
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective synthesis of novel adamantane derivatives with high potency against rimantadine-resistant influenza A virus strains†
Nikolai Yu.
Kuznetsov
 *a,
Rabdan M.
Tikhov
a,
Ivan A.
Godovikov‡
a,
Michael G.
Medvedev§
a,
Konstantin A.
Lyssenko§
a,
Elena I.
Burtseva
b,
Elena S.
Kirillova
b and
Yuri N.
Bubnov
*ac
*a,
Rabdan M.
Tikhov
a,
Ivan A.
Godovikov‡
a,
Michael G.
Medvedev§
a,
Konstantin A.
Lyssenko§
a,
Elena I.
Burtseva
b,
Elena S.
Kirillova
b and
Yuri N.
Bubnov
*ac
aA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, Vavilov 28, 119991, Moscow, Russian Federation. E-mail: nkuznff@ineos.ac.ru
bN.F. Gamaleya Institute of Epidemiology and Microbiology, Russian Academy of Medicinal Sciences, Gamaleya 18, 123098, Moscow, Russian Federation
cN.D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky pr. 47, 119991 Moscow, Russian Federation
First published on 10th March 2017
Abstract
A series of (R)- and (S)-isomers of new adamantane-substituted heterocycles (1,3-oxazinan-2-one, piperidine-2,4-dione, piperidine-2-one and piperidine) with potent activity against rimantadine-resistant strains of influenza A virus were synthesized through the transformation of adamantyl-substituted N-Boc-homoallylamines 8 into piperidine-2,4-diones 11 through the cyclic bromourethanes 9 and key intermediate enol esters 10. Biological assays of the prepared compounds were performed on the rimantadine-resistant S31N mutated strains of influenza A – A/California/7/2009(H1N1)pdm09 and modern pandemic strain A/IIV-Orenburg/29-L/2016(H1N1)pdm09. The most potent compounds were both enantiomers of the enol ester 10 displaying IC50 = 7.7 μM with the 2016 Orenburg strain.
Adamantane derivatives have found numerous applications in various fields of chemistry, but their use in catalysis1 and medicinal chemistry2 has been especially fruitful. With a low-molecular weight and highly symmetric structure, adamantane fragments provide a domain with critical lipophilicity (with a molecular diameter of 6.36 Å)3 when inserted in the structure of known pharmacophores, improving pharmacokinetic profiles of the modified drugs.4 Simple aminoadamantanes (amantadine, rimantadine, tromantadine, memantine) (Fig. 1) have occupied a reliable place in the pharmaceutical market, proving their efficiency for the treatment of viral diseases (influenza A, herpes, hepatitis C, HIV) and neurological disorders (Parkinson and Alzheimer diseases).2
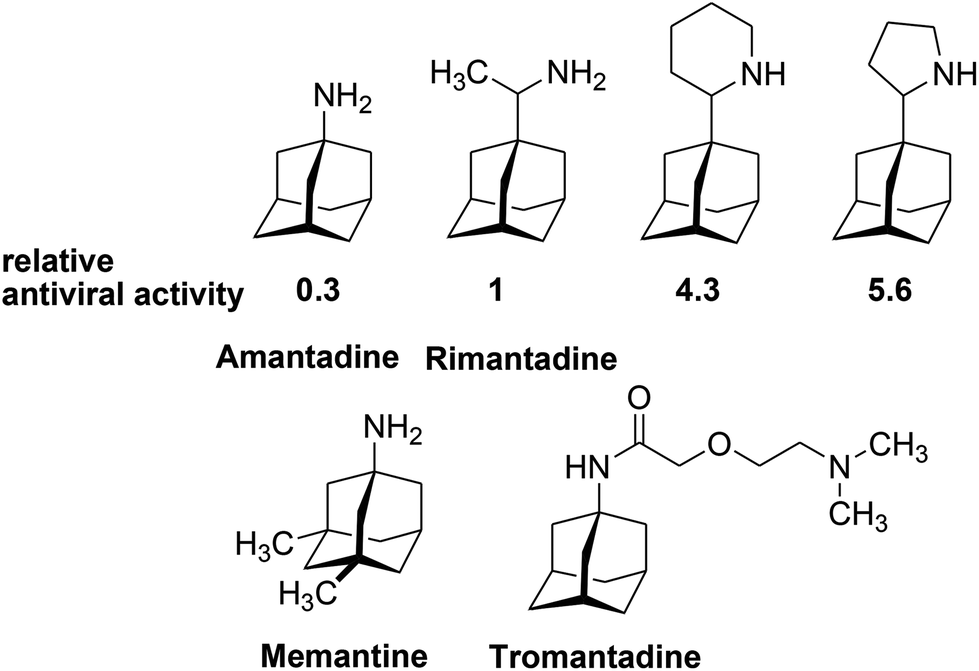 | ||
| Fig. 1 Biologically active aminoadamantanes. | ||
It is known that piperidine and pyrrolidine derivatives of adamantane (Fig. 1) possess high antiviral activity towards the influenza A virus (A2/Japan/305/1957(H2N2)), exceeding rimantadine activity by 4.3 and 5.6 times, respectively.5 Meanwhile, mutations and evolutionary changes in the influenza A virus have led to the development of 45% resistance in all the circulating virus strains towards adamantane-based drugs, and the average resistances of the most frequent H1 and H3 subtypes were 70% (years 1933–2013) and 44% (years 1968–2013), respectively.6 Moreover, the temporal distribution of the virus's resistance is alarming; for example, during 2009–2013, the resistance of the H1N1 subtype reached 100% in many countries. This situation has resulted in a complete ban on the use of rimantadine and amantadine in medical practices. Nevertheless, the comprehensive knowledge concerning the mechanism of the action of aminoadamantanes, as inhibitors of the proton M2 channel of the influenza virus, together with the high pharmaceutical value of adamantane derivatives stimulates research on the synthesis of novel adamantane-containing molecules.
