
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsolation and structure elucidation of natural products of three soft corals and a sponge from the coast of Madagascar†
Marie Pascaline
Rahelivao
a,
Tilo
Lübken
 a,
Margit
Gruner
a,
Olga
Kataeva
b,
Rahanira
Ralambondrahety
c,
Hanta
Andriamanantoanina
c,
Marek P.
Checinski
d,
Ingmar
Bauer
a and
Hans-Joachim
Knölker
*a
a,
Margit
Gruner
a,
Olga
Kataeva
b,
Rahanira
Ralambondrahety
c,
Hanta
Andriamanantoanina
c,
Marek P.
Checinski
d,
Ingmar
Bauer
a and
Hans-Joachim
Knölker
*a
aDepartment Chemie, Technische Universität Dresden, Bergstr. 66, 01069 Dresden, Germany. E-mail: hans-joachim.knoelker@tu-dresden.de; Fax: +49 351 463-37030
bA. M. Butlerov Chemistry Institute, Kazan Federal University, Kremlevskaya Str. 18, Kazan 420008, Russia
cCentre National de Recherche sur l'Environnement, BP 1739, Antananarivo 101, Madagascar
dCreativeQuantum GmbH, Wegedornstr. 32, 12524 Berlin, Germany
First published on 24th February 2017
Abstract
We investigated the three soft corals Sarcophyton stellatum, Capnella fungiformis and Lobophytum crassum and the sponge Pseudoceratina arabica, which have been collected at the coast of Madagascar. In addition to previously known marine natural products, S. stellatum provided the new (+)-enantiomer of the cembranoid (1E,3E,11E)-7,8-epoxycembra-1,3,11,15-tetraene (2). Capnella fungiformis afforded three new natural products, ethyl 5-[(1E,5Z)-2,6-dimethylocta-1,5,7-trienyl]furan-3-carboxylate (6), ethyl 5-[(1E,5E)-2,6-dimethylocta-1,5,7-trienyl]furan-3-carboxylate (7) and the diepoxyguaiane sesquiterpene oxyfungiformin (9a). The extracts of all three soft corals exhibited moderate activities against the malarial parasite Plasmodium falciparum. Extracts of the sponge Pseudoceratina arabica proved to be very active against a series of Gram-positive and Gram-negative bacteria.
Introduction
In continuation of our studies on the chemical constituents of marine organisms from the coast of Madagascar including red algae,1 brown algae,2 and soft corals,3 we herein turned our attention to soft corals of the species Sarcophyton stellatum, Capnella fungiformis, and Lobophytum crassum. A sponge of the species Pseudoceratina arabica has also been included in the present investigation. Soft corals and sponges have been frequently investigated and found to be rich sources of biologically active compounds.4–14 However, only a few reports are available on soft corals and sponges from the coast of Madagascar.3,15–17 This region is of particular interest since it is known that the marine environment of the Southern Africa and Western Indian Ocean region contains a wealth of endemic species, which increases the chance to find new natural products with useful biological activities.18–20 In our previous study on Madagascan soft corals, we have isolated five new compounds including the cadinane-type sesquiterpene vanderlandin, the spatane-type diterpene gravilin and the cembranoid diterpene isodecaryiol.3 These results encouraged us to extend our search for new and potentially bioactive compounds by investigating further soft coral species and a sponge from this region.Results and discussion
Sarcophyton stellatum
Several species of soft corals of the genus Sarcophyton have been investigated for their chemical constituents.6,7,11,13,14,21 However, no such reports are available for the species S. stellatum. We collected samples of this species at the inner reef of Mahambo, Madagascar, at a depth of 5–6 m. The bodies of S. stellatum were sliced and exhaustively extracted with methanol at room temperature. The methanol extract of S. stellatum exhibited moderate antimalarial activity with an IC50 value of 35.20 ± 5.02 μg mL−1 for the inhibition of the FCM29 strain of Plasmodium falciparum. Further solvent partitioning of the crude extract afforded a diethyl ether and a dichloromethane extract. Three known cembranoid diterpenes and the unknown enantiomer of a fourth representative were isolated from the diethyl ether and dichloromethane extracts of S. stellatum. Three of these compounds were isolated from both extracts, namely (+)-(7S,8S)-epoxy-7,8-dihydrocembrene C [(+)-1], (+)-(1E,3E,11E)-7,8-epoxycembra-1,3,11,15-tetraene [(+)-2] and (−)-(2R,7R,8R)-sarcophytoxide [(−)-4] (Fig. 1). The fourth compound, (+)-(7R,8R,14S,1Z,3E,11E)-14-acetoxy-7,8-epoxycembra-1,3,11-triene [(+)-3], was found only in the diethyl ether extract. In addition, the diethyl ether extract of S. stellatum contained a small amount of (S)-3-O-octadecylglycerol (batyl alcohol) (5), the scaffold structure for a variety of ether lipids of the 1-O-alkyl-2,3-O-diacyl-sn-glycerol type (Fig. 2).22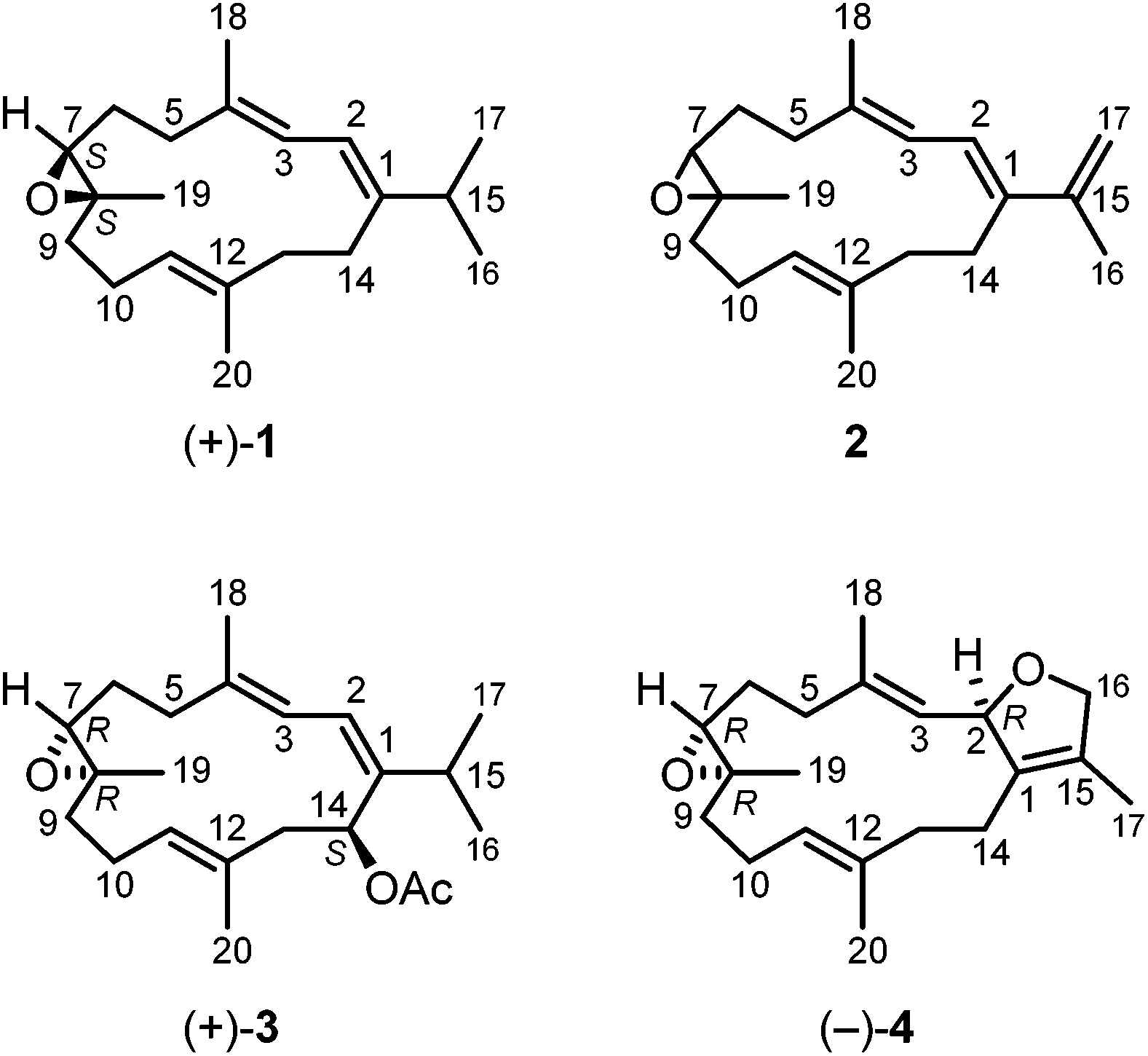 | ||
| Fig. 1 Structures of the cembranoid diterpenes isolated from Sarcophyton stellatum. | ||
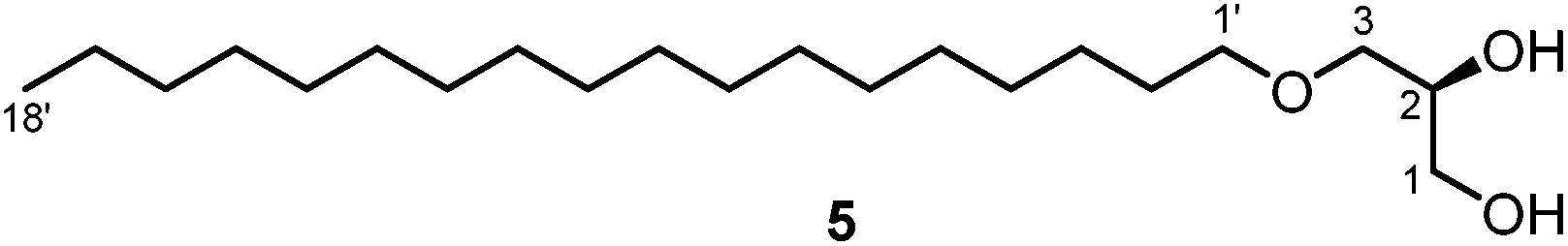 | ||
| Fig. 2 Structure of (S)-3-O-octadecylglycerol (batyl alcohol) (5). | ||
Compound (+)-1 was isolated as a colorless oil with a specific optical rotation of [α]20D = +44.6 (c 0.5, MeOH). CD spectroscopy revealed a molar circular dichroism of Δε = +0.23 (242 nm, MeOH). The molecular formula of (+)-1 was established as C20H32O by the monoisotopic mass of 288 obtained from the EI mass spectrum in combination with the number and intensities of the 1H and 13C NMR signals. The structure of this compound was deduced from extensive 1D and 2D NMR measurements (COSY, HSQC, HMBC, and NOESY). Our NMR data were generally in agreement with those reported for (+)-(7S,8S)-epoxy-7,8-dihydrocembrene C [(+)-1] in the literature (Table S1†),23 except for the presence of an additional signal at δH = 1.51 ppm which we assigned to H-9b. In addition, we revised the assignment of the 13C NMR signals for C-6 and C-9 and of some proton signals. The isolation of (+)-(7S,8S)-epoxy-7,8-dihydrocembrene C [(+)-1] has been reported before by Deng and coworkers from the soft coral S. molle with a specific optical rotation of [α]25D = +99.0 (c 1.9, acetone)24 and by Seifert et al. from S. ehrenbergi with [α]25D = +19 (c 0.5, CHCl3).23 (−)-(7R,8R)-Epoxy-7,8-dihydrocembrene C [(−)-1] has been isolated first from the soft coral S. crassocaule by Bowden et al. with an [α]D value of −22.5 (c 0.19, CHCl3).25 Subsequently, the same compound has been obtained from an unidentified species of a Caribbean gorgonian of the genus Eunicea26 and from the soft coral S. trocheliophorum.27 The absolute stereochemistry of (−)-1 was assigned as (7R,8R) via enantioselective total synthesis in 2000 by Li et al.28 They reported a specific optical rotation of [α]20D = −25.2 (c 0.21, CHCl3) which is in good agreement with the value of the natural product originally isolated by Bowden. We obtained (+)-(7S,8S)-epoxy-7,8-dihydrocembrene C [(+)-1] for the first time from the soft coral S. stellatum. Seifert et al. reported moderate antiproliferative activity of (+)-1 against the cell lines HUVEC and K-562 and a moderate cytotoxicity against the HeLa cell line.23
The structurally related metabolite (+)-2 was isolated as a colorless oil with a specific optical rotation of [α]20D = +12.0 (c 0.5, MeOH). The CD spectrum of (+)-2 in methanol exhibited a molar circular dichroism of Δε = −0.11 (251 nm). The [M + H]+ ion at m/z = 287 in the ESI mass spectrum in combination with the number and intensities of the 1H and 13C NMR signals suggested a molecular formula of C20H30O. The structure of metabolite (+)-2 was elucidated based on the analysis of the 1D and 2D NMR spectra. The NMR data were in reasonable agreement with those reported in the literature (Table S2†) for the enantiomeric diterpene (−)-(1E,3E,11E)-7,8-epoxycembra-1,3,11,15-tetraene [(−)-2], which has been originally obtained from the soft coral S. crassocaule25 with an [α]D value of −14.4 (c 0.1, CHCl3) and subsequently also from a Lobophytum species.29 Thus, we have isolated for the first time the opposite enantiomer, (+)-(1E,3E,11E)-7,8-epoxycembra-1,3,11,15-tetraene [(+)-2]. The absolute configuration of this natural product still remains to be determined.
Compound (+)-3 was isolated as a colorless oil with a specific optical rotation of [α]20D = +171.8 (c 0.1, MeOH). The EI mass spectrum with a molecular ion peak at m/z = 346 and the 1H and 13C NMR data were in agreement with the molecular formula of C22H34O3. The IR spectrum of (+)-3 showed an absorption band for an ester group (1724 cm−1). This assignment was confirmed by the 1H and 13C NMR spectra displaying signals for an acetate moiety [δH = 2.04 ppm (s), δC = 170.19 (C) and 21.37 ppm (CH3)]. The 13C NMR spectrum showed signals for 22 resolved carbon atoms including six methyl groups, five methylene groups, six methine groups, and five quaternary carbon atoms. The analytical data proved to be identical with those of (+)-(7R,8R,14S,1Z,3E,11E)-14-acetoxy-7,8-epoxycembra-1,3,11-triene [(+)-3] (Table S3†), which has been isolated by Bowden et al. from the soft coral S. trocheliophorum.30 Prior to that report, Kobayashi's group obtained (+)-3 as a semi-synthetic oxidation product of sarcophytol A.31 They deduced the (14S)-configuration of (+)-3 from sarcophytol A which had been determined by using Horeau's method. Accordingly, the (7R)-configuration was assigned by isomerization of (+)-3 to the corresponding allyl alcohol (Δ8-7,14-dihydroxy compound) and determination of the absolute configuration by the same method. Bowden et al. also determined a (14S)-configuration for the corresponding 14-hydroxy compound, which was interconvertible with (+)-3, by using Mosher's method.30 They assigned a (7R)-configuration for compound (+)-3 in analogy to a related Δ8-7,14-dihydroxy compound which was isolated along with (+)-3. Knowing the configuration at C-7, the (8R)-configuration was derived following Bowden's reasoning.30 The specific optical rotation values of (+)-3 reported by Kobayashi ([α]D = +150 (c 1.02, CHCl3))31 and Bowden ([α]D = +136 (c 1.1, CHCl3))30 confirm that we have obtained the same enantiomer ([α]D = +171.8 (c 0.1, MeOH)). Thus, we have isolated (+)-(7R,8R,14S,1Z,3E,11E)-14-acetoxy-7,8-epoxycembra-1,3,11-triene [(+)-3] for the first time from the soft coral S. stellatum. It should be noted that (+)-3 exhibited a moderate cytotoxicity against P388 (murine leukaemia), A549 (human lung carcinoma), HT29 (human colon carcinoma) and MEL28 (human melanoma) cell lines.30
Compound (−)-4 was isolated as colorless crystals with a melting point of 60–61 °C and a value for the specific optical rotation of [α]20D = −129.4 (c 0.1, MeOH). The CD spectrum of (−)-4 in MeOH showed molar circular dichroism values of Δε = +0.01 (247 nm) and −0.01 (274 nm). The molecular formula of C20H30O2 was derived from the EI-MS, 1H and 13C NMR data. Analysis of a set of 1D and 2D NMR spectra led to the identification of (−)-4 as (−)-sarcophytoxide. This was confirmed by comparison of the 1H and 13C NMR data with those of (−)-sarcophytoxide isolated by Seifert et al. from S. ehrenbergi23 and Bowden and coworkers32,33 (Table S4†).
Recrystallization of (−)-4 from diethyl ether provided single crystals which were suitable for X-ray diffraction. The X-ray crystal structure determination unequivocally confirmed the constitution and relative configuration of (−)-sarcophytoxide [(−)-4] (Fig. 3). In addition, the absolute configuration of (−)-4 has been assigned by the anomalous dispersion as (2R,7R,8R) (Flack parameter: χ = 0.1(0)). (+)-Sarcophytoxide [(+)-4] with a specific optical rotation of [α]20D = +25 (c 2.2, MeOH) was isolated first in 1974 by Kashman and coworkers from S. glaucum.34 A few years later, Bowden et al. obtained the enantiomer (−)-4 with a specific rotation of [α]20D = −191 (c 0.4, MeOH) from the species S. ehrenbergi32 and S. birklandi.33 The absolute (2S,7S,8S)-configuration of (+)-4 from Sarcophyton sp. with a specific optical rotation of [α]20D = +135 (c 0.93, CHCl3) was assigned by Faulkner et al. by chemical correlation with the structurally known (+)-sarcophine.4,35 Similarly, Bowden et al. correlated the structure of (−)-4 with the known (2R)-sarcophytonin by reductive cleavage of the epoxide with a Zn–Cu couple and secured the absolute configuration at C-2.33 X-ray structural determination of (+)-sarcophytoxide [(+)-4], obtained from Sarcophyton sp. with a specific rotation of [α]D = +157 (c 1.0, MeOH), was reported by Kobayashi and coworkers.36 (+)-Sarcophytoxide [(+)-4] has been isolated in a study of the relationship between soft coral diversity and cembranoid diterpene production of Sarcophyton specimens from Okinawa, Japan.37 A more recent isolation of (−)-sarcophytoxide [(−)-4] has been reported by Seifert and coworkers from S. ehrenbergi with [α]25D = −128 (c 1.0, CHCl3), which corresponds to our value ([α]20D = −129.4 (c 0.1, MeOH)).23 We have isolated (−)-sarcophytoxide [(−)-4] for the first time from the soft coral S. stellatum.
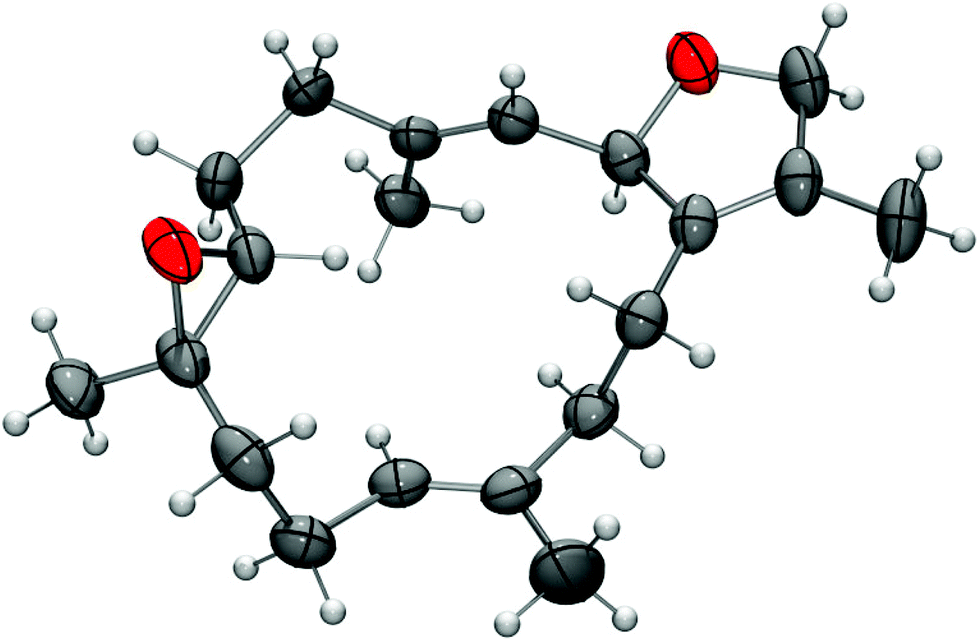 | ||
| Fig. 3 X-Ray crystal structure of (−)-sarcophytoxide [(−)-4] (thermal ellipsoids at the 50% probability level). | ||
Compound 5 (Fig. 2) was isolated as a yellow solid with a melting point of 69–70 °C and a specific optical rotation of [α]20D = +2.6 (c 0.07, MeOH). The analytical studies of compound 5 including 2D NMR experiments suggested that compound 5 is identical with (S)-3-O-octadecylglycerol (batyl alcohol), one of the most common core compounds of 1-O-alkyl-2,3-O-diacyl-sn-glycerols, which are widespread components of the non-polar lipid fractions of aquatic and terrestrial animals.22 Our analytical data are in agreement with those reported by Haraldsson and coworkers for a synthetic sample.38
Capnella fungiformis
So far, soft corals assigned to the species Capnella fungiformis have not been studied for their chemical constituents. We obtained samples of C. fungiformis from the inner reef of Mahambo, Madagascar, at a depth of about 5–6 m. The fresh material of C. fungiformis was cut and soaked in methanol immediately after collection. After removal of the solvent, the crude methanol extract was tested in an antimalarial assay and found to exhibit moderate activity with an IC50 value of 32.80 ± 3.03 μg mL−1 for the inhibition of the FCM29 strain of Plasmodium falciparum. The methanol extract of C. fungiformis was further extracted with diethyl ether and separated by column chromatography over silica gel into two fractions eluting with pentane–diethyl ether (gradient 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 7:3). Fraction F-1 was purified by two additional column chromatographic steps (pentane–diethyl ether, 10:1 and 20:1) affording an inseparable mixture of ethyl 5-[(1E,5Z)-2,6-dimethylocta-1,5,7-trienyl]furan-3-carboxylate (6) and the corresponding (1E,5E)-isomer 7 (subfraction F-1-1-1) along with octadecan-2-one (8) (subfraction F-1-1-2) (Fig. 4).
:1 to 7:3). Fraction F-1 was purified by two additional column chromatographic steps (pentane–diethyl ether, 10:1 and 20:1) affording an inseparable mixture of ethyl 5-[(1E,5Z)-2,6-dimethylocta-1,5,7-trienyl]furan-3-carboxylate (6) and the corresponding (1E,5E)-isomer 7 (subfraction F-1-1-1) along with octadecan-2-one (8) (subfraction F-1-1-2) (Fig. 4).
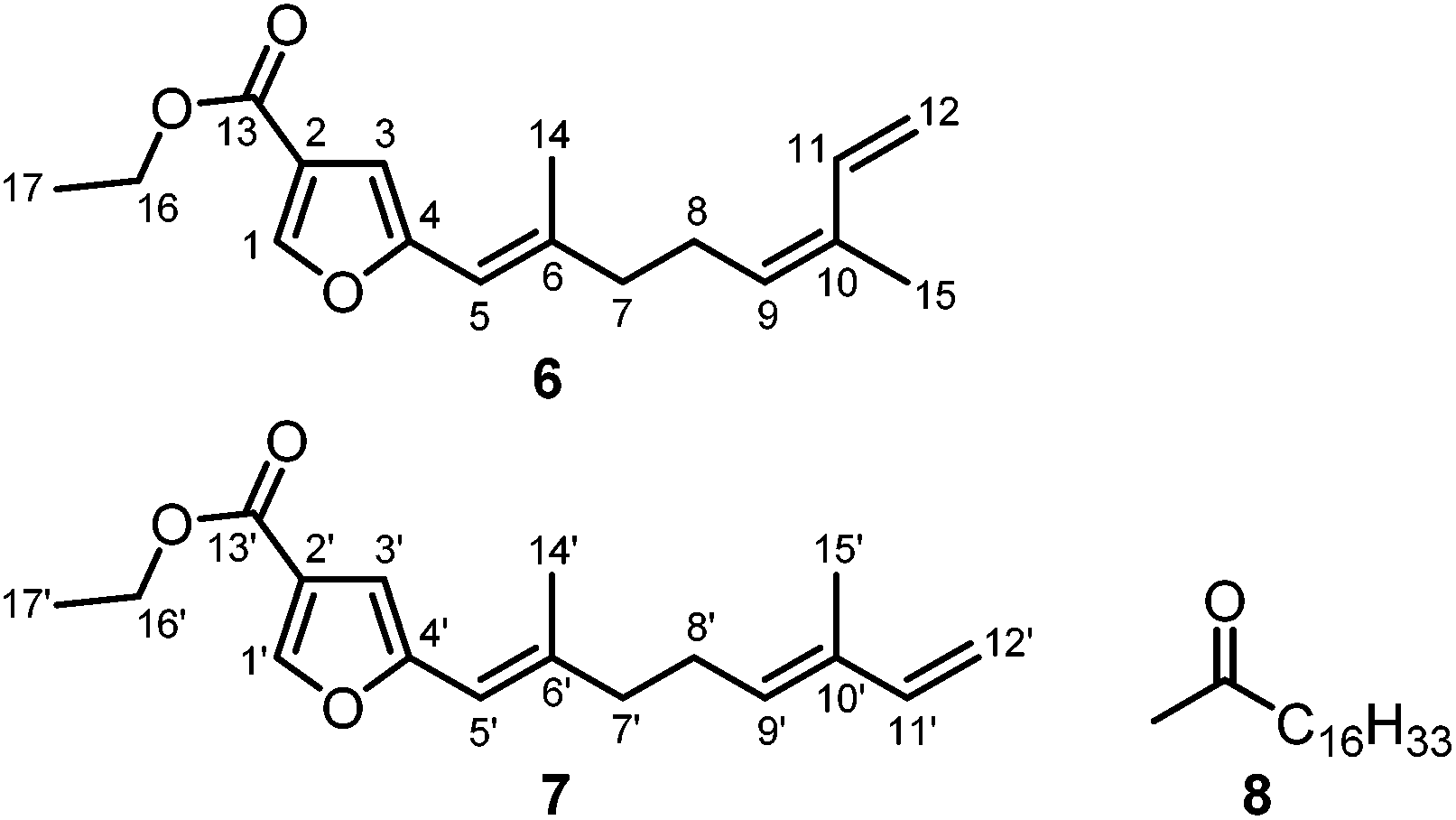 | ||
| Fig. 4 Structures of the ethyl furancarboxylates 6 and 7 (terpene numbering according to Coll et al.39) and octadecan-2-one (8). | ||
The mixture of 6 and 7 was obtained as a yellow solid. The ratio of the Z/E-isomers was determined to be 3:2 (6:7) by GC-MS and NMR spectroscopy. Their molecular formula C17H22O3, corresponding to seven double bond equivalents, was determined on the basis of the molecular ion peak at m/z = 274 in the EI mass spectra in combination with the number and intensities of the 1H and 13C NMR signals. The 1H and 13C NMR data for the isomers 6 and 7 have been assigned unambiguously from the spectra of the mixture. In combination with the DEPT spectrum, signals for three methyl groups, four methylene groups, and five methine groups were identified for each isomer. The 13C NMR spectrum displayed resonances for 17 carbon atoms, including one carbonyl group at δ = 163.38 ppm (C-13/13′) and four quaternary carbon atoms at δ = 120.63 (C-2/2′), 154.61 (C-4/4′), 140.50 (C-6/6′), and 132.84 ppm (C-10/10′). The proton signals at δ = 7.88 (s, H-1/1′) and 6.49 ppm (s, H-3/3′) and the 13C signals at δ = 145.42 (C-1/1′), 120.63 (C-2/2′), 106.74 (C-3/3′), and 154.61 ppm (C-4/4′) were assigned to a 2,4-disubstituted furan ring. Three double bonds were identified in each isomer by signals for olefinic protons at δ = 6.05 (br s, H-5/5′), 5.37 (br t, J = 7.3 Hz, H-9), 5.47 (br t, J = 7.2 Hz, H-9′), 6.76 (ddd, J = 17.3, 10.8, 0.9 Hz, H-11), 6.35 (dd, J = 17.1, 10.7 Hz, H-11′), 5.29 (br d, J = 17.3 Hz, H-12a), 5.09 (d, J = 17.3 Hz, H-12a′), 5.09 (dt, J = 10.5, 1.5 Hz, H-12b), and 4.93 ppm (d, J = 10.9 Hz, H-12b′), and by 13C NMR signals at 113.65 (C-5/5′), 140.50 (C-6/6′), 129.75 (C-9), 131.75 (C-9′), 132.84 (C-10/10′), 133.46 (C-11), 141.34 (C-11′), 113.77 (C-12), and 110.84 ppm (C-12′). The assignment of the protons to the corresponding carbon atoms was achieved by analysis of the HSQC correlations. The positions of the double bonds and the methyl groups at the side chain were confirmed by the HMBC correlations of the olefinic protons H-5/5′ with C-6/6′, C-7/7′, and C-14/14′, of H-9/9′ with C-7/7′, C-8/8′, C-11/11′, and C-15/15′, of H-11/11′ with C-9/C-9′, C-10/10′, and C-15/15′, of H-12a/12a′ with C-10/10′ and C-11/11′, and of H-12b/12b′ with C-10/10′ (Fig. 5, Tables S5 and S6†). Furthermore, the HMBC correlations of H-1/1′ with C-2/2′, C-3/3′, and C-4/4′ and of H-3/3′ with C-1/1′, C-2/2′, C-4/4′, and C-13/13′ established the furan ring and the location of the ester carbonyl group at C-2/2′. The correlation of H3-14/14′ with C-6/6′, C-5/5′, and C-4/4′ suggested that the methyl group is located at C-6/6′ of the Δ5/Δ5′-double bond which is attached to C-4/4′ of the furan ring. The geometry of the double bonds in 6 and 7 was determined by analysis of the NOE correlations (Fig. 5). The protons H2-8 and H-11 of 6 exhibited a strong NOE correlation, whereas the protons H2-8′ and H-11′ in 7 showed no such interaction. Accordingly, NOESY cross-peaks have been observed for the protons H2-8′ and H3-15′ in 7 but not for H2-8 and H3-15 in 6. This fact strongly suggests a Z-configuration of the Δ9-double bond in 6 and an E-configuration for the corresponding double bond in compound 7. The geometry of the Δ5/Δ5′-double bond was determined to be E in both isomers due to strong NOE interactions between H2-7/7′ and H-5/5′ as compared to weak NOE correlations of the protons H3-14/14′ with the protons H-5/5′.
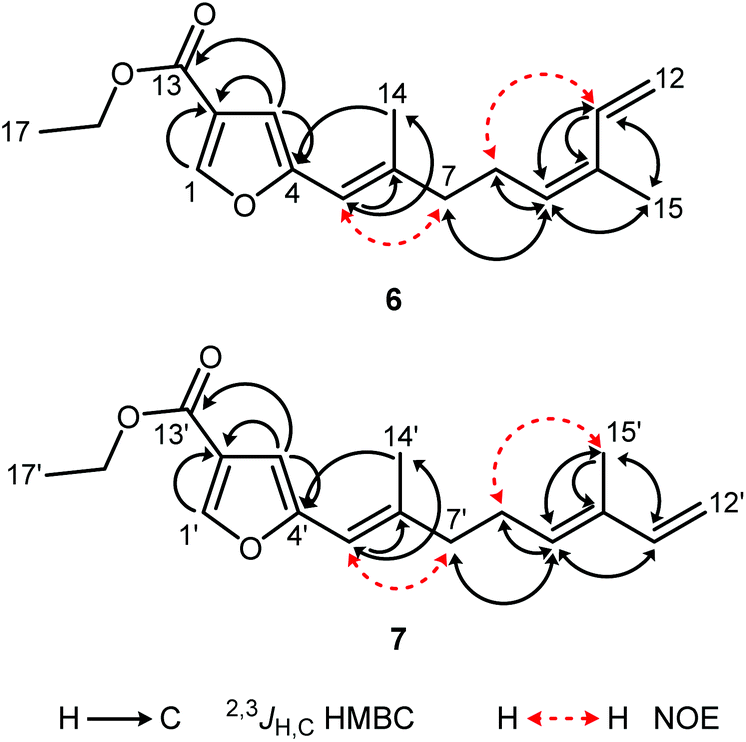 | ||
| Fig. 5 Characteristic HMBC and NOESY correlations of compounds 6 and 7. | ||
The ethyl furancarboxylates 6 and 7 are sesquiterpenes which have not been reported as natural products so far. In 1977, Coll et al. isolated 5-[(1E,5Z)-2,6-dimethylocta-1,5,7-trienyl]furan-3-carboxylic acid, corresponding to compound 6, from the Australian soft coral Sinularia gonatodes Kolonko.39 Derivatization with diazomethane afforded the corresponding methyl ester. Subsequently, Bowden and Coll obtained both, the (1E,5Z) and the (1E,5E)-furan-3-carboxylic acids, along with their methyl esters from Sinularia cappilosa.40 5-[(1E,5Z)-2,6-Dimethylocta-1,5,7-trienyl]furan-3-carboxylic acid exhibited anti-inflammatory activity as shown by inactivation of bee venom phospholipase A2.41 More recent isolations of 5-[(1E,5Z)-2,6-dimethylocta-1,5,7-trienyl]furan-3-carboxylic acid, the corresponding (1E,5E)-isomer, and their methyl esters have been reported by Venkateswarlu and coworkers from Sinularia kavarattiensis.42 A single report for the (1E,5E)-ethyl ester 7 has been published by Faulkner et al. as a synthetic intermediate towards the corresponding furancarboxylic acid.43 The corresponding ethyl 5-[(1E,5Z)-2,6-dimethylocta-1,5,7-trienyl]furan-3-carboxylate (6) has not been reported previously. Since we have not used ethanol during the isolation process, the ethyl furancarboxylates 6 and 7 represent new natural products.
Octadecan-2-one (8) was obtained from subfraction F-1-1-2 of the diethyl ether extract of C. fungiformis as a colorless solid with a melting point of 57–58 °C. Compound 8 has been isolated previously from various natural sources and the analytical data (IR, 1H NMR, EI-MS) are in agreement with those reported in the literature.44
Subfraction F-1-2 of the diethyl ether extract of C. fungiformis afforded compound 9a (Fig. 6) as a colorless solid with a specific optical rotation of [α]20D = +38.0 (c 0.05, MeOH) and a melting point of 66–66.5 °C. The ESI mass spectrum of 9a displayed peaks at 237 and 254 mass units for [M + H]+ and [M + NH4]+ ions, respectively. Detailed analysis of the 2D NMR spectra led to the assignment of all chemical shifts in the 1H and 13C NMR spectra for compound 9a (Table 1). Signals of four methyl groups, four methylene groups, four methine groups, and three quaternary carbon atoms were identified by 1H NMR, 13C NMR, and DEPT measurements. The COSY experiment revealed the presence of four spin systems (Fig. 7). The first one consists of two coupled methylene groups which further interact with the methine proton H-4 and the methyl protons H3-15 to give the fragment –CH2–CH2–CH(CH3)–. The second spin system extends from H-8α/H-8β via H-9α/H-9β and H-10 to the methyl protons H3-14 representing the same structural fragment. A third spin system is formed by the isopropyl group (H-11, H3-12, H3-13) and, finally, the isolated proton H-6 completes the number of observed spin systems. A complete proton to carbon assignment was achieved by analysis of the HSQC spectrum. The presence of a guaiane sesquiterpene skeleton was confirmed by the HMBC spectrum. Characteristic HMBC interactions, which led to the elucidation of the fused seven- and five-membered ring system by connecting the proton spin systems, include those of C-1/H-10, C-1/H-6, C-1/H-4, C-1/H-2α, C-5/H-6, C-5/H-4, C-5/H-2α, C-4/H-6, C-7/H-6, C-7/H-8α, and C-7/H-8β (Fig. 7 and Table 1). The position of the methyl group at C-10 was established based on HMBC cross-peaks between C-10/H3-14, C-14/H-10, C-14/H-9α, C-14/H-9β, C-9/H3-14, and C-1/H3-14. Accordingly, the position of the 4-Me group was derived from HMBC interactions between C-15/H-4, C-15/H-3α, C-15/H-3β, C-5/H3-15, and C-3/H3-15. HMBC cross-peaks between C-7/H-11, C-8/H-11, and C-6/H-11 unambiguously clarified the location of the isopropyl group at C-7. The relative stereochemistry of compound 9a was tentatively assigned by a NOESY experiment (Fig. 8, 9 and Table 1) in combination with the analysis of coupling constants (Tables 1, 2, S7 and S8†) and a comparison of calculated (GIAO) and experimental 1H and 13C NMR shift values (Tables S9 and S10†). NOE interactions have been exploited to determine the positions of the protons of the seven-membered ring and of the 10-Me group (starting point of the assignment, arbitrarily defined as β) relative to those in the five-membered ring. Thus, the observed NOE cross-peaks for H3-14/H-2β and H-10/H-2α are indicative of a cis-arrangement of the corresponding protons (Fig. 8). Moreover, the same signals also suggest an α-position of the 1,5-epoxy group. Support for this assignment is obtained from a weak NOE interaction of H-9β with H-2β and the absence of NOE correlations between α-protons in the five-membered ring with those in the seven-membered ring. Due to ambiguous NOE signals for the interactions of H-2α and H-2β with the protons at C-3, the direct assignment of H-3α and H-3β and the resulting determination of the configuration at C-4 could not be achieved. However, the position of the protons at C-3 relative to H3-15 and H-4 has been unambiguously identified by NOE interactions. The cis-arrangement of H3-15/H-3cis (cis with respect to 4-Me) and of H-4/H-3trans is concluded from distinct NOE cross-peaks. No NOE signal is observed for H3-15/H-3trans and only a weak signal for H-4/H-3cis.
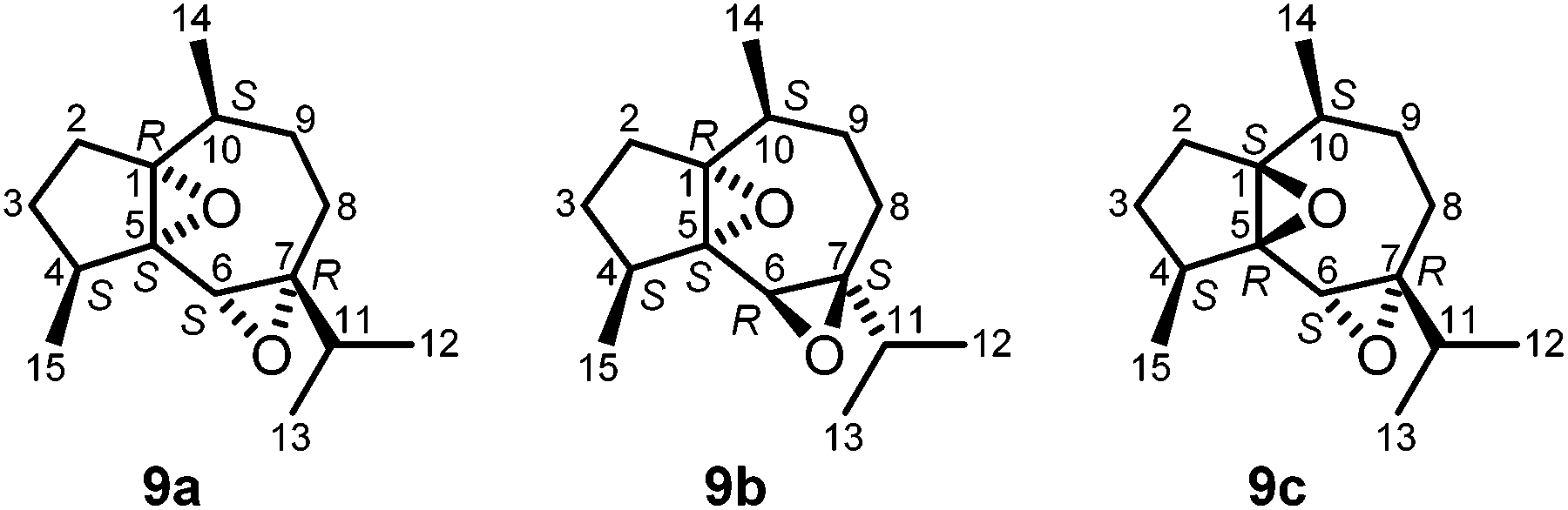 | ||
| Fig. 6 Structures of compound 9a, isolated from Capnella fungiformis, the hypothetical diastereoisomer 9b, and compound 9c, isolated from Sinularia kavarattiensis.42b | ||
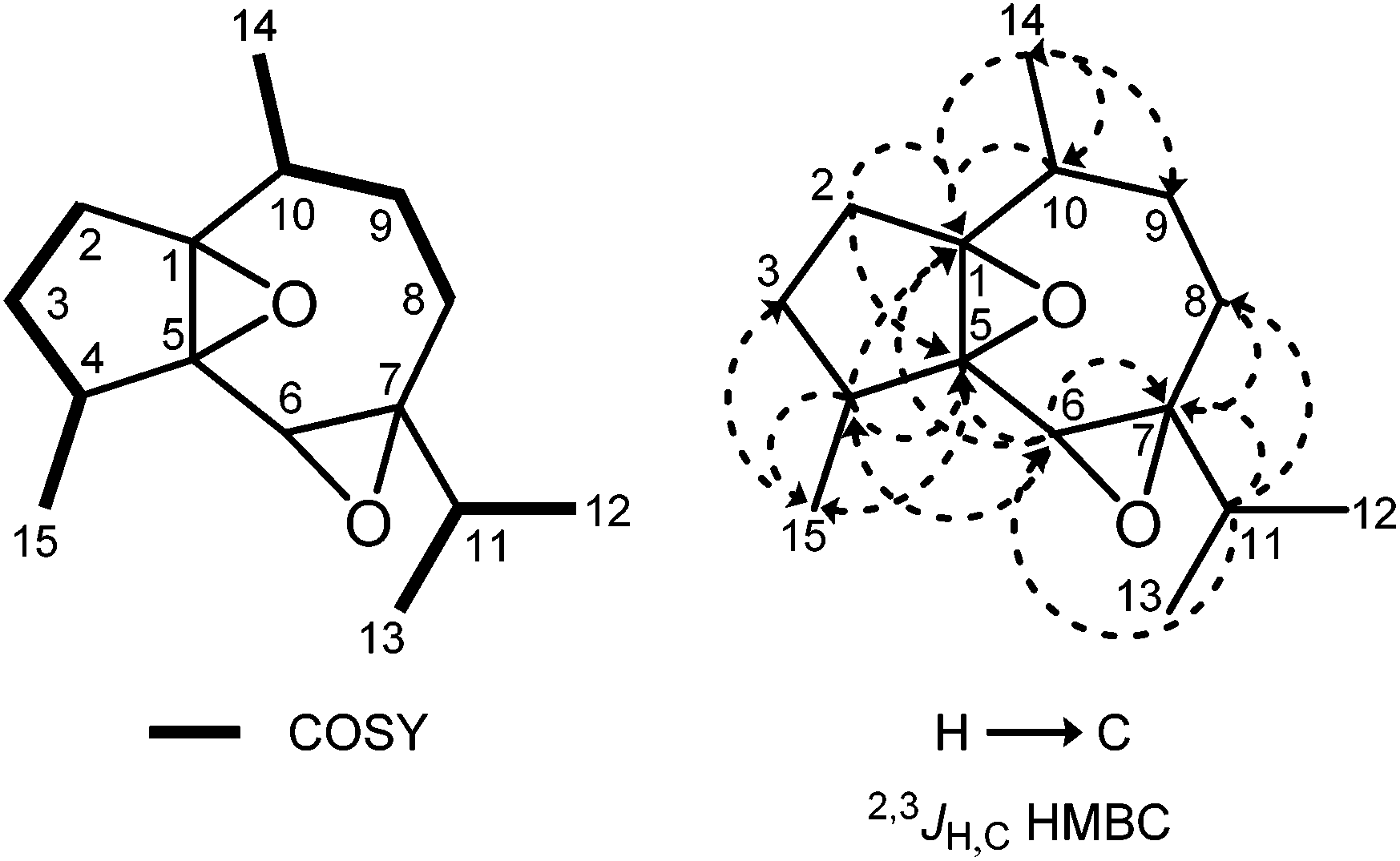 | ||
| Fig. 7 Characteristic COSY and HMBC correlations of compound 9a. | ||
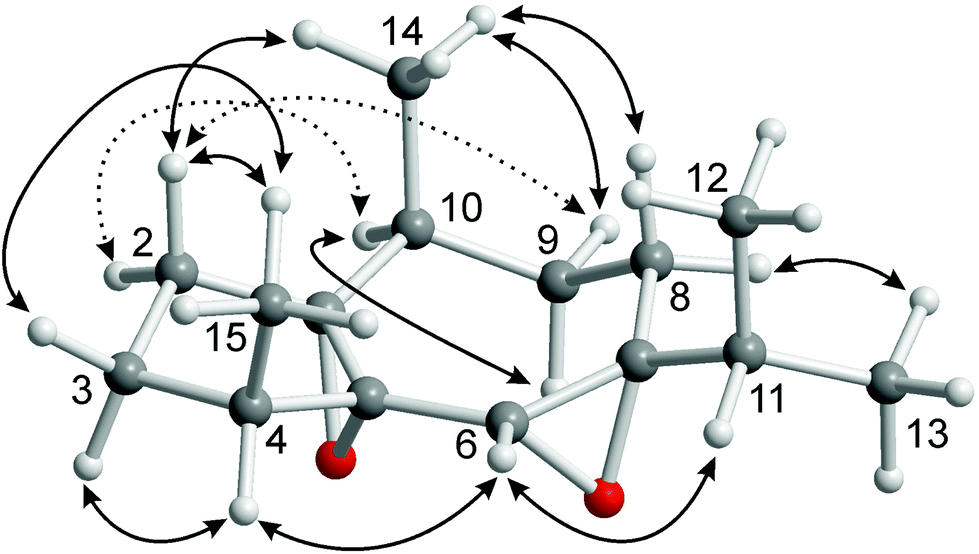 | ||
| Fig. 8 Characteristic NOE correlations of compound 9a shown at a computer-generated model of a minimum energy conformation calculated using PBE0-D3(BJ)/def2-TZVP-F. | ||
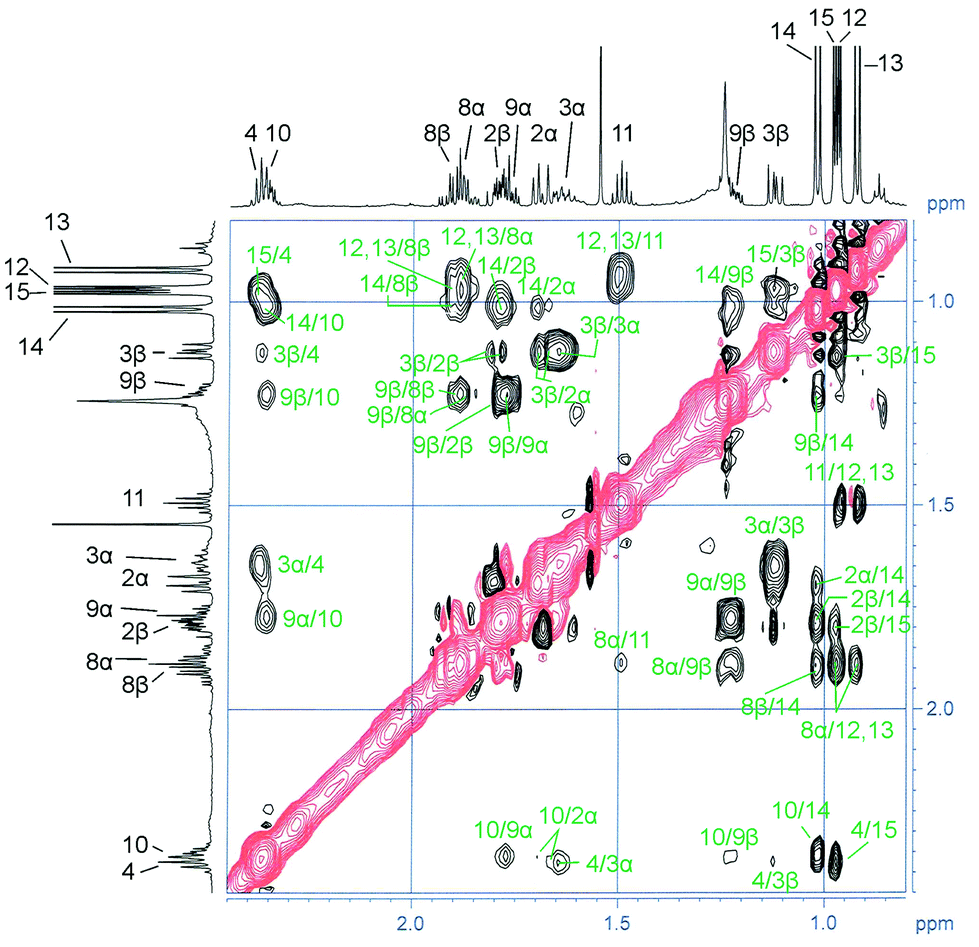 | ||
| Fig. 9 Expansion of the NOESY spectrum of compound 9a. | ||
|
δ
Ha (J in Hz) |
δ
Cb |
HMBCc | NOESYd | |
|---|---|---|---|---|
| a Shift, multiplicity and coupling constants derived from the simulated spectrum (Fig. S11–S17). b The number of attached protons as determined by the DEPT experiment. c HMBC correlations from carbon atoms to protons. d Only the most significant signals are shown. | ||||
| 1 | 73.69 C | 2α, 3α, 4, 6, 9α, 9β, 10, 14 | ||
| 2α | 1.69 dd (13.8, 8.4) | 26.75 CH2 | 3α, 3β | 3β, 10, 14 |
| 2β | 1.79 ddd (13.8, 10.4, 8.3) | 3α, 3β, 9β, 14, 15 | ||
| 3α | 1.64 ddddq (12.2, 10.4, 8.4, 7.6, 0.5) | 26.72 CH2 | 2α, 2β, 4, 15 | 2β, 4 |
| 3β | 1.12 dd (12.0, 8.3) | 2α, 2β, 4, 15 | ||
| 4 | 2.37 dq (7.6, 7.4) | 37.60 CH | 2α, 3α, 3β, 6, 15 | 3α, 3β, 6, 15 |
| 5 | 69.34 C | 2α, 3α, 4, 6, 15 | ||
| 6 | 3.05 s | 58.08 CH | 8α, 8β, 11 | 4, 11, 12, 13 |
| 7 | 68.59 C | 4, 6, 8α, 8β, 9α, 9β, 11, 12, 13 | ||
| 8α | 1.87 dddd (15.6, 6.3, 4.0, 0.9) | 22.44 CH2 | 9α, 9β, 11 | 9α, 9β, 10, 11, 12, 13, 14 |
| 8β | 1.91 ddd (15.6, 10.8, 4.1) | 9β, 12, 13, 14 | ||
| 9α | 1.77 dddd (14.6, 10.8, 4.2, 4.0) | 26.27 CH2 | 8α, 8β, 9, 14 | 8α, 10 |
| 9β | 1.23 dddd (14.6, 7.5, 6.3, 4.1) | 2β, 8α, 8β, 10, 14 | ||
| 10 | 2.36 dqdd (7.5, 7.2, 4.2, 0.9) | 31.45 CH | 8α, 8β, 9α, 9β, 14 | 2α, 8α, 9α, 9β, 14 |
| 11 | 1.49 qq (7.0/6.8) | 36.50 CH | 6, 8α, 12, 13 | 6, 8α, 12, 13 |
| 12 | 0.92 d (7.0) | 17.81 CH3 | 11, 13 | 6, 8α, 8β, 11 |
| 13 | 0.969 d (6.8) | 17.86 CH3 | 11, 12 | 6, 8α, 8β, 11 |
| 14 | 1.02 d (7.2) | 17.30 CH3 | 10, 9α, 9β | 2α, 2β, 8α, 8β, 9β, 10 |
| 15 | 0.974 dd (7.4, 0.5) | 16.01 CH3 | 3α, 3β, 4 | 2β, 3β, 4 |
| Torsion angles | Θ [°] | 3 J HH [Hz] estimatedb | 3 J HH [Hz] experimentalc |
|---|---|---|---|
|
a Torsion angles from a minimum energy conformation obtained by quantum chemical calculations using PBE0-D3(BJ)/def2-TZVP-F (D-PCM, solvent: CDCl3).
b Coupling constants estimated from the torsion angles using the Bothner-By equation (3JHH = 7 − cosθ + 5cos2θ).45
c Coupling constants obtained from the experimental 1H NMR signals (CDCl3) by iterative approximation of simulated signals.
|
|||
| H2α–C2–C3–H3α | −34.3 | 8.0 | 8.4 |
| H2α–C2–C3–H3β | 85.8 | 2.0 | <1 |
| H2β–C2–C3–H3α | −155.2 | 11.1 | 10.4 |
| H2β–C2–C3–H3β | −35.1 | 7.9 | 8.3 |
| H3α–C2–C3–H4 | 32.5 | 8.3 | 7.6 |
| H3β–C2–C3–H4 | −87.3 | 2.0 | <1 |
Coupling constants have been used to correlate the proton assignments of the five-membered ring. For this purpose, the experimental coupling constants in the five-membered ring have been determined from the complex signals in the 1H NMR spectrum by iterative approximation of simulated coupling patterns (Fig. S11–S17†). The minimum energy conformations for all eight possible diastereoisomers of 9 have been obtained by quantum chemical calculations using PBE0-D3(BJ)/def2-TZVP-F (D-PCM, solvent: CDCl3). The envelope conformation of the five-membered ring is identical for all eight diastereoisomers and corresponds to a syn-arrangement of the epoxy moiety and C-3. This is in full agreement with X-ray crystal structures of related epoxyguaiane sesquiterpenes.46 The estimated coupling constants obtained from the torsion angles of the calculated minimum energy conformations (Tables 2 and S7†) using the Bothner-By equation45 have been compared with the experimental data (Tables 2 and S8†). Only the cis-diepoxide 9a and the diastereoisomeric trans-diepoxide 9b (Fig. 6) show good agreement between experimental and estimated data (Table S8†). Both compounds have two very small coupling constants (estimated: 2 Hz, found: <1 Hz, Table 2) for H-2α/H-3cis and H-3cis/H-4 because of torsion angles of approximately 90° between these pseudoequatorial protons (cis and trans denote the orientation of the corresponding proton relative to the 4-Me group). Consequently, the 4-Me group adopts the β-position which is also supported by an NOE signal for the interaction of H-2β with H3-15. At this point, it becomes possible to assign H-3cis as H-3β and H-3trans as H-3α. A distinction between the structures 9a and 9b by NOE interactions proved to be difficult. A strong NOE signal has been observed for H-4/H-6 (Fig. S22†); however, this signal could not be used to assign the relative orientation of the corresponding protons. Ambiguous NOE signals between the protons of the isopropyl group and the adjacent protons H-8α and H8-β prevented a direct assignment of the configuration for the isopropyl group relative to the 10-Me group. Both structures, 9a and 9b, are in agreement with the observed NOE signals and the coupling constants. In order to differentiate between 9a and 9b, we have compared the calculated chemical 1H and 13C NMR shifts (GIAO) with the corresponding experimental data (Tables S9 and S10†). As a result from this comparison, our natural product was assigned as the cis-diepoxide 9a because for this structure we observed smaller deviations between calculated and experimental values, in particular for the 13C NMR shifts. Missing NOE interactions between H-9α and/or H3-12/H3-13 as well as between H-10 and/or H3-12/H3-13 support our assignment.
Based on the spectroscopic data discussed above, the isolated compound was tentatively assigned as (1R/S,4S/R,5S/R,6S/R,7R/S,10S/R)-1,5:6,7-diepoxyguaiane (9a) (Fig. 6) with a cis-configuration of the 4-Me and the 10-Me group as known for the common sesquiterpenoid guaiol. Compound 9a represents a novel diepoxyguaiane sesquiterpene which we have named oxyfungiformin. Biogenetically 9a may derive from a twofold epoxidation of 4αH,10αH-guaia-1(5),6-diene (γ-guaiene)47 or 4βH,10βH-guaia-1(5),6-diene.48 Recently, Venkateswarlu and coworkers reported the isolation of the diastereoisomeric compound 9c (Fig. 6) from Sinularia kavarattiensis (India).42b
Subfraction F-2-2 of the diethyl ether extract of C. fungiformis contained a mixture of sterols. This fraction was additionally purified by reversed-phase HPLC using acetonitrile–water (9:1) as the mobile phase to afford the three known steroids 24-methylenecholesterol (10), (24S)-24-methylcholesterol (11) and gorgosterol (12) (Fig. 10). Compound 10 was obtained as a colorless solid and, based on MS, IR, 1D, and 2D NMR data, identified as 24-methylenecholesterol (10).1,3,49–52 Comparison of the 1H NMR data of 10 with those reported by Su et al.52 for a synthetic sample (Table S11†) and of the 13C NMR shift values with those reported for a sample isolated from Litophyton viridis51 (Table S12†) confirmed the assignment. 24-Methylenecholesterol (10) has been found in various soft corals, for example, Capnella imbricata,53Litophyton viridis,51Sinularia flexibilis,54Sinularia maxima,55 and Sinularia vanderlandi.3 It has not been obtained previously from the soft coral C. fungiformis.
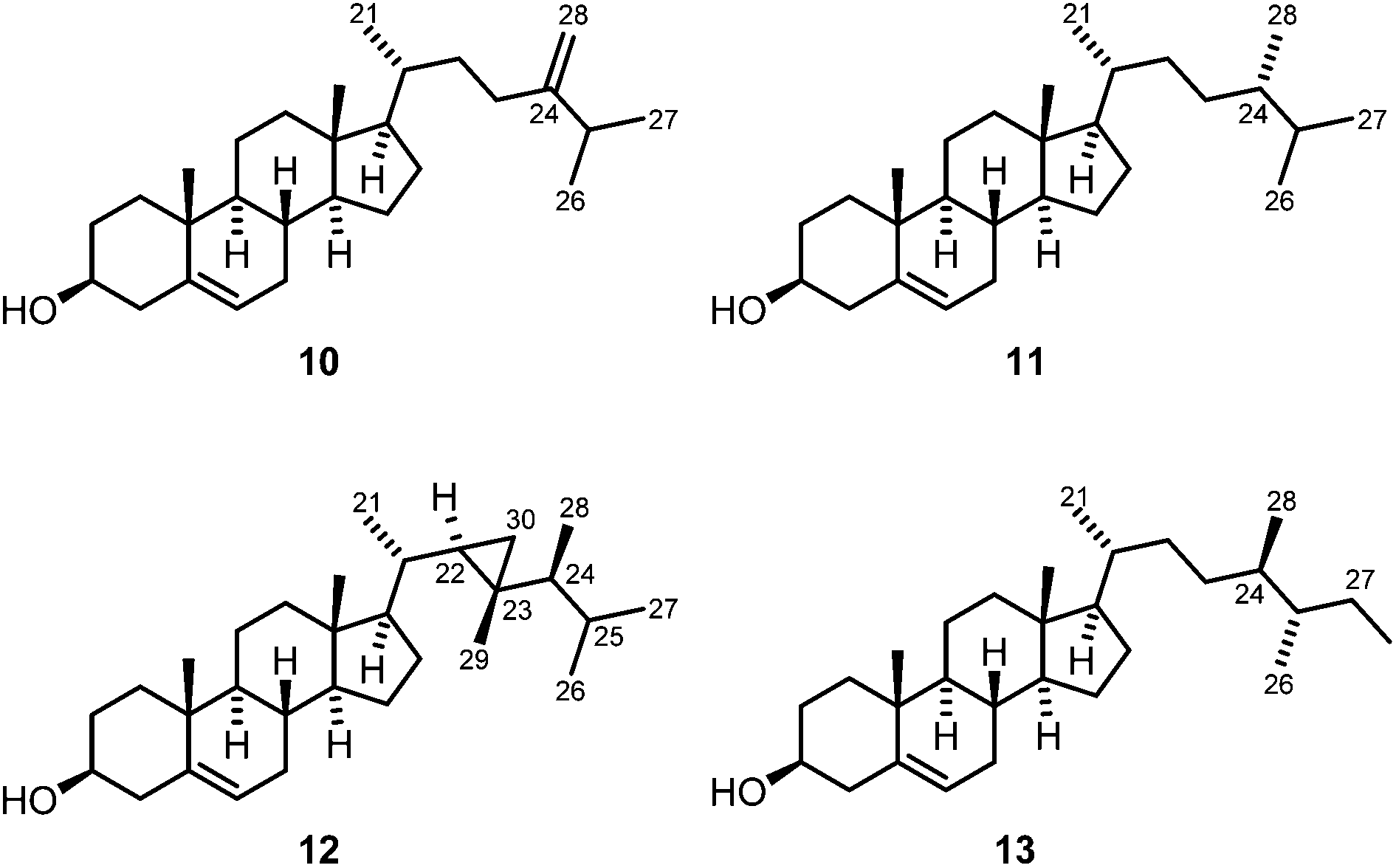 | ||
| Fig. 10 Chemical structures of 24-methylenecholesterol (10), (24S)-24-methylcholesterol (11), gorgosterol (12) and aplysterol (13). | ||
(24S)-24-Methylcholesterol (11) was isolated as a colorless solid and assigned based on comparison of the 1H and 13C NMR data with those reported in the literature (Tables S11 and S12†).56–58 (24S)-24-Methylcholesterol (11), the C-24 epimer of campesterol, has been isolated previously from diverse natural sources, in particular from soft corals (e.g. from Sinularia sp.,59Sinularia dura,60Lobophytum sp.61 and others). The first synthesis of compound 11 has been described in 2012 by McCarthy and coworkers.56 We have isolated (24S)-24-methylcholesterol (11) for the first time from the soft coral C. fungiformis.
The third sterol was identified as gorgosterol (12) (Fig. 10).62–64 It features unusual 1H NMR signals at δH = −0.14 (1H, ddd, J = 5.8, 4.4, 1.3 Hz), 0.13–0.19 (1H, m), and 0.45 ppm (1H, ddd, J = 9.1, 4.3, 2.6 Hz), which are characteristic of the protons of the cyclopropane ring. The structure of the compound was unambiguously confirmed by comparison of the 13C NMR signals with those reported for gorgosterol (12) isolated from the soft coral Alcyonium molle (Table S12†).65 Soft corals are a rich source of gorgosterol and several species have been shown to contain this sterol, for example Sarcophyton glaucum,66Alcyonium molle,65Sinularia kavarattiensis,67 and Asterospicularia laurae.68 The soft coral C. fungiformis investigated herein represents a new source of this sterol.
Lobophytum crassum
Soft corals of the species L. crassum have been frequently studied for their chemical constituents. The first report appeared in 1977 by Bowden et al. who isolated a cembranoid diterpene from a specimen collected in Australia.69 Cembranoids have been repeatedly described as major components of L. crassum.70–72 In the present study, we investigated for the first time the chemical constituents of L. crassum collected at the coast of Madagascar. A specimen of this soft coral was minced and extracted with methanol at room temperature. After evaporation of the solvent, the crude extract was taken up with diethyl ether. Purification of the resulting diethyl ether extract by column chromatography provided a 3.5:1 mixture of (24S)-24-methylcholesterol (11) and cholesterol. The antimalarial assay of the crude methanol extract of L. crassum exhibited an IC50 value of 33.15 ± 2.90 μg mL−1 for the inhibition of the FCM29 strain of Plasmodium falciparum, indicating moderate activity against this pathogen.
Pseudoceratina arabica
Samples of the sponge P. arabica from the Red Sea have already been investigated for their chemical constituents, resulting in the isolation of brominated compounds derived from tyrosine.73–75 Specimens of P. arabica were obtained from the waters around Nosy Be, Madagascar, at a depth of about 18–25 m. The samples were homogenized and then extracted with methanol at room temperature. The combined organic layers were filtered and concentrated in vacuo to obtain the crude methanol extract, which was extracted with hexane and then dichloromethane. The major compound of the hexane extract (F-1) was identified as aplysterol (13) (Fig. 10). Compound 13 was obtained as a colorless solid with a melting point of 125–126 °C and a specific optical rotation of [α]20D = −27.0 (c 0.05, MeOH). The NMR data of 13 were in agreement with those reported in the literature (Tables S11 and S12†).76,77 Aplysterol (13) was previously isolated from other marine sponges, e.g. Aplysina aerophoba78 and Aplysina fistularis,77,79 and also from terrestrial plants, such as Mussaenda macrophylla,80 but not from P. arabica. Further fractionation of the dichloromethane extract (F-2) of P. arabica followed by column chromatography over silica gel afforded additional aplysterol (13) and a mixture of the dibromotyrosine metabolites 14a and 14b (Fig. 11). Compounds 14a and 14b were identified by IR spectroscopy, EI mass spectrometry, 1H and 13C NMR spectroscopy, and a set of 2D NMR methods (COSY, HSQC, HMBC, NOESY). The structure elucidation was confirmed by comparison with the 1H and 13C NMR data of the compounds isolated from Aplysina sp. (Table S13†).81 The GC-MS chromatogram showed only a single peak. However, a double set of signals for H3-9, H2-10 and H3-11 in the 1H NMR spectrum indicated the presence of two diastereoisomers in a ratio of about 3.5:1 in favor of the cis-isomer 14a (cis and trans denote the position of the ethoxy group relative to the hydroxy group), the same ratio as reported by Santalova et al.82 The predominant cis-isomer 14a was identified by a strong NOE correlation between H3-9 and H2-7. The trans-isomer 14b showed NOE cross-peaks between H2-10 and H2-7.
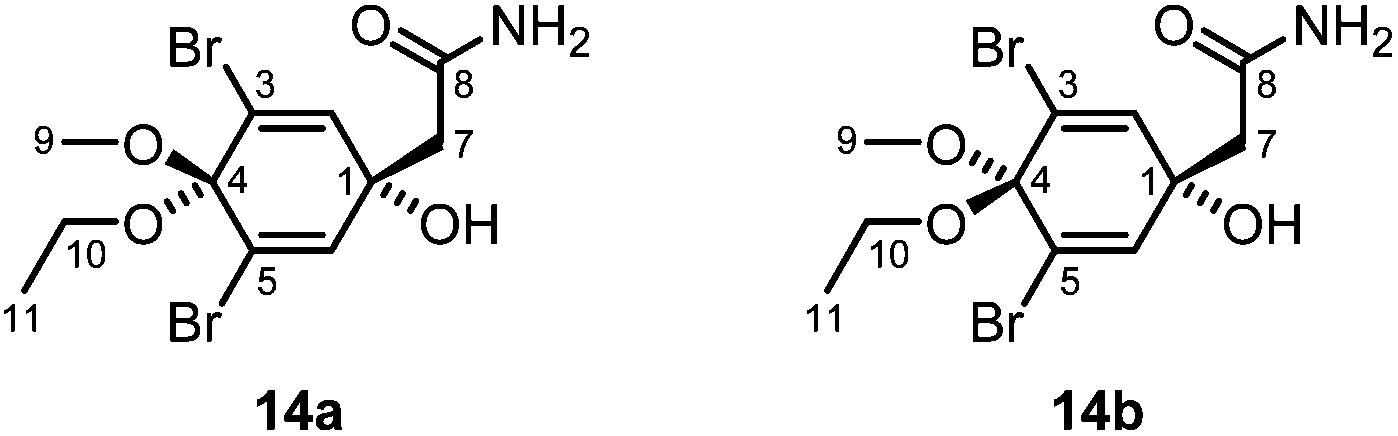 | ||
| Fig. 11 Structures of compounds 14a and 14b. | ||
The diastereoisomeric compounds 14a and 14b have been isolated first from a sponge of the genus Verongia83 and subsequently from Aplysina fistularis,84Aplysina sp.,81,82,85,86Pseudoceratina sp.,87Suberea mollis,88Suberea sp.,89 and Ircinia variabilis.90 Occasionally, they have been described as possible artifacts.83,85 However, since we have not used ethanol during the isolation process, we consider 14a and 14b to be original natural products of P. arabica. Even if not explicitly mentioned, the diastereoisomeric compounds 14a and 14b most likely have always been obtained as a mixture.83,87,89 Separation of the two diastereoisomers by HPLC was achieved by Lin and coworkers91 and Santalova et al.,81 who also provided 1H NMR data for the separated isomers. Even though the sponge P. arabica has already been investigated for its chemical constituents,73–75 compounds 14a and 14b have not been isolated from this species so far.
The dichloromethane fraction (F-2) of P. arabica and the ethyl acetate (F-2-1), n-butanol (F-2-2), and aqueous (F-2-3) fractions derived therefrom (see the Experimental, Extraction and isolation section) have been investigated for their activity against a series of microbes (Table 3). It appeared that the parent dichloromethane (F-2) and the ethyl acetate (F-2-1) extracts of P. arabica are very active against the Gram-negative bacteria Enterobacter cloacae, Klebsiella oxytoca, Shigella boydii, Escherichia coli, and Salmonella enteridis and against the Gram-positive bacteria Bacillus cereus, Staphylococcus aureus, and Streptococcus pneumoniae. Exceptionally high activity was observed for the dichloromethane extract (F-2) of P. arabica against S. boydii. The butanol extract (F-2-2) was very active against S. boydii and significantly active against S. aureus and S. pneumoniae. The aqueous extract (F-2-3) was very active against E. coli and S. enteridis. It should be noted that compounds 14a and 14b, which we isolated from the dichloromethane extract, were reported to show no antimicrobial activity.84,86,88,89 In contrast, the parent ketone and related derivatives are described to be active against a variety of pathogens.73,88,89,92 Some derivatives show antimigratory activity against the highly metastatic MDA-MB-231 human breast cancer cell line,74,75 induce apoptosis in human breast tumor cells,93 or inhibit Na+–K+-ATPase activity.94 It can be assumed that some as yet unidentified components, possibly also dibromotyrosine derivatives, are responsible for the antimicrobial activity of the extracts. None of the four extracts of P. arabica investigated in our antimicrobial assay exhibited activity against the yeast Candida albicans. The crude methanol extract of P. arabica was also inactive against the FCM29 strains of Plasmodium falciparum.
| Microbes | Zone of inhibitionb (Ø in mm) | |||
|---|---|---|---|---|
| Fractions | ||||
| CH2Cl2 (F-2) | EtOAc (F-2-1) | n-BuOH (F-2-2) | H2O (F-2-3) | |
| a Each test was run in triplicate and the mean values are given; the solvent (methanol) was used as the negative control. b Concentration of the extracts: 1 mg mL−1, 10 μL solution per 6 mm disc; Ø < 7 mm: inactive, 7 mm ≤ Ø < 8 mm: slightly active, 8 mm ≤ Ø < 9 mm: significantly active, Ø ≥ 9 mm: very active. | ||||
| Gram-negative bacteria | ||||
| Enterobacter cloacae ATCC 700323 | 12 | 17 | 6.5 | 6.5 |
| Klebsiella oxytoca ATCC 8724 | 14 | 15.5 | 6 | 6 |
| Shigella boydii ATCC 9207 | 22 | 14 | 10 | 6 |
| Escherichia coli | 11 | 15 | 6 | 10 |
| Salmonella enteridis | 12.5 | 13 | 6 | 12 |
| Gram-positive bacteria | ||||
| Bacillus cereus ATCC 13061 | 15 | 11 | 6 | 6 |
| Staphylococcus aureus ATCC 11632 | 14 | 13.5 | 8.5 | 6 |
| Streptococcus pneumoniae ATCC 6301 | 14 | 15 | 8 | 6 |
| Yeast | ||||
| Candida albicans | 6 | 6 | 6 | 6 |
Conclusions
We have investigated the chemical constituents of the Madagascan soft corals Sarcophyton stellatum, Capnella fungiformis, and Lobophytum crassum and the sponge Pseudoceratina arabica. The chemical constituents of S. stellatum and C. fungiformis have been studied for the first time. The crude extracts of the three soft corals showed moderate antimalarial activities (Plasmodium falciparum). The extracts of the sponge P. arabica exhibited strong activity against a series of Gram-positive and Gram-negative bacteria. From S. stellatum we have isolated the previously unknown (+)-enantiomer of the cembranoid diterpene (1E,3E,11E)-7,8-epoxycembra-1,3,11,15-tetraene [(+)-2]. C. fungiformis provided a mixture of the two novel sesquiterpenes ethyl 5-[(1E,5Z)-2,6-dimethylocta-1,5,7-trienyl]furan-3-carboxylate (6) and ethyl 5-[(1E,5E)-2,6-dimethylocta-1,5,7-trienyl]furan-3-carboxylate (7). Moreover, we isolated from C. fungiformis the novel diepoxyguaiane sesquiterpene oxyfungiformin (9a).Experimental
General methods
Thin layer chromatography was performed on aluminum plates from Merck (60 F254) coated with silica gel. For visualization, the plates were analyzed under UV light or treated with a solution of 0.5 g vanillin dissolved in 100 mL of 80/20 (v/v) sulfuric acid/dry ethanol and subsequently heated. Preparative TLC was carried out with glass plates (20 × 20 cm, Merck) coated with a 0.25 mm layer of silica gel (60 F254). Analytical HPLC was carried out using an Agilent 1100 instrument equipped with a G1315B UV-DAD (detection at 215, 260 and 560 nm), a G1321A fluorescence and an evaporative light scattering detector (ELS 1000, Polymer Laboratories) using a Vydac 208TP104 column (reversed-phase C8, 4.6 × 250 mm) under the following conditions: flow rate: 1.0 mL min−1; eluent A: H2O + 0.1% TFA; eluent B: MeCN + 0.1% TFA; gradient from 20 to 90% B in 35 min. Preparative HPLC was performed using a Varian PrepStar system with a Varian ProStar Model 320 UV and an evaporative light scattering detector (ELS 1000, Polymer Laboratories) connected via a Sunchrom Quick-Split splitter. Column chromatography was performed using silica gel from Acros Organics (0.035–0.070 mm). Melting points were measured using a Gallenkamp MPD 350 melting point apparatus. Optical rotations were determined with a PerkinElmer 341 polarimeter at a wavelength of 589 nm (sodium D line) using a 1.0 decimeter cell with a total volume of 1.0 mL. CD spectra were recorded on a JASCO J-815 CD spectrometer. UV spectra were measured with a PerkinElmer Lambda 25 UV-Vis spectrometer. Fluorescence spectra were measured on a Varian Cary Eclipse fluorescence spectrometer. IR spectra were recorded on a Thermo Nicolet Avatar 360 FT-IR spectrometer using the ATR technique (attenuated total reflectance). NMR measurements were performed with a Bruker AVANCE III 600 spectrometer. The chemical shifts δ are reported in ppm using the solvent as the internal standard (1H: δH 7.25 ppm CHCl3; 13C: δC 77.00 ppm CDCl3). The following abbreviations have been used: s: singlet, d: doublet, t: triplet, q: quartet, sp: septet, m: multiplet and br: broad. Assignment of the 1H NMR and 13C NMR signals was achieved using the 2D NMR methods COSY, HSQC, HMBC and NOESY. Mass spectra were measured by GC-MS coupling with an Agilent 6890 N GC System equipped with a 5973 Mass Selective Detector or with HRMS on a Finnigan MAT 95 mass spectrometer (electron impact, 70 eV). ESI-MS spectra were recorded on a Bruker Esquire mass spectrometer with an ion trap mass analyzer; positive and negative ions were detected. Elemental analyses were performed using a EuroVector EuroEA3000 elemental analyzer. X-ray single crystal structure analysis: X-ray diffraction data were collected on a Bruker AXS Kappa APEX Duo diffractometer with a microfocus tube using Cu Kα radiation (λ = 1.54178 Å). The programs used were: for data collection: APEX2,95 for data reduction: SAINT,96 for absorption correction: SADABS version 2.10,97 for structure solution: SIR2002,98 and for structure refinement by full-matrix least-squares against F2: SHELXL.99 Hydrogen atoms were placed in calculated positions and refined as riding atoms at carbon atoms. The figure was generated using the programs ORTEP-III100 and POV-Ray 3.7.Natural sources
Samples of the soft corals C. fungiformis, S. stellatum and L. crassum were collected by hand using scuba in March 2006 in the inner reef of Mahambo (17°29′15.0′′ S; 49°28′32.1′′ E), located in the Tamatave province, at the east coast of Madagascar, at a depth of 5–6 m. The sponge P. arabica was collected using scuba in April 2005 from the waters around Nosy Be, an island off Madagascar located in the Mozambique Channel, near the northwest coast of Madagascar at a depth of 18–25 m. The collection site was determined at Banc des Gorgones 13°25′03.5′′ S and 48°12′25.5′′ E. The species were assigned by Dr Shirley Parker-Nance (Nelson Mandela Metropolitan University, Port Elizabeth, and South African Institute for Aquatic Biodiversity (SAIAB), Grahamstown, South Africa). Voucher specimens of the three soft corals and the sponge investigated in this study have been deposited at the SAIAB (collection numbers: 201734 for S. stellatum, 201730 for C. fungiformis, 201729 for L. crassum and 201727 for P. arabica).Extraction and isolation
A specimen of S. stellatum (1 kg, wet weight) was minced and then extracted with methanol at room temperature. The mixture was filtered and the organic phase was concentrated under reduced pressure to yield the crude extract (29 g), which was partitioned between water/diethyl ether and water/dichloromethane, successively. The diethyl ether extract (11 g) was subjected to column chromatography over silica gel using pentane with increasing proportions of diethyl ether and then diethyl ether–ethyl acetate (10:1) as eluents to afford the three fractions F-1, F-2 and F-3, respectively. Fraction F-1 (2.0 g) eluted with pentane–diethyl ether (10:1) was then separated by column chromatography over silica gel using increasing concentrations of diethyl ether in pentane to give two fractions (F-1-1 and F-1-2) according to TLC detection. Fraction F-1-1 (1.53 g) was further purified by column chromatography over silica gel with pentane–diethyl ether (12:1) to afford (+)-(7S,8S)-epoxy-7,8-dihydrocembrene C [(+)-1] (814 mg) and (+)-(1E,3E,11E)-7,8-epoxycembra-1,3,11,15-tetraene [(+)-2] (316 mg). Fraction F-1-2 (459 mg) was separated by column chromatography over silica gel with pentane–diethyl ether (10:1) as the eluent to obtain (+)-(7R,8R,14S,1Z,3E,11E)-14-acetoxy-7,8-epoxycembra-1,3,11-triene [(+)-3] (207 mg). Fraction F-2 (3.71 g) was subjected to column chromatography over silica gel using pentane–diethyl ether (4:1) as the mobile phase to afford (−)-(2R,7R,8R)-sarcophytoxide [(−)-4] (3.37 g), which crystallized from a small amount of diethyl ether. Fraction F-3 (300 mg) was further purified by preparative HPLC (column: Vydac 208TP1030, reversed phase C8, 30 × 250 mm; flow rate: 20 mL min−1; eluent A: H2O + 0.1% TFA, eluent B: THF + 0.1% TFA; gradient from 50 to 80% B in 20 min) to yield (S)-3-O-octadecylglycerol (batyl alcohol) (5) (11 mg). The dichloromethane extract of S. stellatum (270 mg) was subjected to chromatography over silica gel using pentane with increasing proportions of diethyl ether as the eluent to afford (+)-1 (3 mg), (+)-3 (3 mg) and (−)-4 (10 mg).
The material of C. fungiformis (770 g, wet weight) was cut and then extracted with methanol. The methanol extract was concentrated under reduced pressure and the residue (17 g) was dissolved in dry diethyl ether. The diethyl ether extract (8.0 g) was subjected to column chromatography over silica gel using pentane with increasing proportions of diethyl ether as the eluent to afford two main fractions. The first fraction (F-1) (2.2 g) was separated by column chromatography over silica gel eluting with a gradient of pentane–diethyl ether to obtain three subfractions (F-1-1, F-1-2 and F-1-3) according to TLC detection. Fraction F-1-1 (1.8 g) was purified by column chromatography over silica gel with pentane–diethyl ether (20:1) as mobile phase, affording a mixture of furan sesquiterpenes 6 and 7 (F-1-1-1, 15 mg) in a ratio of about 3:2 (6:7) (GC-MS, 1H NMR) and octadecan-2-one (8) (F-1-1-2, 13 mg). Subfraction F-1-2 (224 mg) was further purified by repeated column chromatography over silica gel with pentane–diethyl ether (10:1) to afford 23 mg of the diepoxyguaiane sesquiterpene 9a. Subfraction F-1-3 (17 mg) contained a mixture of monoalkyldiacylglycerols, which was not investigated in detail. The second fraction of the diethyl ether extract (F-2, 574 mg) was repeatedly subjected to column chromatography over silica gel eluting with pentane–diethyl ether (9:1) to give two fractions (F-2-1 and F-2-2). Fraction F-2-1 (51 mg) contained a mixture of monoalkylmonoacylglycerols. Fraction F-2-2 (156 mg) was further separated by reverse phase HPLC (acetonitrile–water, 9:1) to afford 24-methylenecholesterol (10) (26 mg), (24S)-24-methylcholesterol (11) (77 mg), and gorgosterol (12) (43 mg).
A sample of the fresh soft coral L. crassum (0.5 kg, wet weight) was homogenized and exhaustively extracted with methanol. The combined extracts were concentrated to yield a crude mass of 15 g which was extracted with diethyl ether. The dried diethyl ether extract was concentrated under reduced pressure and the residue (2 g) was subjected to column chromatography over silica gel with pentane–diethyl ether (4:1) to collect a portion (480 mg) showing a main spot on TLC. This portion was subsequently further purified by two column chromatographic steps over silica gel using pentane–ethyl acetate (9:1) and pentane–diethyl ether (7:3) as mobile phases to yield a mixture of (24S)-24-methylcholesterol (11) and cholesterol in a ratio of 3.5:1 according to the GC-MS chromatogram.
The specimens of the sponge P. arabica (957 g, wet weight) were homogenized and extracted with methanol at room temperature. The combined extracts were concentrated to obtain 34 g of a crude extract, which was extracted successively with hexane and dichloromethane to afford 9.6 g of the hexane extract (F-1) and 4.3 g of the dichloromethane extract (F-2). A portion of 782 mg of the hexane extract (F-1) was subjected to column chromatography over silica gel with a stepped solvent gradient from pentane to diethyl ether as the mobile phase. Fractions eluted with pentane–diethyl ether (4:1) were combined to provide 201 mg of aplysterol (13). The dichloromethane extract (F-2) (4.3 g) was partitioned between water and ethyl acetate. After separation of the ethyl acetate layer, the aqueous layer was extracted with n-butanol. Evaporation of the ethyl acetate, n-butanol and the aqueous layers provided residues of 1.3 g (F-2-1), 1.9 g (F-2-2) and 1.0 g (F-2-3), respectively. A 1.0 g portion of the ethyl acetate extract (F-2-1) was extracted with dichloromethane to give 356 mg of a dichloromethane extract (F-2-1-1), which was subjected to column chromatography over silica gel using pentane with increasing proportions of diethyl ether as the eluent. The fractions eluted with pentane–diethyl ether (4:1) were combined to give a fraction of 240 mg, which was subjected to column chromatography over silica gel to provide another 97 mg of aplysterol (13). A 657 mg portion of the n-butanol extract (F-2-2) was further extracted with dichloromethane to afford a residue of 234 mg (F-2-2-1). The dichloromethane extract (F-2-2-1) was subjected to column chromatography over silica gel eluting with: (1) pentane–diethyl ether (4:1) and (2) diethyl ether–dichloromethane (3:2) to afford two main fractions F-2-2-1-1 and F-2-2-1-2. Fraction F-2-2-1-1 (77 mg) was purified by column chromatography over silica gel with pentane–diethyl ether (4:1) providing 40 mg of aplysterol (13). Fraction F-2-2-1-2 (157 mg) was subjected to column chromatography over silica gel using diethyl ether with increasing proportions of dichloromethane as the eluent to collect 100 mg of a fraction showing one main spot on TLC. This fraction was subsequently purified by preparative TLC over silica gel with diethyl ether–dichloromethane (3:2) to afford a 3.5:1 mixture of the acetamides 14a and 14b (26 mg). The aqueous extract (F-2-3, 1.0 g) was extracted with methanol. Column chromatography of the methanol extract (201 mg) over silica gel with ethyl acetate afforded a fraction (116 mg) which showed one main spot on TLC. Another column chromatographic purification over silica gel using diethyl ether–ethyl acetate (3:2) as the mobile phase afforded 49 mg of a mixture of the diastereoisomeric acetamides 14a and 14b.
Biological activity
Molecular modeling
Conformational energy minimization for compound 9a (Fig. 8) and for the seven alternative diastereoisomers 9b–h was achieved by quantum chemical calculations using PBE0-D3(BJ)/def2-TZVP-F (D-PCM, solvent: CDCl3).![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). MS (EI, 70 eV): m/z (%) = 346 [M+] (1), 331 (1), 286 (4), 263 (2), 243 (5), 221 (7), 194 (5), 175 (8), 161 (10), 152 (28), 137 (100), 109 (65), 43 (74).
25). [α]20D = −129.4 (c 0.1, MeOH) (lit.: [α]D = −191 (c 0.4, CHCl3),32 [α]D = −183 (c 0.1),25 [α]25D = −128 (c 1.0, CHCl3)23). CD (MeOH): Δε (λ) +0.01 (247 nm), −0.01 (274 nm). Fluorescence (MeOH): λex = 203 nm, λem = 300 nm. IR (ATR): ν = 2929, 2853, 1755, 1662, 1446, 1384, 1245, 1040, 940, 838 cm−1. 1H NMR (600 MHz, CDCl3): δ = 1.00 (1H, td, J = 13.2, 3.0 Hz, H-9b), 1.26 (3H, s, H3-19), 1.59 (3H, s, H3-20), 1.60–1.65 (2H, m, H-6b, H-14b), 1.64 (3H, s, H3-17), 1.81 (3H, s, H3-18), 1.86–1.94 (4H, m, H-6a, H-10b, H2-13), 2.10 (1H, ddd, J = 13.1, 5.2, 2.8 Hz, H-9a), 2.25 (1H, dddd, J = 14.3, 10.2, 4.9, 3.4 Hz, H-10a), 2.30–2.38 (2H, m, H2-5), 2.51–2.59 (1H, m, H-14a), 2.71 (1H, t, J = 4.1 Hz, H-7), 4.45–4.53 (2H, m, H2-16), 5.09 (1H, dd, J = 9.8, 5.3 Hz, H-11), 5.22 (1H, d, J = 9.8 Hz, H-3), 5.50–5.56 (1H, m, H-2). 13C NMR and DEPT (150 MHz, CDCl3): δ = 10.20 (C-17), 15.06 (C-20), 15.58 (C-18), 16.91 (C-19), 23.52 (C-10), 25.28 (C-6), 26.12 (C-14), 36.66 (C-13), 37.65 (C-5), 39.83 (C-9), 59.84 (C-8), 61.90 (C-7), 78.40 (C-16), 83.63 (C-2), 123.59 (C-11), 126.27 (C-3), 127.86 (C-15), 133.19 (C-1), 136.84 (C-12), 139.30 (C-4). MS (EI, 70 eV): m/z (%) = 302 [M+] (9), 287 (11), 259 (4), 231 (7), 217 (6), 203 (10), 189 (13), 175 (29), 163 (100), 149 (78), 135 (99), 121 (53), 109 (59), 95 (77), 91 (56), 79 (49), 67 (63), 55 (53), 43 (75). ESI-MS (+10 V): m/z = 285 [M + H − H2O]+, 303 [M + H]+, 320 [M + NH4]+. Anal. calcd for C20H30O2: C 79.42, H 10.00 found C 79.15, H 10.34.
136, independent reflections 3258 (Rint = 0.0316), 204 parameters, R1 = 0.0264, wR2 = 0.0747 [I > 2σ(I)]; maximal residual electron density: 0.123 e Å−3. Flack parameter: χ = 0.1 (0). CCDC 1526876 contains the supplementary crystallographic data for this structure.
38). [α]20D = +2.6 (c 0.07, MeOH) (lit.: [α]20D = +2.0 (c 4.40, CHCl3)38). IR (ATR): ν = 3355 (br), 2957, 2916, 2849, 1469, 1330, 1258, 1117, 1061, 1021, 936, 865, 798, 717, 677 cm−1. 1H NMR (600 MHz, CDCl3): δ = 0.87 (3H, t, J = 7.0 Hz, H3-18′), 1.24 (28H, s br, H2-4′–H2-17′), 1.25–1.32 (2H, m, H2-3′), 1.57 (2H, tt, J = 7.6, 6.3 Hz, H2-2′), 3.42–3.50 (2H, m, H2-1′), 3.50 (1H, dd, J = 9.4, 6.0 Hz, H-3b), 3.54 (1H, dd, J = 9.8, 3.8 Hz, H-3a), 3.67 (1H, dd, J = 11.7, 5.3 Hz, H-1b), 3.73 (1H, dd, J = 11.7, 3.8 Hz, H-1a), 3.88 (1H, tt, J = 5.3, 4.1 Hz, H-2). 13C NMR and DEPT (150 MHz, CDCl3): δ = 14.11 (C-18′), 22.68 (C-17′), 26.07 (C-3′), 29.35, 29.44, 29.57, 29.60, 29.65, 29.68 (C-2′, C-4′–C-15′), 31.91 (C-16′), 64.33 (C-1), 70.34 (C-2), 71.88 (C-1′), 72.53 (C-3). MS (EI, 70 eV): m/z (%) = 313 [(M − CH2OH)+] (2), 283 (8), 253 (6), 125 (7), 111 (13), 97 (26), 85 (45), 71 (60), 57 (100), 43 (65). ESI-MS (+25 V): m/z = 345 [M + H]+, 362 [M + NH4]+.
O). MS (EI, 70 eV): m/z (%) = 346 [M+] (1), 331 (1), 286 (4), 263 (2), 243 (5), 221 (7), 194 (5), 175 (8), 161 (10), 152 (28), 137 (100), 109 (65), 43 (74).
25). [α]20D = −129.4 (c 0.1, MeOH) (lit.: [α]D = −191 (c 0.4, CHCl3),32 [α]D = −183 (c 0.1),25 [α]25D = −128 (c 1.0, CHCl3)23). CD (MeOH): Δε (λ) +0.01 (247 nm), −0.01 (274 nm). Fluorescence (MeOH): λex = 203 nm, λem = 300 nm. IR (ATR): ν = 2929, 2853, 1755, 1662, 1446, 1384, 1245, 1040, 940, 838 cm−1. 1H NMR (600 MHz, CDCl3): δ = 1.00 (1H, td, J = 13.2, 3.0 Hz, H-9b), 1.26 (3H, s, H3-19), 1.59 (3H, s, H3-20), 1.60–1.65 (2H, m, H-6b, H-14b), 1.64 (3H, s, H3-17), 1.81 (3H, s, H3-18), 1.86–1.94 (4H, m, H-6a, H-10b, H2-13), 2.10 (1H, ddd, J = 13.1, 5.2, 2.8 Hz, H-9a), 2.25 (1H, dddd, J = 14.3, 10.2, 4.9, 3.4 Hz, H-10a), 2.30–2.38 (2H, m, H2-5), 2.51–2.59 (1H, m, H-14a), 2.71 (1H, t, J = 4.1 Hz, H-7), 4.45–4.53 (2H, m, H2-16), 5.09 (1H, dd, J = 9.8, 5.3 Hz, H-11), 5.22 (1H, d, J = 9.8 Hz, H-3), 5.50–5.56 (1H, m, H-2). 13C NMR and DEPT (150 MHz, CDCl3): δ = 10.20 (C-17), 15.06 (C-20), 15.58 (C-18), 16.91 (C-19), 23.52 (C-10), 25.28 (C-6), 26.12 (C-14), 36.66 (C-13), 37.65 (C-5), 39.83 (C-9), 59.84 (C-8), 61.90 (C-7), 78.40 (C-16), 83.63 (C-2), 123.59 (C-11), 126.27 (C-3), 127.86 (C-15), 133.19 (C-1), 136.84 (C-12), 139.30 (C-4). MS (EI, 70 eV): m/z (%) = 302 [M+] (9), 287 (11), 259 (4), 231 (7), 217 (6), 203 (10), 189 (13), 175 (29), 163 (100), 149 (78), 135 (99), 121 (53), 109 (59), 95 (77), 91 (56), 79 (49), 67 (63), 55 (53), 43 (75). ESI-MS (+10 V): m/z = 285 [M + H − H2O]+, 303 [M + H]+, 320 [M + NH4]+. Anal. calcd for C20H30O2: C 79.42, H 10.00 found C 79.15, H 10.34.
136, independent reflections 3258 (Rint = 0.0316), 204 parameters, R1 = 0.0264, wR2 = 0.0747 [I > 2σ(I)]; maximal residual electron density: 0.123 e Å−3. Flack parameter: χ = 0.1 (0). CCDC 1526876 contains the supplementary crystallographic data for this structure.
38). [α]20D = +2.6 (c 0.07, MeOH) (lit.: [α]20D = +2.0 (c 4.40, CHCl3)38). IR (ATR): ν = 3355 (br), 2957, 2916, 2849, 1469, 1330, 1258, 1117, 1061, 1021, 936, 865, 798, 717, 677 cm−1. 1H NMR (600 MHz, CDCl3): δ = 0.87 (3H, t, J = 7.0 Hz, H3-18′), 1.24 (28H, s br, H2-4′–H2-17′), 1.25–1.32 (2H, m, H2-3′), 1.57 (2H, tt, J = 7.6, 6.3 Hz, H2-2′), 3.42–3.50 (2H, m, H2-1′), 3.50 (1H, dd, J = 9.4, 6.0 Hz, H-3b), 3.54 (1H, dd, J = 9.8, 3.8 Hz, H-3a), 3.67 (1H, dd, J = 11.7, 5.3 Hz, H-1b), 3.73 (1H, dd, J = 11.7, 3.8 Hz, H-1a), 3.88 (1H, tt, J = 5.3, 4.1 Hz, H-2). 13C NMR and DEPT (150 MHz, CDCl3): δ = 14.11 (C-18′), 22.68 (C-17′), 26.07 (C-3′), 29.35, 29.44, 29.57, 29.60, 29.65, 29.68 (C-2′, C-4′–C-15′), 31.91 (C-16′), 64.33 (C-1), 70.34 (C-2), 71.88 (C-1′), 72.53 (C-3). MS (EI, 70 eV): m/z (%) = 313 [(M − CH2OH)+] (2), 283 (8), 253 (6), 125 (7), 111 (13), 97 (26), 85 (45), 71 (60), 57 (100), 43 (65). ESI-MS (+25 V): m/z = 345 [M + H]+, 362 [M + NH4]+.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2).
Yellow solid. ESI-MS (+10 V): m/z = 275 [M + H]+.
:2).
Yellow solid. ESI-MS (+10 V): m/z = 275 [M + H]+.
6: 1H NMR (600 MHz, CDCl3): δ = 1.34 (3H, t, J = 7.2 Hz, H3-17), 1.80 (3H, q, J = 1.1 Hz, H3-15), 1.96 (3H, d, J = 1.1 Hz, H3-14), 2.22 (2H, t, J = 7.9 Hz, H2-7), 2.36 (2H, q, J = 7.5 Hz, H2-8), 4.29 (2H, q, J = 7.1 Hz, H2-16), 5.09 (1H, dt, J = 10.5, 1.5 Hz, H-12b), 5.29 (1H, br d, J = 17.3 Hz, H-12a), 5.37 (1H, br t, J = 7.3 Hz, H-9), 6.05 (1H, br s, H-5), 6.49 (1H, s, H-3), 6.76 (1H, ddd, J = 17.3, 10.8, 0.9 Hz, H-11), 7.88 (1H, s, H-1). 13C NMR and DEPT (150 MHz, CDCl3): δ = 14.11 (C-17), 18.71 (C-14), 19.74 (C-15), 25.80 (C-8), 40.66 (C-7), 60.38 (C-16), 106.74 (C-3), 113.65 (C-5), 113.77 (C-12), 120.63 (C-2), 129.75 (C-9), 132.84 (C-10), 133.46 (C-11), 140.50 (C-6), 145.42 (C-1), 154.61 (C-4), 163.38 (C-13). MS (EI, 70 eV): m/z (%) = 274 [M+] (5), 229 (6), 193 (44), 147 (100), 119 (8), 91 (20), 81 (10), 79 (10), 65 (7), 53 (8), 41 (8).
7: 1H NMR (600 MHz, CDCl3): δ = 1.34 (3H, t, J = 7.2 Hz, H3-17′), 1.74 (3H, s, H3-15′), 1.97 (3H, d, J = 1.1 Hz, H3-14′), 2.24 (2H, t, J = 7.9 Hz, H2-7′), 2.33 (2H, q, J = 7.9 Hz, H2-8′), 4.29 (2H, q, J = 7.1 Hz, H2-16′), 4.93 (1H, d, J = 10.9 Hz, H-12b′), 5.09 (1H, d, J = 17.3 Hz, H-12a′), 5.47 (1H, br t, J = 7.2 Hz, H-9′), 6.05 (1H, br s, H-5′), 6.35 (1H, dd, J = 17.1, 10.7 Hz, H-11′), 6.49 (1H, s, H-3′), 7.88 (1H, s, H-1′); 13C NMR and DEPT (150 MHz, CDCl3): δ = 11.69 (C-15′), 14.32 (C-17′), 18.71 (C-14′), 26.70 (C-8′), 40.27 (C-7′), 60.38 (C-16′), 106.74 (C-3′), 110.84 (C-12′), 113.65 (C-5′), 120.63 (C-2′), 131.75 (C-9′), 132.84 (C-10′), 140.50 (C-6′), 141.34 (C-11′), 145.42 (C-1′), 154.61 (C-4′), 163.38 (C-13′). MS (EI, 70 eV): m/z (%) = 274 [M+] (5), 229 (4), 193 (43), 147 (100), 119 (8), 91 (20), 81 (10), 79 (9), 65 (6), 53 (8), 41 (7).
104). Fluorescence (MeOH): λex = 202 nm, λem = 303 nm. IR (ATR): ν = 2954, 2916, 2848, 1709, 1462, 1372, 1165, 720 cm−1. 1H NMR (600 MHz, CDCl3): δ = 0.87 (3H, t, J = 7.0 Hz, H3-18), 1.22–1.29 (26H, m, H-5–H-17), 1.52–1.58 (2H, m, H2-4), 2.12 (3H, s, H3-1), 2.40 (2H, t, J = 7.5 Hz, H2-3). 13C NMR and DEPT (150 MHz, CDCl3): δ = 14.12 (C-18), 22.69 (C-17), 23.88 (C-4), 29.35, 29.47, 29.60, 29.65, 29.68 (11 CH2, C-5–C-15), 29.85 (C-1), 31.92 (C-16), 43.84 (C-3), 209.43 (C-2). MS (EI, 70 eV): m/z (%) = 268 [M+] (8), 96 (10), 85 (13), 71 (42), 58 (90), 43 (100). ESI-MS (+10 V): m/z = 286 [M + NH4]+, 559 [2M + Na]+.
78). [α]20D = −27.0 (c 0.05, MeOH) (lit.: [α]D = −25 (CHCl3)78). CD (MeOH): Δε (λ) +0.19 (201 nm). Fluorescence (MeOH): λex = 203 nm, λem = 299 nm. IR (ATR): ν = 3339 (br), 2955, 2933, 2866, 1456, 1377, 1056, 1023, 957, 925, 886, 841, 799, 738 cm−1. 1H NMR (600 MHz, CDCl3): δ = 0.67 (3H, s, H3-18), 0.79 (3H, d, J = 6.8 Hz, H3-28), 0.80 (3H, d, J = 7.2 Hz, H3-26), 0.85 (3H, t, J = 7.3 Hz, H3-27-Me), 0.90 (3H, d, J = 6.8 Hz, H3-21), 0.90–0.92 (1H, m, H-9), 0.96–0.99 (1H, m, H-14), 1.00 (3H, s, H3-19), 1.03–1.11 (6H, m, H-1b, H-15b, H-17, H-22b, H-23b, H-27b), 1.14 (1H, td, J = 12.8, 4.9 Hz, H-12b), 1.17–1.20a (1H, m, H-23a), 1.23–1.27 (2H, m, H-16b, H-25), 1.27–1.32 (2H, m, H-22a, H-24), 1.32–1.37 (2H, m, H-20, H-27a), 1.40–1.47 (1H, m, H-8), 1.47–1.57 (4H, m, H-2b, H-7b, H-11a, H-11b), 1.55–1.60 (1H, m, H-15a), 1.80–1.86 (3H, m, H-1a, H-2a, H-16a), 1.96 (1H, dtd, J = 17.3, 4.9, 2.6 Hz, H-7a), 2.00 (1H, dt, J = 12.4, 3.4 Hz, H-12a), 2.19–2.26 (1H, m, H-4b), 2.29 (1H, ddd, J = 13.2, 5.3, 2.3 Hz, H-4a), 3.51 (1H, tt, J = 11.2, 4.7 Hz, H-3), 5.34 (1H, dt, J = 5.0, 2.6 Hz, H-6); aChemical shift derived from the HSQC spectrum. 13C NMR and DEPT (150 MHz, CDCl3): δ = 11.87 (C-18), 12.22 (C-27-Me), 15.90 (C-26), 16.54 (C-28), 18.71 (C-21), 19.40 (C-19), 21.10 (C-11), 24.30 (C-15), 25.78 (C-27), 28.23 (C-16), 29.04 (C-23), 31.70 (C-2), 31.93 (C-7, C-8), 33.90 (C-22), 35.88 (C-20), 36.52 (C-10), 37.27 (C-1), 37.53 (C-24), 39.80 (C-12), 39.86 (C-25), 42.34 (C-4, C-13), 50.17 (C-9), 56.15 (C-17), 56.79 (C-14), 71.83 (C-3), 121.72 (C-6), 140.78 (C-5). MS (EI, 70 eV): m/z (%) = 414 [M+] (95), 399 (36), 396 (49), 381 (34), 354 (9), 329 (52), 303 (49), 273 (27), 255 (32), 231 (20), 213 (39), 145 (51), 105 (57), 57 (100). ESI-MS (+10 V): m/z = 397 [M + H − H2O]+, 432 [M + NH4]+.
:1).
Amorphous solid. Fluorescence (MeOH): λex = 205 nm, λem = 300 nm. IR (ATR): ν = 3417, 3350, 3196, 2979, 2930, 2845, 1662, 1395, 1321, 1214, 1089, 1059, 966, 870, 815, 698 cm−1. MS (EI, 70 eV): m/z (%) = 297 (3), 295 (3), 283 (15), 281 (33), 279 (16), 269 (17), 267 (33), 265 (17), 247 (8), 245 (9), 219 (56), 217 (61), 187 (100), 185 (97), 159 (21), 157 (22), 131 (12), 129 (9), 53 (78). ESI-MS (+10 V): m/z = 401 [M + NH4]+, 403, 405, 789 [2M + Na]+, 791, 793, 795, 797.
14a: 1H NMR (600 MHz, CDCl3): δ = 1.26 (3H, t, J = 7.0 Hz, H3-11), 2.50 (2H, s, H2-7), 3.14 (3H, s, H3-9), 3.34 (2H, q, J = 7.2 Hz, H2-10), 5.20 (1H, s, OH), 5.59 (1H, br s, N-Hb), 5.73 (1H, br s, N-Ha), 6.72 (2H, s, H-2/H-6). 13C NMR and DEPT (150 MHz, CDCl3): δ = 14.94 (C-11), 43.67 (C-7), 50.88 (C-9), 59.74 (C-10), 70.92 (C-1), 96.12 (C-4), 124.41 (C-3/C-5), 139.44 (C-2/C-6), 172.31 (C-8).
14b: 1H NMR (600 MHz, CDCl3): δ = 1.27 (3H, t, J = 6.8 Hz, H3-11), 2.50 (2H, s, H2-7), 3.20 (3H, s, H3-9), 3.24 (2H, q, J = 7.2 Hz, H2-10), 5.16 (1H, s, OH), 5.59 (1H, br s, N-Hb), 5.73 (1H, br s, N-Ha), 6.72 (2H, s, H-2/H-6). 13C NMR and DEPT (150 MHz, CDCl3): δ = 15.07 (C-11), 43.71 (C-7), 51.16 (C-9), 59.34 (C-10), 70.90 (C-1), 96.13 (C-4), 124.42 (C-3/C-5), 139.42 (C-2/C-6), 172.31 (C-8).
Acknowledgements
Marie Pascaline Rahelivao is grateful to the European Commission (Erasmus Mundus Programme), the Gesellschaft von Freunden und Förderern der TU Dresden (GFF TU Dresden), and the TUD Graduate Academy for providing scholarships. We would like to thank Dr Shirley Parker-Nance (Nelson Mandela Metropolitan University, Port Elizabeth, and South African Institute for Aquatic Biodiversity, Grahamstown, South Africa) for the identification of the soft corals and the sponge. We are indebted to Mrs Herivony Onja Andriambeloson (Laboratoire de Microbiologie de l'Environnement, Centre National de Recherche sur l'Environnement, Antananarivo, Madagascar) for the antimicrobial activity tests and Mr Michel Ratsimbason (Centre National d'Application de Recherche Pharmaceutique, Antananarivo, Madagascar) for the antimalarial assay.Notes and references
- M. P. Rahelivao, M. Gruner, H. Andriamanantoanina, B. Andriamihaja, I. Bauer and H.-J. Knölker, Mar. Drugs, 2015, 13, 4197–4216 CrossRef CAS PubMed.
- M. P. Rahelivao, M. Gruner, H. Andriamanantoanina, I. Bauer and H.-J. Knölker, Nat. Prod. Bioprospect., 2015, 5, 223–235 CrossRef CAS PubMed.
- M. P. Rahelivao, M. Gruner, T. Lübken, D. Islamov, O. Kataeva, H. Andriamanantoanina, I. Bauer and H.-J. Knölker, Org. Biomol. Chem., 2016, 14, 989–1001 CAS.
- D. J. Faulkner, Nat. Prod. Rep., 1984, 1, 551–598 RSC.
- W. Fenical, J. Nat. Prod., 1987, 50, 1001–1008 CrossRef CAS.
- A. S. R. Anjaneyulu and G. V. Rao, J. Indian Chem. Soc., 1997, 74, 272–278 CAS.
- Y. Li, L. Peng and T. Zhang, in Medicinal Chemistry of Bioactive Natural Products, ed. X.-T. Liang and W.-S. Fang, Wiley, Hoboken, NJ, USA, 2006, pp. 257–300 Search PubMed.
- P. Radhika, Biochem. Syst. Ecol., 2006, 34, 781–789 CrossRef CAS.
- D. Kelman, Y. Kashman, R. T. Hill, E. Rosenberg and Y. Loya, Pure Appl. Chem., 2009, 81, 1113–1121 CrossRef CAS.
- J. Hu, B. Yang, X. Lin, X. Zhou, X. Yang, L. Long and Y. Liu, Chem. Biodiversity, 2011, 8, 1011–1032 CAS.
- L.-F. Liang and Y.-W. Guo, Chem. Biodiversity, 2013, 10, 2161–2196 CAS.
- R. Y. P. Burhan, E. M. M. Putri, Y. Zetra, R. Herdhiansyah and M. Y. Putra, J. Chem. Pharm. Res., 2014, 6, 585–595 Search PubMed.
- Q. Yu, P. Sun, B.-s. Liu, H. Tang and W. Zhang, Dier Junyi Daxue Xuebao, 2014, 35, 206–215 CAS.
- T. A. Hussien and T. A. Mohamed, Res. J. Pharm., Biol. Chem. Sci., 2015, 6, 1301–1319 CAS.
- A. Longeon, M.-L. Bourguet-Kondracki and M. Guyot, Tetrahedron Lett., 2002, 43, 5937–5939 CrossRef CAS.
- J. J. Poza, R. Fernández, F. Reyes, J. Rodríguez and C. Jiménez, J. Org. Chem., 2008, 73, 7978–7984 CrossRef CAS PubMed.
- Y. Hou and L. Harinantenaina, Curr. Med. Chem., 2010, 17, 1191–1219 CrossRef CAS PubMed.
- C. L. Griffiths, Indian J. Mar. Sci., 2005, 34, 35–41 Search PubMed.
- Drug Discovery in Africa, ed. K. Chibale, M. Davies-Coleman and C. Masimirembwa, Springer, Berlin, Germany, 2012 Search PubMed.
- Vlaams Institute voor de Zee, Assessing potential World Heritage marine sites in the Western Indian Ocean. http://www.vliz.be/projects/marineworldheritage/index.php (accessed 11 January 2017).
- M. S. Zubair, K. O. Al-Footy, S.-E. N. Ayyad, S. S. Al-Lihaibi and W. M. Alarif, Nat. Prod. Res., 2016, 30, 869–879 CrossRef CAS PubMed.
- Ether Lipids, Biochemical and Biomedical Aspects, ed. H. K. Mangold and F. Paltauf, Academic Press, New York, USA, 1983 Search PubMed.
- K. H. Shaker, M. Müller, M. A. Ghani, H.-M. Dahse and K. Seifert, Chem. Biodiversity, 2010, 7, 2007–2015 CAS.
- Z.-G. Rao, S.-Z. Deng, F.-Y. Li, H.-M. Wu and Z.-S. Shi, Chin. J. Org. Chem., 1997, 17, 252–255 CAS.
- B. F. Bowden, J. C. Coll and S. J. Mitchell, Aust. J. Chem., 1980, 33, 879–884 CrossRef CAS.
- J. Shin, W. Fenical, T. J. Stout and J. Clardy, Tetrahedron, 1993, 49, 515–524 CrossRef CAS.
- H. Dong, Y.-L. Gou, R. M. Kini, H.-X. Xu, S.-X. Chen, S. L. M. Teo and P. P.-H. But, Chem. Pharm. Bull., 2000, 48, 1087–1089 CrossRef CAS PubMed.
- Z. Liu, W. Z. Li, L. Peng, Y. Li and Y. Li, J. Chem. Soc., Perkin Trans. 1, 2000, 4250–4257 RSC.
- J. C. Coll, B. F. Bowden, G. M. König, R. Braslau and I. R. Price, Bull. Soc. Chim. Belg., 1986, 95, 815–834 CrossRef CAS.
- G. J. Greenland and B. F. Bowden, Aust. J. Chem., 1994, 47, 2013–2021 CrossRef CAS.
- M. Kobayashi, K. Kondo, K. Osabe and H. Mitsuhashi, Chem. Pharm. Bull., 1988, 36, 2331–2341 CrossRef CAS PubMed.
- B. F. Bowden, J. C. Coll, W. Hicks, R. Kazlauskas and S. J. Mitchell, Aust. J. Chem., 1978, 31, 2707–2712 CrossRef CAS.
- B. F. Bowden, J. C. Coll, A. Heaton, G. König, M. A. Bruck, R. E. Cramer, D. M. Klein and P. J. Scheuer, J. Nat. Prod., 1987, 50, 650–659 CrossRef CAS.
- Y. Kashman, E. Zadock and I. Néeman, Tetrahedron, 1974, 30, 3615–3620 CrossRef CAS.
- J. M. Frincke, D. E. McIntyre and D. J. Faulkner, Tetrahedron Lett., 1980, 21, 735–738 CrossRef CAS.
- J. Kobayashi, Y. Ohizumi, H. Nakamura, T. Yamakado, T. Matsuzaki and Y. Hirata, Experientia, 1983, 39, 67–69 CrossRef CAS PubMed.
- S. Aratake, T. Tomura, S. Saitoh, R. Yokokura, Y. Kawanishi, R. Shinjo, J. D. Reimer, J. Tanaka and H. Maekawa, PLoS One, 2012, 7, e30410 CAS.
- C. D. Magnusson, A. V. Gudmundsdottir and G. G. Haraldsson, Tetrahedron, 2011, 67, 1821–1836 CrossRef CAS.
- J. C. Coll, S. J. Mitchell and G. J. Stokie, Tetrahedron Lett., 1977, 18, 1539–1542 CrossRef.
- B. F. Bowden, J. C. Coll, E. D. de Silva and M. S. L. de Costa, Aust. J. Chem., 1983, 36, 371–376 CrossRef CAS.
- K. J. S. Grace, D. Zavortink and R. S. Jacobs, Biochem. Pharmacol., 1994, 47, 1427–1434 CrossRef CAS PubMed.
- (a) S. Rajaram, U. Ramulu, D. Ramesh, D. Srikanth, P. Bhattacharya, P. Prabhakar, S. V. Kalivendi, K. S. Babu, Y. Venkateswarlu and S. Navath, Bioorg. Med. Chem. Lett., 2013, 23, 6234–6238 CrossRef CAS PubMed; (b) S. Rajaram, D. Ramesh, U. Ramulu, M. Anjum, P. Kumar, U. S. N. Murthy, M. A. Hussain, G. N. Sastry and Y. Venkateswarlu, Indian J. Chem., 2014, 53B, 1086–1090 CAS.
- D. H. Williams and D. J. Faulkner, Tetrahedron, 1996, 52, 4245–4256 CrossRef CAS.
- (a) D. H. R. Barton, H. Togo and S. Z. Zard, Tetrahedron, 1985, 41, 5507–5516 CrossRef CAS; (b) A. Ulubelen and T. Baytop, Phytochemistry, 1973, 12, 1824 CrossRef CAS; (c) J. O. Grimalt, L. Angulo, A. López-Galindo, M. C. Comas and J. Albaigés, Chem. Geol., 1990, 82, 341–363 CrossRef CAS; (d) M. A. Kruge, A. Permanyer, J. Serra and D. Yu, J. Anal. Appl. Pyrolysis, 2010, 89, 204–217 CrossRef CAS.
- (a) A. A. Bothner-By, Adv. Magn. Opt. Reson., 1965, 1, 195–316 Search PubMed; (b) M. J. Minch, Concepts Magn. Res., 1994, 6, 41–56 CrossRef CAS.
- (a) X.-Q. Ma, C.-Z. Liao, J.-Y. Su and L.-M. Zeng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, o889–o890 CAS; (b) A.-C. Huang, C. J. Sumby, E. R. T. Tiekink and D. K. Taylor, J. Nat. Prod., 2014, 77, 2522–2536 CrossRef CAS PubMed.
- (a) R. B. Bates and R. C. Slagel, J. Am. Chem. Soc., 1962, 84, 1307–1308 CrossRef CAS; (b) M. V. Kadival, M. S. R. Nair and S. C. Bhattacharyya, Tetrahedron, 1967, 23, 1241–1249 CrossRef CAS.
- (a) C. Ehret and G. Ourisson, Tetrahedron, 1969, 25, 1785–1799 CrossRef CAS; (b) H. D. Friedel and R. Matusch, Helv. Chim. Acta, 1987, 70, 1616–1622 CrossRef CAS; (c) H. D. Friedel and R. Matusch, J. Chromatogr., A, 1987, 407, 343–348 CrossRef CAS.
- G. W. Patterson, Comp. Biochem. Physiol., 1968, 24, 501–505 CAS.
- G. W. Patterson, Lipids, 1971, 6, 120–127 CrossRef CAS.
- K. Iguchi, S. Saitoh and Y. Yamada, Chem. Pharm. Bull., 1989, 37, 2553–2554 CrossRef CAS.
- W. Lu, C. Zhang, L. Zeng and J. Su, Steroids, 2004, 69, 803–808 CrossRef CAS PubMed.
- W. C. M. C. Kokke, S. Epstein, S. A. Look, G. H. Rau, W. Fenical and C. Djerassi, J. Biol. Chem., 1984, 259, 8168–8173 CAS.
- S. Yu, Z. Deng, L. van Ofwegen, P. Proksch and W. Lin, Steroids, 2006, 71, 955–959 CrossRef CAS PubMed.
- Y. Venkateswarlu, K. V. Sridevi and M. R. Rao, J. Nat. Prod., 1999, 62, 756–758 CrossRef CAS PubMed.
- N. M. O'Connell, Y. C. O'Callaghan, N. M. O'Brien, A. R. Maguire and F. O. McCarthy, Tetrahedron, 2012, 68, 4995–5004 CrossRef.
- D. Sica, V. Piccialli and A. Masullo, Phytochemistry, 1984, 23, 2609–2611 CrossRef CAS.
- J. Zielinski, H. T. Li and C. Djerassi, J. Org. Chem., 1982, 47, 620–625 CrossRef CAS.
- M. Y. Putra, G. Bavestrello, C. Cerrano, B. Renga, C. D′Amore, S. Fiorucci, E. Fattorusso and O. Taglialatela-Scafati, Steroids, 2012, 77, 433–440 CrossRef CAS PubMed.
- M. M. Radwan, S. P. Manly, K. A. El Sayed, V. B. Wali, P. W. Sylvester, B. Awate, G. Shah and S. A. Ross, J. Nat. Prod., 2008, 71, 1468–1471 CrossRef CAS PubMed.
- C. Subrahmanyam, C. Venkateswara Rao, V. Anjaneyulu, P. Satyanarayana, P. V. Subba Rao, R. S. Ward and A. Pelter, Tetrahedron, 1992, 48, 3111–3120 CrossRef CAS.
- W. Bergmann, M. J. McLean and D. Lester, J. Org. Chem., 1943, 8, 271–282 CrossRef CAS.
- N. C. Ling, R. L. Hale and C. Djerassi, J. Am. Chem. Soc., 1970, 92, 5281–5282 CrossRef CAS PubMed.
- R. L. Hale, J. Leclercq, B. Tursch, C. Djerassi, R. A. Gross Jr., A. J. Weinheimer, K. Gupta and P. J. Scheuer, J. Am. Chem. Soc., 1970, 92, 2179–2180 CrossRef CAS.
- B. F. Bowden, J. C. Coll and M. C. Dai, Aust. J. Chem., 1989, 42, 665–673 CrossRef CAS.
- M. Kobayashi, A. Tomioka and H. Mitsuhashi, Steroids, 1979, 34, 273–284 CrossRef CAS PubMed.
- V. Sepe, F. S. Di Leva, C. D'Amore, C. Festa, S. De Marino, B. Renga, M. V. D'Auria, E. Novellino, V. Limongelli, L. D'Souza, M. Majik, A. Zampella and S. Fiorucci, Mar. Drugs, 2014, 12, 3091–3115 CrossRef CAS PubMed.
- B. F. Bowden, B. J. Cusack and A. Dangel, Mar. Drugs, 2003, 1, 18–26 CrossRef CAS.
- B. F. Bowden, J. A. Brittle, J. C. Coll, N. Liyanage, S. J. Mitchell and G. J. Stokie, Tetrahedron Lett., 1977, 18, 3661–3662 CrossRef.
- N. X. Cuong, N. P. Thao, B. T. T. Luyen, N. T. T. Ngan, D. T. T. Thuy, S. B. Song, N. H. Nam, P. V. Kiem, Y. H. Kim and C. V. Minh, Chem. Pharm. Bull., 2014, 62, 203–208 CrossRef PubMed.
- S.-Y. Cheng, S.-K. Wang and C.-Y. Duh, Mar. Drugs, 2014, 12, 6028–6037 CrossRef PubMed.
- C.-Y. Kao, J.-H. Su, M.-C. Lu, T.-L. Hwang, W.-H. Wang, J.-J. Chen, J.-H. Sheu, Y.-H. Kuo, C.-F. Weng, L.-S. Fang, Z.-H. Wen and P.-J. Sung, Mar. Drugs, 2011, 9, 1319–1331 CrossRef CAS PubMed.
- J. M. Badr, L. A. Shaala, M. I. Abou-Shoer, M. K. Tawfik and A.-A. M. Habib, J. Nat. Prod., 2008, 71, 1472–1474 CrossRef CAS PubMed.
- L. A. Shaala, D. T. A. Youssef, M. Sulaiman, F. A. Behery, A. I. Foudah and K. A. El Sayed, Mar. Drugs, 2012, 10, 2492–2508 CrossRef CAS PubMed.
- L. A. Shaala, D. T. A. Youssef, J. M. Badr, M. Sulaiman, A. Khedr and K. A. El Sayed, Tetrahedron, 2015, 71, 7837–7841 CrossRef CAS.
- A. Kelecom, G. J. Kannengiesser and P. M. Baker, An. Acad. Bras. