
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Strained alkynes derived from 2,2′-dihydroxy-1,1′-biaryls; synthesis and copper-free cycloaddition with azides†
Alessandro
Del Grosso
,
Lavrentis-Dimitrios
Galanopoulos
,
Cookson K. C.
Chiu
 ,
Guy J.
Clarkson
,
Peter B.
O′ Connor
and
Martin
Wills
*
,
Guy J.
Clarkson
,
Peter B.
O′ Connor
and
Martin
Wills
*
Department of Chemistry, The University of Warwick, Coventry, CV4 7AL, UK. E-mail: m.wills@warwick.ac.uk
First published on 17th May 2017
Abstract
A series of strained alkynes were prepared from 2,2′-dihydroxy-biaryls. Several were characterised by X-ray crystallography, revealing strained C(sp)–C(sp)–C(sp3) bond angles in the range of 163–167°. Their cycloadditions with azides proceed without a catalyst. Functionalised versions of these reagents have potential applications to materials synthesis and bioconjugations.
Azides react with terminal alkynes to form a triazole in what is commonly referred to as a ‘click’ reaction.1–6 Reactions of this type proceed under mild conditions, in high conversion and their biorthogonal nature allows them to be used to attach functional molecules (e.g. dyes, fluorescent groups, drugs etc.) to biomolecules, and to create antibody/drug conjugates (ADCs) for therapeutic use.7–9 The undesirable requirement for the use of a copper catalyst can be eliminated through the use of reactive strained alkynes in ‘Strain-Activated Alkyne Azide Cycloaddition’.10 Examples of recently-reported strained alkynes are illustrated in Fig. 1.11–21
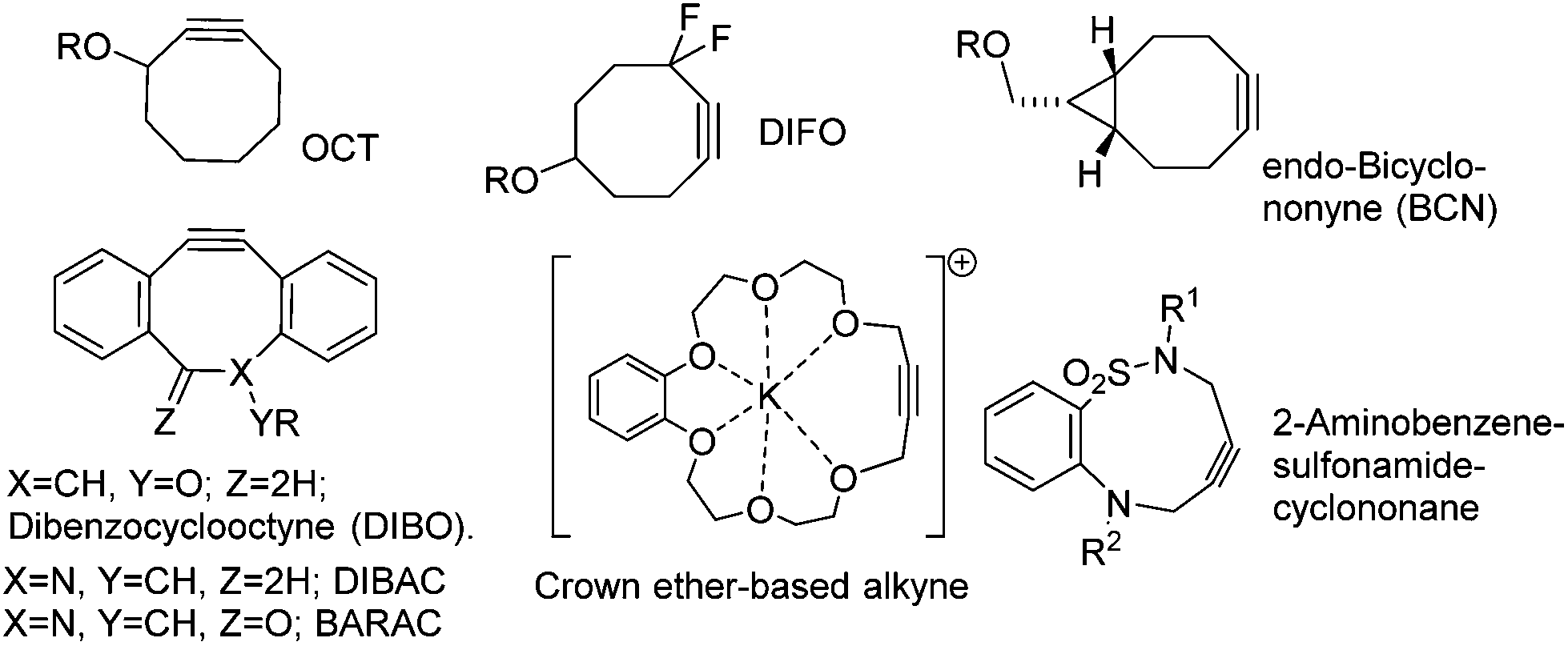 | ||
| Fig. 1 Examples of strained alkynes which react with azides without the requirement for a copper catalyst. R represents a point of attachment to either a biomolecule or other functional group. | ||
The majority of reported strained alkynes contain 8-membered rings, however there are some examples of 9-membered ring reagents20 such as the 2-amidobenzene sulphonamide cyclononane21 and even a 19-membered crown ether,22 both illustrated in Fig. 1. Thiacycloakyne and dibenzoselenacycloheptynes have also been reported.23
Strained alkynes have also been used to conjugate functional polymers, for example to combine24a contrasting chains and to form crosslinks in the formation of hydrogels.24b They have also been used to create modified pharmaceutical agents.25 Of the reported strained reagents, DIBO, DIBAC and BCN, due to their high reactivity, have been particularly widely adopted for bioconjugation reactions.11 In this paper, we describe the synthesis and applications of a class of strained alkyne which can be prepared through a short and facile synthetic sequence from readily available and inexpensive starting materials.
Following a single precedent in the literature,26a we investigated the reaction of biaryl diols with 1,4-ditosyl-but-2-yne, which resulted in the formation of cyclic alkynes 1–4 (Fig. 2). A literature search revealed just two other reports of this heterocyclic structure, both binaphthyl-derived,26b,c whilst their reactions with azides have not been reported. The 2,2′-biphenols and their strained-alkyne derivatives are racemic, however compound 2 was prepared as a single enantiomer derived from (R)-BINOL. Compounds 2–4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27,28 were characterised by X-ray crystallography (Fig. 3) which served to confirm the strained nature of the triple bonds in each product. The sp angles at the triple bonds (C(sp)–C(sp)–C(sp3)) in 3 and 4 were ca. 166–167°, however, due to the presence of three crystallographically independent molecules in the unit cell of 2, the sp bond angles in this molecule ranged from 166.9–163.0° with an average of ca. 165°. The high aryl/aryl torsion angles (Fig. 3) reflect the wide spacing required to accommodate the linear butyne component in the ring. This angle increased to 112.5° in the dibromo 3 and 122.1° in the tetrabromo compound 4.
27,28 were characterised by X-ray crystallography (Fig. 3) which served to confirm the strained nature of the triple bonds in each product. The sp angles at the triple bonds (C(sp)–C(sp)–C(sp3)) in 3 and 4 were ca. 166–167°, however, due to the presence of three crystallographically independent molecules in the unit cell of 2, the sp bond angles in this molecule ranged from 166.9–163.0° with an average of ca. 165°. The high aryl/aryl torsion angles (Fig. 3) reflect the wide spacing required to accommodate the linear butyne component in the ring. This angle increased to 112.5° in the dibromo 3 and 122.1° in the tetrabromo compound 4.
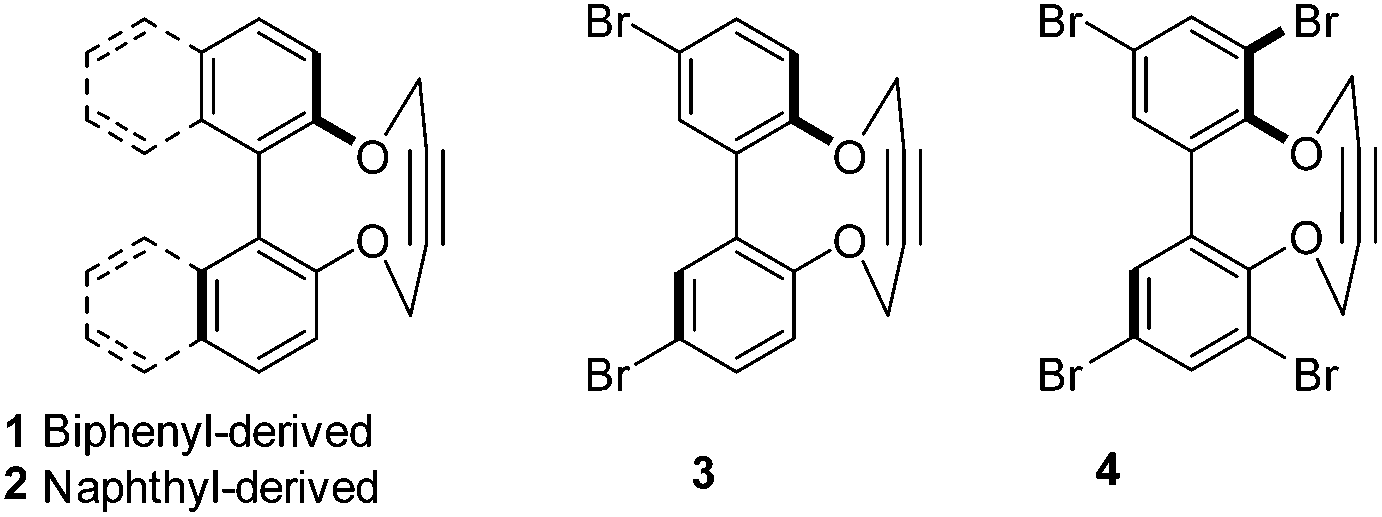 | ||
| Fig. 2 Strained alkynes 1–4 derived from biaryl and binapthyl diols. | ||
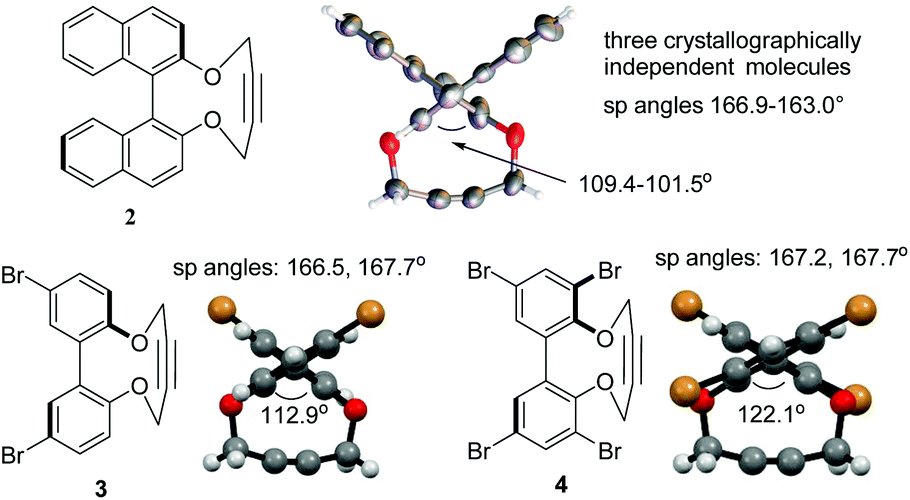 | ||
| Fig. 3 X-ray crystallographic structures of strained alkynes 2–4. | ||
The strain in the alkyne is reflected in the reactivity of 1–4 towards benzyl azide and arylazides (Scheme 1), which resulted in formation of cycloaddition products 5–8 in high yields, at room temperature, and without the need for a metal catalyst. The X-ray crystallographic structure of cycloadduct 5a (Fig. 4) revealed a low torsion angle between the aromatic rings of just 43°, reflecting the greater flexibility of the triazolyldimethylene bridge.
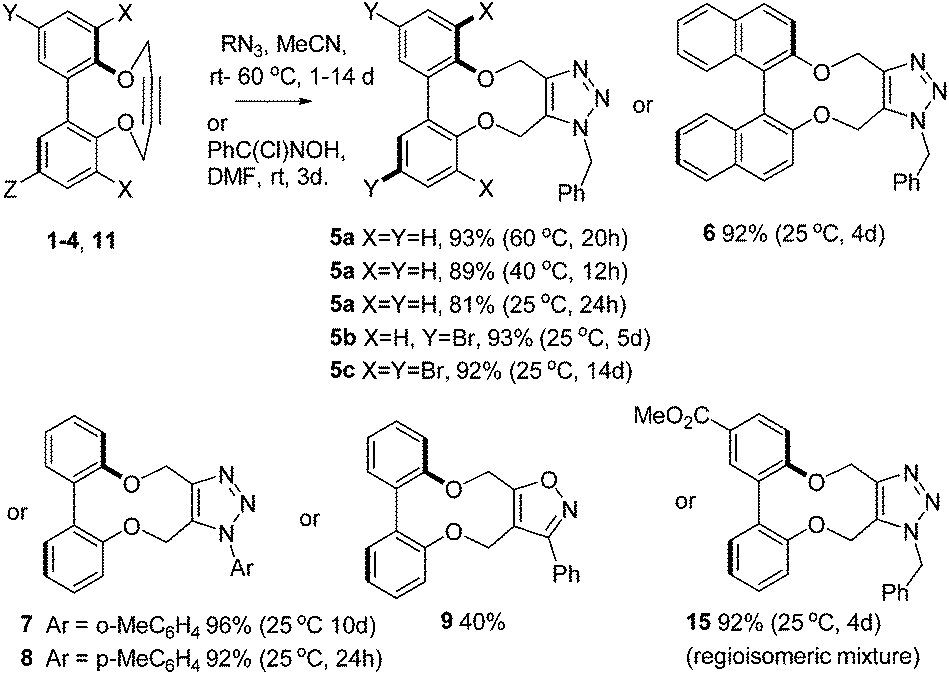 | ||
| Scheme 1 Cycloadditions of strained alkynes with benzyl azide, aryl azides and a nitrile oxide. | ||
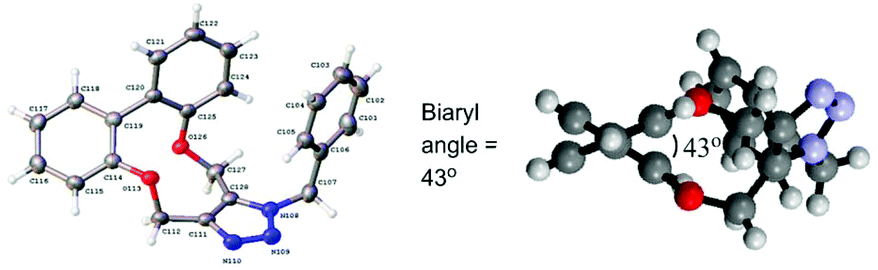 | ||
| Fig. 4 X-ray crystallographic structure of cycloadduct 5a. | ||
Conversions to cycloadducts were typically ca. 90% or higher at 40 °C within 24 h in several cases, however the reactions could be run for several days without substrate decomposition, to generate products in high yield even at the lower temperature. The cycloaddition of a nitrile oxide (generated in situ from the chloride precursor)29 to give adduct 9, was also completed (Scheme 1).
The second-order rate constants for a number of the reactions were determined (Table 1, ESI†). The reactions of the unsubstituted derivative 1 with both benzyl azide and p-tolylazide, were followed over time at three temperatures by 1H NMR to allow calculations of thermodynamic data. For the unsubstituted substrate 1, 81% conversion was observed after 24 h at 25 °C, and 89% after 12 h at 40 °C (Table 1). The rate constants are higher than for a potassium complex of a cyclic 1,4-diether containing a but-2-yne (Fig. 1; 1.3 × 10−5 M−1 s−1), which is a rare example of a strained alkyne based on 1,4-but-2-yne diether structure.22 The reactivity of the dibromide 3 is similar to that of 1, however the tetrabromo 4 was of lower reactivity, possibly due to steric hindrance caused by the large bromine atoms. Arylazide cycloaddition was also achieved without the need for a catalyst, and p-tolylazide was more reactive than o-tolylazide; again this may be due to higher steric hindrance in the ortho-substituted azide.
| Product | Temp./°C | Solvent | [alkyne]a/M | Rate constant |
|---|---|---|---|---|
|
a Alkyne:azide = 1:1.
b 81% conv. in 24 h.
c 89% conv. in 12 h.
d Benzyl azide adduct 1; ΔE‡ = 15.9 kcal mol−1, ΔH‡ = 15.2 kcal mol−1, ΔS‡ = −24.3 cal mol−1 K.
e 96% yield in 10 days.
f 92% yield in 24 h.
g
p-Tolylazide adduct 8: ΔE‡ = 14.6 kcal mol−1, ΔH‡ = 14.0 kcal mol−1, ΔS‡ = −29.2 cal mol−1 K.
|
||||
|
5ab |
25 | MeCN | 0.25 | 1.85 × 10−4 M−1 s−1 |
|
5ac |
40 | MeCN | 0.25 | 7.56 × 10−4 M−1 s−1 |
|
5ad |
60 | MeCN | 0.25 | 3.12 × 10−3 M−1 s−1 |
| 5a | 25 | CDCl3 | 0.10 | 1.28 × 10−4 M−1 s−1 |
| 5b | 25 | CDCl3 | 0.10 | 1.41 × 10−4 M−1 s−1 |
| 5c | 25 | CDCl3 | 0.10 | 3.64 × 10−5 M−1 s−1 |
| 15 | 25 | MeCN | 0.25 | 2.50 × 10−4 M−1 s−1 |
|
7e |
25 | CDCl3 | 0.10 | 3.07 × 10−5 M−1 s−1 |
|
8f |
25 | CDCl3 | 0.10 | 1.24 × 10−4 M−1 s−1 |
| 8 | 40 | CDCl3 | 0.10 | 5.00 × 10−4 M−1 s−1 |
|
8g |
60 | CDCl3 | 0.10 | 1.69 × 10−3 M−1 s−1 |
The rate constants for 1, within the temperature range examined, are similar to that reported for benzyl azide with ‘OCT’ (2.4 × 10−3 M−1 s−1), but lower than those of other cyclooctyne reagents illustrated in Fig. 1 (DIFO; 7.6 × 10−2, DIBO; 0.17, DIBAC; 0.31, BCN; 0.11–0.29, BARAC; 0.96 M−1 s−1). The relative reactivities are also reflected in the distorted sp bond angles of 161/151° (DIFO2) and 153° (BARAC).30 However the reactivity of 1 and its derivatives is sufficient for it to be a practical reagent for use in synthetic chemistry applications under mild, Cu-free conditions.
Whilst following the cycloaddition of 1 with benzylazide at 60 °C, we observed that cycloadduct 5a exhibited significant peak broadening in the 1H-NMR spectrum whilst the starting material 1 did not. A systematic study of their variable temperature 1H-NMR spectra, in d6-DMSO was completed and this indicated that 1 did not undergo helical inversion even up to 100 °C, whereas 5a exhibited coalescence of the OCH2 peaks at ca. 56 °C. The ΔG# required for atropisomer interconversion was calculated to be 16.3 kcal mol−1 (ESI†). The related aliphatic compound 10 was independently synthesised31 and its OCH2 peak signals were found to broaden and coalesce at ca. 74 °C (Fig. 5; biaryl angle 55.6°, ΔG# required for atropisomer interconversion ca. 16.7 kcal mol−1). For 5a in CD3CN solvent, coalescence was at a similar temperature of ca. 60 °C. This confirms a much higher level of rigidity in 1 (and hence 2–4) compared to their more flexible cycloadducts and to compound 10 (Fig. 5, ESI†). Hence the well-defined structures of the cyclic strained alkynes offers the potential for them to be used as the basis of asymmetric ligands, and possibly as building blocks for extended materials.
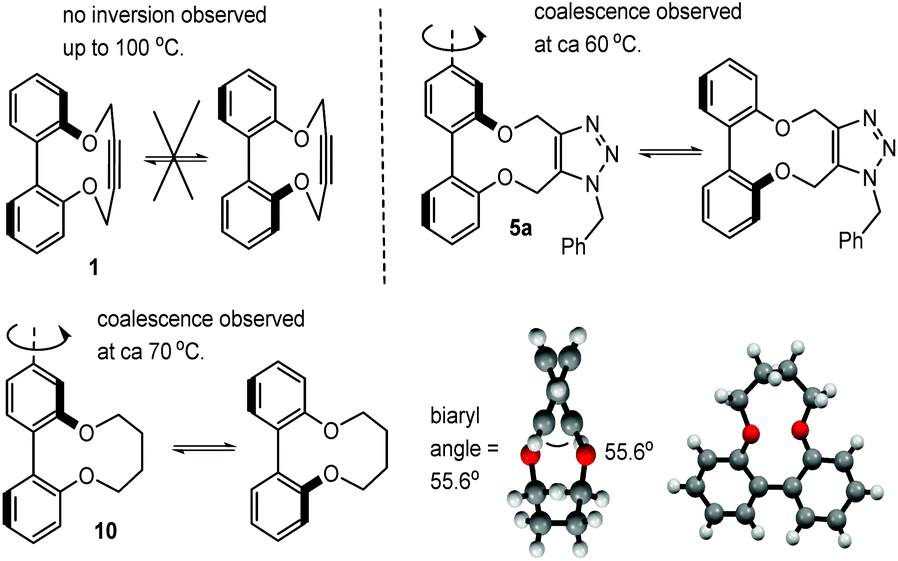 | ||
| Fig. 5 Restricted rotation of strained alkyne 1, adduct 5a and aliphatic-ring compound 10. Also shown is the X-ray crystallographic structure of 10. | ||
There is potential for the attachment of derivatives of 1 to proteins and functional groups through the introduction of a linking functional group. In order to demonstrate the potential for this, the ester-functionalised alkyne 11 was prepared in one step from the diphenol 1232 in an unoptimised 50% yield (some of the [2 + 2] and [3 + 3] products were also isolated; see ESI† for structures). Hydrolysis of the ester to acid 13 was followed by formation of the activated NHS ester 14 (Scheme 2). Fig. 6 illustrates the X-ray crystallographic structures of both 11 and 13, which exhibit very similar biaryl torsion angles and strained sp bond angles to each other and to 1–4. The reaction of 11 with benzyl azide proceeded (Scheme 1) without a catalyst, giving product 15 in 92% yield at room temperature after a reaction period of 4 days The second order rate constant for the cycloaddition was similar to that for 1 (Table 1), indicating that the introduction of the ester had not significantly diminished the reactivity of the alkyne.
