
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards the biodegradation pathway of fosfomycin†
K.
Pallitsch
 *a,
A.
Schweifer‡
,
A.
Roller
b and
F.
Hammerschmidt
a
*a,
A.
Schweifer‡
,
A.
Roller
b and
F.
Hammerschmidt
a
aUniversity of Vienna, Institute of Organic Chemistry, Währingerstrasse 38, 1090, Vienna, Austria. E-mail: katharina.pallitsch@univie.ac.at; Tel: +43 (0) 1 4277 52123 or 52105
bUniversity of Vienna, Institute of Inorganic Chemistry, Währingerstrasse 42, 1090, Vienna, Austria
First published on 21st March 2017
Abstract
Three functionalised propylphosphonic acids were synthesised to study C–P bond cleavage in R. huakuii PMY1. (R)-1-Hydroxy-2-oxopropylphosphonic acid [(R)-5] was prepared by chiral resolution of (±)-dimethyl 1-hydroxy-2-methylallyllphosphonate [(±)-12], followed by ozonolysis and deprotection. The N-(L-alanyl)-substituted (1R,2R)-2-amino-1-hydroxypropylphosphonic acid 10, a potential precursor for 2-oxopropylphosphonic acid (5) in cells, was obtained by coupling the aminophosphonic acid with benzotriazole-activated Z-L-alanine and hydrogenolytic deprotection. (1R*,2R*)-1,2-Dihydroxy-3,3,3-trifluoropropylphosphonic acid, a potential inhibitor of C–P bond cleavage after conversion into its 2-oxo derivative in the cell, was accessed from trifluoroacetaldehyde hydrate via hydroxypropanenitrile 21, which was silylated and reduced to the aldehyde (±)-23. Diastereoselective addition of diethyl trimethylsilyl phosphite furnished diastereomeric α-siloxyphosphonates. The less polar one was converted to the desired racemic phosphonic acid (±)-(1R*,2R*)-9 as its ammonium salt.
Introduction
Phosphonates are characterised by a direct covalent phosphorus to carbon bond, making them extremely stable against enzymatic cleavage, while at the same time being the reason for their potent bioactivity.1,2 Their favourable properties led to the use of both synthetic and biogenic phosphonates in medicine and agriculture. Among the economically most important phosphonates phosphinothricin (1),3 used as a herbicide and fosfomycin (2),4 utilised as a clinical antibiotic should be mentioned (Fig. 1).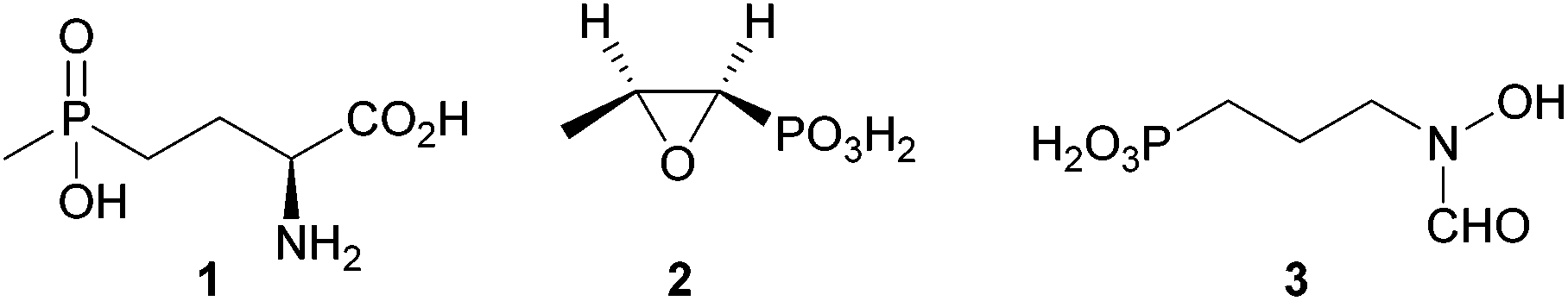 | ||
| Fig. 1 Some biogenic phosphonates of agricultural and medicinal importance. | ||
Fosmidomycin (3) is clinically evaluated for the treatment of malaria alone and in combination therapy, but has not yet reached the market.5 Despite these useful properties, phosphonates were thought to have little environmental importance for a long time. Lately, phosphonate catabolism in marine environments was shown to heavily influence the global phosphate and carbon cycle,6 contradicting this theory. Biodegradation of methylphosphonic acid was shown to be the major source of methane under aerobic conditions in ocean surface waters. In general, phosphonate degradation was shown to be quite common among microbes: studies on the distribution of the genes responsible for phosphonate breakdown in marine environments showed 40% of the studied bacterial genomes to contain one or more pathways for phosphonate degradation.7
This is not surprising regarding the low solubility of inorganic phosphate at ambient temperature and neutral pH8 and the fact that phosphorus is a vital nutrient for all life forms. Dissolved organic phosphates (DOP) often represent the only available source of phosphate in marine ecosystems. Therefore, nature evolved mechanisms to use them as a source of inorganic phosphate. DOPs mainly consist of phosphate esters (which are easy to hydrolyse), but also contain about 25% phosphonates, which can be used as a phosphate source too.9 Very specialised strategies are necessary to cleave the intrinsic C–P bond. This seemingly simple transformation is always the result of very unusual enzymatic pathways, of which three general types are known at present.2,10 While the most important pathway, called the C–P lyase pathway, follows a radical mechanism and has a very broad substrate scope, the two other known pathways (hydrolytic cleavage and oxidative cleavage) have a very narrow substrate specificity. There is also striking evidence for the existence of other, until now unknown pathways.11
In this article we describe the synthesis of three interesting target structures (Fig. 2), which will be used to study the catabolism of fosfomycin by Rhizobium huakuii PMY1, which is a microorganism that is able to use fosfomycin as the sole phosphorus and carbon source.12
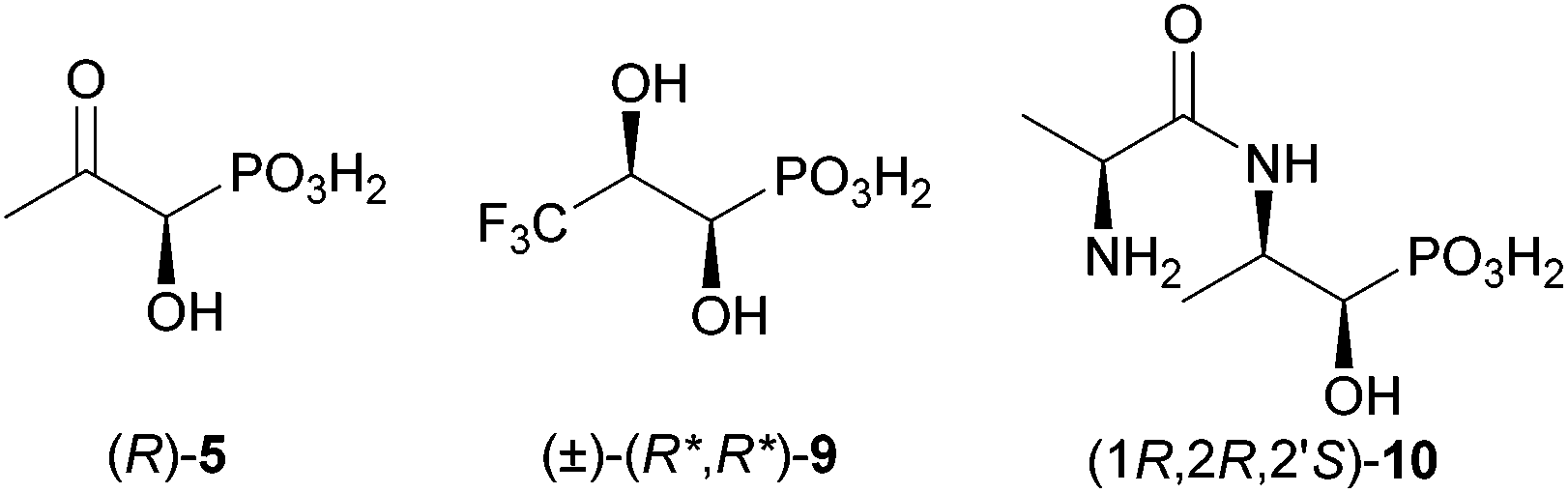 | ||
| Fig. 2 Target structures to study the biodegradation of fosfomycin. | ||
We focus on the possibility of hydrolytic cleavage of the C–P bond by attack of an active site nucleophile of a hydrolase at the phosphorus atom after activation. This hypothesis is based on the findings of Quinn et al. who reported the isolation of Rhizobium huakuii PMY1.12 To unravel the stepwise mineralisation of fosfomycin, they prepared putative intermediates of the catabolic pathway, among those also some isotopically labelled compounds.13,14 The synthesised compounds were tested as phosphorus/carbon source for the bacterium. Furthermore, cell lysates were incubated with the compounds.
These initial studies revealed the first aspects of fosfomycin metabolism by R. huakuii PMY1. It was shown that the first enzymatic step in its breakdown is a region- and stereospecific net opening of the epoxide ring by water at C-2 to give (1R,2R)-1,2-dihydroxypropylphosphonic acid (4) (Scheme 1).
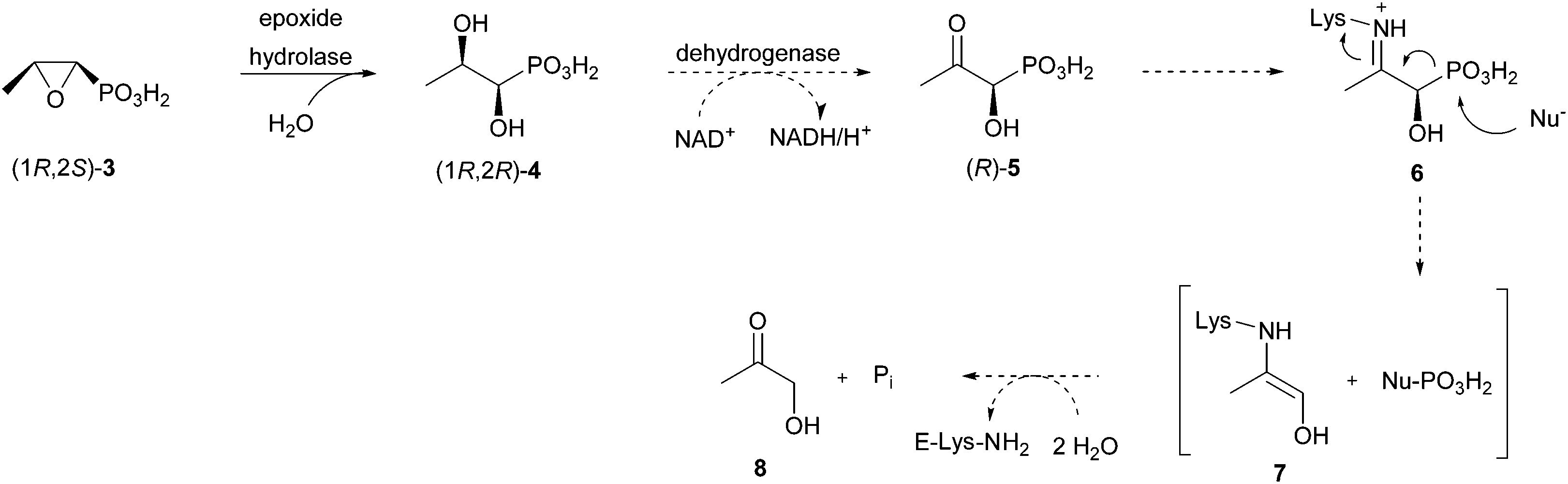 | ||
| Scheme 1 Proposed biodegradation of fosfomycin by R. huakuii PMY1. Known conversions are denoted with full arrows, assumed transformations with dashed arrows. | ||
The detailed mechanism for this process remains unknown. Of all other tested compounds none supported the growth of a culture of R. huakuii PMY1, except hydroxyacetone (8). However, in vitro C–P bond cleavage activity was detected with racemic 1-hydroxy-2-oxopropylphosphonic acid (5, see Fig. 2 for the (R)-enantiomer).14 The experiments with the labelled compounds support the hypothesis that 4 is oxidised to the corresponding 2-oxo compound 5.13 As it has a carbonyl group in the β-position to the phosphonic group, a hydrolytic mechanism for the cleavage of the C–P bond is indicated.7 This hypothesis forms the basis of our synthetic experiments.
Results and discussion
Proposed biodegradation pathway for fosfomycin
Based on the findings from the above described studies,13,14 we suggest a hypothetical biodegradation route for fosfomycin in Rhizobium huakuii PMY1 (Scheme 1) following a hydrolytic mechanism. We assume that dihydroxypropylphosphonic acid (R,R)-4 is formed by opening of the epoxide ring by an NAD+ dependent dehydrogenase to give (R)-1-hydroxy-2-oxopropylphosphonic acid [(R)-5]. The subsequent C–P bond cleavage yielding hydroxyacetone and phosphate is presumed to involve iminium ion 6. The latter is generated as an intermediate from the ω-amino group of an active site lysine of the putative hydrolase and the carbonyl group of (R)-5. This iminium ion is reminiscent of a similar one detected during the C–P bond cleavage of phosphonoacetaldehyde by phosphonatase.15Here we present the synthesis of three compounds that can be used to study the final C–P bond cleavage step in the catabolism of fosfomycin by R. huakuii PMY1.
In previous studies a cell lysate released inorganic phosphate from racemic 1-hydroxy-2-oxopropylphosphonic acid [(±)-5], indicating conversion by an enzyme of R. huakuii PMY1.14 However, (±)-5 did not support the growth of this bacterium. As the (R)-enantiomer is most probably formed in vivo from diol 4 by a dehydrogenase, it was assumed that either cellular uptake did not take place or that the (S)-enantiomer acts as a growth inhibitor.14 Therefore, both enantiomers of 5 will be needed to test them with a cell lysate and a growing culture of the bacterium separately. It is noteworthy that hydroxypropylphosphonate epoxidase (HppE) catalysing the final step of fosfomycin biosynthesis transforms both diastereomeric (1R,2R/S)-1-hydroxy-2-aminopropylphosphonic acids into (R)-5in vitro.16
An alternative to (R)-5 would be the use of a pro-metabolite. We reasoned that (R,R)-2-amino-1-hydroxypropylphosphonic acid [(R,R)-16] could be a potential precursor for (R)-5, as it could be converted by a transaminase. No phosphate was released from (R,R)-16 (for the structure see Scheme 5) as the sole carbon and phosphorus source in growth experiments.17 However, there was a small detectable C–P cleavage activity with cell-extracts. It is likely that the putative precursor did not enter the bacterial cells. Thus, we envisaged a potential biochemical precursor, which will possibly be incorporated more easily by R. huakuii PMY1. Bacteria contain a number of transporters for amino acids, di-, tri- and oligopeptides. For example, the phosphonic acid analogue of L-alanine, a bactericidal compound, was incorporated into di-, tri- and tetrapeptides to extensively study its facilitated uptake and release by bacteria.18 Also, phosphinothricin (1), a widely used herbicide, easily enters bacterial cells as the tripeptide bialaphos with two L-alanines attached to the amino group.19 This tripeptide is split inside the cells by a peptidase to release phosphinothricin, the inhibitor of glutamine synthetase. Similarly, to facilitate the uptake of (R,R)-16 by R. huakuii PMY1,20 it was converted to its dipeptide (1R,2R,2′S)-10 with alanine (Fig. 2). For simplicity, we started with the most simple pro-metabolite, a dipeptide. However, in case this is not incorporated by R. huakuii PMY1, we will aim for the corresponding tripeptide.
As described above, we presume that the cleavage of the C–P bond of 1-hydroxy-2-oxopropylphosphonic acid (5) involves the formation of an imine (see Scheme 1). The latter is probably protonated (6) to facilitate the attack of an active site nucleophile at the phosphorus atom (7) in analogy to phosphonatase-catalysed reactions.21 Thus, C–P bond cleavage is induced by the formation of a Schiff's base. To prove this hypothesis, we decided to prepare a putative enzyme inhibitor. We assume that 3,3,3-trifluoro-1-hydroxy-2-oxopropylphosphonic acid (26, Scheme 2) would allow formation of a similar imine as in the case of the nonfluorinated analogue. However, the resulting Schiff's base (27) will be too electron deficient at the nitrogen (because of the electron withdrawing effect of the CF3 group) to be protonated. The putative hydrolase, covalently modified at the active site lysine, will be blocked. Knowing that (1R,2R)-dihydroxypropylphosphonic acid [(1R,2R)-4] is taken up by R. huakuii PMY1 and metabolised, we opted for the synthesis of its racemic 3,3,3-trifluoromethyl analogue (9) as a stable precursor for 3,3,3-trifluoro-1-hydroxy-2-oxopropylphosphonic acid (26). Oxidation of 9 inside the cell is proposed to occur upon the action of the same dehydrogenase, which is proposed to convert 4 to 5 (compare Scheme 1 and 2).
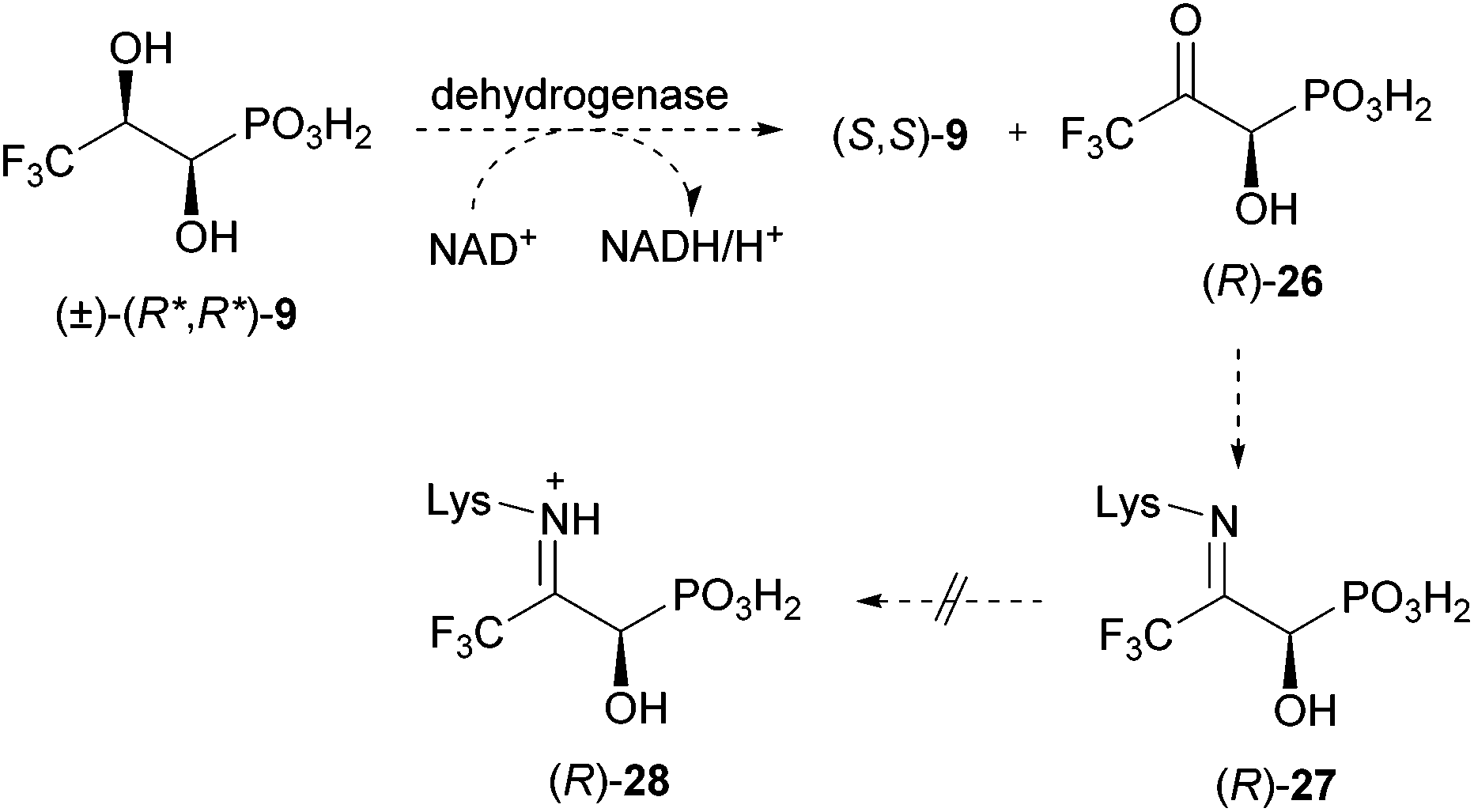 | ||
| Scheme 2 Proposed inhibition mode of fosfomycin catabolism by (±)-(R*,R*)-9 in R. huakuii PMY1. | ||
If the enzyme performing cleavage of the C–P bond is blocked, accumulation of (R)-3,3,3-trifluoro-1-hydroxy-2-oxopropylphosphonic acid (26) would be a possible detectable consequence. This would be additional information pointing towards the presented mechanism of C–P bond cleavage as the final steps in fosfomycin degradation by R. huakuii PMY1.
(R)- and (S)-1-Hydroxy-2-oxopropylphosphonic acid
Here we describe only the synthesis of the (R)-enantiomer of 5, as the (S)-enantiomer can be prepared similarly just before use. The synthesis of (R)-1-hydroxy-2-oxopropylphosphonic acid [(R)-5] was accomplished in 5 steps from racemic dimethyl 1-hydroxy-2-methylallylphosphonate [(±)-12], which was obtained by a literature procedure from methacrolein and dimethyl phosphite (Scheme 3).14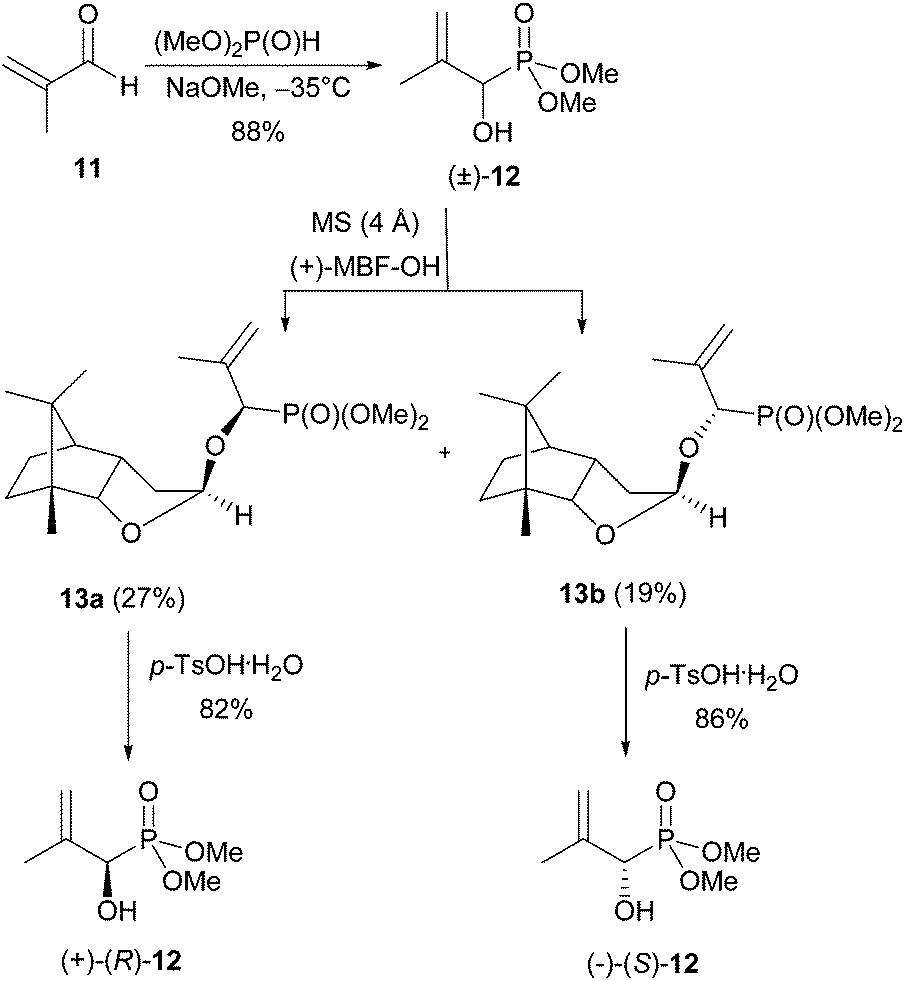 | ||
| Scheme 3 Chiral resolution of (±)-12 using Noe's lactol. | ||
The crude racemate (±)-12 was resolved into its enantiomers using Noe's lactol,22 which reacts with alcohols in the presence of a catalytic amount of pTSA.23 Here, a mixture of three diastereomeric acetals was formed. The major components were the two axially22 substituted acetals 13a and 13b, which are favoured due to the anomeric effect. They were separated by flash chromatography. Only one equatorially substituted acetal was observed in trace amounts. It was easily removed from the major ones.
The yield of the two main products varied slightly depending on whether Noe's lactol was used in its monomeric [(+)-MBF-OH] or dimeric [(+)-MBF-O-MBF] form for the derivatisation. In general the monomer gave slightly higher yields (27 and 19% isolated yield for 13a and 13b, respectively). The enantiomeric hydroxyphosphonates (+)- and (−)-12 were then obtained by deprotection of the two acetals 13a and 13b, with pTSA·H2O in a mixture of methanol and CH2Cl2 (82% and 86% yield).
Esterification of small samples of the enantiomers with (S)-Mosher chloride showed them to be enantiomerically pure (ee > 99%). As the phosphorus atom of the (R)-Mosher ester derived from enantiomer (+)-12 resonated at higher field in the 31P NMR spectrum than that derived from (−)-12, (+)-12 has (R)-configuration.24 The levorotary enantiomer, derived from 13b has therefore (S)-configuration. Hydroxyphosphonate (R)-12 was deprotected at the phosphorus atom at 0 °C using TMSBr in combination with allyltrimethylsilane (Scheme 4) followed by an aqueous workup with neutralisation and lyophilisation.
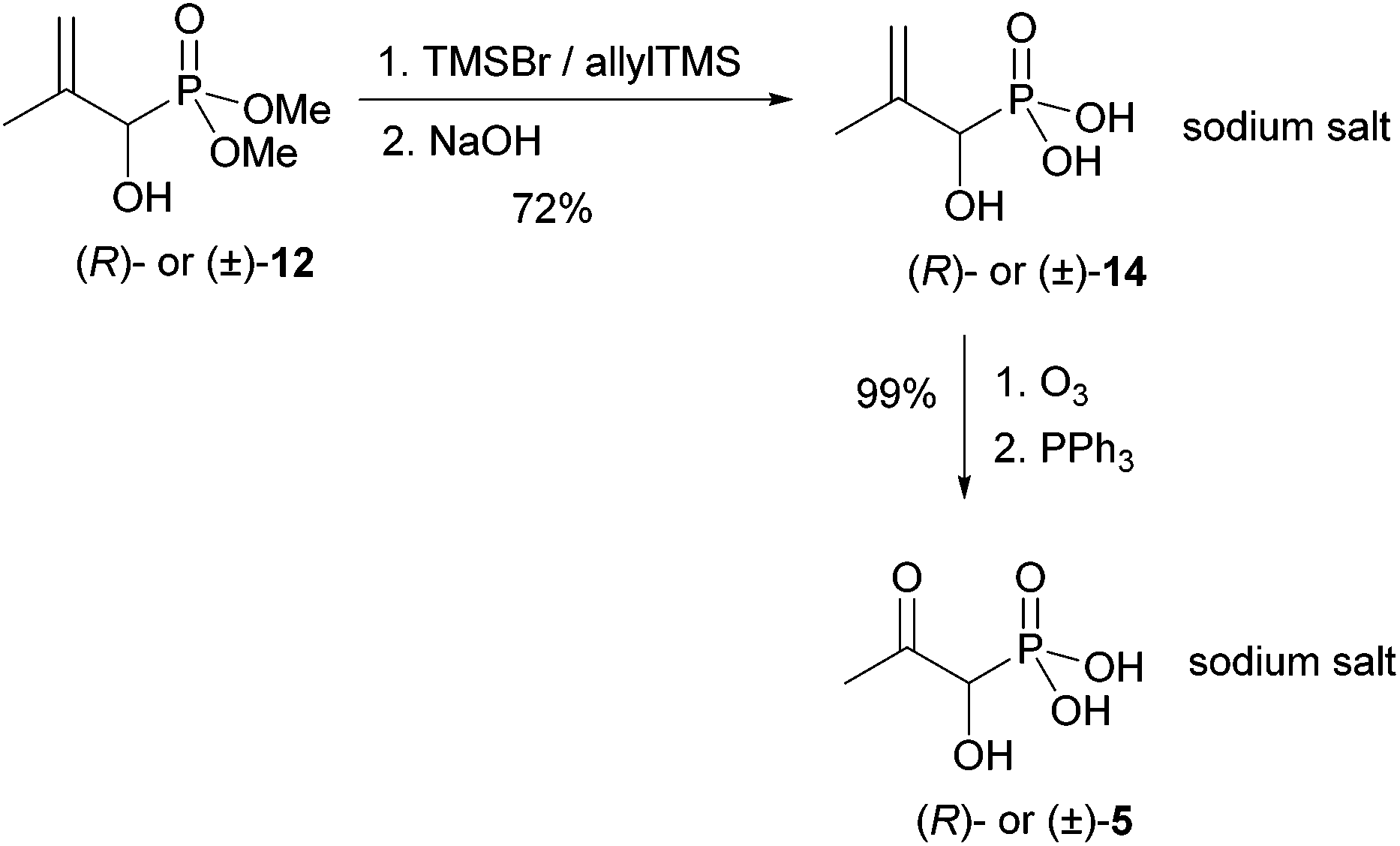 | ||
| Scheme 4 Final transformations for the synthesis of (R)- and (±)-5. | ||
This furnished the sodium salt of (R)-1-hydroxy-2-methylallylphosphonic acid [(R)-14] in 72% yield. This sodium salt of (R)-14 was ozonolysed at −78 °C. Lyophilisation gave the desired sodium salt of (R)-5 as a colourless powder in quantitative yield. Samples of the racemic product (±)-5 were obtained similarly for comparative purposes.
As the sodium salts of (R)- and (S)-1-hydroxy-2-oxopropylphosphonic acid [(R)- and (S)-5] will be used for biodegradation studies, their stability towards racemisation in aqueous media at neutral and basic pH was tested. A sample of racemic (±)-5 was dissolved in D2O and the hydrogen/deuterium exchange at C-1 was monitored as a function of time at neutral and basic pH. Surprisingly, racemisation proceeded very slowly at pH 7 at ambient temperature (10–12% during the first 24 h, complete H/D-exchange within 5 days). However, racemisation was faster (50% H/D exchange after 6 h at room temperature) at pH > 8.5.
Further, the stability of the P–C bond was tested at acidic pH.25 A sample of oxophosphonate 5 was dissolved in D2O and acidified to pH 4.5 by addition of DCl/D2O. The phosphonate/phosphate ratio was determined by 31P NMR spectroscopy, which showed formation of less than 5% phosphate from the phosphonate within the first 24 h. These findings suggest that the enantiomers of 1-hydroxy-2-oxopropylphosphonic acid (5) are stable for biological experiments at or around pH 7.
A phosphonic acid dipeptide
The phosphonodipeptide (1R,2R,2′S)-10 is easily accessible from commercially available fosfomycin (2) (as its disodium salt) in three steps (Scheme 5).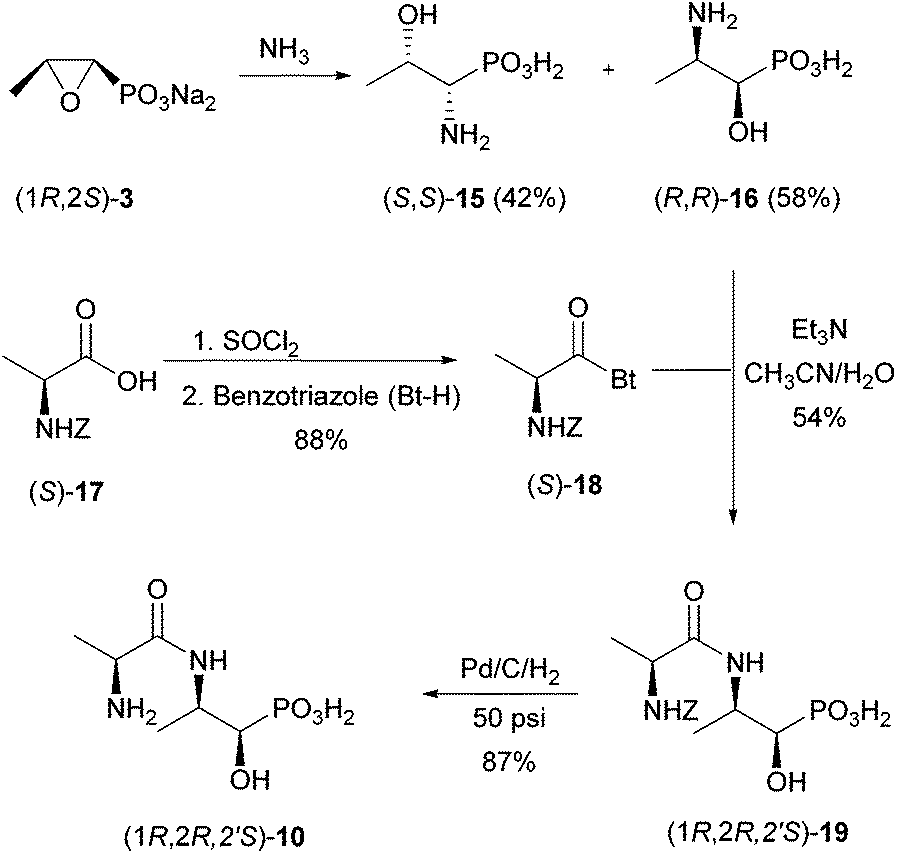 | ||
| Scheme 5 Synthesis of phosphonodipeptide (1R,2R,2′S)-10. | ||
Ammonolysis of fosfomycin at 60 °C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 delivered a mixture of isomeric aminophosphonates (S,S)-15 and (R,R)-16. The needed regioisomer (R,R)-16, which could be easily separated from (S,S)-15 by ion exchange chromatography, was coupled with benzotriazole-activated, Z-protected L-alanine [(S)-18].27 The latter was prepared from Z-L-alanine and benzotriazole in 88% yield.
26 delivered a mixture of isomeric aminophosphonates (S,S)-15 and (R,R)-16. The needed regioisomer (R,R)-16, which could be easily separated from (S,S)-15 by ion exchange chromatography, was coupled with benzotriazole-activated, Z-protected L-alanine [(S)-18].27 The latter was prepared from Z-L-alanine and benzotriazole in 88% yield.
Aminophosphonic acid (R,R)-16 and activated L-alanine (S)-18 were coupled in a mixture of CH3CN/H2O (7:3) in the presence of one equivalent of Et3N to give the Z-protected dipeptide 19 in 54% yield. An unidentified by-product and unreacted aminophosphonic acid were detected in the reaction mixture. The unknown by-product could neither be removed from the Z-protected dipeptide 19 nor could its formation be suppressed.
Finally, the Z-group was removed hydrogenolytically at 50 psi using Pd on charcoal (10% Pd) as a catalyst to give (1R,2R,2′S)-10 as a white solid (87% yield). Crystallisation of the dipeptide from H2O/MeOH removed the major portion of all impurities at the expense of yield.
A potential trifluorinated inhibitor of P–C bond cleavage in fosfomycin biodegradation
Racemic (R*,R*)-3,3,3-trifluoro-1,2-dihydroxypropylphosphonic acid [(±)-(R*,R*)-9] was prepared from commercially available trifluoroacetaldehyde hydrate (20) and NaCN in six steps (Scheme 6).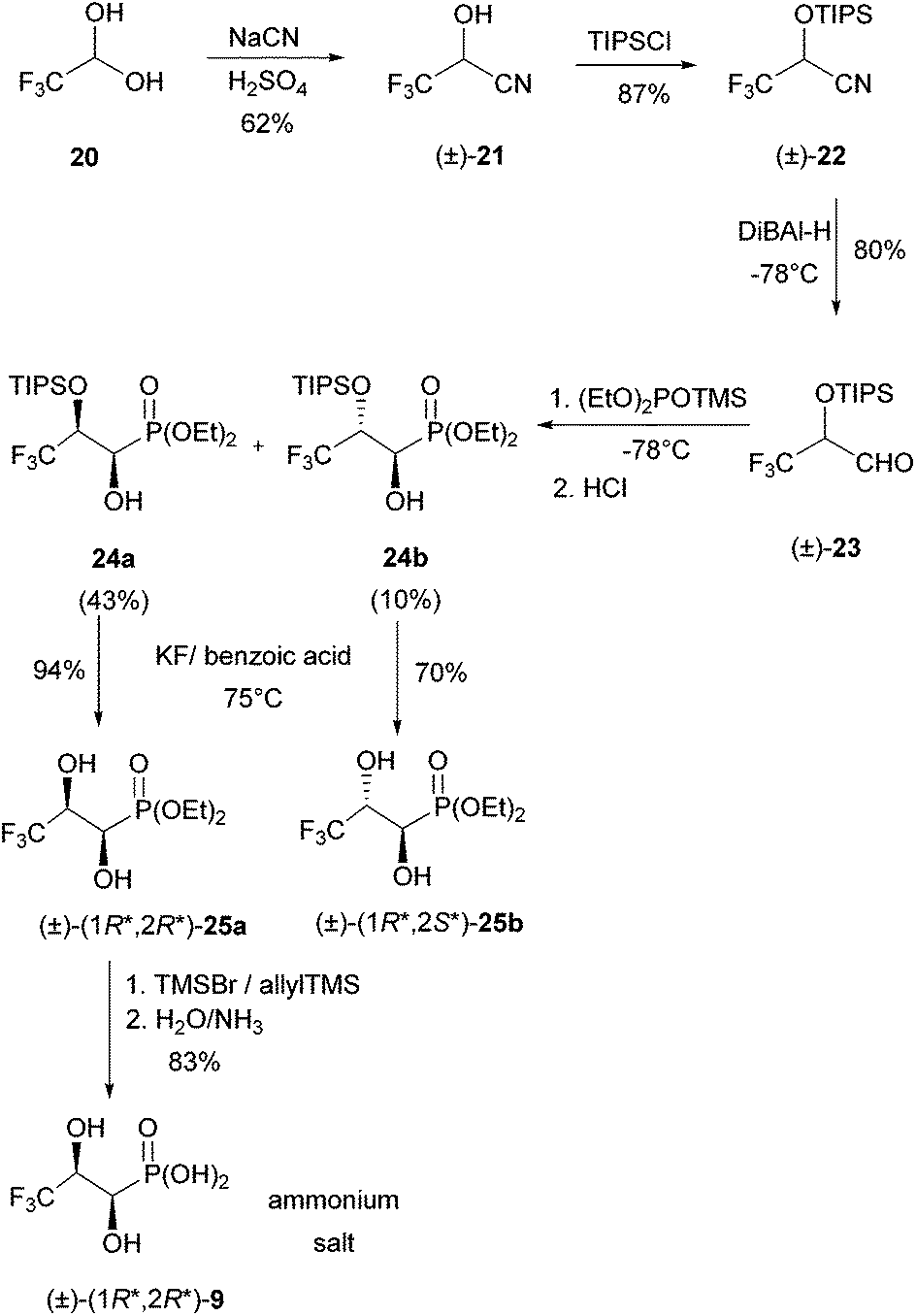 | ||
| Scheme 6 Synthetic sequence towards (±)-(1R*,2R*)-9. | ||
This dihydroxyphosphonate has already been synthesised by dihydroxylation of diethyl E-3,3,3-trifluoroprop-1-enylphosphonate with OsO428 or KMnO4.29 We developed this new access as it will allow the synthesis of the compound labelled at C-1 with 13C derived from Na13CN. First sodium cyanide was added to trifluoroacetaldehyde at 0 °C under acidic conditions. The crude, racemic trifluorinated hydroxynitrile (±)-21 was distilled (62% yield, as judged by 1H NMR spectroscopy) and used for the next step immediately. Due to the low stability of hydroxynitrile (±)-21, the hydroxyl group was protected using chlorotriisopropylsilane/imidazole. Protection proceeded in 87% yield and the resulting product (±)-22 was purified by flash chromatography and stored at room temperature. The TIPS-protected hydroxynitrile was subsequently reduced to the corresponding aldehyde 23 with DiBAl-H at −78 °C followed by acidic workup. The isolated, crude aldehyde 23 was not purified by flash chromatography, as it decomposed on silica gel. Therefore, diethyl trimethylsilyl phosphite was added to a solution of the aldehyde at −78 °C (Abramov reaction). The reaction mixture was allowed to warm to room temperature overnight and the TMS groups of the resulting α-(trimethylsiloxy)phosphonates were removed by addition of a few drops of concentrated HCl to the ethanolic reaction mixture. A mixture of two racemic, diastereomeric phosphonates, 24a (43%, less polar) and 24b (10%, more polar) resulted, which was easily separable by column chromatography. The configurations of 24a and 24b could be assigned after TIPS deprotection (see the following paragraph).
Attempted removal of the TIPS group from the less polar (and major) phosphonate 24a with concentrated HF (40%) in acetonitrile caused its decomposition. Therefore, desilylation was performed under milder conditions with in situ generated HF from a mixture of potassium fluoride and benzoic acid in acetonitrile at 75 °C for 24 h. The diol (1R*,2R*)-25a could be purified by flash chromatography to give a colourless, crystalline solid.
The more polar (and minor) phosphonate 24b was deprotected similarly. The obtained diol 25b could be crystallised from heptanes/dichloromethane to give colourless crystals, which were suitable for X-ray crystallography. It showed that the two hydroxyl groups are anti-orientated (see the ESI†). This findings allowed assignment of the (1R*,2R*)-configuration to 24a and the (1R*,2S*)-configuration to 24b as given in Scheme 6. Satisfyingly, the major isomer 25a is the one needed for studying C–P bond cleavage. Only this phosphonate was deprotected using bromotrimethylsilane and allyltrimethylsilane in 1,2-dichloroethane at room temperature over a period of three days. Aqueous workup with the addition of NH3, followed by lyophilisation gave the ammonium salt of (1R*,2R*)-3,3,3-trifluoro-1,2-dihydroxypropylphosphonic acid [(1R*,2R*)-9], in 83% yield.
Experimental section
General experimental
1H, 13C and 31P NMR spectra were recorded in CDCl3, d4-methanol, d8-toluene or D2O on a Bruker Avance DRX 400 (1H: 400.13 MHz, 13C: 100.61 MHz, 31P: 161.98 MHz), AV 400 (1H: 400.27 MHz, 13C: 100.65 MHz, 31P: 162.03 MHz) or AV600 Avance 3 (1H: 600.25 MHz, 13C: 150.95 MHz, 31P: 242.97 MHz) spectrometer. Proton NMR chemical shifts were referenced to residual CHCl3 (δH 7.24), CHD2OD (δH 3.31), C6D5CD2H (δH 2.09) or HOD (δH 4.80). 13C NMR spectra were referenced to CDCl3 (δC 77.23), CD3OD (δC 49.15) and C6D5CD3 (δC 21.4); 31P NMR spectra to external H3PO4 (85%) (δP 0.0). Chemical shifts (δ) are given in ppm and coupling constants (J) in Hz. IR spectra were run on a Bruker VERTEX 70 IR spectrometer in ATR mode or from solution on a silicon disc.30 Optical rotations were measured at 20 °C on a PerkinElmer 341 polarimeter in a 1 dm cell. [α]D values are given in 10−1 deg cm2 g−1. Melting points were determined on a Leica Galen III Reichert Thermovar instrument and were uncorrected.TLC was carried out on 0.25 mm thick Merck plates coated with silica gel 60 F254. Spots were visualised by UV light and/or dipping the plate into a solution of (NH4)6Mo7O24·4H2O (25.0 g) and Ce(SO4)2·4H2O (1.0 g) in aqueous 10% H2SO4 (500 mL), followed by heating with a heat gun. In case an amino-functionality was present in the molecule, the spots were visualised by dipping the plate into a solution of ninhydrin (0.2 w/v%) in ethanol followed by heating with a heat gun. Flash (column) chromatography was performed with Merck silica gel 60 (230–400 mesh).
Preparation of the sodium salt of (R)-1-hydroxy-2-oxopropylphosphonic acid [(R)-5]
:1), filtered and dried in vacuo to yield α-hydroxyallylphosphonate (±)-12 (1.579 g, 88%) as a colourless oil. The product was used for the following reaction without further purification. The recorded spectra are identical to those reported in the literature;14δP (161.98 MHz, CDCl3) 25.2 (s).
:1 → ethyl acetate, Rf (13a, ethyl acetate–hexanes 1:1) 0.36 and Rf (13b, ethyl acetate–hexanes 1:1) 0.29] to yield the two diastereomeric acetals 13a (1.452 g, 27%) and 13b (1.005 g, 2.8 mmol, 19%) as colourless oils.
The acetalisation with Noe's lactol dimer [(+)-MBF-OH dimer] can be performed similarly, except that only 0.5 equivalents of the dimer were used relative to hydroxyphosphonate (±)-12.
Diastereomer 13a. [α]20D +130.77 (c 0.91, acetone). Found: C, 60.32; H, 8.72; P 8.64. Calc. for C18H31O5P: C, 45.49; H, 8.11; P 8.64. νmax/cm−1 2954, 1451, 1262, 1181, 1040; δH (400.27 MHz, d8-toluene) 5.17 (1 H, broadened d, JHP 4.7, O–CH–O), 5.15–5.10 (1 H, m, CH2
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 5.04–5.00 (1 H, m, CH2), 4.68 (1 H, d, JHH 18.7, CH–O), 4.43 (1 H, dd, JHP 9.5, JHH 1.6, CH–P), 3.53 (3 H, d, JHP 10.4, O–CH3), 3.48 (3 H, d, JHP 10.5, O–CH3), 2.88–2.77 (1 H, m, CH), 1.96–1.91 (3 H, m, CH3–C(CH2)–), 1.90–1.82 (1 H, m, CH), 1.76–1.61 (2 H, m, 2 × CH), 1.50–1.35 (2 H, m, 2 × CH), 1.27 (1 H, ddd, JHH 13.5, JHH 9.2, JHH 4.2, CH), 1.11 (1 H, tdd, JHH 12.2, JHH 3.8, JHH 1.5, CH), 0.90 (3 H, s, CH3), 0.89 (3 H, s, CH3), 0.81 (3 H, s, CH3); δC (100.61 MHz, d8-toluene) 140.9 (s, Cq), 117.0 (d, JCP 12.2, CH2), 106.8 (d, JCP 12.2, O–CH–O), 90.9 (s, CH–O), 75.5 (d, JCP 169.1, CH–P), 54.1 (d, JCP 6.7, P–OCH3), 53.8 (s, Cq), 53.4 (d, JCP 6.3 Hz, P–OCH3), 49.8 (s, Cq), 48.7 (s, CH), 41.5 (s, CH), 33.4 (s, CH2), 27.8 (s, CH2), 21.9 (s, CH3), 21.8 (s, CH2), 20.6 (s, CH3), 19.7 (s, CH3), 15.9 (s, CH3); δP (162.03 MHz, d8-toluene) 22.0 (s).
), 5.04–5.00 (1 H, m, CH2), 4.68 (1 H, d, JHH 18.7, CH–O), 4.43 (1 H, dd, JHP 9.5, JHH 1.6, CH–P), 3.53 (3 H, d, JHP 10.4, O–CH3), 3.48 (3 H, d, JHP 10.5, O–CH3), 2.88–2.77 (1 H, m, CH), 1.96–1.91 (3 H, m, CH3–C(CH2)–), 1.90–1.82 (1 H, m, CH), 1.76–1.61 (2 H, m, 2 × CH), 1.50–1.35 (2 H, m, 2 × CH), 1.27 (1 H, ddd, JHH 13.5, JHH 9.2, JHH 4.2, CH), 1.11 (1 H, tdd, JHH 12.2, JHH 3.8, JHH 1.5, CH), 0.90 (3 H, s, CH3), 0.89 (3 H, s, CH3), 0.81 (3 H, s, CH3); δC (100.61 MHz, d8-toluene) 140.9 (s, Cq), 117.0 (d, JCP 12.2, CH2), 106.8 (d, JCP 12.2, O–CH–O), 90.9 (s, CH–O), 75.5 (d, JCP 169.1, CH–P), 54.1 (d, JCP 6.7, P–OCH3), 53.8 (s, Cq), 53.4 (d, JCP 6.3 Hz, P–OCH3), 49.8 (s, Cq), 48.7 (s, CH), 41.5 (s, CH), 33.4 (s, CH2), 27.8 (s, CH2), 21.9 (s, CH3), 21.8 (s, CH2), 20.6 (s, CH3), 19.7 (s, CH3), 15.9 (s, CH3); δP (162.03 MHz, d8-toluene) 22.0 (s).
Diastereomer 13b. [α]20D +98.80 (c 0.92, acetone). νmax/cm−1 2954, 1458, 1259, 1182, 1099, 1033, 985; δH (400.27 MHz, d8-toluene) 5.65 (1 H, d, JHP 4.8, O–CH–O), 5.23–5.18 (1 H, m, CH2
), 4.97–4.90 (1 H, m, CH2), 4.49 (1 H, d, JHH 13.3, CHO), 4.25 (1 H, dd, JHP 9.5, JHH 1.5, CH–P), 3.49 (3 H, d, JHP 10.3, O–CH3), 3.47 (3 H, d, JHP 10.3 Hz, O–CH3), 2.86–2.76 (1 H, m, CH), 1.97–1.92 (3 H, m, CH3C(CH2)–), 1.90–1.67 (3 H, m, CH3), 1.47–1.33 (2 H, m, 2 × CH), 1.32–1.22 (1 H, m, CH), 1.15–1.05 (1 H, m, CH), 0.85 (3 H, s, CH3), 0.83 (3 H, s, CH3), 0.78 (3 H, s, CH3); δC (100.61 MHz, d8-toluene) 142.6 (d, JCP 1.7, Cq), 114.7 (d, JCP 11.0, CH2), 110.4 (d, JCP 5.5, O–CH–O), 91.3 (s, CH–O), 76.7 (d, JCP 161.8, CH–P), 53.721 (2 C, d, JCP 6.9, 2 × O–CH3), 53.720 (s, Cq), 53.71 (d, JCP 6.9, O–CH3), 49.82 (s, Cq), 48.79 (s, CH), 41.5 (s, CH), 33.8 (s, CH2), 27.8 (s, CH2), 21.9 (s, CH3), 21.8 (s, CH2), 21.0 (s, CH3), 19.8 (s, CH3), 15.9 (CH3); δP (162.32 MHz, d8-toluene) 22.6 (s).
(−)-12 was prepared similarly starting from diastereomer 13b (1.005 g, 2.8 mmol) as a colourless solid (0.424 g, 86%); mp 47–49 °C (from diisopropyl ether); [α]20D −2.80 (c 0.965 in acetone), [α]20365–9.95 (c 0.965 in acetone). The measured spectroscopic data of both enantiomers were identical to those of the racemic compound.
The absolute configuration of the obtained enantiomeric α-hydroxyphosphonates was determined by esterification using (S)-Mosher chloride, following a literature procedure to give the corresponding (R)-Mosher esters.24 Relevant NMR signals are denoted below for both diastereomeric Mosher esters.
CH2)–); δP (161.98 MHz, CDCl3) 19.27 (s).
CH2)–); δP (161.98 MHz, CDCl3) 19.73 (s).
These NMR data allow assignment of the (R)-configuration to (+)-12 and (S) to (−)-12.
), 5.04 (1 H, br. s, CH2), 4.28 (1 H, d, JHP 13.7, CH–P), 1.90 (3 H, s, CH3); δC (100.65 MHz, D2O) 143.8 (d, JCP 3.5, Cq), 112.3 (d, JCP 9.8, CH2), 73.9 (d, JCP 149.7, CH–P), 19.2 (d, JCP 1.5, CH3); δP (162.02 MHz, D2O) 16.4 (s).
O), 80.0 (d, JCP 120.4, CH–P), 27.1 (s, CH3); δP (162.03 MHz, D2O) 9.4 (s, 93 mol%, desired product), 3.7 (s, 2 mol%, unknown byproduct), 1.17 (s, 5 mol%, phosphate). The product has to be stored at −25 °C.
Preparation of (+)-N-[(S)-alanyl]-(1R,2R)-2-amino-1-hydroxypropylphosphonic acid [(1R,2R,2′S)-10]
:3:1; Rf (product) 0.64, Rf (unknown impurity) 0.70, Rf (16) 0.24, Rf (18) 0.90, Rf (benzotriazole) 0.87, Rf (17) 0.79).
The reaction mixture was concentrated in vacuo and dissolved in water (20 mL), extracted with CH2Cl2 (4 × 30 mL) and the aqueous phase was concentrated to about 5 mL under reduced pressure. The residual solution was applied to a column filled with Dowex 50 W × 8, H+ (90 mL) and eluted with water. The fractions containing the product (TLC: silica gel, eluent: iPrOH/H2O/NH3 (25 w/w%) 6:3:1, product moves with the front) were collected and concentrated in vacuo to give the crude Z-protected peptide 19 (0.389 g, 1.1 mmol, 54%; and two impurities of an unknown structure), which was used for the next step without further purification. δH (400.27 MHz, D2O) 7.55–7.36 (5 H, m, 5 × CH from Z), 5.18 (2 H, s, CH2), 4.33–4.08 (2 H, m, 2 × CH), 3.84 (1 H, br. s, CH), 1.40 (3 H, d, JHH 7.0, CH3 alanyl residue), 1.24 (3 H, d, JHH 5.3, CH3 phosphonic acid); δP (162.03 MHz, D2O) 19.6 (s, 91 mol%, product), 11.7 (s, 7 mol%, unknown impurity), 10.1 (s, 2 mol%, unknown impurity).
:1). The filtrate was concentrated in vacuo to give a white solid (0.196 g, 87%) which was purified by crystallisation. The product was suspended in methanol (2.0 mL) and drops of water were added until a clear solution was obtained, which was stored at 4 °C to give colourless crystals of the desired phosphonodipeptide (1R,2R,2′S)-10 [TLC: iPrOH/H2O/NH3 (25 w/w%) 6:3:1, Rf 0.35]; mp 256–260 °C (decomp.); [α]20D +21.57 (c 0.830 in H2O); νmax/cm−1 3264, 3121, 2813, 1666, 1576, 1503, 1447, 1131, 1056; δH (400.27 MHz, D2O) 4.34–4.23 (1 H, m, CH–C–P), 4.11 (1 H, q, JHH 7.1, CH of the alanyl residue), 3.76 (1 H, dd, JHP 10.1, JHH 4.7, CH–P), 1.56 (3 H, d, JHH 7.1, CH3 of the alanyl residue), 1.31 (d, JHH 6.8, CH3 of phosphonic acid); δC (100.65 MHz, D2O) 169.9 (s, CO), 70.8 (d, JCP 155.5 Hz, CH–P), 49.3 (s, CH–CO), 47.4 (d, JCP 3.6 Hz, CH–C–P), 17.3 (d, JCP 8.0, CH3 of phosphonic acid), 16.4 (s, CH3 of the alanyl residue); δP (162.03 MHz, D2O) 16.62 (s, 97 mol%, product), 15.53 (s, 3 mol%, unknown impurity).
Preparation of the ammonium salt of (±)-(1R*,2R*)-3,3,3-trifluoro-1,2-dihydroxypropylphosphonic acid [(±)-9]
:1, Rf 0.67) to give silyl ether (±)-22 (1.779 g, 6.32 mmol, 87%) as a colourless oil; found: C, 51.36; H, 7.92; N, 4.88. Calc. for C12H22F3NOSi: C, 51.22; H, 7.88; N, 4.98; νmax/cm−1 3407, 2949, 1703, 1347, 1266, 1204, 1153, 1109; δH (400.27 MHz, CDCl3) 4.88 (1 H, q, JHF 5.2, CH), 1.25–1.14 (3 H, m, 3 × (CH3)2CH–Si), 1.10, 1.08 (18 H, overlapping doublets, J 5.7, J 5.6, 6 × CH3 of TIPS); δC (100.65 MHz, CDCl3) 121.3 (q, JCF 283.0, CF3), 113.8 (s, CN), 63.3 (q, JCF 38.3, CH–CF3), 17.70 (3 C, s, 3 × CH3 of TIPS), 17.65 (3 C, s, 3 × CH3 of TIPS), 12.1 (3 C, s, 3 × (CH3)2CH–Si).
The diastereomers were separated by flash chromatography [hexanes–acetone 3:1, Rf (1R*,2R*-24a) 0.21, Rf (1S*,2R*-24b) 0.13] to yield (±)-(1R*,2R*)-24a (0.858 g, 43% calculated starting from 22) and (±)-(1S*,2R*)-24b (0.154 g, 10% calculated starting from 22).
(±)-(1R*,2R*)-24a. Mp 63–64 °C (from hexanes); found: C, 45.53; H, 8.05. Calc. for C16H34F3O5PSi: C, 45.49; H, 8.11; νmax/cm−1 3268, 2945, 2869, 1462, 1264, 1164, 1140, 1025, 971; δH (400.13 MHz, CDCl3) 4.63–4.53 (1 H, m, CF3CH), 4.25–4.11 (4 H, m, 2 × OCH2CH3), 4.07 (1 H, m, CH–P), 2.84 (1 H, d, JHH 9.8, OH), 1.33 (6 H, t, JHH 7.1, 2 × OCH2CH3), 1.28–1.17 (3 H, m, 3 × (CH3)2CHSi), 1.11 and 1.09 (18 H, 2 × d, JHH 3.5, 6 × CH3 of TIPS); δC (100.61 MHz, CDCl3) 124.3 (qd, JCF 284.9, JCP 21.9, CF3), 69.6 (qd, JCF 31.1, JCP 3.2, CF3–C), 66.1 (dd, JCP 169.3, JCF 1.9, CH–P), 63.6 (d, JCP 7.6, OCH2CH3), 62.8 (d, JCP 7.6, OCH2CH3), 18.3 (3 C, s, 3 × CH3 of TIPS), 18.1 (3 C, s, 3 × CH3 of TIPS), 16.7 (d, JCP 5.7, OCH2CH3), 16.6 (d, JCP 6.0, OCH2CH3), 13.0 (3 C, s, 3 × (CH3)2CHSi); δP (162.03 MHz, CDCl3) 20.9 (s).
(±)-(1S*,2R*)-24b. Mp 51–52 °C (from hexanes); found: C, 45.70; H, 7.97. Calc. for C16H34F3O5PSi: C, 45.49; H, 8.11; νmax/cm−1 3266, 2946, 2870, 1467, 1262, 1177, 1140, 1054, 1026; δH (400.13 MHz, CDCl3) 4.56–4.46 (1 H, m, CF3CH), 4.25–4.11 (5 H, m, 2 × OCH2CH3 and CH–P), 2.90 (1 H, br. s, OH), 1.33 (6 H, td, JHH 7.0, JHP 0.3, 2 × OCH2CH3), 1.20–1.03 (21 H, m, 3 × (CH3)2CHSi + 3 × (CH3)2CHSi); δC (100.61 MHz, CDCl3) 124.1 (qd, JCF 284.1, JCP 3.4, CF3), 72.3 (qd, JCF 30.9, JCP 6.2, CF3–C), 70.5 (d, JCP 161.1, CH–P), 63.4 (d, JCP 6.4, OCH2CH3), 63.3 (d, JCP 6.4, OCH2CH3), 18.1 (3 C, s, 3 × CH3 of TIPS), 18.0 (3 C, s, 3 × CH3 of TIPS), 16.6 (2 C, d, JCP 5.6, 2 × OCH2CH3), 12.7 (3 C, s, 3 × (CH3)2CHSi); δP (162.03 MHz, CDCl3) 19.9 (s).
(±)-(1R*,2R*)-25a. Mp 38–40 °C (from hexanes–CH2Cl2) (lit.,29 38 °C); found: C, 31.43; H, 5.18. Calc. for C7H14F3O5F3P: C, 31.59; H, 5.30; νmax/cm−1 3255, 2985, 1276, 1259, 1166, 1137, 1029; δH (400.13 MHz, CDCl3) 5.40 (1 H, br. s, OH), 4.75 (1 H, br. s, OH), 4.32–4.14 (6 H, m, 2 × OCH2CH3, CF3CH and CH–P), 1.34 (6 H, t, JHH 7.1, 2 × OCH2CH3); δC (100.61 MHz, CDCl3) 124.4 (qd, JCF 284.0, JCP 22.8, CF3), 69.3 (qd, JCF 31.1, JCP 3.1, CF3–C), 66.8 (dd, JCP 165.8, JCF 1.0, CH–P), 64.3 (d, JCP 7.0, OCH2CH3), 63.9 (d, JCP 7.4, OCH2CH3), 16.6 (d, JCP 5.3, OCH2CH3), 16.5 (d, JCP 5.3, OCH2CH3); δP (161.98 MHz, CDCl3) 21.8 (q, JPF 4.3).
(±)-(1R*,2S*)-25b. δ H (600.25 MHz, CDCl3) 4.68 (1 H, br. d, JHH 6.4, OH), 4.31–4.10 (6 H, m, 2 × OCH2CH3, CF3CH and CH–P), 3.33 (1 H, br. t, JHH 7.5, OH), 1.37 (3 H, t, JHP 7.1, OCH2CH3), 1.34 (3 H, t, JHP 7.1, OCH2CH3); δC (150.95 MHz, CDCl3), 124.3 (qd, JCF 282.8, JCP 14.5, CF3), 71.46 (qd, JCF 30.9, JCP 1.5, CF3–C), 67.11 (d, JCP 162.0, CH–P), 64.1 (d, JCP 7.1, OCH2CH3), 63.80 (d, JCP 7.1, OCH2CH3), 16.6 (d, JCP 5.7, OCH2CH3), 16.5 (d, JCP 5.8, OCH2CH3); δP (242.97 MHz, CDCl3) 20.9 (br s).
