
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimisation of the dibromomaleimide (DBM) platform for native antibody conjugation by accelerated post-conjugation hydrolysis†
Maurício
Morais
 ,
João P. M.
Nunes
,
Kersti
Karu
,
Nafsika
Forte
,
Irene
Benni
,
Mark E. B.
Smith
,
Stephen
Caddick
,
Vijay
Chudasama
* and
James R.
Baker
*
,
João P. M.
Nunes
,
Kersti
Karu
,
Nafsika
Forte
,
Irene
Benni
,
Mark E. B.
Smith
,
Stephen
Caddick
,
Vijay
Chudasama
* and
James R.
Baker
*
Department of Chemistry, UCL, 20 Gordon St, London, UK WC1H 0AJ. E-mail: j.r.baker@ucl.ac.uk; v.chudasama@ucl.ac.uk
First published on 14th March 2017
Abstract
Disulfide bridging offers a convenient approach to generate site-selective antibody conjugates from native antibodies. To optimise the reagents available to achieve this strategy, we describe here the use of dibromomaleimides designed to undergo accelerated post-conjugation hydrolysis. Conjugation and hydrolysis, which serve to ‘lock’ the conjugates as robustly stable maleamic acids, is achieved in just over 1 h. This dramatic acceleration is also shown to infer significant improvements in homogeneity, as demonstrated by mass spectrometry analysis.
Introduction
Antibody bioconjugation is a key technological challenge in chemical biology, enabling a broad range of applications from targeted therapeutics (e.g. antibody–drug conjugates; ADCs) to radioimmunoconjugates and immunoassays.1 Conjugation to antibodies is most readily achieved by exploiting the reactivity of lysine and cysteine residues to specific electrophilic reagents. There are >80 lysine residues in a typical antibody, and it has been estimated for ADCs that an average loading of 3–4 drugs per antibody leads to around 106 molecular species present in the resulting heterogeneous conjugates.2 The use of such complex mixtures in therapeutics is far from optimal, as each component will have a different pharmacological profile.2b,3 An alternative strategy is to release cysteine residues as sites for attachment by reduction of the interchain disulfide bonds, of which there are four in the major therapeutically relevant isotype (IgG1). Loading each of the cysteines with a reactive reagent, most commonly achieved using classical maleimides, still affords a heterogeneous mixture of products with drug-to-antibody ratios (DARs) of 0, 2, 4, 6 and 8 as the major components.4 The higher loaded species present have been identified as having reduced stability,5 and poorer outcomes in vivo due to their accelerated clearance.4,6There is significant evidence that increased homogeneity in conjugates will afford improved therapeutic windows,2b,6,7 and thus approaches for site-selective conjugation are gaining momentum and can be expected to form a key component of future generations of ADCs.8 These include the use of genetic engineering to incorporate cysteine mutants, non-natural amino-acids or enzymatic recognition sequences as handles for controlled drug loading.7a,8,9 Site-selective methods which avoid this requirement for engineering can offer the further advantage of being directly applicable to native antibodies. To this end disulfide bridging is a highly promising strategy, in which the interchain disulfide bonds are targeted with reagents which reconnect the two cysteine residues.8b,10 This approach aims to generate antibody conjugates with a controlled loading of one drug per disulfide bond, and thus a DAR of 4 in IgG1s.
We have recently described the use of a class of modified maleimide reagents, known as next generation maleimides (NGMs),11 which are able to achieve efficient disulfide bridging and thus represent a platform for site-selective ADC development (Fig. 1).12 We have shown that dithiophenolmaleimide–monomethyl auristatin E (DTPM–MMAE) reagents can afford ADCs which offer potent and selective tumor cell killing activity in vitro12d and in vivo.13 In addition to affording access to a narrow range of DAR species, with an average close to four, the NGMs provide a route to serum stable conjugates. This is achieved by hydrolysis of the maleimide ring to ‘lock’ the products as maleamic acids (see the structure shown in Fig. 1).12d
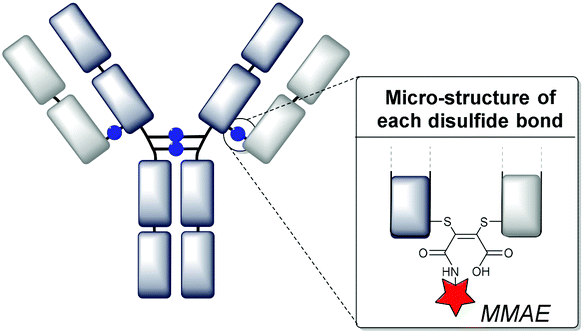 | ||
| Fig. 1 NGM–ADC conjugates constructed by disulfide bridging.12d | ||
Jackson et al. have also applied alternative NGMs, dibromomaleimides (DBMs), to generate analogous ADCs using MMAF as the payload, and have confirmed that the resultant ADCs offer improved in vivo efficacy compared to classical maleimide conjugates.14 Notably no hydrolysis step is included in this DBM–MMAF conjugation, and the resultant conjugates can be expected to cleave over several days in serum due to thiol exchange reactions.12d
These approaches to NGM–ADCs, whilst highly promising, require optimisation. Post-conjugation hydrolysis is key to ensure robust serum stability, to prevent the loss of free drug leading to off-site toxicity; however the current prolonged conditions described, 72 h at pH 8.4,12d are sub-optimal. This will serve to increase process times for downstream product development and can even introduce undesirable heterogeneity as shown herein. In this work we describe an investigation into accelerated hydrolysis, by means of linker design and conjugation conditions, and show that a dramatic increase in the rate of the reaction sequence is facilitated along with improved homogeneity. Whilst this optimisation is carried out in the context of ADCs, it also has implications on the design of NGM reagents for bioconjugation more widely, as these reagents are finding increasing application on diverse scaffolds ranging from peptides and proteins to polymers and surfaces.15
Results and discussion
Dibromomaleimide (DBM) reagent selection and synthesis
We chose to employ DBMs, which represent the simplest and most accessible disulfide bridging reagents, with varying linkers attached to the maleimide nitrogen. By varying from the C-6 (caproyl) linker which has been used in previous work to a C-2 (glycine derived) or an aryl linker, we expected to effect an increase in the rate of hydrolysis (due to increased electron withdrawal from the imide).16 Previous work has shown that aryl linkers on classical maleimides and DTPMs afford antibody conjugates which undergo accelerated hydrolysis.12c,17The synthesis of DBM reagents is commonly achieved by treatment of dibromomaleic anhydride (or commercially available diacid18) with an amine, refluxing in acetic acid to induce ring closure. Alternatively, an N-methoxy carbonyl activated DBM can be employed when a milder, room temperature, approach is required.19 Here we favoured a different strategy, in which the linker was pre-installed with the DBM, and an appended carboxylic acid could then be coupled with an amine. Notably the use of an NHS ester (comparable to ubiquitously employed heterobifunctional maleimide reagents) led to competing attack on the DBM to generate aminomaleimides. This highlights the increased electrophilicity of these substituted maleimides. To overcome this, a more active acylating agent is required, which can be achieved by the use of an EEDQ coupling (Scheme 1). Using readily synthesised acids 1–3 (see the ESI†) this strategy gave access to three model reagents 4–6, incorporating an alkyne handle suitable for subsequent functionalisation if desired.
