
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the pharmacokinetics of cucurbit[7, 8 and 10]uril: and a dinuclear ruthenium antimicrobial complex encapsulated in cucurbit[10]uril†
Fangfei
Li
a,
Anil K.
Gorle
a,
Marie
Ranson
b,
Kara L.
Vine
b,
Robert
Kinobe
cd,
Marshall
Feterl
cd,
Jeffrey M.
Warner
cd,
F. Richard
Keene
 *de,
J. Grant
Collins
*a and
Anthony I.
Day
*a
*de,
J. Grant
Collins
*a and
Anthony I.
Day
*a
aSchool of Physical, Environmental and Mathematical Sciences, University of New South Wales, Australian Defence Force Academy, Canberra, ACT 2600, Australia. E-mail: g.collins@adfa.edu.au; a.day@adfa.edu.au
bSchool of Biological Sciences, Illawarra Health and Medical Research Institute, University of Wollongong, Wollongong, NSW 2522, Australia
cCollege of Public Health, Medical and Veterinary Sciences, James Cook University, Townsville, QLD 4811, Australia
dCentre for Biodiscovery and Molecular Development of Therapeutics, James Cook University, Townsville, QLD 4811, Australia
eSchool of Physical Sciences, University of Adelaide, Adelaide, SA 5000, Australia. E-mail: richard.keene@adelaide.edu.au
First published on 20th April 2017
Abstract
The relatively non-toxic family of cucurbit[n]uril, Q[n], have shown considerable potential in vitro as drug delivery agents, with only a few examples of pharmacokinetic (PK) studies for drug⊂Q[n]. Drug-free Q[n] PK studies are the next step in determining the pharmacological applicability in their drug delivery potential. The results for the first PK and bio-distribution of drug-free 14C-Q[7] are described for administration via intravenous (i.v.) and intraperitoneal (i.p.) dosing. A study of oral administration of drug-free 14C-Q[8] has also been undertaken to determine the time course for the gastrointestinal tract (GIT), absorption and subsequent bio-distribution. Q[10], a potential drug carrier for larger drugs, was evaluated for its effect on the PK profile of a dinuclear ruthenium complex (Rubb12), a potential antimicrobial agent. The Rubb12⊂Q[10] complex and free Rubb12 were administered by i.v. to determine differences in Rubb12 plasma concentrations and organ accumulation. Interestingly, the PK profiles and bio-distribution observed for Q[7] showed similarities to those of Rubb12⊂Q[10]. Drug-free Q[7] has a relatively fast plasma clearance and a generally low organ accumulation except for the kidneys. Drug-free Q[8] showed a low absorption from the GIT into the blood stream but the small percentage absorbed reflected the organ accumulation of Q[7]. These results provide a better understanding of the probable PK profile and bio-distribution for a drug⊂Q[n] through the influence of the drug delivery vehicle and the positive clearance of drug-free Q[n] via the kidneys supports its potential value in future drug delivery applications.
Introduction
The family of macrocyclic host molecules known as cucurbit[n]uril (Q[n]); (see Fig. 1) has recently attracted considerable attention through the realisation of a wide-range of potential applications – including catalysis, the modification of electrochemical properties, the enhancement of some analytical processes, the synthesis of nano-materials and supramolecular polymers, for drug delivery and the modification of various biological processes.1–16 | ||
| Fig. 1 The structure of cucurbit[n]uril (left-hand side) and Rubbn (right-hand side). | ||
The Q[n] are available in a range of cavity sizes prescribed by the number of glycoluril moieties for a particular member (n = 5–8 and 10).1–3 Other non-classical Q[n] members are also now available, including Q[13–15], which have limited cavities as a consequence of their twisted structures, in the form of Möbius strips.17,18 The three Q[n] members that are of prime interest in the area of drug delivery are Q[7, 8 and 10], which have cavities large enough to accommodate drug molecules or parts of a drug molecule with cross-sections within the portal size ranges of 5.4–10.6 Å.4–11,19 The hydrophobic regions of a drug are encapsulated within the hydrophobic cavity and electropositive groups can further stabilise encapsulation by electrostatic interactions with the carbonyl-rimmed portals. As encapsulation in Q[n] can increase the aqueous solubility of a drug, or reduce its toxicity or its degradation into non-active metabolites, Q[n] have been proposed as delivery vehicles for a number of therapeutic drugs.4–11,19–21 The drug delivery potential has been demonstrated experimentally in vitro, and in a few cases in vivo.6,12,22,23 These are important as first steps in a pharmacological evaluation but the effect of the Q[n]-encapsulation on the pharmacokinetic (PK) profile (plasma concentration as a function of time and organ accumulation) of the drug in vivo is the next important determinant of the clinical potential of Q[n] as a drug delivery vehicle.24–27 The Q[n] vehicle itself has already been shown to be relatively non-toxic.28–31 To date there are few studies of the PK effect of encapsulated drugs in Q[n] and no studies on the PK of free Q[n]. The few examples of PK effects with drug⊂Q[n] include those by Ma et al. (oral administration)32 and Plumb et al. (intraperitoneal administration)33 of drugs encapsulated in Q[7] and have demonstrated improvements in plasma drug concentrations. A third example by Nguyen et al. (oral administration) of a drug/Q[6] nanoparticle also showed improved plasma drug concentrations.34 However, it is not clear in these examples, whether it was the free drug after it was released from the host that diffuses into the blood stream in a sustained manner, or whether the drug encapsulated in Q[7] or Q[6] was carried into the plasma. It was anticipated that the study described herein, would help to answer this question through the PK profiling of free Q[7 and 8].
The increasing importance of Q[n] demonstrated by in vivo trials raises the significance of PK studies of free Q[n], especially for n = 7 and 8 as these cavity sizes have recently seen increasing potential as drug delivery hosts. In this context we present our results on the PK profiling of drug-free 14C-labelled Q[7] and Q[8] with a focus on plasma clearance and organ accumulation. In addition, to determining the fate of Q[7 and 8] we also report a comparative PK profile of a relatively new potential antimicrobial agent encapsulated in Q[10], the largest available Q[n] cavity.19 This antimicrobial agent is the inert dinuclear ruthenium(II) complex, [{Ru(phen)2}2(μ-bb12)]4+ {phen = 1,10-phenanthroline; bb12 = 1,12-bis[4(4′-methyl-2,2′-bipyridyl)]dodecane} (Rubb12 – see Fig. 1, n = 12). The association complexes Rubbn⊂Q[10] have Ka > 109 M−1,32 and are therefore ideal candidates to evaluate the PK of Q[10] through the ready detection of ruthenium in plasma and organ samples by ICP-MS quantification.
The PK profiling of Rubb12⊂Q[10] compared to free Rubb12 provided data on the PK changes asserted upon the encapsulated drug and served to demonstrate parallels to the PK findings of a drug⊂Q[n] relative to free Q[7] and Q[8].
Results and discussion
Drug-free 14C-labelled Q[7] and Q[8]
The few examples of published PK studies of Q[n] carrying a drug involved a determination of the fate of the drug but not the function (delivery potential and drug retention) or fate of the drug carrier. A delivered drug will inevitably leave an empty Q[n], which is a phenomenon that also needs to be understood in terms of PK.Drug-free Q[7] has the highest intrinsic aqueous solubility of any of the classical Q[n] family (here “classical” refers to the original Q[n] without substituents) and drug-free Q[8] has an intrinsic solubility of <0.1 mM,36 while the least soluble was drug-free Q[10] (∼25 μM).37 Drug-free Q[7] in clinical saline is soluble to ∼35 mM, which makes this Q suitable for intravenous (i.v.) or intraperitoneal administration (i.p.) and PK profiling while carrying a 14C-label. However, drug-free Q[8] with its lower solubility was considered only suitable for an oral PK study. The high stability of the Q[n], also supports its potential applicability to an oral route for drug delivery.32,34 Q[8] and Q[10] are only very slightly increased in solubility in saline, although drug⊂Q[8 or 10] often have considerably higher aqueous solubility compared to the free host, with observed increases up to 100-fold for both.35,38
The radiolabelled 14C-Q[7 and 8] were prepared by the acid-catalysed reaction of 14C-paraformaldehyde with glycoluril under previously established unlabelled conditions.39 Purification was achieved by a combination of dissolution and chromatography on cation exchange resin and each Q[n] verified by 1H NMR.
The 14C-Q[7] was assessed for its plasma clearance properties and organ uptake in Balb/c mice after i.p. administration, and in Sprague Dawley rats after i.v. administration. The i.p. plasma clearance in mice (Fig. 2a) appears to fit a 2-phase (2-compartment model) exponential decay curve, giving clearances of t1/2α = 17.0 min and t1/2β = 270.7 min (half-lives for the two phases). Similarly i.v. administration in Sprague Dawley rats also fitted a multiphasic clearance model, where a t1/2β = 12.8 h, calculated from the linear 6–48 h post-dose interval (see ESI, plasma clearance Table S1, Fig. S1 and urine excretion Table S2†). It should be noted that the percentage of the injected i.p. dose was <30%, at maximum plasma concentration, and decreased relatively quickly.
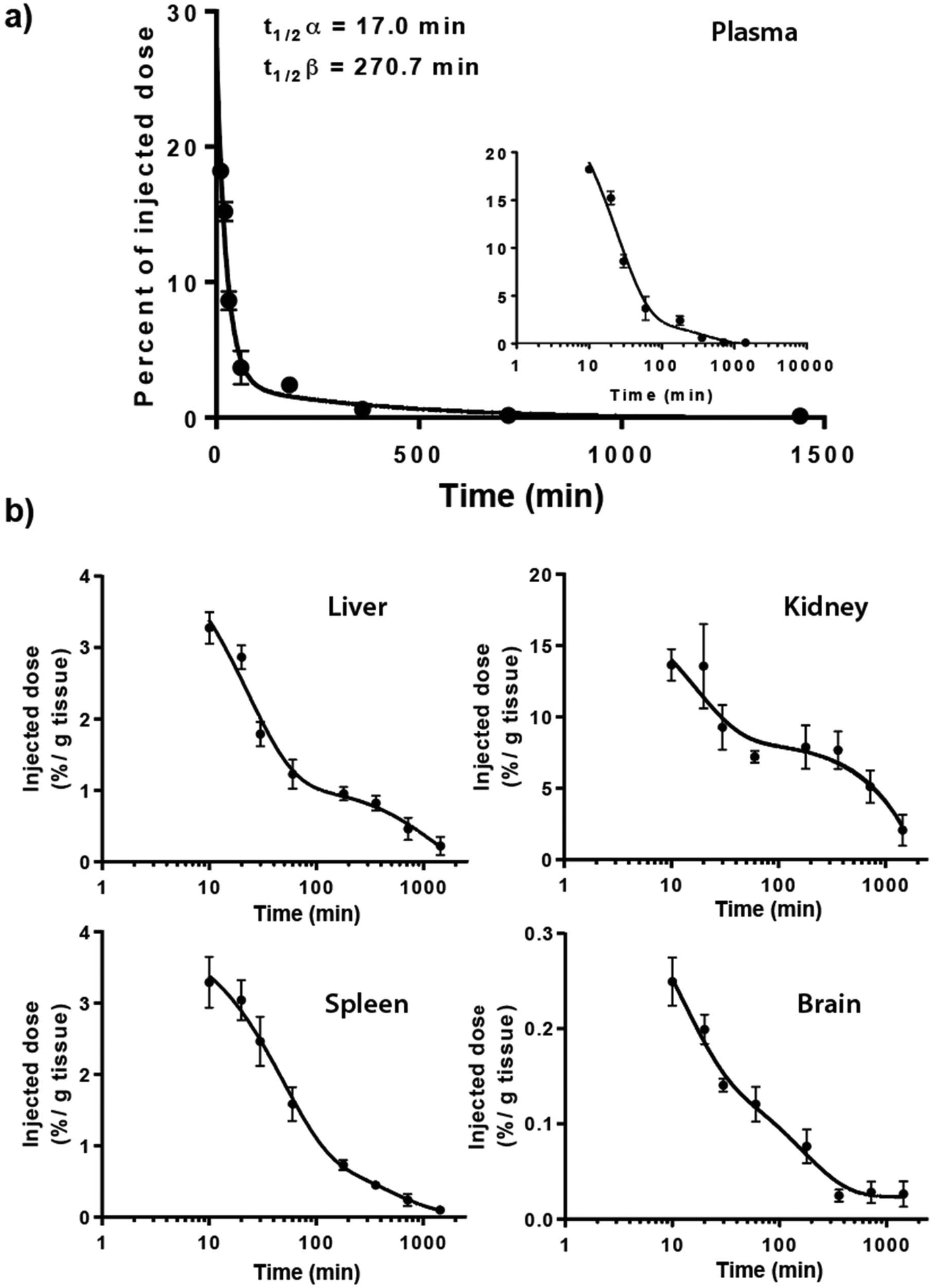 | ||