:1 (10 mL) and stirred for 3 h at room temperature. The solution was concentrated again to give a white powdery solid. Water (3 mL) and ammonia solution (5 drops, 25 w/w% in water) were added. The solution was lyophilised to give the ammonium salt of racemic (±)-(1R*,2R*)-9 (0.219 g, 83%, calculated yield for the diammonium salt); mp 172–175 °C; νmax/cm−1 3019, 2816, 1437, 1264, 1114, 1038, 970; δH (400.13 MHz, D2O) 4.42 (1 H, qdd, JHF 7.6, JHP 4.8, JHH 0.9, CF3–CH), 3.99 (1 H, dd, JHP 13.0, JHH 0.9, CH–P); δC (100.61 MHz, D2O), 125.9 (qd, JCF 282.4, JCP 19.2, CF3), 69.7 (qd, JCF 30.0, JCP 1.5, CF3–C), 66.6 (d, JCP 147.6, CH–P); δP (161.98 MHz, D2O) 15.7 (s).
Conclusions
In order to unravel the biodegradation pathway of fosfomycin, which is a biogenic phosphonate of high medicinal importance a set of three phosphonic acids was synthesised. These will help to either underpin or rule out our hypothesis for the final step in fosfomycin metabolism: the cleavage of the C–P bond. The synthesised compounds will be used as probes in future experiments with cell lysates and growing cultures of R. huakuii PMY1, a bacterium which is able to use fosfomycin as the sole phosphorus and carbon source.Acknowledgements
We greatly acknowledge the financial support by the Austrian Science Fund (FWF): P27987-N28. We thank S. Felsinger for recording NMR spectra and J. Theiner for performing combustion analyses.References
- R. L. Hilderbrand, The Role of Phosphonates in Living Systems 1983, CRC Press Inc., Boca Raton, 1983 Search PubMed.
- (a) G. P. Horsman and D. L. Zechel, Chem. Rev., 2016 DOI:10.1021/acs.chemrev.6b00536; (b) J. P. Chin, J. W. McGrath and J. P. Quinn, Curr. Opin. Chem. Biol., 2016, 31, 50 CrossRef CAS PubMed.
- A. K. White and W. W. Metcalf, Annu. Rev. Microbiol., 2007, 61, 379 CrossRef CAS PubMed.
- F. M. Kahan, J. S. Kahan, P. J. Cassidy and H. Kropp, Anal. N. Y. Acad. Sci., 1974, 235, 364 CrossRef CAS.
- (a) J. Wiesner, S. Borrmann and H. Jomaa, Parasitol. Res., 2003, 90(Suppl. 2), 71 CrossRef PubMed; (b) S. Sooriyaarachchi, R. Chofor, M. D. P. Risseeuw, T. Bergfors, J. Pouyez, C. S. Dowd, L. Maes, J. Wouters, T. A. Jones, S. Van Calenbergh and S. L. Mowbray, ChemMedChem, 2016, 11, 1 CrossRef PubMed.
- (a) S. S. Kamat, H. J. Williams, L. J. Dangott, M. Chakrabarti and F. M. Raushel, Nature, 2013, 497, 132 CrossRef CAS PubMed; (b) W. W. Metcalf, B. M. Griffin, R. M. Cicchillo, J. Gao, S. C. Janga, H. A. Cooke, B. T. Circello, B. S. Evans, W. Martens-Habbena, D. A. Stahl and W. A. van der Donk, Science, 2012, 337, 1104 CrossRef CAS PubMed; (c) S. C. Peck and W. A. van der Donk, Curr. Opin. Chem. Biol., 2013, 17, 580 CrossRef CAS PubMed; (d) D. M. Karl, L. Beversdorf, K. M. Björkman, M. J. Church, A. Martinez and E. F. DeLong, Nat. Geosci., 2008, I, 473 CrossRef.
- J. F. Villarreal-Chiu, J. P. Quinn and J. W. McGrath, Front. Microbiol., 2012, 3, 19 CAS.
- M. A. Pasek, J. M. Sampson and Z. Atlas, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15468 CrossRef CAS PubMed.
- L. C. Kolowith, E. D. Ingall and R. Brenner, Limnol. Oceanogr., 2012, 46, 309 CrossRef.
- J. P. Chin, J. W. McGrath and J. P. Quinn, Curr. Opin. Chem. Biol., 2016, 31, 50 CrossRef CAS PubMed.
- (a) J. L. Ford, N. O. Kaakoush and G. L. Mendz, Antonie van Leeuwenhoek, 2010, 97, 51 CrossRef CAS PubMed; (b) E. M. Fox and G. L. Mendz, Enzyme Microb. Technol., 2006, 40, 145 CrossRef CAS; (c) G. L. Mendz, F. Megraud and V. Korolik, Arch. Microbiol., 2005, 183, 113 CrossRef CAS PubMed.
- J. W. McGrath, F. Hammerschmidt and J. P. Quinn, Appl. Environ. Microbiol., 1998, 64, 356 CAS.
- J. W. McGrath, F. Hammerschmidt, H. Kählig, F. Wuggenig, G. Lamprecht and J. P. Quinn, Chem. – Eur. J., 2011, 17, 13341 CrossRef CAS PubMed.
- J. W. McGrath, F. Hammerschmidt, W. Preusser, J. P. Quinn and A. Schweifer, Org. Biomol. Chem., 2009, 7, 1944 CAS.
- S.-L. Lee, T. W. Hepburn, W. H. Swartz, H. L. Ammon, P. S. Mariano and D. Dunaway-Mariano, J. Am. Chem. Soc., 1992, 114, 7346 CrossRef CAS.
- W. Chang, M. Dey, P. Liu, S. O. Mansoorabadi, S. Moon, Z. K. Zhao, C. L. Drennan and H. Liu, Nature, 2013, 496, 114 CrossRef CAS PubMed.
- J. Quinn and F. Hammerschmidt, unpublished results.
- F. A. Atherton, M. J. Hall, C. H. Hassall, R. W. Lambert and P. S. Ringrose, Antimicrob. Agents Chemother., 1979, 15, 677 CrossRef CAS PubMed.
- F. Hammerschmidt and H. Kählig, J. Org. Chem., 1991, 56, 2364 CrossRef CAS.
- N. Simon, Biochim. Biophys. Acta, 2015, 1850, 488 CrossRef PubMed.
- S. S. Kamat and F. M. Raushel, Curr. Opin. Chem. Biol., 2013, 17, 589 CrossRef CAS PubMed.
- (a) C. R. Noe, Chem. Ber., 1982, 115, 1576 CrossRef CAS; (b) C. R. Noe, Chem. Ber., 1982, 115, 1591 CrossRef CAS; (c) C. R. Noe, M. Knollmüller, B. Oberhauser, G. Steinbauer and E. Wagner, Chem. Ber., 1986, 119, 729 CrossRef CAS.
- F. Hammerschmidt and H. Völlenke, Liebigs Ann. Chem., 1989, 577 CrossRef CAS.
- F. Hammerschmidt and Y. Li, Tetrahedron, 1994, 50, 10253 CrossRef CAS.
- B. L. Wanner, Biodegradation, 1994, 5, 175 CrossRef CAS PubMed.
- L. M. van Staalduinen, F. R. McSorley, K. Schiessl, J. Séguin, P. B. Wyatt, F. Hammerschmidt, D. L. Zechel and Z. Jia, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 5171 CrossRef CAS PubMed.
- A. R. Katritzky, K. Suzuki and S. K. Singh, Synthesis, 2004, 2645 CrossRef CAS.
- W. Cen, X. Dai and Y. Shen, J. Fluorine Chem., 1993, 65, 49 CrossRef CAS.
- C. Yuan, Z. Jinfeng, Y. Yuan, C. Li and W. Zhang, J. Fluorine Chem., 2006, 127, 44 CrossRef CAS.
- W. Mikenda, Vib. Spectrosc., 1992, 3, 327 CrossRef CAS.
- K. K. Ogilvie, E. A. Thompson, M. A. Quilliam and J. B. Westmore, Tetrahedron Lett., 1974, 33, 2865 CrossRef.
- P. Kumar, K. Ohkura, D. Beiki, L. I. Wiebe and K. Seki, Chem. Pharm. Bull., 2003, 51, 399 CrossRef CAS PubMed.
- A. R. Katritzky, A. A. Shetopalov and K. Suzuki, ARKIVOC, 2005, (vii), 36 Search PubMed.
- F. Hammerschmidt, G. Bovermann and K. Bayer, Liebigs Ann. Chem., 1990, 1055 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 13C and 31P NMR spectra of the synthesised compounds, X-ray data. CCDC 1522706 for 25b. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ob00546f |
| ‡ Deceased. |
| This journal is © The Royal Society of Chemistry 2017 |