
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Expanding the substrate scope of phenylalanine ammonia-lyase from Petroselinum crispum towards styrylalanines†
László Csaba
Bencze‡
a,
Alina
Filip‡
a,
Gergely
Bánóczi
b,
Monica Ioana
Toşa
a,
Florin Dan
Irimie
a,
Ákos
Gellért
c,
László
Poppe
 *abd and
Csaba
Paizs
*a
*abd and
Csaba
Paizs
*a
aBiocatalysis and Biotransformation Research Centre, Faculty of Chemistry and Chemical Engineering, Babeş-Bolyai University of Cluj-Napoca, Arany János Str. 11, RO-400028 Cluj-Napoca, Romania. E-mail: paizs@chem.ubbcluj.ro
bDepartment of Organic Chemistry and Technology, Budapest University of Technology and Economics, Műegyetem rkp. 3, H-1111 Budapest, Hungary. E-mail: poppe@mail.bme.hu
cAgricultural Institute, Centre of Agricultural Research, Hungarian Academy of Sciences, Brunszvik u. 2, H-2462 Martonvásár, Hungary
dSynBiocat Ltd, Lövőház u. 19/1, H-1024 Budapest, Hungary
First published on 4th April 2017
Abstract
This study focuses on the expansion of the substrate scope of phenylalanine ammonia-lyase from Petroselinum crispum (PcPAL) towards the L-enantiomers of racemic styrylalanines rac-1a–d – which are less studied and synthetically challenging unnatural amino acids – by reshaping the aromatic binding pocket of the active site of PcPAL by point mutations. Ammonia elimination from L-styrylalanine (L-1a) catalyzed by non-mutated PcPAL (wt-PcPAL) took place with a 777-fold lower kcat/KM value than the deamination of the natural substrate, L-Phe. Computer modeling of the reactions catalyzed by wt-PcPAL indicated an unproductive and two major catalytically active conformations and detrimental interactions between the aromatic moiety of L-styrylalanine, L-1a, and the phenyl ring of the residue F137 in the aromatic binding region of the active site. Replacing the residue F137 by smaller hydrophobic residues resulted in a small mutant library (F137X-PcPAL, X being V, A, and G), from which F137V-PcPAL could transform L-styrylalanine with comparable activity to that of the wt-PcPAL with L-Phe. Furthermore, F137V-PcPAL showed superior catalytic efficiency in the ammonia elimination reaction of several racemic styrylalanine derivatives (rac-1a–d) providing access to D-1a–d by kinetic resolution, even though the D-enantiomers proved to be reversible inhibitors. The enhanced catalytic efficiency of F137V-PcPAL towards racemic styrylalanines rac-1a–d could be rationalized by molecular modeling, indicating the more relaxed enzyme–substrate complexes and the promotion of conformations with higher catalytic activities as the main reasons. Unfortunately, ammonia addition onto the corresponding styrylacrylates 2a–d failed with both wt-PcPAL and F137V-PcPAL. The low equilibrium constant of the ammonia addition, the poor ligand binding affinities of 2a–d, and the non-productive binding states of the unsaturated ligands 2a–d within the active sites of either wt-PcPAL or F137V-PcPAL – as indicated by molecular modeling – might be responsible for the inactivity of the PcPAL variants in the reverse reaction. Modeling predicted that the F137V mutation is beneficial for the KRs of 4-fluoro-, 4-cyano- and 4-bromostyrylalanines, but non-effective for the KR process of 4-trifluoromethylstyrylalanine.
Introduction
The synthesis of unnatural aromatic amino acids in an enantiopure form is still a challenge in synthetic chemistry, highlighted by the significant interest in these building blocks for the development of therapeutic peptides and proteins.1–3Since phenylalanine – as a pharmacophore element – has an important role in biologically active peptides and proteins, the development of synthetic methods leading to sterically more demanding analogues of this amino acid has gained increased attention.4–7 An attractive enzymatic route for the synthesis of non-natural phenylalanine analogues is provided by aromatic ammonia-lyases and aminomutases which show high structural and sequence similarities, and have a common electrophilic prosthetic group, the autocatalytically forming 3,5-dihydro-5-methylidene-4H-imidazol-4-one (MIO).8,9,10–12
Recent advances in the improvement of the functional properties of the MIO-enzymes initiated their application as biocatalysts for the synthesis of non-natural amino acids, well exemplified by the phenylalanine ammonia-lyase (PAL)-based process, developed by DSM (Netherlands), for the multiton scale production of (S)-2,3-dihydro-1H-indole-2-carboxylic acid.13
Due to its broad substrate tolerance, phenylalanine ammonia-lyase from Petroselinum crispum (PcPAL) was one of the most frequently used aromatic amino acid ammonia-lyases applied as biocatalyst.14–16 Immobilization of isolated PcPAL led to increased operational stability and enabled the employment of PcPAL for the synthesis of non-natural substrates in continuous-flow packed-bed reactors,17 and in magnetic nanoparticle-filled microfluidic systems,18,19 and provided new perspectives in the development of stable and efficient catalysts for industrial applications.
Numerous lines of evidence suggest that interactions between the amino group of the substrate L-Phe and the electrophilic MIO of PAL facilitate the ammonia-lyase reaction.20 The crystal structures of phenylalanine 2,3-aminomutases from Pantoea agglomerans (PaPAM) complexed with L-phenylalanine21 and from Taxus chinensis (TchPAM) complexed with L-α,α-difluoro-β-phenylalanine,22 and the tyrosine 2,3-aminomutase from Streptomyces globisporus (SgTAM) co-crystallized with L-α,α-difluoro-β-tyrosine,23 all with a covalent bond between the amino group of the substrate/inhibitor and the exocyclic methylene carbon of the MIO, supported a reaction mechanism proceeding through the so called N-MIO intermediate. This is also in agreement with QM/MM calculations on TAL24 and PAL19 supporting ammonia elimination via an N-MIO intermediate and suggesting the formation of similar N-MIO complexes as a common feature of the mechanisms for all MIO-enzymes.20
Proceeding from the N-MIO intermediate of PAL with the aid of the catalytically essential Tyr110,25 the elimination is either stepwise (i) through a benzylic carbanion (E1cB) or (ii) through a carbocation (E1) or (iii) it is a concerted process (E2). A thorough kinetic analysis of the PAL reaction,26 based on kinetic isotope effects of [15N]- and/or [3-2H2]-phenylalanine, favored the mechanism via a carbanionic intermediate (E1cB). In a recent study,27 a previously unrevealed, competing MIO-independent reaction pathway was reported with PALs from Anabaena variabilis (AvPAL), Rhodotorula glutinis (RgPAL), and parsley (PcPAL) which proceeds in a non-stereoselective manner and results in the generation of both L- and D-phenylalanine derivatives. Isotope-labeling studies and mutagenesis of key active site residues were consistent with amino acid deamination occurring by a stepwise E1cB elimination mechanism in this case as well.