| Fig. 2 Bio-distribution of 14C-Q[7] in mice after i.p. administration. Percent of injected dose in plasma determined from t = 0–1440 min with the insert highlighting the period from 10 min, x-axis log10 and y-axis as percentage of injected dose corrected for the entire plasma volume (a); organ distribution (liver, kidneys, spleen and brain) t = 10–1440 min (b). | ||
In addition to plasma clearance, the bio-distribution of 14C-Q[7] in mice following i.p. administration was also examined. Maximum organ accumulation of the 14C-Q[7] was detected 10 min post-administration, in the spleen, liver and brain, while the kidneys saw two activity phases at ∼20 and ∼180 min (Fig. 2b). The large amount of activity detected in the kidneys (as opposed to the liver) suggests that Q[7] is renally excreted unchanged, possibly through glomerular filtration (in a fashion similar to β-cyclodextrin and hydroxypropyl-β-cyclodextrin).40 Similarly, i.v. administration of 14C-Q[7] in rats and urine sampling, at intervals within the periods 0–4, 4–8, 8–24 and 24–48 h, showed that approximately 50% of the excretion of 14C-Q[7] occurred within the first 4 h. The total 14C-Q[7] excreted over 48 h was 71.6% ± 16.5 of the total administered.
Low levels of 14C-Q[7] were found accumulated in the spleen and liver (<4%, 10 min post-administration) in comparison to the plasma and kidneys (18.2% and 13.7% respectively, 10 min post-administration). The brain accumulated the least activity (<0.1% ± 0.06, over the entire duration of the study) suggesting that the 14C-Q[7] does not cross the blood brain barrier.
Q[n] is potentially applicable for oral drug delivery in order to protect the drug from the harsh conditions of the gastrointestinal tract (GIT) and/or provide a mechanism for controlled adsorption into the blood stream. In this context we undertook a preliminary PK study of orally administered drug-free 14C-Q[8]. A single bolus of a mix of labelled and unlabelled Q[8] was administered to Sprague Dawley rats and the bio-distribution established. Three time points of 2, 6 and 24 h were measured for the stomach, small and large intestines, liver, kidneys spleen, brain and bladder. Activity in the plasma was measured at 11 time points up to 48 h. The plasma activity vs. time profile showed limited absorption with a maximum concentration (∼0.001% of the dose), obtained at ∼2 h, followed by an elimination phase (t1/2β = 20 h) (Fig. 3). However, the total activity detected in the plasma was very low, consistent with the low urine recovery of 3.6% (of the total dose administered). Hence organ distribution was also low but proportionally similar to the results found in Fig. 2.
 | ||
| Fig. 3 Plasma concentrations of 14C-Q[8] up to 48 h, following oral administration. Activity measured as disintegrations per minute (DPM) and maximum activity is ∼0.001% of the dose. | ||
In contrast, a relatively high recovery of activity was measured from excreted faeces, 44.7% ± 20.4 of the total dose, with the largest proportion occurring in the 24–48 h period. The time course of activity for the passage through the GIT showed a peak at 2 h for stomach and the small intestine, followed by a peak at 6 h for the large intestine (see ESI Fig. S3†).
The low plasma activity and relatively high activity observed in the faeces and the GIT following oral dosing are consistent with a relatively poor absorption via this route. While drug free Q[8] may have low absorption from the GIT this does not exclude the value of the drug delivery vehicle from providing protection to the drug prior to release and absorption nor does it eliminate the possibility that a drug⊂Q[7 or 8] may have a different absorption profile, dependent upon the drug being carried.
PK profile of a Rubb12⊂Q[10]
Rubb12 (Fig. 1) as a potential antimicrobial agent is highly active against both Gram-positive and Gram-negative bacteria with retention of activity against drug-resistant strains, and it is non-toxic against healthy eukaryotic cells at concentrations well above its minimum inhibitory concentration (MIC).41 As we have previously demonstrated Rubb12 can form a high affinity, stable, water soluble complex with Q[10], and as such is suitable to evaluate the PK of Rubb12⊂Q[10] and to compare this with the PK of the ruthenium complex.19The maximum single dose tolerance (MTD) for free Rubb12 and Rubb12⊂Q[10] was established in mice following i.v. administration. It was found that encapsulation resulted in a two-fold decrease in toxicity for Rubb12 (free – 1 mg kg−1, encapsulated – 2 mg kg−1). Using these dose limits, free Rubb12 (1 mg kg−1) was administered to mice by i.v. to establish the serum concentration profiles over a 12 h period post injection. A comparative group of mice were also administered with Rubb12⊂Q[10] (equal quantity of Rubb12) and serum concentrations of Rubb12 were quantified using Inductively Coupled Plasma Mass Spectrum (ICP-MS) determination of the Ru concentrations. The results of the 12 h sampling of serum concentrations are shown in Fig. 4. After the initial equally rapid plasma clearance for both free and Q[10] bound Rubb12, within the first 15 min of the exponential decay curve, there is a second clearance phase, which demonstrates a slightly slower clearance for Rubb12⊂Q[10], t1/2β ∼150 min (Fig. 4).
 | ||
| Fig. 4 The comparison of the serum level of free Rubb12 and Rubb12⊂Q[10] (1 mg kg−1 of drug) over 12 h after i.v. administration. The inset highlights the second phase of elimination over the period 0.5–12 h. | ||
Of greater significance was the bio-distribution of the free Rubb12 compared to Rubb12⊂Q[10] in a range of organs, 6 h after the ruthenium complex had been administered by i.v. (Fig. 5). The primary site of accumulation for free Rubb12 was the liver, with the kidneys, spleen, heart and lungs also exhibiting measurable quantities of ruthenium (∼35% of injected dose). Interestingly, while free Rubb12 accumulated predominantly in the liver, when administered as Rubb12⊂Q[10] it was found to be distributed in comparable amounts in both the liver and kidneys. A substantial reduction (∼2-times less) in accumulated complex in the liver was reflected by an increase in the kidneys (∼4-fold). The other organs – spleen, heart lungs and brain – all showed lower levels of accumulated Rubb12⊂Q[10] with the most notable differences found in the spleen and heart.
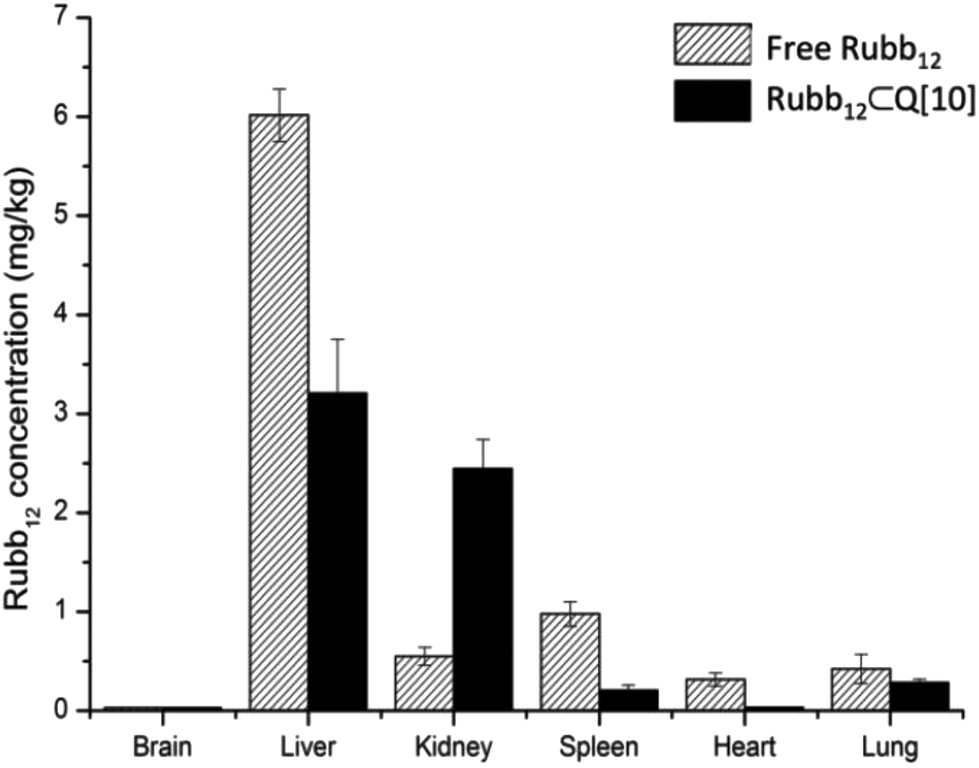 | ||
| Fig. 5 Tissue accumulation of Rubb12 after 6 h of a single dose at 1 mg kg−1 by i.v. injection of free Rubb12 and Rubb12⊂Q[10] in mice. | ||
The very low accumulation in the heart as a result of encapsulation in Q[10] may have future significance in the case of drugs that have deleterious effects on heart tissue, as this finding may point to a method for limiting such negative effects of drugs delivered without encapsulation in Q[n].
Given the significant accumulation in the liver and the kidneys of Rubb12, both free and encapsulated, a longer time course of up to 1 week was evaluated following a single i.v. injection. The results are shown in Fig. 6 reflecting a large percentage of the injected dose (∼70%). The accumulation of ruthenium in the liver (Fig. 6a) reached a maximum level at 48 h for Rubb12⊂Q[10] from which point the accumulated amount remained relatively constant up to 1 week. For free Rubb12 it continues to accumulate from 6 h onwards, with levels doubling after 1 week.
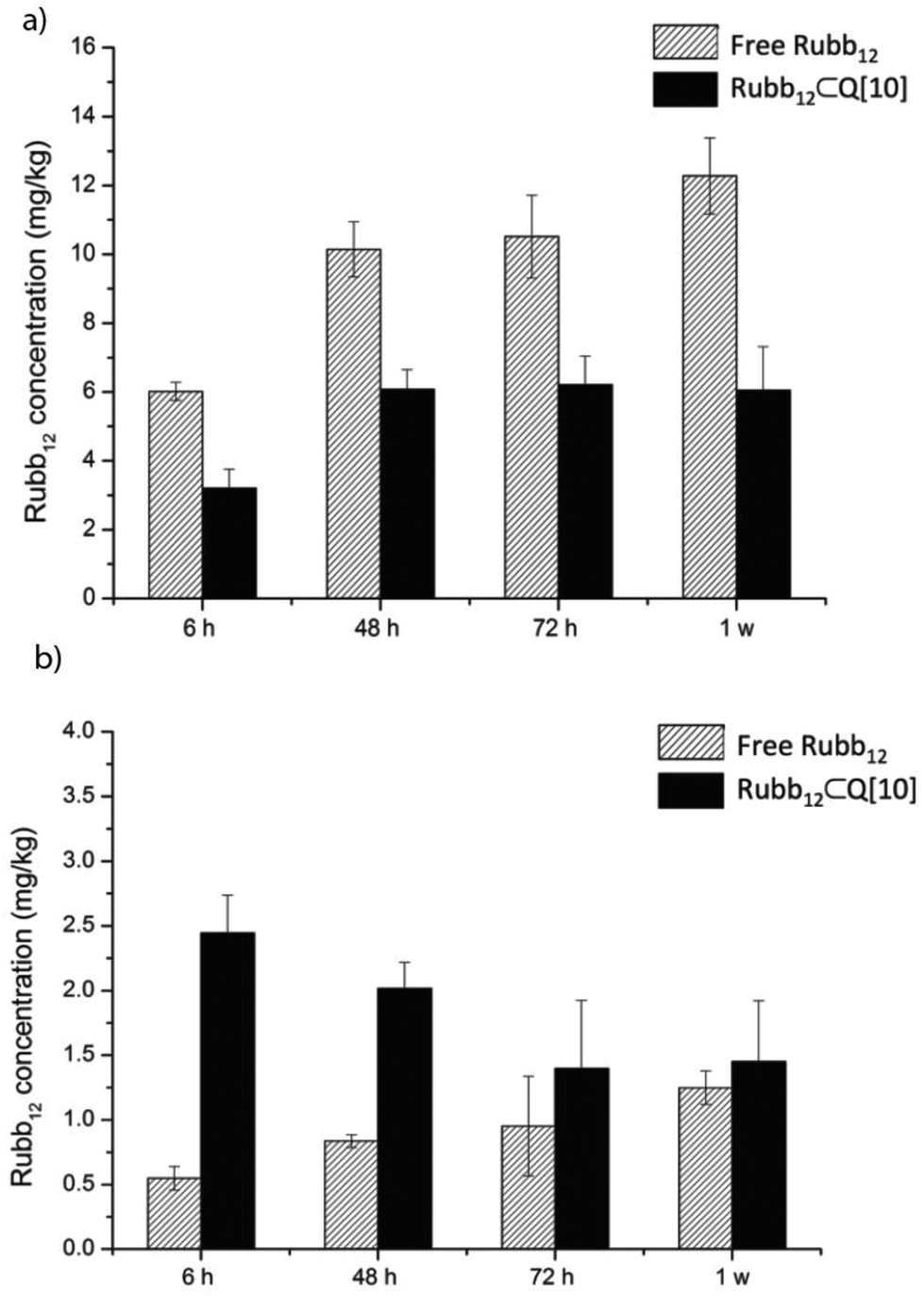 | ||
| Fig. 6 The accumulation of Rubb12 in the liver (a) and the kidneys (b) 6, 48, 72 h and 1 week after a single 1 mg kg−1 dose of free or Rubb12⊂Q[10] by i.v. injection. | ||
Compared to the accumulation in the liver, the quantity of ruthenium found in the kidneys when delivered as the Rubb12⊂Q[10] complex was maximal at 6 h and then steadily declined to half this level by 72 h up to 1 week, indicating a constant elimination (Fig. 6b). For the mice administered with free Rubb12, accumulation of ruthenium in the kidneys slowly increased over time to a level similar to that of Rubb12⊂Q[10] after 1 week following dosing.
These results demonstrate a higher accumulation and faster clearance of Rubb12 through the kidneys when administered as the Rubb12⊂Q[10] complex and a significant reduction in liver accumulation compared to administration of free Rubb12. For an antimicrobial drug, a sufficient resident time within the body is desirable to complete its function but it is also important that it can be cleared from the body after serving its purpose. The positive clearance for Rubb12⊂Q[10] from the body as shown in Fig. 6, demonstrates a desirable attribute imparted by Q[10] in its role as a potential drug delivery vehicle.
Conclusions
This study represents the first PK profile of the promising class of drug delivery vehicles Q[7 and 8] in the absence of a drug. In addition, we report the PK profiling of Rubb12⊂Q[10] as a promising antimicrobial agent with improved PK when Rubb12 is encapsulated in Q[10].The acute toxicity study demonstrated that the MTD of Rubb12 can be doubled when encapsulated as Rubb12⊂Q[10], increasing the dose size from 1 to 2 mg kg−1. This study provides the first data on the organ accumulation for a Rubbn complex.
The results with the 14C-Q[7] provide strong evidence that the significant increase in kidney accumulation is due to the renal excretion of Q[n], rather than a specific effect of a drug⊂Q[n] complex. Consequently, it can be assumed that any drug administered as drug⊂Q[n] will show significant levels of accumulation in the kidneys driven largely by the Q[n] and less by the drug that it contains. However, the level of accumulation in other organs is likely to decrease also driven by the host Q[n]. The results of this study indicate that free Q[7] is relatively rapidly eliminated from the plasma when administered by either i.p. or i.v. routes, and from the study with Rubb12, encapsulated drugs will be eliminated from the plasma at a comparable rate to the free Q[n]. Encapsulation in Q[n] will affect the rate a drug enters the plasma after administration by non-intravenous routes. However, and importantly, encapsulation in Q[n] will modify the level of drug accumulation in various organs. This has potentially important implications for the toxicity of the encapsulated drug.
The results from oral administration of 14C-Q[8] demonstrated that absorption of Q from the GIT into the blood stream is very low, and that the most likely application of drug⊂Q[n] is for protection and increased aqueous solubility to facilitate intestinal absorption. This suggestion is also supported by the reported improvements achieved for the drug triamterene⊂Q[7], which showed improved bioavailability and sustained plasma concentrations.32
Encapsulation in Q[10] with respect to plasma concentrations only led to a small increase in the concentration of Rubb12, following the initial rapid concentration decrease to a linear elimination phase at ∼30 min. After this point there was a slightly higher concentration of Rubb12. Similarly, a reported, i.p. administered cisplatin⊂Q[7] complex decreased the maximum concentration in plasma, but slightly increased the concentration after the initial rapid build up of the drug following i.p. administration.33
The tissue distribution study demonstrated that the main site of accumulation for Rubb12 is the liver, with considerable quantities also being found in the spleen and kidneys. The concentrations of Rubb12 in the liver and kidneys were found to continually increase over time, up to one week after administration. This implies that the rapid clearance from the blood stream did not lead to a rapid excretion of Rubb12 from the body, instead the complex concentrated in tissue as shown by the organ accumulation. Interestingly, Rubb12⊂Q[10] exhibited a considerable lowering of accumulated Rubb12 in most of the organs except for the kidneys. Significantly, the amount of Rubb12 accumulating in the liver decreases, approximately 2-fold when administrated as Rubb12⊂Q[10] and as the MTD was also improved, this may suggest that the main mechanism of toxicity of the Rubb12 complexes was liver damage.
There are PK profile similarities between Q[7 and 8] and even without following the fate of free Q[10] directly, the Rubb12⊂Q[10] complex exhibited a profile comparable to free Q[7 and 8]. Assuming that there was no significant chemical instability due to 14C labelling,42 these results suggest that the PK and bio-distribution will be dominated by the host for drugs⊂Q[7,8 and 10] where drug binding is relatively high.
Experimental
All the animal experiments were carried out in accordance with the institutional guidelines following approval from the respective animal ethics committees (University of Queensland Animal Ethics Committee Group 5) Project code TetraQ/059/09/NEWSOUTH; (James Cook University Animal Ethics Committee) App. no. A1753; (University of Wollongong Animal Ethics Committee) App. no. AE08/14.Animals
Mixed-sex in-bred BALB/c mice aged 9–12 weeks were obtained from the small animal house located at the College of Public Health, Medical and Veterinary Sciences, James Cook University, Townsville, Australia.Male Sprague Dawley rats 9–13 weeks were obtained from the Australian Institute of Bioengineering and Nanotechnology Animal House, University of Queensland.
In all studies food and water were available ad libitum throughout the life of the animals.
Synthesis of 14C-labelled Q[7] and Q[8]
14C-labelled Q[7] and Q[8] were prepared by our general cucurbit[n]uril synthetic method39 on a scale where solid 14C-paraformaldehyde (186.0 mg, 1 mCi, 37 MBq) was used in a reaction vial with HCl 32% (400 μL). After the reaction was complete the acid was completely removed and the dry solid residue treated with water (16 mL) at 90 °C cooled and centrifuged. The clear aqueous solution was separated from the solid plug. The water was evaporated at 60 °C and the residue purified on a short column of Dowex 50WX2 cation exchange resin, eluting with 0.5 M HCl/50% formic acid. The appropriate fractions yielded 14C-cucurbit[7]uril 120 mg. The water insoluble material was suspended in 60% formic acid heated to 60 °C and then cooled. The remaining suspension was centrifuged. The solution was removed from the solid plug, which was then washed with a small volume of 60% formic acid and the centrifugation process repeated. The remaining solid was dried at 50 °C to give 14C-cucurbit[8]uril (35 mg).All samples of radiolabelled Q were stored as solids at −20 °C and used within a period of several months (the i.p. experiments were conducted with samples stored for <3 weeks) to minimise radiolytic decomposition.42
Intraperitoneal (i.p.) experiments with 14C-Q[7]in mice
The intraperitoneal (i.p.) experiments with the free Q[7] were carried out using 14C-Q[7] with BALB/c mice. Saturated solutions of 14C-Q[7] were prepared in phosphate buffer saline (pH 7.4) by sonication, then heating to 95 °C cooled and filtered through 22 μm nylon syringe filters into sterile plastic sample tubes. Mice were injected (i.p.) with a single bolus dose (50 μL) of 14C-Q[7] then sacrificed at 10, 20, 30, 60, 180, 360, 720 and 1440 min after injection. The total amount of activity injected per mouse was 4 μCi. Blood was immediately collected into tubes containing EDTA and plasma samples taken after centrifugation. Major organs were collected and weighed for further tissue/organ analysis. Samples were accurately weighed (between 0.1–0.5 g) and processed for liquid scintillation counting by incubation for 24 h at 60 °C with tissue solubiliser. Radioactivity was measured following mixing of the denatured homogenate sample with Packard Ultima Gold liquid scintillation counting cocktail (approximately 10 mL).Intravenous (i.v.) experiments 14C-Q[7] in rats
The i.v. administration experiments with 14C-labelled Q[7] were carried out using Sprague Dawley rats. Blood samples were collected at 0, 2, 5, 10, 30 min, 1, 2, 4, 6, 12, 24 and 48 h post-dose into samples tubes containing the anti-coagulant lithium heparin. Samples were kept cold and centrifuged and the plasma transferred to a clean, polypropylene tubes. A 50–100 μL plasma aliquot was used to determine radioactivity.Urine samples were collected at intervals pre-dose, 0–4 h, 4–8 h, 8–24 h and 24–48 h. Urine volume was determined by weighing. At the end of the 48 h period the collecting funnel was washed and the activity measured added to the total.
Oral experiments 14C-Q[8] in rats
14C-Q[8] was administered as a single bolus dose (2 mL kg−1) as a water/PEG 400 solution (5 μCi of 14C-Q[8] in 20 mg kg−1 cold Q[8]) by oral gavage. The animals were euthanased at three post-dose time points of 2, 6 24 h. The organs (stomach, small and large intestines, liver, kidneys, spleen whole brain and bladder) were excised, weighed and processed. Homogenated samples were accurately weighed (between 0.1–0.5 g) and processed for scintillation counting by incubation for 24 h at 60 °C with tissue solubiliser. Radioactivity was measured following mixing of the denatured homogenate sample with Packard Ultima Gold liquid scintillation-counting cocktail (approximately 10 mL).Faeces collected during each time period were weighed and homogenised in 4 volumes of deionised water using a mechanical homogeniser. Approximately 0.1 to 1 g (accurately weighed) of this slurry was transferred to a 20 mL glass scintillation vial, 2 mL of tissue solubiliser added and the vials capped and incubated at 60 °C for at least 24 h.
The level of radioactivity in dose formulation, plasma (50 or 100 μL), urine (100 μL), faeces (0.1 to 1 g) and washout solution (100 μL) was added to scintillation vials containing Packard Ultima Gold liquid scintillation-counting cocktail (2.0 mL for plasma and dose, 5.0 mL for urine and cage washings, 10 mL for faeces).
Rubb12 and Rubb12⊂Q[10] in vivo experiments
Rubb12 and Q[10] were prepared as previously described.19Pharmokinetic analysis of Rubb12
Blood was collected from the mice by cardiac puncture immediately after they were sacrificed and then transferred to 1.7 mL eppendorf micro-centrifuge tubes. The samples were kept in the fridge at 4 °C until the blood was completely clotted and then the serum was separated by centrifugation at 3000g for 5 min. Tissues were washed with sterile PBS and weighed. Serum and tissue samples were stored at −20 °C until analysis.
Acknowledgements
We thank TetraQ, University of Queensland, for the 14C-Q[7 and 8] i.v. and oral animal data collection, NewSouth Innovations for funding support through a COMET Grant 2009 and a NHMRC Development Grant application 514644 to M. Ranson.References
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC.
- L. Isaacs, Acc. Chem. Res., 2014, 47, 2052–2062 CrossRef CAS PubMed.
- E. Masson, X. Ling, R. Joseph, L. Kyeremeh-Mensah and X. Lu, RSC Adv., 2012, 2, 1213–1247 RSC.
- J. Liu, Y. Lan, Z. Yu, C. S. Y. Tan, R. M. Parker, C. Abell and O. A. Scherman, Acc. Chem. Res., 2017, 50, 208–217 CrossRef CAS PubMed.
- J. Liu, C. S. Y. Tan, Y. Lan and O. A. Scherman, Macromol. Chem. Phys., 2016, 217, 319–332 CrossRef CAS.
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed.
- N. Saleh, I. Ghosh and W. M. Nau, in Supramolecular Systems in Biomedical Fields, ed. H.-J. Schneider, Royal Society of Chemistry, 2013, ch. 7, pp. 164–212 Search PubMed.
- V. Saluja and B. S. Sekhon, J. Pharm. Educ. Res., 2013, 4, 16–25 CAS.
- I. Ghosh and W. M. Nau, Adv. Drug Delivery Rev., 2012, 64, 764–783 CrossRef CAS PubMed.
- A. I. Day and J. G. Collins, in Supramolecular Chemistry from molecules to nanomaterials, ed. J. W. Steed and P. A. Gale, John Wiley & Sons Ltd, Chichester, UK, 2012, vol. 3, pp. 983–1000 Search PubMed.
- S. Walker, R. Oun, F. J. McInnes and N. J. Wheate, Isr. J. Chem., 2011, 51, 616–624 CrossRef CAS.
- D. H. Macartney, Future Med. Chem., 2013, 5, 2075–2089 CrossRef CAS PubMed.
- A. A. Elbashir and H. Y. Aboul-Enein, Crit. Rev. Anal. Chem., 2015, 45, 52–61 CrossRef CAS.
- A. E. Kaifer, Acc. Chem. Res., 2014, 47, 2160–2167 CrossRef CAS PubMed.
- J. P. Da Silva, R. Choudhury, M. Porel, U. Pischel, S. Jockusch, P. C. Hubbard, V. Ramamurthy and A. V. Canario, ACS Chem. Biol., 2014, 9, 1432–1436 CrossRef CAS PubMed.
