), 16626 CrossRef CAS PubMed; (i) J. P. Patel, A.-H. Li, H. Dong, V. L. Korlipara and M. J. Mulvihill, Tetrahedron Lett., 2009, 50(44), 5975 CrossRef CAS.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed.
- (a) D. Chusov and B. List, Angew. Chem., Int. Ed., 2014, 53, 5199 CAS; (b) P. N. Kolesnikov, D. L. Usanov, E. A. Barablina, V. I. Maleev and D. Chusov, Org. Lett., 2014, 16, 5068 CrossRef CAS PubMed; (c) P. N. Kolesnikov, N. Z. Yagafarov, D. L. Usanov, V. I. Maleev and D. Chusov, Org. Lett., 2015, 17, 173 CrossRef CAS PubMed; (d) N. Z. Yagafarov, P. N. Kolesnikov, D. L. Usanov, V. V. Novikov, Y. V. Nelyubina and D. Chusov, Chem. Commun., 2016, 52, 1397 RSC; (e) O. I. Afanasyev, A. A. Tsygankov, D. L. Usanov and D. Chusov, Org. Lett., 2016, 18(22), 5968 CrossRef CAS PubMed.
- For the recent examples of using carbon monoxide for nitro group reductive transformation see: H.-Q. Li, X. Liu, Q. Zhang, S.-S. Li, Y.-M. Liu, H.-Y. He and Y. Cao, Chem. Commun., 2015, 51, 11217 RSC.
- For the state-of-the-art examples of reductive application CO in the water-gas shift reaction in organic chemistry see: (a) S. E. Denmark, M. Y. S. Ibrahim and A. Ambrosi, ACS Catal., 2017, 7(1), 613 CrossRef CAS; (b) A. Ambrosi and S. E. Denmark, Angew. Chem., Int. Ed., 2016, 55, 12164 CrossRef CAS PubMed; (c) J. W. Park and Y. K. Chung, ACS Catal., 2015, 5, 4846 CrossRef CAS; (d) S. E. Denmark and Z. D. Matesich, J. Org. Chem., 2014, 79, 5970 CrossRef CAS PubMed; (e) S. E. Denmark and S. T. Nguyen, Org. Lett., 2009, 11, 781 CrossRef CAS PubMed; (f) L. He, L. C. Wang, H. Sun, J. Ni, Y. Cao, H. Y. He and K. N. Fan, Angew. Chem., Int. Ed., 2009, 48, 9538 CrossRef CAS PubMed; (g) J. Huang, L. Yu, L. He, Y. M. Liu, Y. Cao and K. N. Fan, Green Chem., 2011, 13, 2672 RSC; (h) J. Dong, M. Zhu, G. Zhang, Y. Liu, Y. Cao, S. Liu and Y. Wang, Chin. J. Catal., 2016, 37, 1669 CrossRef CAS; (i) J. Ni, L. He, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, Chem. Commun., 2011, 47, 812 RSC; (j) L. He, F.-J. Yu, X.-B. Lou, Y. Cao, H.-Y. He and K. N. Fan, Chem. Commun., 2010, 46, 1553 RSC.
- For the recent examples of metal carbonyl as the source of carbon monoxide see: (a) N. Jana, F. Zhou and T. G. Driver, J. Am. Chem. Soc., 2015, 137(21), 6738 CrossRef CAS PubMed; (b) F. Zhou, D. S. Wang and T. G. Driver, Adv. Synth. Catal., 2015, 357(16–17), 3463 CrossRef CAS.
- A. R. Kudinov, D. A. Loginov, Z. A. Starikova and P. V. Petrovskii, J. Organomet. Chem., 2002, 649, 136 CrossRef CAS.
- C. White, A. Yates and P. M. Maitlis, Inorg. Synth., 1992, 29, 228 CrossRef CAS.
- D. A. Loginov, A. M. Miloserdov, Z. A. Starikova, P. V. Petrovskii and A. R. Kudinov, Mendeleev Commun., 2012, 22, 192 CrossRef CAS.
- A. A. Chamkin, A. M. Finogenova, Yu. V. Nelyubina, J. Laskova, A. R. Kudinov and D. A. Loginov, Mendeleev Commun., 2016, 26, 491 CrossRef CAS.
- C. White, S. J. Thompson and P. M. Maitlis, Dalton Trans., 1978, 10, 1305 RSC.
- J. F. Frazier and J. S. Merola, Polyhedron, 1992, 11, 2917 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1536381. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ob01005b |
| This journal is © The Royal Society of Chemistry 2017 |