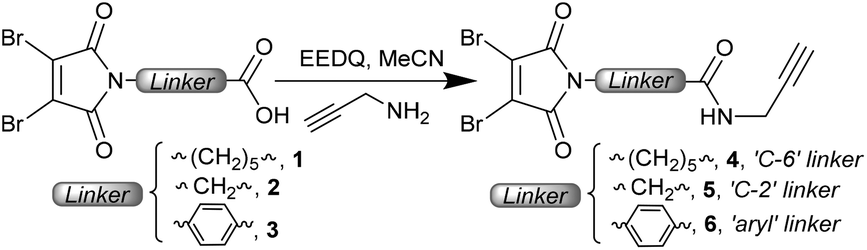 | ||
| Scheme 1 Synthesis of dibromomaleimide reagents 4–6 with C-6, C-2 and aryl linkers. | ||
LC-MS validation and propensity of IgG1 to form disulfide isomers
We chose clinically relevant IgG1 trastuzumab20 as a model antibody for our conjugation study, and sought to evaluate the LC-MS method that would be crucial to assessing the homogeneity of the conjugates. Trastuzumab was thus deglycosylated using PNGase F, and analysed by capillary electrospray ionisation on a Q-TOF LC-MS. The result was a single main species, corresponding to the deglycosylated full antibody (Fig. 2A, see the ESI† for raw data). Upon reduction with TCEP free heavy and light chains are observed (Fig. 2B), due to the denaturing conditions of the LC-MS. Finally we reoxidised this sample, using Ellman's reagent, to represent a control for the data that could be expected upon reformation of the covalent attachments between the chains (Fig. 2C). Intriguingly, whilst reoxidation was highly efficient, the product was formed as two isomers; the full antibody (minor) and the ‘half-antibody’ (major). This half-antibody is simply the product in which the disulfides in the hinge region of the antibody have reformed in an intrachain, rather than interchain fashion, and is thus observed by the denaturing LC-MS method. This competitive inter- and intramolecular reactivity matches the outcome of disulfide bridging reagents12c,14 and suggests that IgG1s are susceptible to such disulfide scrambling. It should be noted that oxidation under other conditions, such as dehydroascorbic acid,21 is known to generate the native disulfide configuration. We postulate that the difference is due to Ellman's reagent's high reactivity, trapping the intrachain connectivity as a kinetic outcome. Indeed treatment of the isomeric mixture with an alkyl thiol (cysteamine, 10 equiv.) re-equilibrates the system to generate the native connectivity (ESI Fig. S8†).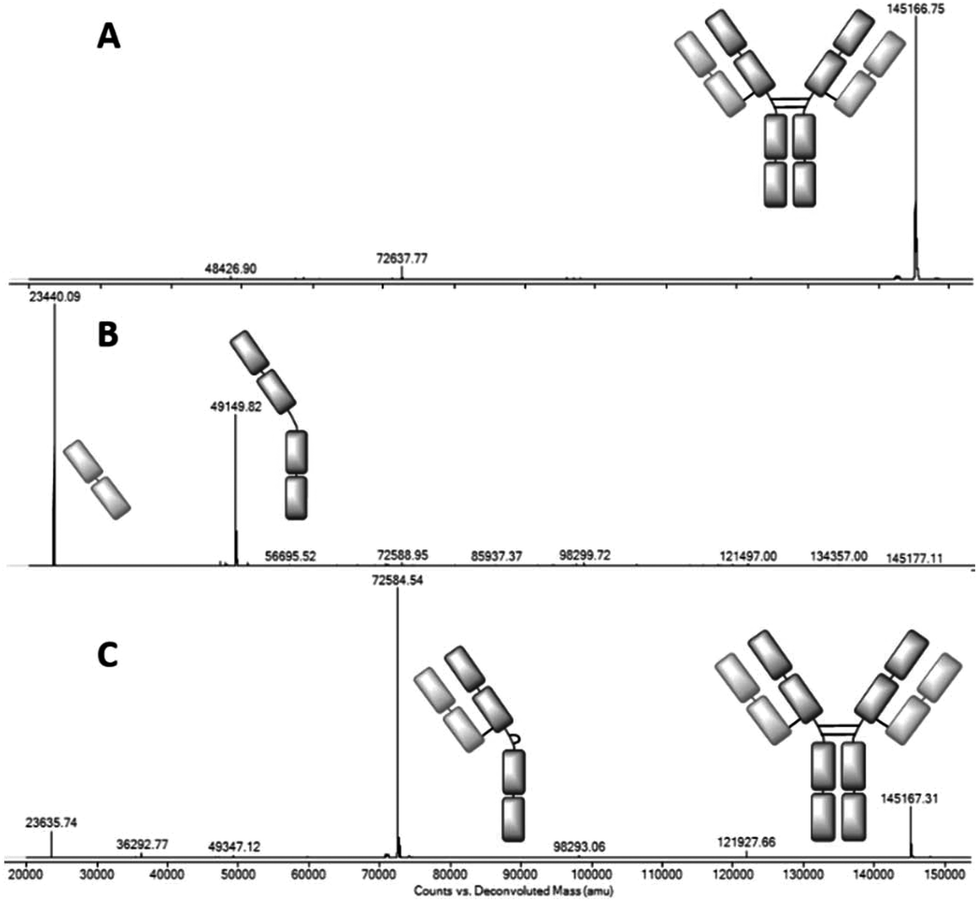 | ||
Fig. 2 Deconvoluted LC-MS data on: A – trastuzumab (mass observed 145![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 167, expected 145167); B – reduced trastuzumab (mass observed 23440 and 49150, expected 23440 and 49150); C – reoxidised trastuzumab using Ellman's reagent (observed 72585 and 145167, expected 72584 and 145167). 167, expected 145167); B – reduced trastuzumab (mass observed 23440 and 49150, expected 23440 and 49150); C – reoxidised trastuzumab using Ellman's reagent (observed 72585 and 145167, expected 72584 and 145167). | ||