Recently, an efficient stereoselective synthesis route for various substituted piperidin-2,4-diones and related piperidinone-type molecules starting from readily available homoallylamines was developed by our team (Scheme 1).7 This approach is based on the anionic enolate-type rearrangements of the cyclic enol esters (E), which are disclosed as the reactive intermediates (A) enolate-isocyanate or enolate-carbodiimide and further cyclized into the corresponding piperidinone-type molecules.
 | ||
| Scheme 1 General scheme and conditions of anionic enolate-type rearrangements. | ||
Regarding applications of adamantane chemistry, we were interested in the synthesis of new enantiomerically pure molecules with joint adamantane and piperidinone skeletons in order to evaluate their antiviral activity, which is expected to be higher than that of rimantadine. Previous data obtained in vivo (mice) indicates that both enantiomers of rimantadine are equipotent with the racemic mixture;8 however, the recent NMR study of stereoisomers of rimantadine shows their different binding profiles with the M2 virus channel.9 The effect of chirality will strengthen with the growth in size of the substituents at the chiral center.
Results and discussion
Though many adamantane-containing molecules have been synthesized to date, there is still a lack of low-molecular weight adamantanes for modifications and biological tests, especially in an enantiomerically pure form. The presented approach allows access to new chiral adamantanes that were unavailable before. Synthesis of the substituted adamantanes started from the condensation of 1-adamantylcarbaldehyde 1 and Ellman's (R)- and (S)-sulfinamides 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 in a solution of neat Ti(OiPr)4 (5 equiv.) that produced the corresponding imines 3 (Scheme 2). Subsequent allylation of the imines 3 was carried out using two different metals, Zn and In, in THF because the different Lewis acidity of the zinc and indium allylic reagents usually affected the stereoselectivity of the allylation reaction.11,7b The obtained diastereomeric ratio in the allylated product 4 could not be determined by 1H NMR because only one set of signals with few impurities was observed. Therefore, the sulfinamides 4 through the consecutive desulfination with HCl and acylation with Cbz-Cl, were converted to N-Cbz-protected amines (S)- and (R)-5, which were suitable for chiral HPLC analysis. For the racemate preparation, chiral sulfoximine (RS)-3 was transformed to achiral sulfonimine 6 through the mCPBA mediated oxidation. Allylation of the latter with AllZnBr gave sulfonamide 7, which was deprotected by treatment with TfOH upon mild heating (40 → 50 °C). The deprotected racemic amine without isolation was acylated with CbzCl in a DCM solution in the presence of 20% NaOH, which afforded rac-5. HPLC analysis of the isomers 5 shows >99% ee with Zn and 97% ee with In, which evidently corresponds to the diastereoselectivity of the allylation reaction. Sulfinamide 4 forms a very stable complex with the In salt, which does not decompose in the conventional workup with a NH4Cl solution, and washing the reaction mixture with Trilon B gives an excellent isolated yield of 4 (ca. 100%). The description of the diastereoselectivity models in the allylation reactions can be found in paper7b and the references therein. Removal of Ellman's auxiliary by treatment of 4 with HCl in MeOH followed by in situ acylation of the homoallylamine with Boc-anhydride leads to N-Boc-derivatives 8 (80%). The absolute (S)-configuration of the chiral center in one of the isomers 8 was established by X-ray single crystal analysis ((S)-8) obtained from ((RS)-2) (Fig. S1†).12 The interaction of 8 with NBS in DCM gave rise to isomeric urethanes 9a (Scheme 3) in the ratio cis/trans 4:1 (1H NMR, CDCl3), and they were not separable by FC. Therefore, the major isomer cis-9a was isolated from the mixture by crystallization from EtOAc, and the relative configurations of the chiral centers were determined by NOESY experiment.