It was already shown that phenylalanine 2,3-aminomutase from Taxus canadensis (TcaPAM) could transform (S)-α-styrylalanine (L-1a) into (2E,4E)-5-phenylpenta-2,4-dienoic acid (2a) as the major product.28 Considering the high structural and sequence similarities between TcaPAM and PcPAL, it seemed commendable to explore the potential of PcPAL in the kinetic resolution of α-styrylalanines and stereoselective amination of (2E,4E)-styrylacrylates in the reverse reaction (Scheme 1).
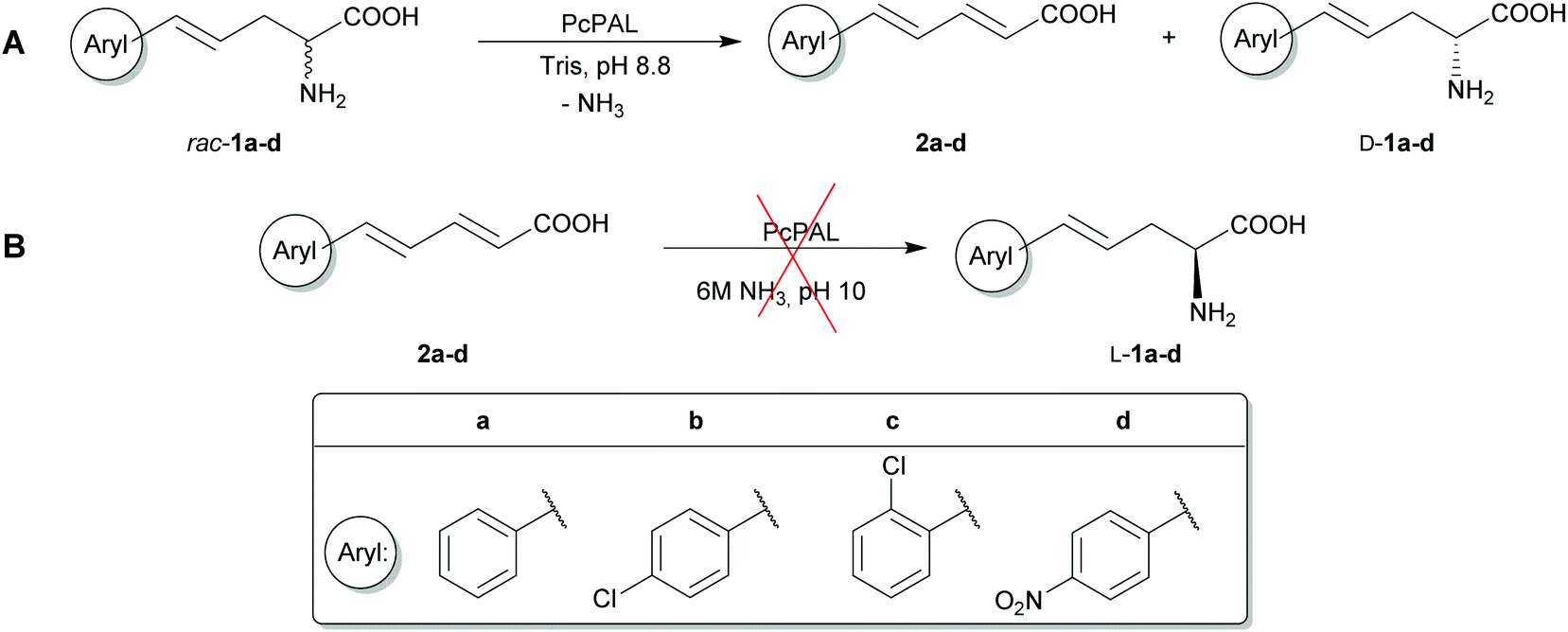 | ||
| Scheme 1 (A) Ammonia elimination from racemic styrylalanines 1a–d and (B) attempted ammonia addition onto styrylacrylates 2a–d catalyzed by wt- and mutant PcPALs. | ||
The present study – with the aim of extending the substrate scope of PcPAL towards sterically demanding phenylalanine analogues – created mutant variants of recombinant PcPAL with an enlarged aromatic binding pocket enabling smooth fit of styrylalanines with increased distance between the aromatic moiety and the α-stereogenic carbon into the active site.
The results of this study were rationalized by molecular modeling in the framework of a straightforward physics-based approach based on ligand/N-MIO intermediate poses/conformation sets and several molecular descriptors to approximate changes on the potential energy surface and thus characterize certain phenomena of the catalysis.
Results and discussion
Initial studies and F137X-PcPAL mutant set construction
First, to assess the applicability of wt-PcPAL to the styrylalanine compound family, the commercially available L-styrylalanine (L-1a) was tested as the substrate in the ammonia elimination reaction (Scheme 1), monitored by HPLC and UV-based assays. The 1H-NMR spectra of the reaction mixtures confirmed the formation of (2E,4E)-5-phenylpenta-2,4-dienoic acid (2a) as the product. Kinetic studies of the ammonia elimination catalyzed by wt-PcPAL showed 14.7-fold lower kcat and 777-fold lower kcat/KM values for the deamination of L-1a in comparison with the deamination of the natural substrate, L-Phe (Table 1).| PcPAL | K M (μM) | L-Phenylalanine | L-Styrylalanine | |||
|---|---|---|---|---|---|---|
| k cat (s−1) | k cat/KM (M−1 s−1) | K M (μM) | k cat (s−1) | k cat/KM (M−1 s−1) | ||
| wt | 83 ± 5 | 0.694 ± 0.020 | 8361 ± 291 | 4384 ± 158 | 0.0471 ± 0.0003 | 10.7 ± 0.4 |
| F137V | 86 ± 10 | 0.173 ± 0.001 | 2011 ± 131 | 186 ± 6 | 0.4220 ± 0.0280 | 2269 ± 168 |
| F137A | 1732 ± 15 | 0.283 ± 0.001 | 163 ± 2 | 1173 ± 70 | 0.1320 ± 0.0026 | 112.5 ± 7.2 |
| F137G | 4969 ± 153 | 0.052 ± 0.003 | 10.4 ± 0.9 | 4120 ± 270 | 0.0345 ± 0.0038 | 8.3 ± 0.2 |
The low enzyme activity, observed with L-styrylalanine (L-1a), was rationalized by molecular modeling. Evaluation of energetics indicated as the most disadvantageous interaction the one between the phenyl ring of the N-MIO intermediate from L-1a and the phenyl ring of the residue F137 in the aromatic binding pocket of wt-PcPAL (Fig. 1). Moreover, enumeration of the possible conformations indicated three major species. In panel A of Fig. 1, two apparently catalytically active conformations, resembling the active arrangement of phenylalanine in PcPAL,19 are depicted. These two N-MIO intermediate conformations can result in two different arrangements of the diene product (2a), the s-cis [referred to as pro-s-cis (psc)] or the s-trans [referred to as pro-s-trans (pst)] conformations. Panel B of Fig. 1 depicts a third conformation adopting a somewhat different arrangement with significantly less stress exerted by F137. However, this specimen would lead to (2Z,4E)-5-phenylpenta-2,4-dienoic acid which was not detected by NMR in the reaction mixture. Thus, conformation L-1au could be considered an unproductive state. These modeling results implied that the reaction rate might be improved by reducing the clash between the aromatic moieties of L-1a in its N-MIO state and F137 within the active site of wt-PcPAL, and simultaneously by relocating the population of the unproductive conformation. It is notable that, earlier, in the case of 4-substituted cinnamates as substrates in the reverse amination reaction with PcPAL, mutations of the F137 site were efficacious.29 Therefore, to provide more space for the phenyl ring of L-1a, the residue Phe137 in the hydrophobic pocket of wt-PcPAL was changed to smaller hydrophobic residues Val, Ala, and Gly in mutants F137V-, F137A-, and F137G-PcPAL, respectively. The kinetic parameters of the wild-type and the mutant PcPALs were determined in the ammonia elimination reaction for the natural substrate L-Phe and for L-1a (Table 1).