Conjugation of DBM-linker reagents 4–6 with optimisation
This initial MS data served as a benchmark for our conjugation experiments. Thus trastuzumab was reduced with TCEP and treated with DBM reagent 4, which contains the C-6 linker and is expected to hydrolyse slowly post-conjugation. The LC-MS analysis (Fig. 3A) revealed that the conjugate was a mixture of species – the major components being the half antibody (with two attached NGM linkers), and non-crosslinked light and heavy chains. A closer inspection of the light chain mass reveals that the major species (23361 Da) has undergone a decarboxylative cleavage of the C-terminal cysteine. This reaction, which has previously been observed to occur on trastuzumab–thiomaleimide conjugates under photochemical conditions,22 occurs even if light is excluded.
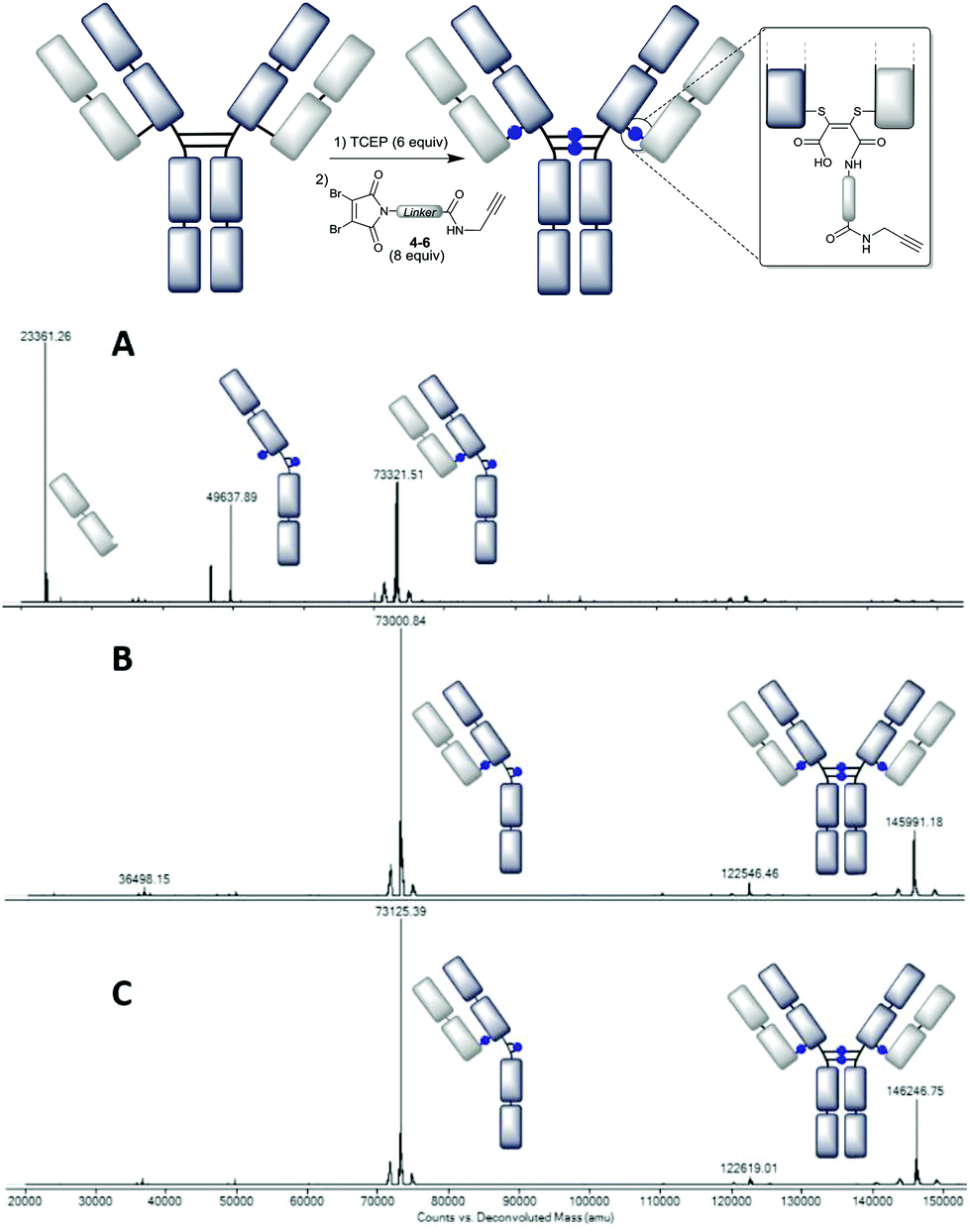 | ||
| Fig. 3 Deconvoluted LC-MS data on the trastuzumab–NGM conjugates: A – using DBM-C6-alkyne 4 at pH 8.0 (mass observed 73119* and 73322, expected 73117); B – using DBM-C2-alkyne 5 at pH 8.5 (mass observed 73001 and 145991, expected 73005 and 146008); C – using DBM-aryl-alkyne 6 at pH 8.5 (mass observed 73125 and 146247, expected 73125 and 146249). *See the ESI† for the expanded region in LC-MS. | ||
To preclude this undesirable heterogeneity, we proposed that rapid hydrolysis post-conjugation would be beneficial as it would remove the reactive dithiomaleimide functional group from the conjugate. Indeed the use of the C-2 and aryl linker (4 and 5) avoided this competing decarboxylative side-reaction, generating much improved homogeneity (ESI Fig. S11 and S13†). An additional improvement was achieved by switching the pH from 8.0 to 8.5, to further accelerate hydrolysis (Fig. 3B and C).
For both linkers the LC-MS data confirm that two isomeric products are formed almost exclusively, with one containing inter-heavy chain bridges and the other intrachain bridges; both represent antibody conjugates loaded with 4 DBMs. The raw data (see the ESI†) also give further insights, confirming this major improvement in homogeneity and showing that the half-antibody conjugate envelope (1400–2200 m/z) and the full antibody conjugate envelope (2600–3400 m/z) approach a 1:1 ratio in the optimised conjugates. SDS-PAGE analysis (ESI Fig. S2 and S3†) further confirms this improvement in homogeneity, and indicates that the full antibody conjugate now represents the major species. Overall these data reveal that these accelerated hydrolysis DBM reagents offer efficient and highly homogeneous conjugation with a loading of 4. Notably the LC-MS profile is similar to that obtained from the Ellman's reoxidation (Fig. 1C), indicating that the DBM bridging offers a comparably efficient mode of reactivity to disulfide reformation.
Rate of hydrolysis post-conjugation
To investigate the rate of hydrolysis of these conjugates we exploited the convenient absorbance of the dithiomaleimides at ∼400 nm23 which is lost upon conversion to the corresponding maleamic acid (see the ESI†). The kinetic data at pH 8.5 confirm the dramatic acceleration inferred by the C-2 and aryl linkers (Fig. 4); the hydrolysis half-lives of 16–19 min (cf. 48 h for C-6 linker) are comparable to classical maleimides containing aryl linkers.17 These data confirmed that the conjugates using C-2 and aryl linkers are almost completely hydrolysed in an hour.
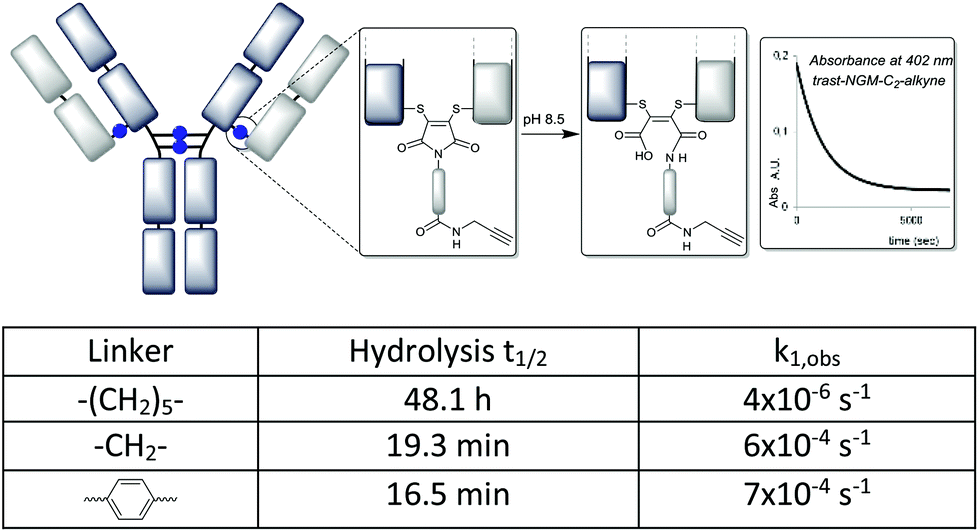 | ||
| Fig. 4 Hydrolysis of antibody conjugates monitored by disappearance of dithiomaleimide absorbance (λmax = 402–406 nm). | ||
Rapid hydrolysis of DBM reagents
Given the acceleration in hydrolysis afforded the dithiomaleimide bridges by the electron withdrawing linkers, we were intrigued to compare the hydrolytic lability of the dibromomaleimide reagents themselves. Using a similar approach, we monitored the disappearance of absorbance associated with dibromomaleimide; in this case at 325 nm. The C-2 and C-6 linkers (reagents 4 and 5) were selected for the study, as they offered clearly distinguishable absorbances at this wavelength, which are again lost upon hydrolysis to generate the maleamic acids (see the ESI†). Intriguingly the dibromomaleimides were found to hydrolyse extremely rapidly, with a half-life of under a minute for the C-2 linker, even at the slightly lower pH of 8.0 (Fig. 5). This reveals that bromines serve to further activate the imide to hydrolysis, through inductive electron-withdrawal. For comparison, dithiophenolmaleimides were found to hydrolyse much more slowly (t1/2 = 30–60 min, ESI Fig. S25†), again highlighting the tuneable reactivity of NGMs.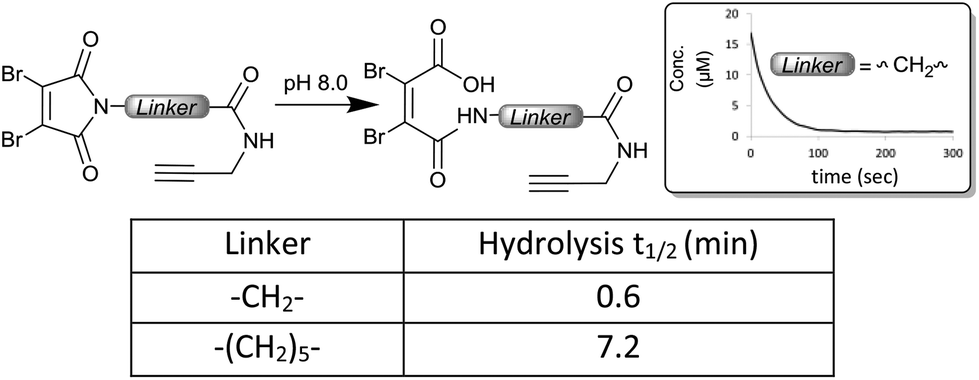 | ||
| Fig. 5 Hydrolysis of the DBM reagents 4 and 5 monitored by the loss of absorbance at 325 nm. | ||
The significance of this rapid reagent hydrolysis is that the DBM reagents must conjugate at sub-1 min timescales to affect efficient conjugation, which is clearly the case in the antibody conjugations reported herein. Indeed this is confirmed by the UV analysis of the conjugation, which reveals the maximum dithiomaleimide absorbance to be observed immediately upon taking the first reading (<1 min). It is also notable that any unreacted DBM reagent post-conjugation will be rapidly deactivated by hydrolysis. This suggests that there is unlikely to be any requirement to remove excess reagent post-conjugation. Indeed we trialled a one-pot conjugation and hydrolysis in 1 h using reagents 4 and 5, and found that the outcome by LC-MS was unchanged, efficient conjugation to generate DAR 4 conjugates.
Maleamic acid stability of C-2 and aryl linkers at low pH
Having previously shown that maleamic acid conjugates are robustly stable in serum,12d we focused here on further profiling their known susceptibility to cleave at low pH.12a We were keen to compare the C-2 and aryl linkers described in this work, in case the nature of the linker affected the rate of cleavage, and as a result the stability of the conjugates. Thus Alexa Fluor 488® was attached by a CuAAC reaction onto the pendant alkynes12d and the conjugates monitored over 24 h by fluorescence SEC-HPLC (Fig. 6). We chose to compare pH 7.4 which would approximate the pH in circulation, with pH 5.5 representing acidic conditions approximating endosomal pH24 (and thus the most acidic conditions a recycling antibody is likely to experience, prior to lysosomal degradation). The conjugates with C-2 and aryl linkers showed complete stability at pH 7.4 (even up to 10 days, see ESI Fig. S32†), whilst only the N-aryl conjugate showed a hint of cleavage at pH 5.5. Overall it can be summarised that maleamic acid antibody conjugates with various linkers will have robust stability in circulation in vivo.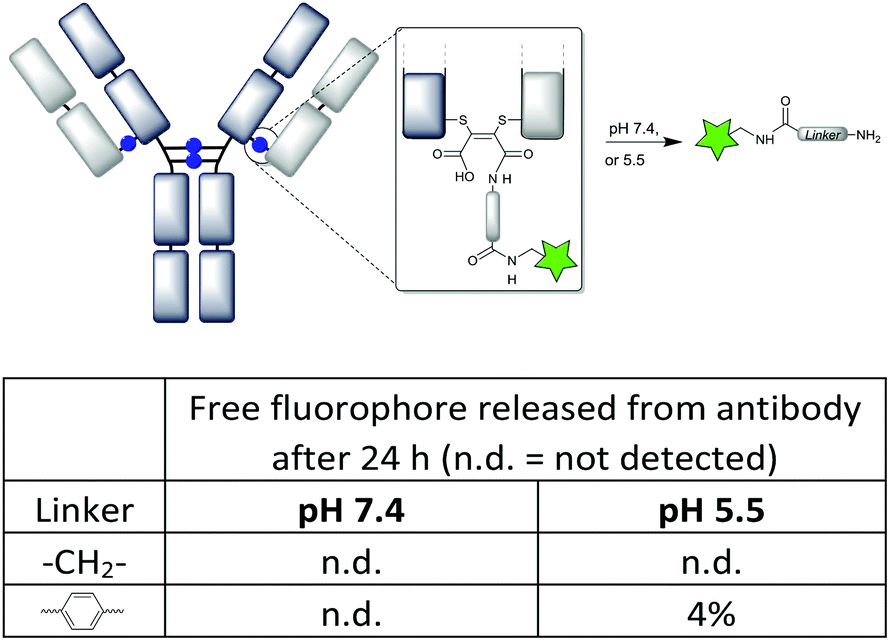 | ||
| Fig. 6 Stability of the maleamic acid conjugates with C-2 and aryl linkers tested at pH 7.4 and 5.5, measured by fluorescence SEC-HPLC (see the ESI† for traces). | ||
Functional DBM conjugation using optimised linkers and protocols
With improved linkers in hand, and robust stability further demonstrated, we constructed functionalised antibody conjugates using the optimised conditions. The C-2 linker was selected to avoid the unnecessary incorporation of extra hydrophobicity by an aryl ring, whilst dansyl and doxorubicin were chosen as a model fluorophore and drug respectively. The conjugations were carried out using the optimised protocol established, 5 min conjugation at pH 8.5 with a further 1 h for hydrolysis. The LC-MS analysis revealed that the two isomeric DAR 4 species were again successfully generated (Fig. 7). This was also consistent with the UV read-out for the doxorubicin conjugate, which indicated a DAR of 4.1.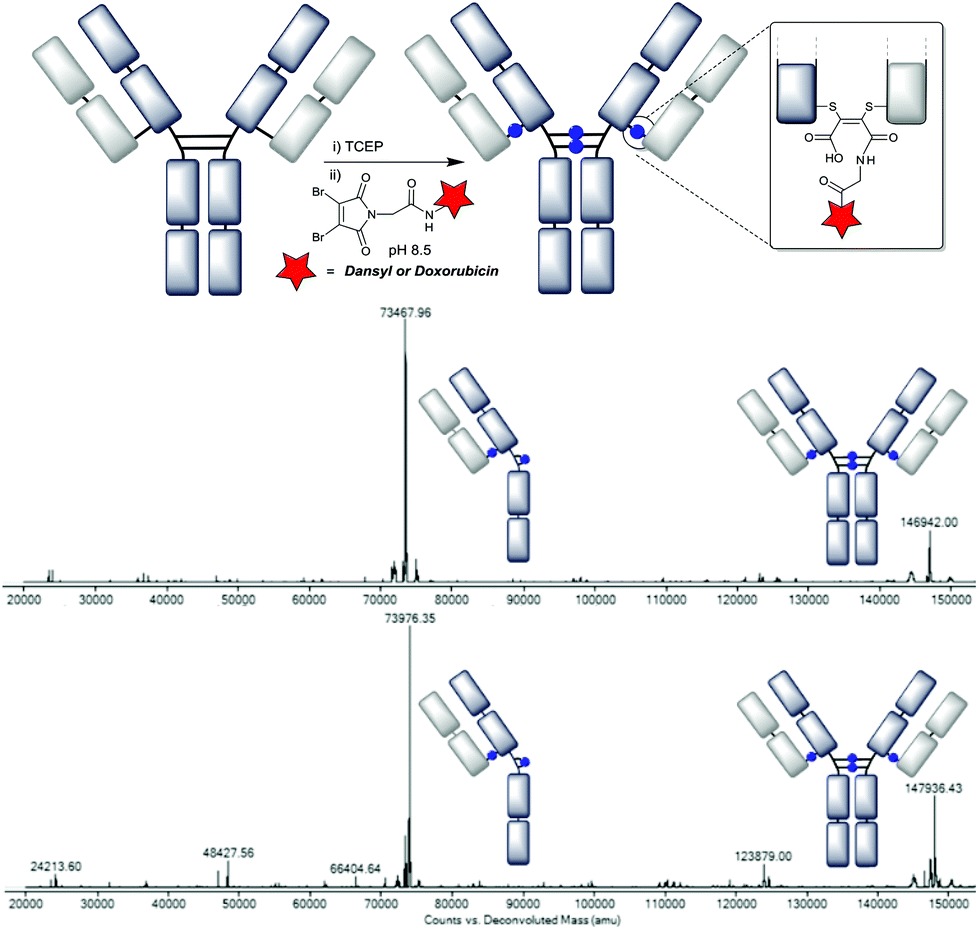 | ||
| Fig. 7 Deconvoluted LC-MS data on the trastuzumab–NGM conjugates: A – using DBM-C2-dansyl (mass observed 73468 and 146942, expected 73480 and 146959); C – using DBM-C2-doxorubicin (mass observed 73976 and 147936, expected 73980 and 147959). | ||
Conclusions
Dibromomaleimides with electron-withdrawing C-2 linkers are demonstrated to offer an optimised bioconjugation platform for the construction of highly homogeneous and robustly stable antibody conjugates from native antibodies. These reagents enable a protocol which takes just over 1 hour for conjugation and ‘locking’ by hydrolysis, to generate an antibody loaded with a DAR of 4. This represents a substantial improvement in the 72 h protocol previously described for NGM reagents. By the combined use of these ‘fast-hydrolysing’ linkers and a pH of 8.5, we also show that improved homogeneity is inferred on the conjugates. This is due to the rapid hydrolysis of the dithiomaleimide bridges preventing undesired side-reactions, such as C-terminal decarboxylation of the light chain. It is notable that antibodies themselves are stable to such short exposure to alkaline conditions. Indeed they have been reported to only undergo degradation, by partial asparagine deamidation, under much harsher conditions (pH 9.2, 45 °C, 48 h), and even then full binding and structural integrity is retained.25Whilst other reports in the literature have focused on succinimide hydrolysis as a means to improve the stability of classical maleimide conjugates from native antibodies, the corresponding ADCs remain a heterogeneous mixture. The DBM technology combines stabilisation through hydrolysis with a significant improvement in homogeneity and controlled drug loading delivered by disulfide bridging, as demonstrated by LC-MS characterisation. We suggest that these readily available DBM reagents offer a convenient approach to antibody bioconjugation more widely, and can also be applicable to other peptide, protein and polymer scaffolds.