10 in a solution of neat Ti(OiPr)4 (5 equiv.) that produced the corresponding imines 3 (Scheme 2). Subsequent allylation of the imines 3 was carried out using two different metals, Zn and In, in THF because the different Lewis acidity of the zinc and indium allylic reagents usually affected the stereoselectivity of the allylation reaction.11,7b The obtained diastereomeric ratio in the allylated product 4 could not be determined by 1H NMR because only one set of signals with few impurities was observed. Therefore, the sulfinamides 4 through the consecutive desulfination with HCl and acylation with Cbz-Cl, were converted to N-Cbz-protected amines (S)- and (R)-5, which were suitable for chiral HPLC analysis. For the racemate preparation, chiral sulfoximine (RS)-3 was transformed to achiral sulfonimine 6 through the mCPBA mediated oxidation. Allylation of the latter with AllZnBr gave sulfonamide 7, which was deprotected by treatment with TfOH upon mild heating (40 → 50 °C). The deprotected racemic amine without isolation was acylated with CbzCl in a DCM solution in the presence of 20% NaOH, which afforded rac-5. HPLC analysis of the isomers 5 shows >99% ee with Zn and 97% ee with In, which evidently corresponds to the diastereoselectivity of the allylation reaction. Sulfinamide 4 forms a very stable complex with the In salt, which does not decompose in the conventional workup with a NH4Cl solution, and washing the reaction mixture with Trilon B gives an excellent isolated yield of 4 (ca. 100%). The description of the diastereoselectivity models in the allylation reactions can be found in paper7b and the references therein. Removal of Ellman's auxiliary by treatment of 4 with HCl in MeOH followed by in situ acylation of the homoallylamine with Boc-anhydride leads to N-Boc-derivatives 8 (80%). The absolute (S)-configuration of the chiral center in one of the isomers 8 was established by X-ray single crystal analysis ((S)-8) obtained from ((RS)-2) (Fig. S1†).12 The interaction of 8 with NBS in DCM gave rise to isomeric urethanes 9a (Scheme 3) in the ratio cis/trans 4:1 (1H NMR, CDCl3), and they were not separable by FC. Therefore, the major isomer cis-9a was isolated from the mixture by crystallization from EtOAc, and the relative configurations of the chiral centers were determined by NOESY experiment.
 | ||
| Scheme 2 Synthesis of both (S)- and (R)-8 and standards 5 for HPLC analysis. | ||
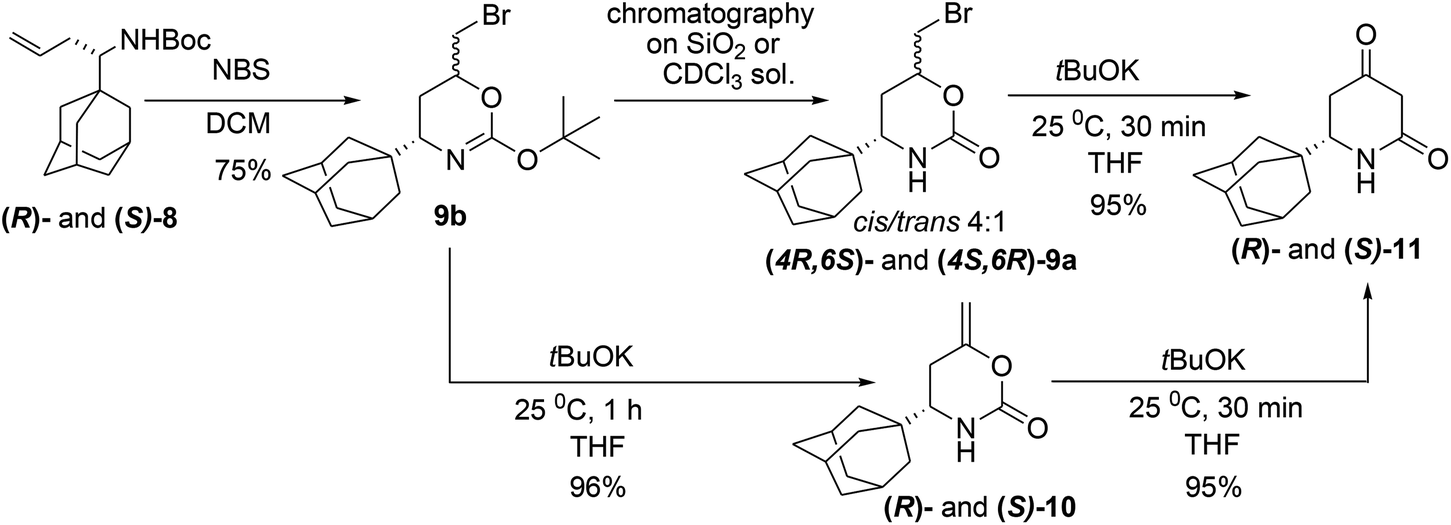 | ||
| Scheme 3 Synthesis of of both (S)- and (R)-isomers of enol ester 10 and dione 11. | ||
Upon treatment of the mixture of cis/trans-isomers 9a with tBuOK (25 °C, 30 min), an expected dione 11 was formed in a high yield. However, during repetition of the experiments, we observed that together with 11, a cyclic enol ester 10 was formed, and its amount changed widely in a range of 20–90%. Actually, 10 is an intermediate and cannot be isolated under usual conditions because the transformation of 10 into 11 occurs even at −50 °C (Scheme 1)7a and is much faster than the elimination of HBr from 9a. In the preceding papers devoted to enolate-carbodiimide rearrangement,7c we observed the formation of some enol esters as intermediates with substrates bearing N-EWG-groups, but in the case of the enolate-carbodiimide rearrangement, this is possible because of the low rate of the reaction (Scheme 1). Considering the possible methods of ester 10 survival, we concluded that the reactivity can be affected by an intermolecular association of the molecules into dimeric complexes where the NH-protons are shielded from the attack of tBuOK by voluminous adamantane groups. The XRD of (S)-10 supported our hypothesis (Fig. 2).12 In the crystals, two independent molecules (S)-10 are assembled into homochiral dimers due to the typical N–H⋯O (N⋯O 2.935(2)–2.973(2) Å) hydrogen bonding of the lactame