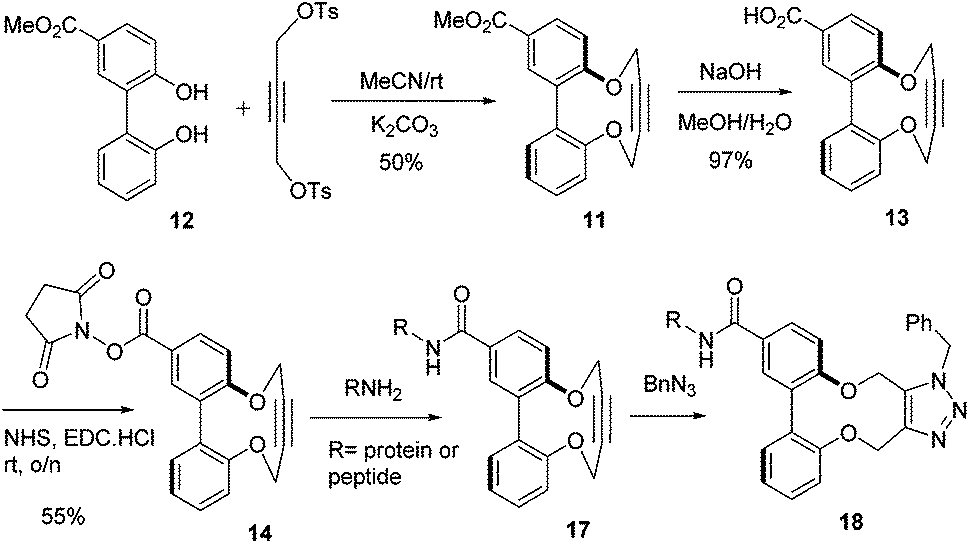 | ||
| Scheme 2 Synthesis of activated NHS ester 14 and subsequent reaction with peptides and a protein. | ||
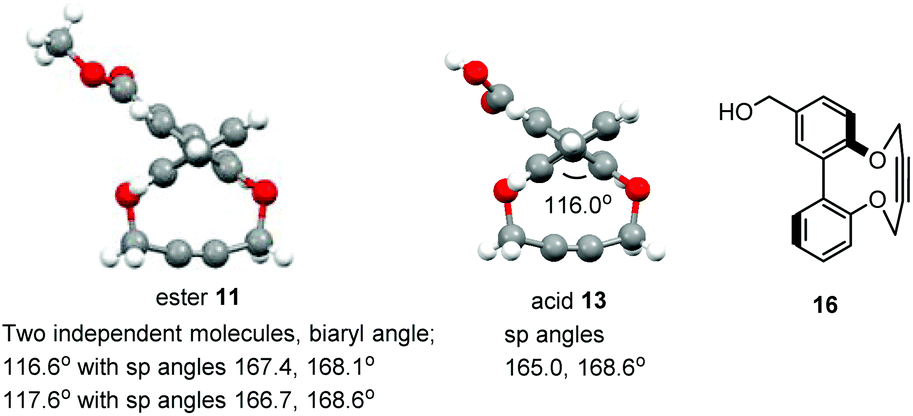 | ||
| Fig. 6 X-ray crystallographic structure of ester 11 and acid 13, and structure of alcohol 16. | ||
Reduction of 14 with DIBAL resulted in formation of alcohol 16, which also has the potential to be converted to a conjugation reagent. Although lower in reactivity than the most active strained alkynes (Fig. 1), we sought to obtain preliminary evidence of its potential for bioconjugation. To obtain a proof of principle in this respect, 14 was reacted with the small pep-tides substance P, vasopressin and bombesin, and the protein myoglobin, the reactions being followed using FTICR mass spectrometry (Scheme 2). The reactions of peptides with the activated ester 14 in water/methanol solution resulted in partial functionalization to give adducts 17 (see ESI†). MS/MS studies confirmed that the lysine residue in each peptide had been functionalised. The subsequent reaction with an excess of benzyl azide in each case occurred with >95% conversion to the ‘clicked’ product 18.
Solutions of myoglobin and 14 in methanol/water were pre-pared, and after standing for one day at rt were analysed by FTICR. Since myoglobin contains multiple lysine residues, the protein was only partly functionalised (ca. 10%) in order to avoid formation of a range of functionalised proteins, which would be more difficult to analyse. An excess of benzyl azide in the same solvent was then added and after 7 days the solutions were analysed by FTICR (Fig. 7 features an expansion of one charged form at each stage). Significantly, the subsequent cycloaddition reaction of the fraction of functionalised protein had undergone >95% conversion to the ‘click’ cycloadduct.
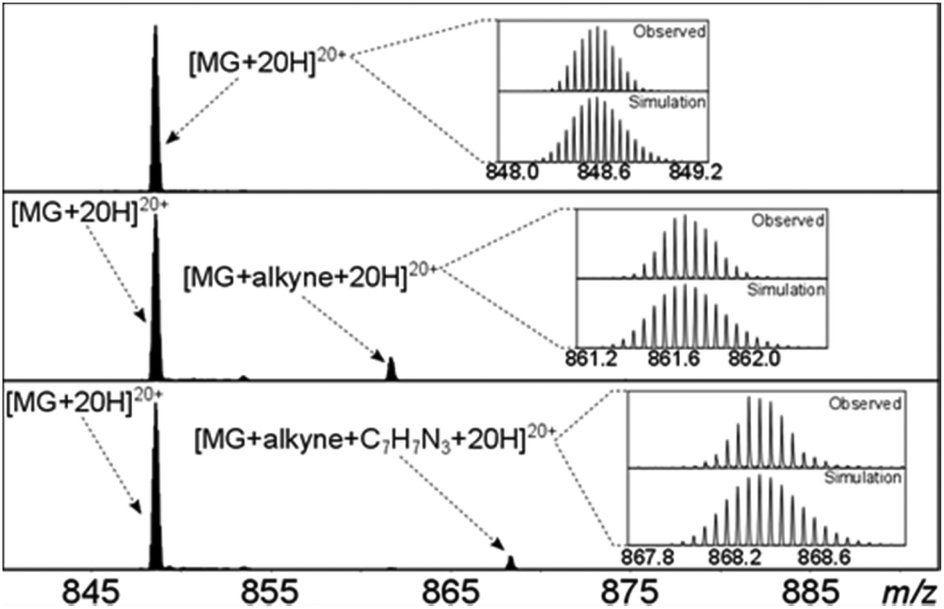 | ||
| Fig. 7 The FTICR-MS spectra of one charge state of myoglobin. Top; without 14, middle; following addition of 14, bottom; following addition of PhCH2N3. | ||
In conclusion, we have demonstrated that strained alkynes based on diether derivatives of 2,2′-diphenol react with azides without the need for a metal catalyst. The strained alkyne can be attached to a biomolecule or other group, hence demonstrating the potential for the reagents to be used broadly in synthetic reactions where a Cu catalyst must be avoided, and with potential for use in bioconjugation applications. Importantly, the strained alkynes described herein benefit from extreme ease of synthesis, from readily available and inexpensive starting materials.
Acknowledgements
The EPSRC are thanked for funding ADG (project grant ref no. EP/M006670/1) and CKCC (DTG PhD Studentship). Bruker (UK) Ltd are also thanked for support of CKCC through a CASE collaboration. Warwick University are thanked for supporting LG through the Analytical Sciences MSc programme. The X-ray diffraction instrument was obtained through the Science City Project with support from the AWM and part funded by the ERDF.Notes and references
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed
.