Acknowledgements
The authors gratefully acknowledge BBSRC, BRC, HEFCE, and EPSRC for funding. We also acknowledge Prof. Erik Arstad and Dr Kerstin Sanders for assistance with the fluorescence SEC-HPLC and EPSRC UK National Mass Spectrometry Facility (NMSF), Swansea.Notes and references
- (a) J. R. Adair, P. W. Howard, J. A. Hartley, D. G. Williams and K. A. Chester, Expert Opin. Biol. Ther., 2012, 12, 1191–1206 CrossRef CAS PubMed; (b) A. M. Wu, Methods, 2014, 65, 139–147 CrossRef CAS PubMed.
- (a) L. T. Wang, G. Amphlett, W. A. Blattler, J. M. Lambert and W. Zhang, Protein Sci., 2005, 14, 2436–2446 CrossRef CAS PubMed; (b) J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. M. Lu, Y. G. Meng, C. Ng, J. H. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. L. Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed.
- J. R. Junutula, K. M. Flagella, R. A. Graham, K. L. Parsons, E. Ha, H. Raab, S. Bhakta, T. Nguyen, D. L. Dugger, G. M. Li, E. Mai, G. D. L. Phillips, H. Hiraragi, R. N. Fuji, J. Tibbitts, R. Vandlen, S. D. Spencer, R. H. Scheller, P. Polakis and M. X. Sliwkowski, Clin. Cancer Res., 2010, 16, 4769–4778 CrossRef CAS PubMed.
- K. J. Hamblett, P. D. Senter, D. F. Chace, M. M. C. Sun, J. Lenox, C. G. Cerveny, K. M. Kissler, S. X. Bernhardt, A. K. Kopcha, R. F. Zabinski, D. L. Meyer and J. A. Francisco, Clin. Cancer Res., 2004, 10, 7063–7070 CrossRef CAS PubMed.
- (a) N. S. Beckley, K. P. Lazzareschi, H. W. Chih, V. K. Sharma and H. L. Flores, Bioconjugate Chem., 2013, 24, 1674–1683 CrossRef CAS PubMed; (b) Y. T. Adem, K. A. Schwarz, E. Duenas, T. W. Patapoff, W. J. Galush and O. Esue, Bioconjugate Chem., 2014, 25, 656–664 CrossRef CAS PubMed.
- C. A. Boswell, E. E. Mundo, C. Zhang, D. Bumbaca, N. R. Valle, K. R. Kozak, A. Fourie, J. Chuh, N. Koppada, O. Saad, H. Gill, B. Q. Shen, B. Rubinfeld, J. Tibbitts, S. Kaur, F. P. Theil, P. J. Fielder, L. A. Khawli and K. Lin, Bioconjugate Chem., 2011, 22, 1994–2004 CrossRef CAS PubMed.
- (a) P. Dennler, E. Fischer and R. Schibli, Antibodies, 2015, 4, 197–224 CrossRef; (b) D. Jackson, J. Atkinson, C. I. Guevara, C. Y. Zhang, V. Kery, S. J. Moon, C. Virata, P. Yang, C. Lowe, J. Pinkstaff, H. Cho, N. Knudsen, A. Manibusan, F. Tian, Y. Sun, Y. C. Lu, A. Sellers, X. C. Jia, I. Joseph, B. Anand, K. Morrison, D. S. Pereira and D. Stover, PLoS One, 2014, 9, 14 Search PubMed.
- (a) P. Akkapeddi, S. A. Azizi, A. M. Freedy, P. Cal, P. M. P. Gois and G. J. L. Bernardes, Chem. Sci., 2016, 7, 2954–2963 RSC; (b) D. Y. Jackson, Org. Process Res. Dev., 2016, 20, 852–866 CrossRef CAS.
- V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 113–118 CrossRef PubMed.
- (a) S. L. Kuan, T. Wang and T. Weil, Chem. – Eur. J., 2016, 22, 17112–17129 CrossRef CAS PubMed; (b) P. Bryant, M. Pabst, G. Badescu, M. Bird, W. McDowell, E. Jamieson, J. Swierkosz, K. Jurlewicz, R. Tommasi, K. Henseleit, X. B. Sheng, N. Camper, A. Manin, K. Kozakowska, K. Peciak, E. Laurine, R. Grygorash, A. Kyle, D. Morris, V. Parekh, A. Abhilash, J. W. Choi, J. Edwards, M. Frigerio, M. P. Baker and A. Godwin, Mol. Pharm., 2015, 12, 1872–1879 CrossRef CAS PubMed; (c) G. Badescu, P. Bryant, M. Bird, K. Henseleit, J. Swierkosz, V. Parekh, R. Tommasi, E. Pawlisz, K. Jurlewicz, M. Farys, N. Camper, X. B. Sheng, M. Fisher, R. Grygorash, A. Kyle, A. Abhilash, M. Frigerio, J. Edwards and A. Godwin, Bioconjugate Chem., 2014, 25, 1124–1136 CrossRef CAS PubMed; (d) A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS PubMed.
- (a) L. M. Tedaldi, M. E. B. Smith, R. Nathani and J. R. Baker, Chem. Commun., 2009, 6583–6585 RSC; (b) M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed; (c) C. P. Ryan, M. E. B. Smith, F. F. Schumacher, D. Grohmann, D. Papaioannou, G. Waksman, F. Werner, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 5452–5454 RSC.