groups that are additionally stabilized by the C–H⋯O (H(9AA)⋯O(1) 2.36, H(14A)⋯O(1A) 2.46 Å) interactions formed with the adamantyl substituents occupying the pseudoequatorial position. The distances between the nonbonded atoms, H(9AA)⋯HN(1A) 2.15 Å, H(14A)⋯HN(1) 2.23 Å, are generally less than the sum of the van der Waals radii of the hydrogen atoms, thus, the NH-protons can be shielded by adamantane from the base attack. For the analysis of the intermolecular interactions, QTAIM theory13 was used. This theory gives an opportunity to locate all the bonding interactions by a critical point search as well as estimating the energy of the interactions by means of the Espinosa correlation.14 The electron density function in the crystal was obtained by means of a recently introduced concept of invarioms,15 and the accuracy of this approach for the estimation of weak and moderate strength hydrogen bonds was recently verified.16
 | ||
| Fig. 2 The general view of hydrogen bonded molecules in the crystal of (S)-10 in the representation of atoms by thermal ellipsoids (p = 50%). | ||
However, our attempts to reveal the existence of the dimers in a DMSO-D6 solution by 1H DOSY experiments were unsuccessful. Finally, we disclosed that the enol ester 10 arises only if the reaction mixture of 8 and NBS was not purified by conventional chromatography and was only filtered through the short pad of the silica gel or not purified at all. We proposed the formation of the cyclic tert-butyl ether 9b, in which HBr-elimination would proceed more readily.7a An analysis of the proton spectrum of the crude reaction mixture in C6D6, after passing through the 0.5 cm thickness layer of silica gel, has shown that the mixture consists of two isomers of tert-butyl ether 9b. For analytical purposes, the major isomer cis-9b was isolated by crystallization from cooled hexane, and its structure was studied by XRD (Fig. S2†). This product was very sensitive to acids and immediately decomposed into 9a on silica gel TLC, FC or in a solution of CDCl3, so its discovery was a lucky circumstance. In fact, using two types of isolation procedures for bromides 9a,b, flash chromatography or short pad filtration, we were able to synthesis both enantiomers of either enol ester 10 or dione 11 in preparative quantities. The molecular structure of (R)-11 was also studied by XRD.12 In the crystal of 11 in contrast to 10, the amide groups assemble molecules into infinite chains in which molecules are assembled by N–H⋯O (7.12 kcal mol−1) and C–H⋯O (0.7–1.91 kcal mol−1) bonds (Fig. S3†). The 4-keto-group in 11 was diastereoselectively reduced with NaBH4 (Scheme 4), which is known to produce excellent cis/trans selectivity dr = 32:1 for the reduction of the 6-phenyl-substituted dione.17
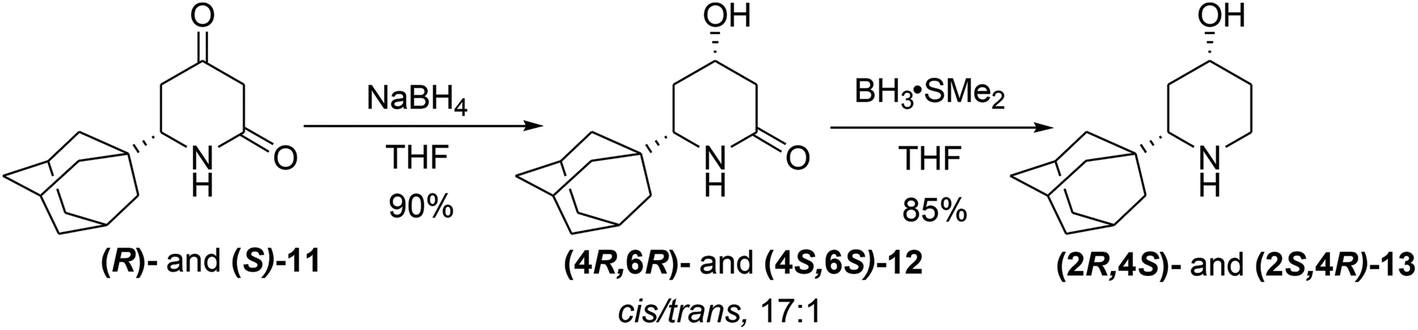 | ||
| Scheme 4 Stereoselective synthesis of hydroxypiperidines. | ||
In our case, the diastereomeric ratio of the 6-(1-adamantyl)-substituted alcohols 12 formed upon the action of NaBH4 at −78 °C was only 10:1, and upon decreasing the reaction temperature to −90 °C, alcohols 12 with dr = 17:1 were obtained in the best experiment (because of the heterogeneity of the reaction mixture, diastereoselectivity value was vary in different experiments at the same conditions). In the crystal of alcohol (4R,6R)-12, the presence of the OH-group leads to principally different crystal packing patterns with participation of OH-groups of two independent molecules in hydrogen bonding with the NH(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) function (Fig. S4†).12 The mixture of diastereomeric alcohols 12 was further subjected to a reduction with 5 equiv. of LiAlH4; however, surprisingly, even after prolonged heating (24 h) in THF, 12 was returned without changes. Adamantane-derivative 12 has demonstrated distinct behavior from other 6-substituted piperidine-2-ones, which are usually readily reduced with LiAlH4 to piperidines.18 The reduction of the lactame group in 12 can be blocked by the formation of some aggregates similar to the dimeric complexes observed in crystals of 10. On the other hand, the BH3*SMe2 complex, which is less sterically demanding than the solvated LiAlH4, reduces lactame 12 easily to give 4-hydroxypiperidine 13 in a high yield (85%) after FC. Aminoalcohols 13 were converted to hydrochlorides by treatment with ethereal HCl followed by recrystallization from the EtOAc/MeOH mixture that gave diastereomerically pure hydrochlorides 13.
O) function (Fig. S4†).12 The mixture of diastereomeric alcohols 12 was further subjected to a reduction with 5 equiv. of LiAlH4; however, surprisingly, even after prolonged heating (24 h) in THF, 12 was returned without changes. Adamantane-derivative 12 has demonstrated distinct behavior from other 6-substituted piperidine-2-ones, which are usually readily reduced with LiAlH4 to piperidines.18 The reduction of the lactame group in 12 can be blocked by the formation of some aggregates similar to the dimeric complexes observed in crystals of 10. On the other hand, the BH3*SMe2 complex, which is less sterically demanding than the solvated LiAlH4, reduces lactame 12 easily to give 4-hydroxypiperidine 13 in a high yield (85%) after FC. Aminoalcohols 13 were converted to hydrochlorides by treatment with ethereal HCl followed by recrystallization from the EtOAc/MeOH mixture that gave diastereomerically pure hydrochlorides 13.
Having in hand a set of enantiomerically pure adamantyl substituted piperidines 9a–13, we undertook a study of their antiviral properties. A mechanism of anti-influenza A activity of amantadine and rimantadine was studied in detail and it was found to be mainly related to blocking of the proton channel M2 of the influenza A virus. A comprehensive study of a protein complex of the tetrameric M2 channel with amantadine and rimantadine by high-resolution NMR as well as crystallographic methods has revealed two types of blocking mechanisms. A main mechanism is considered to be “sterical blocking”, wherein a drug molecule acts as a stopper in a bottle neck inside the channel of M2, and this mechanism operates with a minimal amount of the aminoadamantane inhibitor (ratio Ad:M2 = 1:1).19 Another possibility for the channel binding is the interaction of aminoadamantane with the outer side of the tetrameric channel M2 (ratio Ad:M2 = 4:1) in pockets formed by the hydrophilic part of Asp 44 and hydrophobic residues of Leu 40, Ile 42 and Leu 43. Such binding reduces the mobility of the protein chains and stabilizes the closed form of the M2 channel. In the case of amantadine, the position in the channel lumen is more preferable (40 fold greater affinity) than the peripheral site.19 As we mentioned above, a majority of the contemporary virus strains is resistant to the adamantane-type M2 channel inhibitors because of the so-called S31N mutation.20 In recent years, considerable progress was achieved in designing potent inhibitor molecules that target the M2 channel of drug resistant influenza A viruses.21,22 The most potent inhibitors of M2/S31N mutants were aminoadamantanes-based molecules, such as 2-propyl-2-aminoadamantane,22a pyrimidine-(pyridine-)substituted adamantanes22b and isoxazole-containing molecules.22c,d Since the A/M2-S31N mutant is one of the most conserved viral proteins among the current influenza A viruses, with more than 95% of the virus carriers, S31N mutated etalon pandemic strain of influenza virus A/California/7/2009(H1N1)pdm09 and modern A/IIV-Orenburg/29-L/2016(H1N1)pdm09 were selected as targets for tests. The tests were performed in the infected cells of MDCK tissue, and the values on the suppression of the virus reproduction are presented in Table 1.
