
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-pericyclic cycloaddition of gem-difluorosubstituted azomethine ylides to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) O bond: computational study and synthesis of fluorinated oxazole derivatives†
O bond: computational study and synthesis of fluorinated oxazole derivatives†
Karina R.
Gaisina
,
Alexander F.
Khlebnikov
 * and
Mikhail S.
Novikov
* and
Mikhail S.
Novikov
St. Petersburg State University, Institute of Chemistry, 7/9 Universitetskaya nab., St. Petersburg, 199034 Russia. E-mail: a.khlebnikov@spbu.ru; Fax: +7 812-428-6939; Tel: +7 812-428-9344
First published on 4th May 2017
Abstract
The cycloaddition of arenecarbaldehydes and α,α,α-trifluoroacetophenones with gem-difluoro-substituted azomethine ylides, generated from N-benzhydrylideneamines and difluorocarbene, occurs regioselectively to give, after hydrolysis, oxazolidin-4-ones. The primary cycloadducts of trifluoroacetophenones, 4,4-difluoro-5-trifluoromethyloxazolidine derivatives, are sufficiently stable to be isolated in reasonable to excellent yields. The results of correlation analysis and DFT calculations reveal a non-pericyclic step-wise mechanism of the reaction. The replacement of the two geminal hydrogen atoms in the azomethine ylide intermediate for fluorine atoms results in a dramatic change in the reaction mechanism from pericyclic to step-wise, proceeding via a zwitterion-like transition state in which no C–O bonding is observed.
Introduction
The 1,3-dipolar cycloaddition of nitrogen ylides to carbon–heteroatom multiple bonds is an attractive synthetic approach to a great variety of five-membered heterocycles.1 This strategy has usually the advantage of synthetic efficiency, and high regio- and stereoselectivities. The main features of this type of pericyclic reaction were formulated by Huisgen.1a,2 Stereoselectivity and stereospecificity of the cycloaddition of nitrogen ylides to multiple bonds is typical of the reactions proceeding via a concerted pericyclic mechanism.1–3 This mechanism is usually characteristic of reactions of azomethine ylides with multiple carbon–carbon bonds.1,2 A switch from a concerted to a non-concerted pathway can, however, occur in the presence of certain combinations of substituents. Thus, strong electron-withdrawing substituents in dipolarophiles, like in dialkyl dicyanobutenedioates, can significantly reduce the barrier for the formation of zwitterionic intermediates in the reaction with azomethine ylides so that a stepwise cycloaddition can become competitive with a concerted cycloaddition.4 At the same time, substituents in the azomethine ylide, that destabilize the positive charge in the intermediate zwitterion, favour the concerted cycloaddition, even with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C dipolarophiles with strong electron-withdrawing substituents.4b Because of the importance of fluorinated heterocycles in medicinal chemistry the cycloaddition of azomethine ylides and dipolarophiles containing fluorine atoms attracts a lot of attention.5 The introduction of a fluorine atom, the most electronegative element, into a dipole or dipolarophile can have an influence on the mechanism of cycloaddition. Earlier it was found that the 1,3-dipolar cycloaddition of difluoro azomethine ylides to fumaronitrile and maleonitrile is stereoselective, consistent with a concerted mechanism for the reaction.6
C dipolarophiles with strong electron-withdrawing substituents.4b Because of the importance of fluorinated heterocycles in medicinal chemistry the cycloaddition of azomethine ylides and dipolarophiles containing fluorine atoms attracts a lot of attention.5 The introduction of a fluorine atom, the most electronegative element, into a dipole or dipolarophile can have an influence on the mechanism of cycloaddition. Earlier it was found that the 1,3-dipolar cycloaddition of difluoro azomethine ylides to fumaronitrile and maleonitrile is stereoselective, consistent with a concerted mechanism for the reaction.6
The cycloaddition of iminiodifluoromethanides to the CO double bond of aldehydes and ketones proceeds regioselectively but nonstereoselectively to give the corresponding oxazolidinones after hydrolysis of the unstable intermediate difluorooxazolidines.7 The reaction of azomethine ylide 2, generated from ethyl benzhydrylidene glycinate 1, proceeds in a more complex manner providing aziridines 8 and 10 along with the expected oxazolidinone 5 (Scheme 1). Aziridine 10 is the product of ylide 2–ylide 9 isomerization followed by 1,3-cyclization.8 The formation of aziridine 8 can proceed via zwitterion 6, resulting from the nucleophilic addition of ylide 2, to the carbonyl carbon of benzaldehyde, followed by a 1,5-prototropic shift to give ylide 7, which then undergoes a 1,3-cyclization. This implies that the formation of oxazolidine 4 can be the result of a nonconcerted cycloaddition of the fluorinated ylide to the CO double bond of benzaldehyde.