- (a) L. Castaneda, A. Maruani, F. F. Schumacher, E. Miranda, V. Chudasama, K. A. Chester, J. R. Baker, M. E. B. Smith and S. Caddick, Chem. Commun., 2013, 49, 8187–8189 RSC; (b) F. F. Schumacher, V. A. Sanchania, B. Tolner, Z. V. F. Wright, C. P. Ryan, M. E. B. Smith, J. M. Ward, S. Caddick, C. W. M. Kay, G. Aeppli, K. A. Chester and J. R. Baker, Sci. Rep., 2013, 3, 1525 Search PubMed; (c) F. F. Schumacher, J. P. M. Nunes, A. Maruani, V. Chudasama, M. E. B. Smith, K. A. Chester, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2014, 12, 7261–7269 RSC; (d) J. P. Nunes, M. Morais, V. Vassileva, E. Robinson, V. S. Rajkumar, M. E. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, Chem. Commun., 2015, 51, 10624–10627 RSC.
- E. Robinson, J. P. Nunes, V. Vassileva, A. Maruani, J. Nogueira, M. E. B. Smith, S. Caddick, J. R. Baker and V. Chudasama, RSC Adv., 2017, 7, 9073–9077 RSC.
- C. R. Behrens, E. H. Ha, L. L. Chinn, S. Bowers, G. Probst, M. Fitch-Bruhns, J. Monteon, A. Valdiosera, A. Bermudez, S. Liao-Chan, T. Wong, J. Melnick, J.-W. Theunissen, M. R. Flory, D. Houser, K. Venstrom, Z. Levashova, P. Sauer, T.-S. Migone, E. H. van der Horst, R. L. Halcomb and D. Y. Jackson, Mol. Pharm., 2015, 12, 3986–3998 CrossRef CAS PubMed.
- (a) S. Y. Long, Q. Q. Tang, Y. Wu, L. X. Wang, K. Zhang and Y. M. Chen, React. Funct. Polym., 2014, 80, 15–20 CrossRef CAS; (b) S. Ramesh, P. Cherkupally, T. Govender, H. G. Kruger, F. Albericio and B. G. de la Torre, Org. Lett., 2015, 17, 464–467 CrossRef CAS PubMed; (c) A. Ross, H. Durmaz, K. Cheng, X. P. Deng, Y. W. Liu, J. Oh, Z. Chen and J. Lahann, Langmuir, 2015, 31, 5123–5129 CrossRef CAS PubMed; (d) Z. L. Li, L. H. Sun, J. M. Ma, Z. Zeng and H. Jiang, Polymer, 2016, 84, 336–342 CrossRef CAS; (e) M. P. Robin and R. K. O'Reilly, Chem. Sci., 2014, 5, 2717–2723 RSC; (f) M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 1847–1852 CrossRef CAS PubMed; (g) M. S. S. Palanki, A. Bhat, B. Bolanos, F. Brunel, J. Del Rosario, D. Dettling, M. Horn, R. Lappe, R. Preston, A. Sievers, N. Stankovic, G. Woodnut and G. Chen, Bioorg. Med. Chem. Lett., 2013, 23, 402–406 CrossRef CAS PubMed.
- (a) S. D. Fontaine, R. Reid, L. Robinson, G. W. Ashley and D. V. Santi, Bioconjugate Chem., 2015, 26, 145–152 CrossRef CAS PubMed; (b) R. P. Lyon, J. R. Setter, T. D. Bovee, S. O. Doronina, J. H. Hunter, M. E. Anderson, C. L. Balasubramanian, S. M. Duniho, C. I. Leiske, F. Li and P. D. Senter, Nat. Biotechnol., 2014, 32, 1059 CrossRef CAS PubMed.
- R. J. Christie, R. Fleming, B. Bezabeh, R. Woods, S. Mao, J. Harper, A. Joseph, Q. L. Wang, Z. Q. Xu, H. Wu, C. S. Gao and N. Dimasi, J. Controlled Release, 2015, 220, 660–670 CrossRef CAS PubMed.
- U. Muus, C. Hose, W. Yao, T. Kosakowska-Cholody, D. Farnsworth, M. Dyba, G. T. Lountos, D. S. Waugh, A. Monks, T. R. Burke Jr. and C. J. Michejda, Bioorg. Med. Chem., 2010, 18, 4535–4541 CrossRef CAS PubMed.
- L. Castaneda, Z. V. F. Wright, C. Marculescu, T. M. Tran, V. Chudasama, A. Maruani, E. A. Hull, J. P. M. Nunes, R. J. Fitzmaurice, M. E. B. Smith, L. H. Jones, S. Caddick and J. R. Baker, Tetrahedron Lett., 2013, 54, 3493–3495 CrossRef CAS PubMed.
- H. A. Burris III, H. S. Rugo, S. J. Vukelja, C. L. Vogel, R. A. Borson, S. Limentani, E. Tan-Chiu, I. E. Krop, R. A. Michaelson, S. Girish, L. Amler, M. Zheng, Y.-W. Chu, B. Klencke and J. A. O'Shaughnessy, J. Clin. Oncol., 2011, 29, 398–405 CrossRef PubMed.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. L. Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed.
- D. A. Richards, S. A. Fletcher, M. Nobles, H. Kossen, L. Tedaldi, V. Chudasama, A. Tinker and J. R. Baker, Org. Biomol. Chem., 2015, 14, 455–459 Search PubMed.
- L. M. Tedaldi, A. E. Aliev and J. R. Baker, Chem. Commun., 2012, 48, 4725–4727 RSC.
- C. J. Choy, J. J. Geruntho, A. L. Davis and C. E. Berkman, Bioconjugate Chem., 2016, 27, 824–830 CrossRef CAS PubMed.
- L. N. Tumey, M. Charati, T. He, E. Sousa, D. S. Ma, X. G. Han, T. Clark, J. Casavant, F. Loganzo, F. Barletta, J. Lucas and E. I. Graziani, Bioconjugate Chem., 2014, 25, 1871–1880 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob00220c |
| This journal is © The Royal Society of Chemistry 2017 |