 | ||
| Fig. 1 Active site models of the three major conformations of the N-MIO complex of L-1a with wt-PcPAL [colors refer to the atomic contributions to the total energy ranging from green (beneficial, ≤−5 kcal mol−1) to red (detrimental, ≥2 kcal mol−1)]. (A) The two presumably catalytically active conformations L-1apsc and L-1apst leading to the product 2a in s-cis (synperiplanar) or s-trans (antiperiplanar) arrangements, respectively. (B) The apparently catalytically inactive conformation L-1au compared to the apparently catalytically active L-1apsc conformation. | ||
The ratio of kcat and KM – often referred to as the “catalytic efficiency” – is a useful index for comparing the efficiency of an enzyme acting on alternative substrates.30 Not surprisingly, all mutants showed decreased – declining with side chain volume – catalytic efficiencies in the ammonia elimination reaction of the natural substrate, L-Phe, in comparison with wt-PcPAL. However, it can be clearly seen that F137V has superior catalytic efficiency with L-1a as substrate, with nearly 240-fold increased kcat/KM value compared to wt-PcPAL. The F137A mutation also provided an increased kcat/KM value with L-1a (10.5-fold enhancement), while the F137G mutation showed no beneficial effect on the transformation of L-1a. Interestingly, kcat values showed also substantial variances. The trends and maxima were almost the same as those in the case of the kcat/KM values. Regarding L-Phe, kcat declined with decreasing side chain volume, except for a surprising spike with F137A-PcPAL. Similarly, F137V-PcPAL with L-1a showed a tremendous, almost 9-fold increase; F137A showed only a moderate 2.8-fold increase; and F137G showed no improvement in kcat compared to wt-PcPAL.
A comparison of the covalent pro-s-cis intermediates from L-1a within wt-PcPAL and F137V-PcPAL is shown in panel A of Fig. 2. The aromatic moiety of the N-MIO intermediate from L-1a is forced towards I460 within the active site of wt-PcPAL, unlike in the case of the more relaxed complex within F137V-PcPAL; meanwhile, the alanyl substructure of the substrate – which is directly involved in bond rearrangement – is kept relatively unchanged in both cases. It is worth noting that upon the binding of L-1a to wt-PcPAL the active site supposedly undergoes a conformational change at the I460-side chain which reshapes the active site and alters the energetics of distinct substrate conformations.
 | ||
| Fig. 2 Molecular models of various N-MIO intermediates in PcPALs. (A) Arrangement of the covalent intermediate L-1apsc in wt-PcPAL (relevant residues are in red) and F137V-PcPAL (relevant residues are in cyan). Note the different side chain conformations of I460. (B) Arrangement of the unproductive covalent complex from D-1a (yellow) compared to the productive intermediate L-1apsc (red) in wt-PcPAL. | ||
Synthetic applications in kinetic resolution
Next, we wanted to explore the potential of F137V-PcPAL in the synthetically valuable ammonia elimination and addition reactions of various styrylalanines and their acrylic counterparts. Therefore, racemic styrylalanines rac-1a–d as well as the corresponding styrylacrylates 2a–d were synthesized (see Schemes S1 and S2 in the ESI†) and tested with F137V-PcPAL and wt-PcPAL as biocatalysts (Scheme 1).In the ammonia elimination reaction all styrylalanines (rac-1a–d) were accepted as substrates by wt-PcPAL as well as by F137V-PcPAL. However, the F137V mutant showed significantly improved catalytic activities (kcat and kcat/KM) in comparison with the wild-type enzyme in all cases (Table 2).
| Substrate | wt-PcPAL | F137V-PcPAL | ||||
|---|---|---|---|---|---|---|
| K M (μM) | k cat (s−1) | k cat/KM (M−1 s−1) | K M (μM) | k cat (s−1) | k cat/KM (M−1 s−1) | |
| rac-1a | 395 ± 6 | 0.00620 ± 0.00070 | 15.6 ± 0.9 | 201 ± 12 | 0.2760 ± 0.0130 | 1373 ± 97 |
| rac-1b | 154 ± 7 | 0.00024 ± 0.00001 | 1.5 ± 0.03 | 78.3 ± 2 | 0.0786 ± 0.0044 | 1004 ± 86 |
| rac-1c | 28 ± 1 | 0.00034 ± 0.00002 | 12.1 ± 0.6 | 94.7 ± 2 | 0.1570 ± 0.0020 | 1657 ± 10 |
| rac-1d | 287 ± 3 | 0.00320 ± 0.00030 | 11.1 ± 0.5 | 326 ± 10 | 0.0099 ± 0.0001 | 30.2 ± 1.9 |
The synthetic usefulness of F137V-PcPAL towards styrylalanines in the ammonia elimination reactions was further tested by kinetic resolutions (KRs) with 0.1 mmol rac-1a–d at 5 mM concentration using 0.5–1.0 mg of pure PcPALs. The conversion of the PcPAL-catalyzed reactions from racemic styrylalanines rac-1a–d (Fig. 3), and the enantiomeric excess of the residual substrates D-1a–d were both monitored (Table 3). In the case of longer reaction times (>48 h), a fresh enzyme batch was added to the reaction mixture at the end of each 48 h period to overcome enzyme inactivation during long incubation times.
 | ||
| Fig. 3 Comparison of the time profiles of the conversions in the ammonia elimination reactions from rac-1a–d catalyzed by F137V-PcPAL (continuous lines) and wt-PcPAL (dashed lines). | ||
| Entry | PcPAL | Substrate | Time (h) | c (%) | eetheora (%) | eeobs (%) |
|---|---|---|---|---|---|---|
| a eetheor = c/(1 − c) for a fully selective kinetic resolution. | ||||||
| 1 | F137V | rac-1a | 24 | 50 | 100 | >99 |
| 2 | rac-1b | 274 | 50 | 100 | >99 | |
| 3 | rac-1c | 134 | 50 | 100 | >99 | |
| 4 | rac-1d | 300 | 50 | 100 | >99 | |
| 5 | Wild-type | rac-1a | 274 | 50 | 100 | >98 |
| 6 | rac-1b | 504 | 29 | 41 | 41 | |
| 7 | rac-1c | 600 | 36 | 56 | 55 | |
| 8 | rac-1d | 600 | 31 | 45 | 41 | |
Time course analysis of the conversion of the KRs of rac-1a–d (Fig. 3) also indicated the beneficial catalytic properties of F137V-PcPAL compared to that of wt-PcPAL. The theoretically possible conversion in a kinetic resolution (KR) with exclusive selectivity (50%) could be almost perfectly approached (c > 49%) with the F137V mutant in reactions of rac-1a,c after moderate reaction times (24 h and 134 h, respectively) and in the case of rac-1b,d after longer incubation times (274 h and 300 h, respectively). The unusually long time required for the compounds rac-1b,d can be significantly shortened by using higher enzyme to substrate ratios. This might be easily performed by applying immobilized forms of PcPAL in microreactors.17
On the other hand, in reactions of racemic styrylalanines rac-1a–d with wt-PcPAL, almost complete conversion of the L-enantiomer could be obtained only with rac-1a (c > 49% after 274 h), while the conversions of rac-1b–d could not approach the theoretically possible value within 500–600 h.
KRs of rac-1a–c with wt-PcPAL showed that enantiomeric excess of residual D-1a–c fractions were in close agreement with the theoretical values of a fully selective KR indicating excellent enantiomer selectivity and preference of the process to digest the L-enantiomers, L-1a–c (Table 3). Although the enantiomeric excess of the residual D-1d fractions was somewhat smaller than that expected for a fully selective KR, it was not possible to determine whether this was due to experimental errors of the HPLC process or due to the non-perfect enantiomer selectivity of the KR from rac-1d.
KRs of rac-1a–d with F137V-PcPAL, however, showed that the residual D-1a–d fractions had enantiomeric excess values over 99% indicating unquestionably exclusive enantiomer selectivity in all cases. This was also obvious from the time course curves of conversions approaching asymptotically the 50% value when F137V-PcPAL was applied as the biocatalyst in the KRs (Fig. 3). Time course profiles of the reactions and the comparison of the kinetic constants with L-1a (Table 1) and with rac-1a (Table 2) suggest that D-styrylalanine inhibits the ammonia elimination of L-1a with both wt-PcPAL and F137V-PcPAL. If D-1a had no interaction with wt-PcPAL, then only a 2-fold higher apparent KM of the racemate rac-1a would be observed as compared to the pure L-enantiomer L-1a without affecting the kcat value. However, when rac-1a was applied as the substrate instead of the pure L-enantiomer L-1a, the Vmax (thus implicitly the apparent kcat value) decreased 7.6-fold, while the KM value decreased 11-fold. With F137V-PcPAL, the differences became less significant, kcat decreased only 1.5-fold, and KM showed only a minor variation, within the range of experimental errors. Because the residual fractions from the KRs of rac-1a–d had quite high enantiomeric excess at 50% conversion (ee > 99%, see Table 3), the D-enantiomers D-1a–d were apparently not converted. This implied that D-1a is a reversible inhibitor with considerable affinity forming an unproductive enzyme–D-substrate complex. The covalent, unproductive N-MIO complex from D-1a showed astonishing overlap with the productive intermediate L-1apst within the active site of wt-PcPAL (panel B in Fig. 2).
Rationalization of the reactions of styrylalanines rac-1a–d catalyzed by the PcPAL variants
To obtain a more detailed view of the catalytic process, a quantitative model was created and validated. Because N-MIO complexes were assumed to be a common feature of the mechanism for all MIO-enzymes,20 the enzyme–substrate complexes within the active site of the PcPAL variants were constructed as N-MIO intermediates of L- and D-1a–d applying an induced-fit covalent docking protocol which was previously used to rationalize the reactions of various substrates within another MIO enzyme, the phenylalanine 2,3-aminomutase from Pantoea agglomerans.31The non-covalent substrate-binding states of L- and D-1a–d – directly preceding their N-MIO intermediate states – were also modeled. Because the calculations did not reveal any unproductive states for these non-covalent complexes, they will not be discussed hereafter.
The occurrence of the non-stereoselective, MIO-independent reaction pathway27 was ruled out experimentally. The attempted ammonia elimination from rac-1a–d by wt-PcPAL with the inactivated MIO group (obtained by treatment with sodium borohydride)32 showed no product formation. The conversion of 4-nitrophenylalanine with the sodium borohydride treated wt-PcPAL served as a positive control for the integrity of the residual parts of the active site.
To investigate the catalytic activities quantitatively with all substrate and enzyme mutant combinations, the apparent kcat values – as determined from the maximum reaction rates according to the left-hand side of eqn (1) – were decomposed into their constituents.
At substrate saturation, when the concentration of the free enzyme is negligible, four species of enzyme–substrate intermediate complexes (EL-psc, EL-psc, EL-u, and ED) can be present in thermal equilibrium (allosteric inhibition was not considered in this study). As discussed previously, from the L-enantiomer two catalytically active enzyme–substrate intermediate complexes (the s-cis and s-transL-1a–d–N-MIO states, abbreviated as EL-psc, EL-pst) and an unproductive one (the unproductive L-1a–d–N-MIO states, abbreviated as EL-u) could be formed, while another unproductive complex could be formed from the D-enantiomers (the unproductive D-1a–d–N-MIO states, abbreviated as ED). Thus, according to eqn (1), the catalytic activity is the sum of the activities of EL-psc and EL-pst.
| Vmax = kcat[E*] = kcat, L-psc[Epsc] + kcat, L-pst[Epst] | (1) |
Expressing the substrate concentrations as molar fractions and substituting the Eyring equation yields eqn (2):
 | (2) |
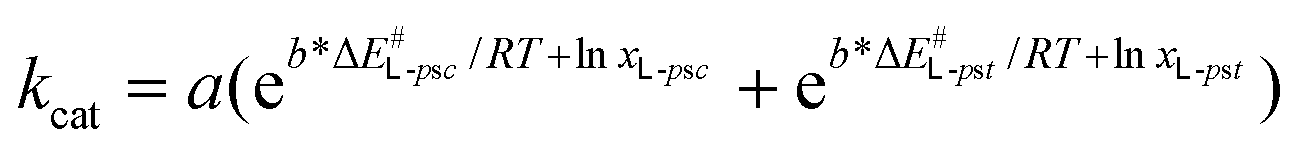 | (3) |
| PcPAL | Substrate | b R ΔEL-pscb (kcal mol−1) | x L-psc (%) | x L-pst (%) | x D (%) | x L-u (%) | Strain R ΔEL-pscb (kcal mol−1) | Strain R ΔEL-pstb (kcal mol−1) | cB R ΔEL-pscb (kcal mol−1) | cB R ΔEL-pstb (kcal mol−1) | cB R ΔEL-pst − cBRΔEL-pscb (kcal mol−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a In these cases multiple psc and pst conformations were obtained, but only the lowest energy representatives were taken into account. b Quantities are related to the same property of L-Phe with wt-PcPAL, in the form of RΔE = ΔE − ΔEL-Phe;wt-PcPAL. For a detailed description of the calculation method, see the ESI. For the reasoning of the necessary relativization, see ref. 30. c Molar fractions were calculated based on Boltzmann probabilities incorporating all substrate conformations. | |||||||||||
| Wild-type | 1a | 13.0 | 99.7 | 0.2 | 0.1 | 0.0 | 11.0 | 16.2 | −2.2 | 3.8 | 6.1 |
| 1b | 22.5 | 2.1 | 0.0 | 0.0 | 97.9 | 22.5 | 28.5 | −1.5 | −1.8 | −0.3 | |
| 1c | 13.6 | 41.5 | 0.0 | 0.7 | 57.8 | 12.9 | 20.4 | 11.9 | 8.2 | −3.6 | |
| 1d | 33.7 | 93.4 | 0.0 | 0.0 | 6.6 | 33.9 | 42.4 | −11.4 | −11.5 | 0.1 | |
| F137V | 1a | −3.3 | 95.2 | 4.8 | 0.0 | 0.0 | −0.7 | 1.1 | −4.6 | −10.7 | −6.2 |
| 1b | −3.2 | 99.9 | 0.1 | 0.0 | 0.0 | 1.4 | 5.7 | −5.4 | −7.7 | −2.4 | |
| 1c | 1.4 | 60.9 | 5.0 | 0.8 | 0.0 | 5.3 | 6.8 | 11.3 | 0.5 | −10.8 | |
| 1d | 22.7 | 0.3 | 18.1 | 0.9 | 80.7 | 25.3 | 22.9 | −31.6 | −20.4 | 11.3 | |
Non-linear estimation of the constants in eqn (3) gave statistically significant regression (R2 = 0.8, p = 0.002) and parameters (a = 90.63, p = 0.023; b = 0.0791, p = 0.014). The complete derivation of eqn (3) is described in the ESI.†
Evaluation of the F137V mutation based on computed data (Table 4) clearly demonstrated that the affinities of L-1a–d in the pro-s-cis conformation (bRΔEL-psc) increased substantially, as was assumed. Moreover, the affinities of L-1a–d in the pro-s-trans conformation increased even more, as their population increased relative to the pro-s-cis conformation (xL-pstvs. xL-psc), with the exception of L-1d with F137V-PcPAL. The unproductive complexes of 1a–d (xD and xL-u), however, showed a more complex situation. By the F137V-mutation, in some cases unproductive states were displaced (e.g.1c), and in some other cases they were introduced (e.g.1d). These results indicated that a simple increase of the active site volume is not sufficient from the standpoint of enzyme design, and a more sophisticated active site shape altering scheme – taking the relative spatial requirements of the intermediate states and unproductive states into account – should be employed. Inspection of the descriptors for the acidity of the pro-S β-hydrogen (cBRΔE) – which was derived from single point DFT calculations on a truncated part of the intermediate model – revealed that in almost all cases the acidity of the pro-s-trans conformations compared to the pro-s-cis conformations (cBRΔEL-pst − cBRΔEL-psc) was comparable or higher, except for 1b/wt-PcPAL and 1d/F137V-PcPAL. Thus, the higher proportion of the pro-s-trans conformations (xL-pst), enabling more facilitated reaction due to the more acidic pro-S β-hydrogens, could contribute to increased activity of F137V-PcPAL. Moreover, the acidity descriptors cBRΔEL-psc and cBRΔEL-pst varied remarkably in wt- and F137V-PcPAL, mostly due to changes in dihedral angle between the phenyl ring and the conjugated double bond of the styryl moiety. The more planar the skeleton was, the better the dispersion of negative charge due to the higher degree of conjugation. This implied that in an efficient enzyme design protocol, the substrate affinity and electronic effects should be monitored simultaneously.
This exploratory model validates the very concept of our computations and can be used as a tool to analyze various factors of importance in catalysis and to provide valuable input for further studies. Although this model was not expected to predict the apparent kcat with high accuracy, it enabled us to predict the order of magnitude of the catalytic activity for several further congeneric substrates.
With our model, we examined the behavior of further substituted styrylalanines bearing bromo, cyano, fluoro, and trifluoromethyl substituents at position 4 (Table 5). Calculations with wt-PcPAL predicted rac-4-fluorostyrylalanine and rac-4-cyanostyrylalanine to be poor substrates behaving similarly to rac-styrylalanine, rac-1a. Modeling forecast rac-4-bromo- and rac-4-trifluorostyrylalanines to be extremely poor substrates or reversible inhibitors (kcat-pred ≤ 0.0001 s−1).
| PcPAL | 4-Substituenta | b R ΔEL-pscb (kcal mol−1) | x L-psc (%) | x L-pst (%) | x D (%) | x L-u (%) | Strain R ΔEL-pscb (kcal mol−1) | Strain R ΔEL-pstb (kcal mol−1) | cB R ΔEL-pscb (kcal mol−1) | cB R ΔEL-pstb (kcal mol−1) | k cat-pred (s−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Substituent of the further 4-substituted styrylalanines. b Quantities are related to the same property of L-Phe with wt-PcPAL, in the form of RΔE = ΔE − ΔEL-Phe;wt-PcPAL. For the detailed description of the calculation method, see the ESI. For the reasoning of the necessary relativization, see ref. 30. c Molar fractions were calculated based on Boltzmann probabilities incorporating all substrate conformations. | |||||||||||
| Wild-type | F– | 13.2 | 88.0 | 0.0 | 0.0 | 12.0 | 13.5 | 20.3 | −0.3 | −1.5 | 0.014 |
| CF3– | 38.7 | 0.2 | 0.0 | 0.0 | 99.8 | 40.1 | 48.0 | −3.7 | −3.8 | <0.0001 | |
| Br– | 23.3 | 2.1 | 0.0 | 0.0 | 97.9 | 24.2 | 32.3 | −1.7 | −2.4 | 0.0001 | |
| NC– | 33.7 | 93.4 | 0.0 | 0.0 | 6.6 | 33.9 | 42.4 | −11.4 | −11.5 | 0.004 | |
| F137V | F– | −4.3 | 99.3 | 0.7 | 0.0 | 0.0 | 0.6 | 3.6 | −5.7 | −6.9 | 0.180 |
| CF3– | 15.5 | 0.0 | 0.0 | 0.0 | 100.0 | 21.5 | 15.5 | −6.8 | −8.2 | <0.0001 | |
| Br– | -2.6 | 95.9 | 0.0 | 0.0 | 4.1 | 2.9 | 8.2 | −5.4 | −1.6 | 0.120 | |
| NC– | 8.3 | 100.0 | 0.0 | 0.0 | 0.0 | 13.1 | 17.8 | −13.7 | −15.0 | 0.098 | |
The calculations predicted that the F137V mutant of PcPAL would be inefficient only in KR of rac-4-trifluorostyrylalanine (kcat-pred ≤ 0.0001 s−1). Modeling within this mutant indicated comparable high activity for rac-4-fluorostyrylalanine (kcat-pred = 0.18 s−1) as for rac-styrylalanine (rac-1a: kcat = 0.28 s−1). Somewhat lower catalytic activities were predicted for rac-4-bromo- and rac-4-cyanostyrylalanines (kcat-pred = 0.120 s−1 and 0.098 s−1, respectively) being in the range of the activity of rac-4-chlorostyrylalanine (rac-1c: kcat = 0.16 s−1).
Study of ammonia addition onto styrylacrylates 2a–d with PcPALs
Regrettably, no 1a–d could be detected by HPLC in the attempted ammonia addition reactions of styrylacrylates 2a–d with either wt-, F137V-, F137A-, or F137G-PcPAL even after 20 days (Scheme 1, for details see section 5.6 of the ESI†). The possible reasons for these results were analyzed by computer modeling in the case of wt- and F137V-PcPAL. It was not surprising that the relative binding energies of 2a–d indicated poor binding to wt-PcPAL, which was remarkably improved with F137V-PcPAL (Table 6, bRΔEmono). Modeling revealed presumably unproductive ligand poses for 2a–d with both PcPAL variants in almost all cases with higher binding affinity as compared to the possibly reactive arrangements of 2a–d. The case of 2a within F137V-PcPAL is shown in Fig. 4. Panel A depicts the lower energy arrangement of the putatively inactive conformation of 2a with a bidentate salt bridge between R354 and the carboxylate group (in blue) compared to the putatively catalytically active conformation forming a monodentate salt bridge (in cyan). | ||
| Fig. 4 Models of possible styrylacrylate 2a-arrangements within amino-enzyme (NH2-N-MIO) forms of PcPAL variants. (A) Hydrogen bonds forming among the carboxylate moiety, a water molecule and R354, N384 of the bidentate and monodentate salt bridge forming poses of 2a within F137V-PcPAL. (B) The distances of the N atom of NH2 of the NH2-N-MIO from the C2 atom of the two lowest energy mono- (2amono) or bidentate (2abi) carboxylate binding poses of 2a within F137V-PcPAL. In both panels, the model depicting 2amono (in cyan) corresponds to a product from the productive N-MIO intermediate (L-1apsc), and the model depicting 2abi (in blue) shows a low energy inactive state. | ||
| PcPAL | wt-PcPAL | F137V-PcPAL | ||||
|---|---|---|---|---|---|---|
| Compound | b R ΔEmonoa kcal mol−1 | x mono (%) | x bi (%) | b R ΔEmonoa kcal mol−1 | x mono (%) | x bi (%) |
| a Quantities are related to the same property of (E)-cinnamate with wt-PcPAL, in the form of RΔE = ΔE − ΔE(E)-cin;wt-PcPAL. For the detailed description of the calculation method, see the ESI. For the reasoning of the necessary relativization, see ref. 30. b Molar fractions were calculated based on Boltzmann probabilities incorporating all ligand poses. | ||||||
| 2a | 17.2 | 1.0 | 97.5 | 2.2 | 15.8 | 71.8 |
| 2b | 25.8 | 18.0 | 81.9 | −0.2 | 77.9 | 14.4 |
| 2c | 20.4 | 0.1 | 75.8 | 2.0 | 0.6 | 91.3 |
| 2d | 41.8 | 0.0 | 92.6 | 10.4 | 6.3 | 93.3 |
Panel B of Fig. 4 depicts models of the possible arrangements of 2a within F137V-PcPAL with an elongated distance between the N atom of NH2 of the NH2-N-MIO and the C2 atom in the model for putatively inactive conformation of 2a (in blue) as compared to the corresponding distance in the model including 2a in reactive conformation (in cyan).
Grand canonical Monte Carlo simulations indicated a water molecule for both binding poses of 2a forming a hydrogen bond with the carboxylate group of the ligand, albeit in remarkably different positions. Irrespective of the monodentate or bidentate nature of the carboxylate moiety regarding the arginine R354, the three oxygen atoms of the carboxylate group and the water always formed hydrogen bonds with residues R354 and N384 (Fig. 4, panel A). These findings agree with the experimental results found in X-ray structure of a TchPAM mutant (PDB ID: 4CQ5).22 In the structure of this enzyme homologous to PcPAL, a continuous electron density between R354 and N384 and the carboxylate of the ligand was observed in the active site complexed with (E)-cinnamic acid and a water which was consistent with multiple ligand poses in a similar arrangement to that found in our models for 2a in PcPAL.
Modeling results suggest that low binding affinities of 2a–d (especially to wt-PcPAL) and substrate inhibition by the inactive substrate poses of 2a–d in both PcPAL variants could contribute to the failure of the ammonia addition. Modeling, however, could not represent results which could completely exclude the possibility of successful ammonia addition, especially in the case of 2a,b with F137V-PcPAL. Therefore, further reasons for the failure of ammonia addition can be proposed: (i) the thermodynamic equilibrium of ammonia addition can be too low to result in detectable amounts of L-1a–d, (ii) the low water solubility and rather poor affinity of 2a–d result in non-detectable reaction rates/product concentrations, and (iii) probably the enzyme adopts a significantly different conformation in 6 M ammonia that makes the computational results invalid under the conditions of the ammonia addition.
Experimental
The detailed description of the materials, analytical methods, chemical synthesis of substrates rac-1a–d, 2a–d, curves from kinetic measurements, the HPLC chromatograms used for the determination of conversion and enantiomeric excess values, as well as the details of the methods used for molecular modeling are given in the ESI.†Site-directed mutagenesis
Site-directed mutagenesis was performed using the protocol described by Naismith and Liu.33The PCR reaction contained 10 ng of template DNA (recombinant plasmid encoding PcPAL34), a 1 μM solution of primer pairs (Table 7), 200 μM dNTPs and 3 units of Pfu DNA polymerase, filled to 50 μL with water. The PCR cycles were initiated at 95 °C for 5 min, followed by 15 amplification cycles. Each amplification cycle consisted of 95 °C for 1 min, at a temperature Tnom of 5 °C for 1 min and 72 °C for 15 min. The PCR cycles were finished with an annealing step at a Tppm of 5 °C for 1 min and the final extension step at 72 °C for 30 min. The PCR products were treated with 5 units of DpnI at 37 °C for 2 h and then 10 μL of each PCR reaction was analyzed by agarose gel electrophoresis. An aliquot of 3 μL from the above PCR product was transformed into a 100 μL suspension of E. coli XL1-Blue competent cells (OD600 2.2) by heat shock. The transformed cells were spread on a Luria–Bertani (LB) plate containing carbenicillin (34 μg mL−1) and chloramphenicol (50 μg mL−1) and incubated at 37 °C for 16 h. Two colonies from each plate were grown and the plasmid DNA was isolated. To verify the mutations, 400 ng of plasmid DNA was mixed with 50 pmol of T7-for, T7-rev and PcPAL-for1, PcPAL-for2 sequencing primers (Table 6) in a final volume of 15 μL. DNA sequencing was carried out by Genomed (Debrecen, Hungary).