- P. Thirumurugan, D. Matosiuk and K. Jowiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed
- J. M. Palomo, Org. Biomol. Chem., 2012, 10, 9309–9318 CAS
- X. Zhang and Y. Zhang, Molecules, 2013, 18, 7145 CrossRef CAS PubMed
- E. Haldon, M. C. Nicasio and P. J. Perez, Org. Biomol. Chem., 2015, 13, 9528–9550 CAS
- Y. Gong and L. Pan, Tetrahedron Lett., 2015, 56, 2123–2332 CrossRef CAS
- H. Li, R. Aneja and I. Chaiken, Molecules, 2013, 18, 9797–9817 CrossRef CAS PubMed
- D. Y. Jackson, Org. Process. Res. Dev., 2016, 20, 852–866 CrossRef CAS
- J. D. Thomas, H. Cui, P. J. North, T. Hofer, C. Rader and T. R. Burke Jr., Bioconjugate Chem., 2012, 23, 2007–2013 CrossRef CAS PubMed
- G. Wittig and A. Krebs, Chem. Ber., 1961, 94, 3260–3275 CrossRef CAS
- J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem. (Z), 2016, 374, 16 CrossRef PubMed
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC
- M. F. Debets, S. S. van Berkel, J. Dommerholt, A. J. Dirks, F. P. J. T. Rutjes and F. L. van Delft, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS PubMed
- OCT: N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed
- DIFO: J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS PubMed
- DIBO: X. Ning, J. Guo, M. Wolfert and G.-J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS PubMed
- BCN: J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hedriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS PubMed
- BCN: G. Stefanetti, Q.-Y. Hu, A. Usera, Z. Robinson, M. Allan, A. Singh, H. Imase, J. Cobb, H. Zhai, D. Quinn, M. Lei, A. Saul, R. Adamo, C. A. Maclennan and F. Micoli, Angew. Chem., Int. Ed., 2015, 54, 13198–13203 CrossRef CAS PubMed
-
(a) DIBAC: M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97–99 RSC
- J. Tummatorn, P. Batsomboon, R. J. Clark, I. V. Alabugin and G. B. Dudley, J. Org. Chem., 2012, 77, 2093–2097 CrossRef CAS PubMed
- Aminobenzene sulphonamide: K. Kaneda, R. Naruse and S. Yamamoto, Org.
Lett., 2017, 19, 1096–1099 CrossRef CAS PubMed
- Crown ether: B. Gold, P. Batsomboon, G. B. Dudley and I. V. Alabugin, J. Org. Chem., 2014, 79, 6221–6232 CrossRef CAS PubMed
-
(a) Thiacycloalkyenes: G. de Almeida, E. M. Sletten, H. Nakamura, K. K. Palaniappan and C. R. Bertozzi, Angew. Chem., Int. Ed., 2012, 51, 2443–2447 CrossRef CAS PubMed
-
(a) J. C. Brendel, G. Gody and S. Perrier, Polym. Chem., 2016, 7, 5536–5543 RSC
-
(a) L. Qian, C.-J. Zhang, J. Wu and S. Q. Yao, Chem. – Eur J., 2017, 23, 360–369 CrossRef CAS PubMed
-
(a) U. Koch-Pomeranz, H.-J. Hansen and H. Schmid, Helv. Chim. Acta, 1971, 56, 2981–3004 CrossRef
- S. Facchetti, D. Losi and A. Iuliano, Tetrahedron: Asymmetry, 2006, 17, 2993–3003 CrossRef CAS
- A. Alexakis, D. Polet, S. Rosset and S. March, J. Org. Chem., 2004, 69, 5660–5667 CrossRef CAS PubMed
- F. Heaney, Eur. J. Org. Chem., 2012, 3043–3058 CrossRef CAS
- C. G. Gordon, J. L. Mackey, J. C. Jewett, E. M. Sletten, K. N. Houk and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 9199–9208 CrossRef CAS PubMed
-
(a) D. W. Allen, P. N. Braunton, I. T. Millar and J. C. Tebby, J. Chem. Soc. C, 1971, 3454–3468 RSC
- B. Schmidt, M. Riemer and M. Karras, J. Org. Chem., 2013, 78, 8680–8688 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, NMR spectra, X-ray crystallographic structures and kinetic data (pdf). CCDC 1515150–1515156. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ob00991g |
| This journal is © The Royal Society of Chemistry 2017 |