- H. H. Lee, T. S. Choi, S. J. Lee, J. W. Lee, J. Park, Y. H. Ko, W. J. Kim, K. Kim and H. I. Kim, Angew. Chem., Int. Ed., 2014, 53, 7461–7465 CrossRef CAS PubMed.
- Q. Li, S.-C. Qiu, J. Zhang, K. Chen, Y. Huang, X. Xiao, Y. Zhang, F. Li, Y.-Q. Zhang, S.-F. Xue, Q.-J. Zhu, Z. Tao, L. F. Lindoy and G. Wei, Org. Lett., 2016, 18, 4020–4023 CrossRef CAS PubMed.
- X.-J. Cheng, L.-L. Liang, K. Chen, N.-N. Ji, X. Xiao, J.-X. Zhang, Y.-Q. Zhang, S.-F. Xue, Q.-J. Zhu, X.-L. Ni and Z. Tao, Angew. Chem., Int. Ed., 2013, 52, 7252–7255 CrossRef CAS PubMed.
- F. Li, M. Feterl, J. M. Warner, A. I. Day, F. R. Keene and J. G. Collins, Dalton Trans., 2013, 42, 8868–8877 RSC.
- H. Chen, J. Y. W. Chan, S. Li, J. J. Liu, I. W. Wyman, S. M. Y. Lee, D. H. Macartney and R. Wang, RSC Adv., 2015, 5, 63745–63752 RSC.
- Y. Xue, Z. Wang, Y. Niu, X. Chen, S. M. Y. Lee and R. Wang, Chem. Med. Comm., 2016, 7, 1392–1397 RSC.
- Y. Chen, Z. Huang, H. Zhao, J.-F. Xu, Z. Sun and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 8602–8608 CAS.
- S. Li, J. Y.-W. Chan, Y. Li, D. Bardelang, J. Zheng, W. W. Yew, D. P.-C. Chan, S. M. Y. Lee and R. Wang, Org. Biomol. Chem., 2016, 14, 7563–7569 CAS.
- M. J. Webber, E. A. Appel, B. Vinciguerra, A. B. Cortinas, L. S. Thapa, S. Jhunjhunwala, L. Isaacs, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14189–14194 CrossRef CAS PubMed.
- X. Miao, Y. Li, I. Wyman, S. M. Y. Lee, D. H. Macartney, Y. Zheng and R. Wang, MedChemComm, 2015, 6, 1370–1374 RSC.
- Q.-L. Li, Y. Sun, Y.-L. Sun, J. Wen, Y. Zhou, Q.-M. Bing, L. D. Isaacs, Y. Jin, H. Gao and Y.-W. Yang, Chem. Mater., 2014, 26, 6418–6431 CrossRef CAS PubMed.
- R. Oun, J. A. Plumb and N. J. Wheate, J. Inorg. Biochem., 2014, 1100–134105 Search PubMed.
- R. Oun, R. S. Floriano, L. Isaacs, E. G. Rowan and N. J. Wheate, Toxicol. Res., 2014, 3, 447–455 RSC.
- G. Hettiarachchi, D. Nguyen, J. Wu, D. Lucas, D. Ma, L. Isaacs and V. Briken, PLoS One, 2010, 5, e10514 Search PubMed.
- V. D. Uzunova, C. Cullinane, K. Brix, W. M. Nau and A. I. Day, Org. Biomol. Chem., 2010, 8, 2037–2042 CAS.
- H. Chen, J. Y. W. Chan, X. Yang, I. W. Wyman, D. Bardelang, D. H. Macartney, S. M. Y. Lee and R. Wang, RSC Adv., 2015, 5, 30067–30074 RSC.
- W.-J. Ma, J.-M. Chen, L. Jiang, J. Yao and T.-B. Lu, Mol. Pharmaceutics, 2013, 10, 4698–4705 CrossRef CAS PubMed.
- J. A. Plumb, B. Venugopal, R. Oun, N. Gomez-Roman, Y. Kawazoe, N. S. Venkataramanan and N. J. Wheate, Metallomics, 2012, 4, 561–567 RSC.
- T. H. Nguyen, P. T. T. Dao, Q. N. Phu, D. D. Le, T. A. Nguyen, T. C. Nguyen and M. C. Dang, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2012, 3, 045004 CrossRef.
- M. J. Pisani, Y. Zhao, L. Wallace, C. E. Woodward, F. R. Keene, A. I. Day and J. G. Collins, Dalton Trans., 2010, 39, 2078–2086 RSC.
- L. M. Heitmann, A. B. Taylor, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2006, 128, 12574–12581 CrossRef CAS PubMed.
- This solubility was determined for acid free Q[10] in pure water.
- Y. Zhao, M. H. Pourgholami, D. L. Morris, J. G. Collins and A. I. Day, Org. Biomol. Chem., 2010, 8, 3328–3337 CAS.
- A. I. Day, A. P. Arnold, R. J. Blanch and B. Snushall, J. Org. Chem., 2001, 66, 8094–8100 CrossRef CAS PubMed.
- Y. Kubota, M. Fukuda, M. Muroguchi and K. Koizumi, Biol. Pharm. Bull., 1996, 19, 1068–1072 CAS.
- A. K. Gorle, X. Li, S. Primrose, F. Li, M. Feterl, R. T. Kinobe, K. Heimann, J. M. Warner, F. R. Keene and J. G. Collins, J. Antimicrob. Chemother., 2016, 71, 1547–1555 CrossRef CAS PubMed.
- A. Fredenhagen, J. Labelled Compd. Radiopharm., 2003, 46, 211–220 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob00724h |
| This journal is © The Royal Society of Chemistry 2017 |