 | ||
| Scheme 1 Reaction of fluorinated ylide 2 with benzaldehyde.7 | ||
Results and discussion
To shed some light on the mechanism of the reaction of iminiodifluoromethanides with carbonyl compounds we turned to the correlation analysis and the quantum-chemical calculations for cycloadditions of iminiodifluoromethanides 2 and 12 with arenecarbaldehydes. The information on the structure of the transition state of the cycloaddition of gem-difluorinated ylide can be retrieved from the ρ value of the Hammett equation for reactions of a series of arenecarbaldehydes with ylide 12 derived from imine 11 and difluorocarbenes. We carried out the reactions of imine 11 with para-substituted benzaldehydes 3a–e in the presence of a difluorocarbene source. Heating a mixture of amine 11, aldehyde, dibromodifluoromethane, lead filings and Bu4NBr in dichloromethane at 40 °C under ultrasound irradiation gave a mixture containing difluorooxazolidines 13. Due to the fast hydrolysis of compounds 13 during the work-up of the reaction mixture, the difluorooxazolidines could not be isolated. Instead oxazolidinones 14a–e were isolated by column chromatography in 52–73% yield (Table 1).|
|
|||
|---|---|---|---|
| Entry | Ar | 3 | 14, yield, % |
| 1 | Ph | a | a, 64 |
| 2 | 4-MeC6H4 | b | b, 52 |
| 3 | 4-MeOC6H4 | c | c, 70 |
| 4 | 4-ClC6H4 | d | d, 73 |
| 5 | 4-NCC6H4 | e | e, 72 |

The obtained relative reaction constants from Hammett competition reaction experiments for aldehydes 3a–e are listed in Table 2 (for details see the ESI†) and were used for a Hammett correlation with σ-constants. A plot of Hammett σ-values vs. log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kR/kH for reactions of aldehydes 3a–e gave a satisfactory linear correlation: logkR/kH = ρσ = (0.08 ± 0.05) + (1.56 ± 0.14)σ (R = 0.988, SD = 0.105, N = 5).
kR/kH for reactions of aldehydes 3a–e gave a satisfactory linear correlation: logkR/kH = ρσ = (0.08 ± 0.05) + (1.56 ± 0.14)σ (R = 0.988, SD = 0.105, N = 5).