| Compound | % of suppression of virus strains of (H1N1)pdm09a | TCD50, μM | |||||
|---|---|---|---|---|---|---|---|
| A/California/7/2009 | IC50, μM | A/IIV-Orenburg/29-L/2016 | IC50, μM | ||||
| 30 μMc | 40 μMc | 30 μMc | 40 μMc | ||||
| a The values obtained upon simultaneous addition of inhibitors and virus. b No activity. c % of suppression of the reproduction of the influenza A virus at 30 and 40 μM (average value). d Half-life time of 10 in the MEM medium; T1/2 = 7.44 h at 37 °C (see ESI file). | |||||||
| (±)-Rimantadine | NAb | NAb | — | NAb | NAb | — | >300 |
| (4S,6R)-9a | 62.7 | 70 | 19.8 | 67.8 | 69 | 21.9 | >300 |
| (4R,6S)-9a | 71.6 | 73 | 11.3 | 72.2 | 74 | 20.1 | >300 |
| (R)-10d | 97.9 | 99.9 | 8.1 | 94.7 | 95.9 | 7.7 | 80 |
| (S)-10d | 99.2 | 100 | 13.7 | 94 | 91.8 | 7.7 | 80 |
| (R)-11 | 68 | 72.5 | 20.6 | 57.5 | 69.9 | 27.1 | 80 |
| (S)-11 | 58 | 82.5 | 26.7 | 46.3 | 54.9 | 34.0 | 160 |
| (4R,6R)-12 | 21.9 | 39.5 | >40.0 | 38.6 | 42.6 | >40.0 | >300 |
| (4S,6S)-12 | 16.4 | 17.4 | >40.0 | 36.6 | 40.2 | >40.0 | >300 |
| (2R,4S)-13 | 51 | 53 | 18.4 | 55 | 57 | 17.7 | 150 |
| (2S,4R)-13 | 32 | 27 | >40.0 | 52 | 53 | 26.9 | 150 |
The activity of all the synthesized compounds considerably exceeds that of rimantadine. The most expected 4-hydroxy-substituted analogues of adamantyl-piperidine (Fig. 1) show moderate activity (2R,4S)-13 with an IC50 = 18.4 and 17.6 μM, and its enantiomer (2S,4R)-13 is almost two times less active with an IC50 > 40.0 μM (California) and 26.9 μM (Orenburg). Both isomers of the lactames 12 have low activities, close to the activity of (2S,4R)-13. Both isomers of dione (R)- and (S)-11 have lower values of IC50 = 20.6, 26.7 μM (California) and 27.1, 34.0 μM (Orenburg) than those of (2R,4S)-13; however, they have a more effective inhibition profile because the % of suppression of virus replication reaches higher values, 72.5 and 82.5% relative to 53 and 27% for (2R,4S)-13 (Table 1). The most potent were isomers of the enol ester 10 with IC50 = 8.1 for (R)-10 and 13.7 for (S)-10 with complete suppression of virus reproduction from 30 μM. Interestingly, that Orenburg mutant strain became less sensitive to all compounds except the isomers of the enol ester 10 with an IC50 = 7.7 μM and a high suppression activity reaching 94–96% at C 30/40 μM (Table 1). Enol ester 10 can be even more potent than we observed because the performed stability test in the MEM medium disclosed that the half-life of 10 is 7.44 hours at a temperature of virus incubation (37 °C). Though the MEM medium has a pH around neutral, 10 can rearrange into dione 11 (Schemes 1 and 3) under alkaline pH in aqueous solution. Bromo-derivatives 9a were also quite active in the suppression of the California strain with an IC50 = 19.8 and 11.3 μM for (4S,6R)- and (4R,6S)-9a, respectively. Generally (R)-isomers were more potent as inhibitors of M2 channels independent of the other structural variations that support the recent NMR study on the rimantadine enantiomers.9 The cytotoxicities of the synthesized compounds are comparable to the value of rimantadine (Table 1), and the most active isomers of the enol ester 10 were also more toxic to MDCK cells. However, the values of IC50 for the isomers 10 are considerably lower than the corresponding values of TCD50.
One conclusion about the mechanism of antiviral action can be drawn from our data. Different from known aminoadamantanes with antiviral activity to A/M2-S31N mutants,21,22 our compounds are not basic (except of 13) and cannot act through the suggested alternative mechanisms connected with the changing pH value. At the same time, all molecules follow the uniform dependence of activity on concentration. It is very probable that the mechanism of their antiviral action is also similar and related to the binding of the M2 channel.
In conclusion, we have demonstrated the antiviral effect on the mutated virus strains by action of very simple molecules with close structures to the known aminoadamantanes with known binding places in and on the M2 channel. It is not known in which site the enol ester 10 binds, but acquiring this knowledge is important for the development of next generation anti-influenza A agents. A compact and rigid structure of the adamantane-derived 6-membered heterocycles has the potential for modification and provides an excellent opportunity for further research in various fields of adamantane chemistry, including drug discovery.
Acknowledgements
This work was financially supported by the Russian Science Foundation (grant no. 15-13-00109). The authors are grateful to Dr S. Lubimov for chiral HPLC analysis.References
- K. A. Agnew-Francisa and C. M. Williams, Adv. Synth. Catal., 2016, 358, 675–700 CrossRef.
- (a) L. Wanka, K. Iqbal and P. R. Schreiner, Chem. Rev., 2013, 113, 3516–3604 CrossRef CAS PubMed; (b) T. P. Stockdale and C. M. Williams, Chem. Soc. Rev., 2015, 44, 7737–7763 RSC.
- N. Moreldesrosiers and J. P. Morel, J. Solution Chem., 1979, 8, 579–592 CrossRef CAS.
- Brain-directing properties: (a) N. Tsuzuki, T. Hama, M. Kawada, A. Hasui, R. Konishi, S. Shiwa, Y. Ochi, S. Futaki and K. Kitagawa, J. Pharm. Sci., 1994, 83, 481–484 CrossRef CAS PubMed; (b) Z. Kazimierczuk, A. Gorska, T. Switaj and W. Lasek, Bioorg. Med. Chem. Lett., 2001, 11, 1197–1200 CrossRef CAS PubMed; (c) K. Kitagawa, N. Mizobuchi, T. Hama, T. Hibi, R. Konishi and S. Futaki, Chem. Pharm. Bull., 1997, 45, 1782–1787 CrossRef CAS PubMed. Gene delivery: (d) M. Manoharan, K. L. Tivel and P. D. Cook, Tetrahedron Lett., 1995, 36, 3651–3654 CrossRef CAS. Enzyme activity: (e) N. D. Igumnova, E. Lemina, I. I. Bitiukova, N. V. Klimova and A. P. Skoldinov, Farmakol. Toksikol., 1988, 51, 38–41 CAS; (f) D. J. Augeri, J. A. Robl and D. A. Betebenner, et al. , J. Med. Chem., 2005, 48, 5025–5037 CrossRef CAS PubMed.
- G. Stamatiou, G. B. Foscolos, G. Fytas, A. Kolocouris, N. Kolocouris, C. Pannecouque, M. Witvrouw, E. Padalko, J. Neyts and E. De Clercq, Bioorg. Med. Chem., 2003, 11, 5485–5492 CrossRef CAS PubMed.
- G. Dong, C. Peng, J. Luo, C. Wang, L. Han, B. Wu, G. Ji and H. He, PLoS One, 2015, 10, e0119115 Search PubMed.
- (a) N. Yu. Kuznetsov, V. I. Maleev, V. N. Khrustalev, A. F. Mkrtchyan, I. A. Godovikov, T. V. Strelkova and Yu. N. Bubnov, Eur. J. Org. Chem., 2012, 334–344 CrossRef CAS; (b) N. Yu. Kuznetsov, V. N. Khrustalev, T. V. Strelkova and Yu. N. Bubnov, Tetrahedron: Asymmetry, 2014, 25, 667–676 CrossRef CAS; (c) N. Yu. Kuznetsov, R. M. Tikhov, I. A. Godovikov, V. N. Khrustalev and Yu. N. Bubnov, Org. Biomol. Chem., 2016, 14, 4283–4298 RSC; (d) N. Yu. Kuznetsov, R. M. Tikhov, T. V. Strelkova, Yu. N. Bubnov and K. A. Lyssenko, Tetrahedron Lett., 2016, 57, 4525–4528 CrossRef CAS.
- P. E. Aldrich, E. C. Hermann, W. E. Meier, M. Paulshock, W. W. Prichard, J. A. Snyder and J. C. Watts, J. Med. Chem., 1971, 14, 535–543 CrossRef CAS PubMed.
- A. K. Wright, P. Batsomboon, J. Dai, I. Hung, H.-X. Zhou, G. B. Dudley and T. A. Cross, J. Am. Chem. Soc., 2016, 138, 1506–1509 CrossRef CAS PubMed.
- M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600–3740 CrossRef CAS PubMed.
- X.-W. Sun, M.-H. Xu and G.-Q. Lin, Org. Lett., 2006, 8, 4979–4982 CrossRef CAS PubMed.
- CCDC 1440627, (S)-8; 1530899, 9b; 1517135, (S)-10; 1517136, (R)-11 and 1517137, (4R,6R)-12. Experimental data contain in the supplementary crystallographic data for this paper. The principal bond lengths and angles in complexes studied are also given in ESI.†.
- (a) E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS; (b) E. Espinosa, I. Alkorta, I. Rozas, J. Elguero and E. Molins, Chem. Phys. Lett., 2001, 336, 457–461 CrossRef CAS.
- R. F. W. Bader, Atoms In molecules. A Quantum Theory, Clarendron Press, Oxford, 1990 Search PubMed.
- B. Dittrich, T. S. Koritsansky and P. Luger, Angew. Chem., Int. Ed., 2004, 43, 2718–2721 CrossRef CAS PubMed.
- (a) Y. V. Nelyubina, A. A. Korlyukov and K. A. Lyssenko, Chem. – Eur. J., 2014, 20, 6978–6984 CrossRef CAS PubMed; (b) Y. V. Nelyubina and K. A. Lyssenko, Chem. – Eur. J., 2015, 21, 9733–9741 CrossRef CAS PubMed.
- F. A. Davis, T. Fang, B. Chao and D. M. Burns, Synthesis, 2000, 2106–2112 CrossRef CAS.
- (a) A. J. Burke, S. G. Davies, A. C. Garner, T. D. McCarthy, P. M. Roberts, A. D. Smith, H. Rodriguez-Solla and R. J. Vickers, Org. Biomol. Chem., 2004, 2, 1387–1394 RSC; (b) Y. Yang, D. P. Phillips and S. Pan, Tetrahedron Lett., 2011, 52, 1549–1552 CrossRef CAS; (c) Y. Xie, A. Raffo, M. Ichise, S. Deng, P. E. Harris and D. W. Landry, Bioorg. Med. Chem. Lett., 2008, 18, 5111–5114 CrossRef CAS PubMed.
- S. D. Cady, K. Schmidt-Rohr, J. Wang, C. S. Soto, W. F. DeGrado and M. Hong, Nature, 2010, 463, 689–693 CrossRef CAS PubMed.
- (a) R. J. Garten, et al. , Science, 2009, 325, 197–201 CrossRef CAS PubMed; (b) A. Antóna, F. Pozoc, J. Niubód, I. Casasc and T. Pumarolaa, Enferm. Infecc. Microbiol. Clin., 2012, 30(Supl. 4), 10–17 CrossRef.
- Reviews: (a) J. Wang, F. Li and C. Ma, Biopolymers, 2015, 104, 291–309 CrossRef CAS PubMed; (b) Z. Shena, K. Loua and W. Wang, Acta Pharm. Sin. B, 2015, 5, 419–430 CrossRef PubMed.
- (a) A. Kolocouris, C. Tzitzoglaki, F. B. Johnson, R. Zell, A. K. Wright, T. A. Cross, I. Tietjen, D. Fedida and D. D. Busath, J. Med. Chem., 2014, 57, 4629–4639 CrossRef CAS PubMed; (b) F. Li, C. Ma, Y. Hu, Y. Wang and J. Wang, ACS Infect. Dis., 2016, 2, 726–733 CrossRef CAS PubMed; (c) F. Li, C. Ma, W. F. DeGrado and J. Wang, J. Med. Chem., 2016, 59, 1207–1216 CrossRef CAS PubMed; (d) F. Li, Y. Hu, Y. Wang, C. Ma and J. Wang, J. Med. Chem., 2017, 60, 1580–1590 CrossRef CAS PubMed; (e) V. A. Shibnev, T. M. Garaev, M. P. Finogenova, E. S. Shevchenko and E. I. Burtseva, Pharm. Chem. J., 2012, 46, 3–7 Search PubMed; (f) P. G. Deryabin, T. M. Garaev, M. P. Finogenova, A. G. Botikov and V. A. Shibne, Bull. Exp. Biol. Med., 2014, 157, 62–65 CrossRef CAS PubMed. Examples of inhibitors A/M2 ineffective against S31N mutants: (g) S. Wu, et al. , ChemMedChem, 2015, 10, 1837–1845 CrossRef CAS PubMed; (h) M. Rey-Carrizo, E. Torres, C. Ma, M. Barniol-Xicota, J. Wang, Y. Wu, L. Naesens, W. F. DeGrado, R. A. Lamb, L. H. Pinto and S. Vázquez, J. Med. Chem., 2013, 56, 9265–9274 CrossRef CAS PubMed; (i) M. Rey-Carrizo, M. Barniol-Xicota, C. Ma, M. Frigolé-Vivas, E. Torres, L. Naesens, S. Llabrés, J. Juárez-Jiménez, F. J. Luque, W. F. DeGrado, R. A. Lamb, L. H. Pinto and S. Vázquez, J. Med. Chem., 2014, 57, 5738–5747 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, copies of the 1H and 13C NMR spectra, HPLC traces, antiviral activity assay and selected crystallographic data. CCDC 1517135–1517137, 1440627 and 1530899. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ob00331e |
| ‡ NMR experiments. |
| § X-Ray diffraction analysis. |
| This journal is © The Royal Society of Chemistry 2017 |