| Primer | Sequence (5′–3′) |
T
ppm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (°C) a (°C) |
T
noma (°C) |
|---|---|---|---|
| a T ppm and Tnom represent the melting temperatures for the primer–primer overlapping sequence and for the primer sequence matched with the template. | |||
| F137V-for | GATTAGAGTCTTGAACGCCGGAATTTTCGGAAACGGATC | 56 | 64 |
| F137V-rev | GGCGTTCAAGACTCTAATCAATTCCTTCTGCAAGGCTCC | 56 | 64 |
| F137A-for | GATTAGAGCCTTGAACGCCGGAATTTTCGGAAACGGA | 58 | 67 |
| F137A-rev | GCGTTCAAGGCTCTAATCAATTCCTTCTGCAAGGCTCCTC | 58 | 67 |
| F137G-for | GATTAGAGGCTTGAACGCCGGAATTTTCGGAAACGGA | 58 | 67 |
| F137G-rev | GCGTTCAAGCCTCTAATCAATTCCTTCTGCAAGGCTCCTC | 58 | 67 |
| PcPAL-for1 | CAGAACATTACTCCCTGCTTG | — | — |
| PcPAL-for2 | TTGACGTATCCAGAAACAAGG | — | — |
Protein expression
The expression and purification of the wild-type PcPAL and the F137 V, F137A, F137G mutants were performed using the protocol described earlier.34 The protein purity and homogeneity were determined using SDS-PAGE and size-exclusion chromatography (Fig. S37 and S38†).Enzyme activity measurements
Enzyme assays were carried out in triplicate at 30 °C in a 1 mL quartz cuvette using 0.1 M Tris and 0.12 M NaCl (pH 8.8) as a buffer at a constant concentration of wt-PcPAL or F137V-PcPAL (for details, see the ESI†), varying the substrate concentration from 0.025 and 7.8 mM until substrate saturation occurred. The kinetic measurements were based on UV-spectroscopy by monitoring the production of the acrylic derivatives 2a–d at wavelengths where the corresponding amino acids rac-1a–d showed no absorption (Table S1†). Kinetic constants (KM, vmax, and kcat) were obtained from the Michaelis–Menten curves by non-linear fitting obtained by using MATLAB (see the ESI†).Preparative scale ammonia elimination reactions from rac-1a–d
The enzymatic reactions were performed in 20 mL of Tris (0.1 M, 0.12 M NaCl, pH 8.8), containing styrylalanines (rac-1a–d, 5 mM) and purified PcPAL (0.5 mg of F137V-PcPAL or 1 mg of wt-PcPAL). Conversion and the enantiomeric excess of the residual D-1a–d were monitored using reverse phase high performance liquid chromatography (HPLC) analysis (see the ESI†). Samples (30 μL) were taken from the reaction mixture after different time intervals, quenched by adding an equal volume of MeOH, vortexed and centrifuged (13400 rpm, 12100 g, 2 min). The supernatant was used directly for HPLC analysis after passing it through a 0.22 μm nylon membrane filter. In the case of enzyme reactions over longer reaction times, another batch of enzyme was added after every 48 h period.
Conclusions
In this study, the substrate scope of PcPAL was extended by a small library of mutants, towards styrylalanine derivatives, a useful class of synthetically challenging phenylalanine analogues.Although L-styrylalanine, L-1a, was accepted as the substrate by wt-PcPAL in the ammonia elimination reaction, low reaction rates were observed with the wild-type enzyme. Molecular modeling suggested that the most serious clash between the aromatic moiety of L-styrylalanine, L-1a, and the side chain of the residue F137 in the hydrophobic binding pocket can be reduced by introducing residues smaller than F137 by point mutations.
Single point mutations of F137 resulted in a small mutant library (F137X-PcPAL, X being V, A, and G), from which F137V-PcPAL showed significantly increased catalytic efficiency and could transform L-styrylalanine, L-1a, with a comparable activity to that of the wt-PcPAL with L-Phe. Furthermore, F137V-PcPAL could be used as an effective biocatalyst in the synthetically valuable kinetic resolutions by ammonia elimination from racemic styrylalanine analogues rac-1a–d providing access to D-styrylalanines D-1a–d, with highly improved catalytic efficiency compared to the wt-PcPAL in the same reactions.
The reverse ammonia addition reactions onto (E)-styrylacrylates 2a–d – which would be useful for the synthesis of L-styrylalanines L-1a–d – failed with either the wild-type or the mutant enzyme variants (F137 V, F137A, F137G), presumably due to the formation of energetically favorable unproductive binding states of the styrylacrylates 2a–d.
Molecular modeling – used as an aid to explain the results of this study – revealed unusually high affinities of the D-enantiomers D-1a–d towards wt-PcPAL, but proved to be unproductive. Moreover, an unproductive and two productive conformations were revealed for the L-enantiomers L-1a–d. Thus, the proportion of the unproductive complexes acting as potent reversible inhibitors compared to the active arrangements of the L-enantiomers L-1a–d turned out to be important.
By the mutation F137V in PcPAL, the less strained arrangement of substrates L-1a–d within the active site along with the more pronounced populations of conformations with higher catalytic activities simultaneously led to the beneficial catalytic efficiency of this mutant. Modeling the behavior of further 4-substituted styrylalanines with PcPAL variants predicted the F137V mutation to be beneficial for the KRs of 4-fluoro-, 4-cyano- and 4-bromostyrylalanines, but non-effective for the KR process of 4-trifluoromethylstyrylalanine.
The results of this study open up new perspectives for the substrate scope expansion of PcPAL towards synthetically challenging phenylalanine analogues with increased length of the carbon skeleton between the aromatic moiety and the α-stereogenic carbon.
Acknowledgements
LCB acknowledges financial support from the Romanian National Authority for Scientific Research and Innovation, CNCS-UEFISCDI (PN-II-RU-TE-2014-4-1668). LP acknowledges financial support from the Hungarian OTKA Foundation (NN-103242), the Hungarian Research and Technology Innovation Fund (KMR 12-1-2012-0140) and the New Hungary Development Plan (TÁMOP-4.2.1/B-09/1/KMR-2010-0002). The licensing of the Schrödinger Suite software package was financed by the Hungarian OTKA Foundation (K 108793).Notes and references
- J. A. Robinson, S. J. DeMarco, F. Gombert, K. Moehle and D. Obrecht, Drug Discovery Today, 2008, 13, 944–951 CrossRef CAS PubMed.
- N. Srinivas, P. Jetter, B. J. Ueberbacher, M. Werneburg, K. Zerbe, J. Steinmann, B. Van der Meijden, F. Bernardini, A. Lederer, R. L. A. Dias, P. E. Misson, H. Henze, J. Zumbrunn, F. O. Gombert, D. Obrecht, P. Hunziker, S. Schauer, U. Ziegler, A. Kach, L. Eberl, K. Riedel, S. J. DeMarco and J. A. Robinson, Science, 2010, 327, 1010–1013 CrossRef CAS PubMed.
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CAS.
- S. E. Gibson, N. Guillo and M. J. Tozer, Tetrahedron, 1999, 55, 585–615 CrossRef CAS.
- I. Berezowska, N. N. Chung, C. Lemieux, B. C. Wilkes and P. W. Schiller, J. Med. Chem., 2009, 52, 6941–6945 CrossRef CAS PubMed.
- X. Yu, P. Talukder, C. Bhattacharya, N. E. Fahmi, J. A. Lines, L. M. Dedkova, J. LaBaer, S. M. Hecht and C. Shengxi, Bioorg. Med. Chem. Lett., 2014, 24, 5699–5703 CrossRef CAS PubMed.
- C. Solanas, B. G. de la Torre, M. Fernandez-Reyes, C. M. Santiveri, M. A. Jimenez, L. Rivas, A. I. Jimenez, D. Andreu and C. Cativiela, J. Med. Chem., 2009, 52, 664–674 CrossRef CAS PubMed.
- N. Turner, Curr. Opin. Chem. Biol., 2011, 15, 234–240 CrossRef CAS PubMed.
- M. M. Heberling, B. Wu, S. Bartsch and D. B. Janssen, Curr. Opin. Chem. Biol., 2013, 17, 250–260 CrossRef CAS PubMed.
- L. Poppe and J. Rétey, Curr. Org. Chem., 2003, 7, 1297–1315 CrossRef CAS.
- L. Poppe and J. Rétey, Angew. Chem., Int. Ed., 2005, 44, 3668–3688 CrossRef CAS PubMed.
- (a) L. Poppe, Curr. Opin. Chem. Biol., 2001, 5, 512–524 CrossRef CAS PubMed; (b) J. Rétey, Biochim. Biophys. Acta, 2003, 1647, 179–184 CrossRef.
- B. de Lange, D. J. Hyett, P. J. D. Maas, D. Mink and F. B. J. van Assema, ChemCatChem, 2011, 3, 289–292 CrossRef CAS.
- A. Gloge, J. Zon, A. Kővári, L. Poppe and J. Rétey, Chem. – Eur. J., 2000, 6, 3386–3390 CrossRef CAS.
- C. Paizs, A. Katona and J. Rétey, Chem. – Eur. J., 2006, 12, 2739–2744 CrossRef CAS PubMed.
- C. Paizs, M. I. Toşa, L. C. Bencze, J. Brem and F. D. Irimie, Heterocycles, 2011, 82, 1217–1228 CAS.
- (a) J. H. Bartha-Vári, M. I. Toşa, F. D. Irimie, D. Weiser, Z. Boros, B. G. Vértessy, C. Paizs and L. Poppe, ChemCatChem, 2015, 7, 1122–1128 CrossRef PubMed; (b) J. H. Bartha-Vári, L. C. Bencze, E. Bell, L. Poppe, G. Katona, F. D. Irimie, C. Paizs and M. I. Toşa, Period. Polytech., Chem. Eng., 2017, 61(1), 59–66 Search PubMed.
- F. Ender, D. Weiser, B. Nagy, L. C. Bencze, C. Paizs, P. Pálovics and L. Poppe, J. Flow Chem., 2016, 6, 43–52 CrossRef CAS.
- D. Weiser, L. C. Bencze, G. Bánóczi, F. Ender, R. Kiss, E. Kókai, A. Szilágyi, B. G. Vértessy, Ö. Farkas, C. Paizs and L. Poppe, ChemBioChem, 2015, 16, 2283–2288 CrossRef CAS PubMed.
- H. A. Cooke, C. V. Christianson and S. D. Bruner, Curr. Opin. Chem. Biol., 2009, 13, 460–468 CrossRef CAS PubMed.
- S. Strom, U. Wanninayake, N. D. Ratnayake, K. D. Walker and J. H. Geiger, Angew. Chem., Int. Ed., 2012, 51, 2898–2902 CrossRef CAS PubMed.
- G. G. Wybenga, W. Szymanski, B. Wu, B. L. Feringa, D. B. Janssen and B. W. Dijkstra, Biochemistry, 2014, 53(19), 3187–3198 CrossRef CAS PubMed.
- C. V. Christianson, T. J. Montavon, G. M. Festin, H. A. Cooke, B. Shen and S. D. Bruner, J. Am. Chem. Soc., 2007, 129, 15744–15745 CrossRef CAS PubMed.
- S. Pilbák, Ö. Farkas and L. Poppe, Chem. – Eur. J., 2012, 18, 7793–7802 CrossRef PubMed.
- (a) D. Röther, L. Poppe, G. Morlock, S. Viergutz and J. Rétey, Eur. J. Biochem., 2002, 269, 3065–3075 CrossRef; (b) S. Pilbák, A. Tomin, J. Rétey and L. Poppe, FEBS J., 2006, 273(5), 1004–1019 CrossRef PubMed.
- J. D. Hermes, C. A. Roeske, M. H. O′Leary and W. W. Cleland, Biochemistry, 1982, 21, 5106–5114 CrossRef CAS PubMed.
- S. L. Lovelock, R. C. Lloyd and N. J. Turner, Angew. Chem., Int. Ed., 2014, 53, 4652–4656 CrossRef CAS PubMed.
- U. Wanninayake, Y. DePorre, M. Ondari and K. D. Walker, Biochemistry, 2011, 50, 10082–10090 CrossRef CAS PubMed.
- S. Bartsch and U. T. Bornscheuer, Protein Eng., Des. Sel., 2010, 23, 929–933 CrossRef CAS PubMed.
- R. Eisenthal, M. J. Danson and D. W. Hough, Trends Biotechnol., 2007, 25(6), 247–249 CrossRef CAS PubMed.
- A. Varga, G. Bánóczi, B. Nagy, L. C. Bencze, M. I. Toşa, Á. Gellért, F. D. Irimie, J. Rétey and L. Poppe, RSC Adv., 2016, 6, 56412–56420 RSC.
- B. Schuster and J. Rétey, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 8433–8437 CrossRef CAS.
- H. Liu and J. H. Naismith, BMC Biotechnol., 2008, 8, 91–101 CrossRef PubMed.
- N. A. Dima, A. Filip, L. C. Bencze, M. Oláh, P. Sátorhelyi, B. G. Vértessy, L. Poppe and C. Paizs, Stud. Univ. Babes-Bolyai Ser. Chem., 2016, 61, 21–34 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c7ob00562h |
| ‡ These authors contributed equally to this work |
| This journal is © The Royal Society of Chemistry 2017 |