| Entry | Ar | 3 | k R/kH ± sm | σ |
|---|---|---|---|---|
| 1 | 4-MeOC6H4 | c | 0.235 ± 0.003 | −0.27 |
| 2 | 4-MeC6H4 | b | 0.481 ± 0.029 | −0.17 |
| 3 | Ph | a | 1.0 | 0 |
| 4 | 4-ClC6H4 | d | 2.23 ± 0.15 | 0.23 |
| 5 | 4-NCC6H4 | e | 7.56 ± 0.75 | 0.66 |
A positive ρ value in the Hammett equation indicates an increase of electron density on the carbon atom of the dipolarophile in the transition state. It can occur either within a step-wise mechanism of the reaction involving a zwitterion intermediate of type 6 or within an asynchronous concerted cycloaddition in which the C–C bond is formed ahead of the C–O bond. Analysis of the ρ value in the Hammett equation may be based on a comparison with the corresponding constants of related reaction series. Unfortunately, not so many studies deal with a Hammett analysis of the interaction of ylides with benzaldehydes, and such information related to the reaction of nitrogen ylides is absent in the literature. Some data were published for the Wittig reaction of substituted benzaldehydes with alkylidenetriphenylphosphoranes, for which the corresponding correlation with the σ-values gives a positive ρ value in the range 0.2–3.2.10 A positive ρ value, 1.6–2.1, in the corresponding Hammett equations was found for Wittig–Horner reactions of phosphonates and substituted benzaldehydes.11 Substituent effects on the reaction rate of the epoxidation of benzaldehydes with sulfur ylides, proceeding via a zwitterionic transition state, were studied, and a Hammett ρ of +2.50 was found.12 This means that data from relative rates and Hammett correlation are not enough to make a conclusion about the mechanism of the cycloaddition. Calculations of transition states of the reactions of ylide 12 with benzaldehydes 3 and ylide 16 with benzaldehyde 3a at the DFT B3LYP/6-31G(d) level13 using the PCM solvent model14 were therefore performed. There are four possible ways ylide 12 can approach the carbonyl group of benzaldehyde 3a (Fig. 1). The first two approaches involve the formation of the C–C bond between the carbonyl carbon and CF2-group and lead to the formation of oxazolidine 13: (1) H of the formyl group syn-oriented to the N–Me bond of the ylide (exo-approach, TS1) and (2) H of the formyl group anti-oriented to the N–Me bond of the ylide (endo-approach, TS2). The second two approaches involve the formation of the C–C bond between the carbonyl carbon and CPh2-group and lead to the formation of oxazolidine 13′: (3) H of the formyl group syn-oriented to the N–Me bond of the ylide (exo-approach, TS1′) and (4) H of the formyl group anti-oriented to the N–Me bond of the ylide (endo-approach, TS2′).
 | ||
| Fig. 1 Energy profiles for the reactions of benzaldehyde 3a with fluorinated ylide 12. Relative free Gibbs energies (in kcal mol−1, 298 K, PCM model for DCM) computed at the DFT B3LYP/6-31G(d) level. | ||
According to the calculations the energies of TS1′ and TS2′ are much higher than the energies of TS1 and TS2. This ensures the regioselectivity of the reaction with the formation of oxazolidine 13a as a sole product. The pathway (1) leads directly to the difluorooxazolidine 13via the transition state TS1, and although formation of the new bonds is quite asynchronous (F2C–CO distance is 2.484 Å and Ph2C–O distance is 3.674 Å for the reaction of 3a) it is impossible to locate any minimum on the energy surface that could correspond to a zwitterion. In contrast, the pathway (2) leads via the transition state TS2 (CF2–CO distance is 2.412 Å and Ph2C–O distance is 3.933 Å) to zwitterion 15 (CF2–CO distance is 1.625 Å and Ph2C–O distance is 3.522 Å), and then to difluorooxazolidine 13avia the transition state TS3 (F2C–CO distance is 1.623 Å and Ph2C–O distance is 2.958 Å) (Fig. 1). This means that the pathway (1) gives a cycloaddition product as a result of a concerted, non-pericyclic very asynchronous process, whereas the pathway (2) by a step-wise cycloaddition with the formation of a zwitterionic intermediate.
The calculations, using dispersion-inclusive functional wB97XD15 and the MPWB1K functional16 together with the 6-311G(d,p) basis set, which was recently used for study of the [3 + 2] cycloaddition reaction of nitrones with ketenes,17 were additionally performed for the reaction of ylide 12 with benzaldehyde 3a in order to instill confidence in the calculation results (Fig. 2). The results obtained show that the change in the functional and the basis set has little effect on the relative energies of the transition states TS1 and TS2, and thus does not affect the conclusions made.
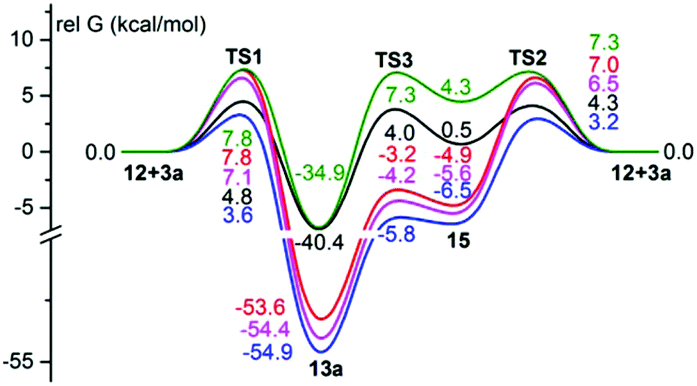 | ||
| Fig. 2 Energy profiles for the reaction of ylide 12 and benzaldehyde 3a. Relative free Gibbs energies (in kcal mol−1, 298 K, PCM model for DCM) computed: black – optimization of geometry at the DFT B3LYP/6-31G(d) level; green – single point calculation at the DFT B3LYP/6-311G(d,p) level with B3LYP/6-31G(d) optimized geometries; red – single point calculation at the DFT MPWB1K/6-311G(d,p) level with B3LYP/6-31G(d) optimized geometries; magenta – optimization of geometry at the DFT MPWB1K/6-311G(d,p) level; blue – single point calculation at the DFT wB97XD/6-311G(d,p) level with B3LYP/6-31G(d) optimized geometries. | ||
Nevertheless, for all energy profiles presented in Fig. 3–5, in addition to the calculations performed at the DFT B3LYP/6-31G(d) level of theory, the single point calculations were also made at the wB97XD/6-311G(d,p) and MPWB1K/6-311G(d,p) levels with B3LYP/6-31G(d) optimized geometries. A comparison of the experimental relative rate constants of the reactions of the ylide 12 and substituted benzaldehydes 3a–e with the corresponding relative free Gibbs energies of the transition states of TS2 showed that the best correlation is observed for energies obtained at the B3LYP/6-31G(d) level: ΔG# = 6.6 ± 0.2 − (2.3 ± 0.3)lgkR/kH [R = 0.925, SD = 0.391, N = 5; single point calculations at the MPWB1K/6-311G(d,p) level with B3LYP/6-31G(d) optimized geometries]; ΔG# = 3.0 ± 0.1 − (2.5 ± 0.3)lgkR/kH [R = 0.954, SD = 0.279, N = 5; single point calculations at the wB97XD/6-311G(d,p) level with B3LYP/6-31G(d) optimized geometries]; ΔG# = 4.4 ± 0.1 − (2.4 ± 0.2)lgkR/kH [R = 0.976, SD = 0.191, N = 5; at the B3LYP/6-31G(d) level]. The calculation results obtained at the B3LYP/6-31G(d) level are presented, therefore, in Fig. 3–5, whereas the results obtained at other levels can be found in Fig. S3–S5 (ESI).†
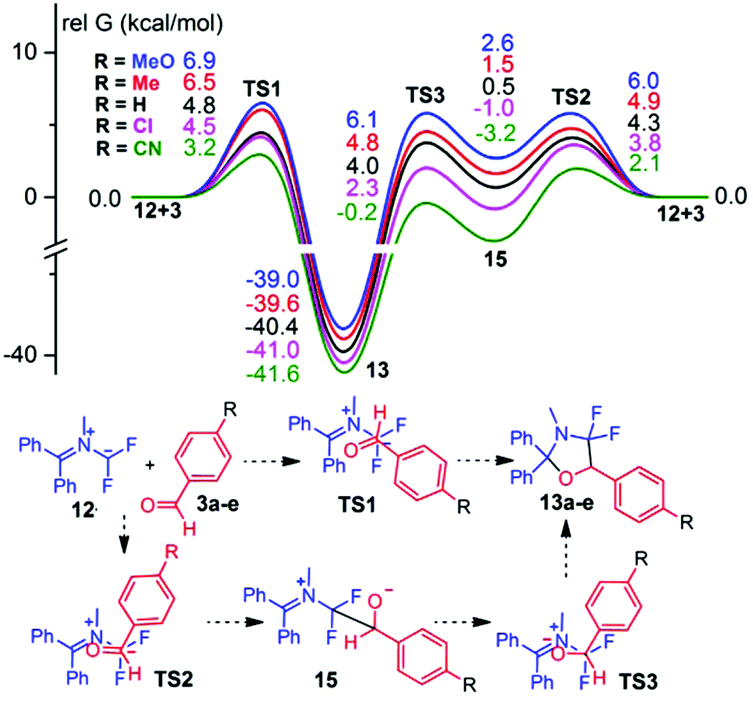 | ||
| Fig. 3 Energy profiles for the reaction of ylide 12 and benzaldehydes 3a–e. Relative free Gibbs energies (in kcal mol−1, 298 K, PCM model for DCM) computed at the DFT B3LYP/6-31G(d). | ||
 | ||
| Fig. 4 Energy profiles for the transformations of 16. Relative free Gibbs energies (in kcal mol−1, 298 K, PCM model for DCM) computed at the DFT B3LYP/6-31G(d) level. | ||
 | ||
| Fig. 5 Energy profiles for the reactions of benzaldehyde 3a with fluorinated ylide 12 and non-fluorinated ylide 21. Relative free Gibbs energies (in kcal mol−1, 298 K, PCM model for DCM) computed at the DFT B3LYP/6-31G(d) level. | ||
The free Gibbs energies of both transition states of the step-wise mechanism (TS2 by 0.5–1.6 kcal mol−1 and TS3 by 0.8–3.0 kcal mol−1) are lower than the energies of the transition state (TS1) for a concerted mechanism for all substituted benzaldehydes 3a–e. The formation of the product therefore proceeds mostly via the zwitterionic intermediate. In accordance with the experimental data the electron-withdrawing substituents in the benzene ring of aldehydes 3a–e reduce the barriers for addition of the ylide 12 to the CO bond, while the donating substituents increase them.
These data assume that the formation of a complex mixture of products for the reaction of ylide 2 with benzaldehyde could really be a result of the non-concertedness of the cycloaddition of the fluorinated ylide to the CO bond (Scheme 1). To evaluate the relative energies of the transition states of the reaction leading to products 5 and 8 we performed DFT calculations of the transformations of the nearest analogue of 2, the corresponding methyl ester 16 (Fig. 4).
According to the calculation, the barrier for a concerted cycloaddition of ylide 16 to the CO bond of benzaldehyde 3a is higher by 0.7 kcal mol−1 than the barrier for the formation of the zwitterion 17, the intermediate of the step-wise route to oxazolidine 18. It is noteworthy that the barrier for the 1,5-prototropic shift in the zwitterion leading to ylide 19, the isomer of starting ylide 16, is lower than the barrier for cyclization of the zwitterion to oxazolidine 18 by 1.3 kcal mol−1. This means that the formation of aziridine 20 can indeed compete with the formation of oxazolidine 18 due to the preference of the step-wise addition of the ylide 16 to the CO bond. Such a low barrier for the transformation of zwitterion 17 into ylide 19 (3.2 kcal mol−1) is largely due to the influence of the electron-withdrawing alkoxycarbonyl group.
In fact, the barrier for the transformation of zwitterion 15a without such an activating group into the corresponding ylide 24 is much higher (TS9, 10.1 kcal mol−1) and that, together with the low barrier for cyclization of 15a (TS3), is preventing the formation of the corresponding aziridine in this case (Fig. 5).
In order to evaluate how the introduction of fluorine atoms into the ylidic carbon effects the concertedness of the cycloaddition to the CO bond the calculation of the interaction of non-fluorinated ylide 21 with benzaldehyde 3a was performed (Fig. 5). According to calculations the concerted cycloaddition (TS10) is, in this case, much more preferred than the step-wise one (TS11, TS12). Moreover, the formation of C–C and C–O bonds becomes more synchronous (F2C–CO distance is 2.484 Å and Ph2C–O distance is 3.674 Å (TS1); H2C–CO distance is 2.316 Å and Ph2C–O distance is 2.804 Å (TS10)). Analysis of the isodesmic equations shows that the replacement of both fluorine atoms in ylide 12 with hydrogens (ylide 21) leads to the stabilization of the ylide species (Scheme 2, reaction (1)). This can be explained by the strong pyramidalization of the NCF2 moiety (pyramidalization angle in ylide 12 is 21.3° versus 1.8° for ylide 21), resulting in the decrease of conjugation in the π-system of the fluorinated 1,3-dipole.
 | ||
| Scheme 2 The isodesmic equations for ylides 12/21, zwitterions 15a/23 and transition states TS1/TS10 and TS2/TS11. Relative free Gibbs energies (in kcal mol−1, 298 K, PCM model for DCM) computed at the DFT B3LYP/6-31G(d) level. | ||
The destabilizing effect of fluorine atoms vanishes in transition state TS1 and becomes a stabilizing effect in TS2 and even more in zwitterion 15a. Thus, the strong pyramidalization of the NCF2 moiety of ylide 12, most probably, is the main cause of the two-stage mechanism of the cycloaddition.
As mentioned above, fluorinated oxazolidines 13 very easily undergo hydrolysis, losing fluorine. As fluorinated heterocycles are widely used as drugs, agrochemicals and materials with special properties18 it would be useful to use the reaction described above to prepare fluorinated derivatives of oxazolidine of type 13. Some perfluorinated oxazolidines have been prepared by the electrochemical fluorination of derivatives of aminosubstituted carboxylic acids in low yield.19 The intramolecular cycloaddition of difluorinated azomethine ylides to an ester carbonyl group gives heterocycles with difluorinated oxazolidine as a part of the bridged system.1c,20 Analysis of these data allows us to conclude that strong electron-withdrawing substituents, like the CF3-group, and a substitution pattern which prevents dehydrofluorination, will stabilise the fluorinated oxazolidine. Taking into account the above results, showing that electron-withdrawing substituents accelerate the reaction of fluorinated ylides with a carbonyl group, we decided that CF3-substituted ketones are good starting materials for the preparation of oxazolidines with fluoro- and trifluoromethyl-substituents. The experimental results confirmed the correctness of our assumptions: 4,4-difluoro-5-trifluoromethyl-substituted oxazolidines were obtained in synthetically significant yields (Table 3). The oxazolidines 28 were, however, still acid sensitive, therefore to preserve fluorine at the ring the isolation of the products should be performed without chromatography on silica. The addition of a catalytic amount of HCl to the DCM solution of the oxazolidines 28 leads to their quantitative transformation into the corresponding oxazolidinones 29 (Table 3).

It is notable that the formation of aziridine 10 in the reaction of imine 1 with trifluoroacetophenone under difluorocarbene generation conditions was not observed. This fact can be explained by the higher electrophilicity of trifluoroacetophenone compared with benzaldehyde, resulting in the inhibition of the isomerization of ylide 2 to ylide 9. The second competing pathway of the reaction, a 1,5-prototropic shift in the zwitterionic intermediate leading to an aziridine product of type 8, is not realized as well. This is probably due to the reduced basicity of the olate oxygen of the corresponding CF3-substituted zwitterionic intermediate which results in an increase of the barrier for the 1,5-prototropic shift.
Conclusions
We have shown that the cycloaddition of arenecarbaldehydes with gem-difluoro-substituted azomethine ylides, generated from benzhydrylidene amines and difluorocarbenes, occurs regioselectively to give after hydrolysis oxazolidin-4-ones. The change of a benzaldehyde for an α,α,α-trifluoroacetophenone as a dipolarophilic agent results in (a) an increase of the stability of the primary difluoropyrrolidine cycloadduct, and (b) inhibition of the competing 1,5-prototropic shift in the zwitterion intermediate. This allowed the isolation of 4,4-difluoro-5-trifluoromethylpyrrolidine cycloadducts in reasonable to excellent yields. Correlation analysis and DFT calculations reveal a non-pericyclic step-wise mechanism of the reaction. The exchange of the two geminal hydrogen atoms in an azomethine ylide intermediate with fluorines results in a dramatic change in the reaction mechanism from pericyclic to step-wise, via a zwitterion-like transition state, in which no C–O bonding is observed.Experimental
General
Melting points were determined on a hot stage microscope and are uncorrected. 1H (300 MHz, 400 MHz) and 13C (75 MHz, 100 MHz) NMR spectra were determined in CDCl3 with a Bruker DPX 300 and a Bruker AVANCE III 400. Chemical shifts (δ) are reported in parts per million downfield from tetramethylsilane (TMS δ = 0.00); 1H NMR spectra were calibrated according to the residual peak of CDCl3 (1H 7.26 ppm, 13C 77.00 ppm). 19F NMR spectra were recorded at 376 MHz; chemical shifts are reported in ppm from CFCl3 as an internal standard. Elemental analysis was performed on a Hewlett-Packard 185B CHN-analyser. Silica gel Merck 60 was used for column chromatography. Thin-layer chromatography (TLC) was conducted on alumina sheets precoated with SiO2 ALUGRAM SIL G/UV254.Calculation details
All calculations were performed with the density functional method by using the Gaussian 09 suite of programs. Geometry optimizations of intermediates, transition states,21 reactants, and products in dichloromethane were performed at the DFT B3LYP/6-31G(d) level13 using the PCM solvent model.14 Stationary points on the respective potential-energy surfaces were characterized at the same level of theory by evaluating the corresponding Hessian indices. Careful verification of the unique imaginary frequencies for transition states was carried out to check whether the frequency indeed pertains to the desired reaction coordinate. Single-point calculations with B3LYP/6-31G(d) optimized geometries, using dispersion-inclusive functional wB97XD15 and the MPWB1K16 functional together with the 6-311G(d,p) basis set were additionally performed.Acknowledgements
We gratefully acknowledge the financial support from the Russian Science Foundation (Grant No. 16-13-10036). This research was carried out using resources of the Centre for Magnetic Resonance, the Computer Centre, and the Centre for Chemical Analysis and Materials of St. Petersburg State University.Notes and references
- (a) 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, Wiley, New York, 1984 Search PubMed; (b) Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, ed. A. Padwa and W. H. Pearson, John Wiley & Sons, New York, 2003 Search PubMed Nitrile oxides, nitrones and nitronates in organic synthesis: novel strategies in synthesis, ed. H. Feuer, John Wiley & Sons, Hoboken, 2008 Search PubMed (c) A. G. Meyer and J. H. Ryan, Molecules, 2016, 21, 935–988 CrossRef PubMed; (d) J. H. Ryan, ARKIVOC, 2015,(i), 160–183 Search PubMed; (e) T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS PubMed; (f) A. F. Khlebnikov and M. S. Novikov, Chem. Heterocycl. Compd., 2012, 48, 179–190 CrossRef CAS.
- (a) R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef; (b) R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 633–645 CrossRef.
- (a) Pericyclic Reactions, ed. A. P. Marchand and R. E. Lehr, Academic Press, New York, 1977, vol. I and II Search PubMed; (b) A. Williams, Concerted organic and Bio-organic mechanisms, CRC Press, Boca Raton, 2000 Search PubMed.
- (a) G. Mlostoń, K. Urbaniak, M. Domagała, A. Pfitzner, M. Zabel and H. Heimgartner, Helv. Chim. Acta, 2009, 92, 2631–2642 CrossRef; (b) A. F. Khlebnikov, A. S. Konev, A. A. Virtsev, D. S. Yufit, G. Mlostoń and H. Heimgartner, Helv. Chim. Acta, 2014, 97, 453–470 CrossRef CAS; (c) S. Emamian, RSC Adv., 2016, 6, 75299–75314 RSC.
- (a) Fluorine in Heterocyclic Chemistry, ed. V. Nenajdenko, Springer International Publishing, Switzerland, 2014, vol. 1 Search PubMed; (b) P. He and S. Z. Zhu, Mini-Rev. Org. Chem., 2004, 1, 417–435 CrossRef CAS.
- M. S. Novikov, A. F. Khlebnikov, M. V. Shevchenko, R. R. Kostikov and D. Vidovic, Russ. J. Org. Chem., 2005, 41, 1496–1506 CrossRef CAS.
- M. S. Novikov, A. F. Khlebnikov, A. Krebs and R. R. Kostikov, Eur. J. Org. Chem., 1998, 133–137 CrossRef CAS.
- A. F. Khlebnikov, M. S. Novikov and R. R. Kostikov, Mendeleev Commun., 1997, 127–128 Search PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 97, 165–195 CrossRef.
- (a) N. S. Isaacs and O. H. Abed, Tetrahedron Lett., 1986, 27, 995–996 CrossRef CAS; (b) L. Donxia, W. Dexian, L. Yaozhong and Z. Huaming, Tetrahedron, 1986, 42, 4161–4167 CrossRef; (c) H. Yamataka, K. Nagareda, A. Ando and T. Hanafusa, J. Org. Chem., 1992, 57, 2865–2869 CrossRef CAS; (d) H. Yamataka, K. Nagareda, T. Takatsuka, K. Ando, T. Hanafusa and S. Nagase, J. Am. Chem. Soc., 1993, 115, 8570–8576 CrossRef CAS; (e) H. Yamataka, T. Takatsuka and T. Hanafusa, J. Org. Chem., 1996, 61, 722–726 CrossRef CAS PubMed.
- Z.-K. Li, C. He, M. Yang, C.-Q. Xia and X.-Q. Yu, ARKIVOC, 2005,(i), 98–104 Search PubMed.
- D. R. Edwards, P. Montoya-Peleaz and C. M. Crudden, Org. Lett., 2007, 9, 5481–5484 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS; (c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 6908–6918 CrossRef CAS.
- M. Ríos-Gutiérrez, A. Darù, T. Tejero, L. R. Domingo and P. Merino, Org. Biomol. Chem., 2017, 15, 1618–1627 Search PubMed.
- (a) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. V. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (b) Fluorine in Heterocyclic Chemistry, ed. V. Nenajdenko, Springer Verlag, Berlin, 2013, vol. 1 and 2 Search PubMed; (c) Fluorinated Heterocyclic Compounds, ed. V. A. Petrov, J. Wiley & Sons Inc., Hoboken, 2009 Search PubMed.
- (a) J. A. Young, T. C. Simmons and F. W. Hoffmann, J. Am. Chem. Soc., 1956, 78, 5637–5639 CrossRef CAS; (b) T. Abe, E. Hayashi, H. Baba and H. Fukaya, J. Fluorine Chem., 1990, 48, 257–279 CrossRef CAS.
- (a) M. S. Novikov, I. V. Voznyi, A. F. Khlebnikov, J. Kopf and R. R. Kostikov, J. Chem. Soc., Perkin Trans. 1, 2002, 1628–1630 RSC; (b) I. V. Voznyi, M. S. Novikov, A. F. Khlebnikov, J. Kopf and R. R. Kostikov, Russ. J. Org. Chem., 2004, 40, 199–205 CrossRef CAS; (c) I. V. Voznyi, M. S. Novikov, A. F. Khlebnikov and R. R. Kostikov, Russ. Chem. Bull., 2004, 53, 1087–1091 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data for all new compounds, competition reaction experiments and computational details. See DOI: 10.1039/c7ob00521k |
| This journal is © The Royal Society of Chemistry 2017 |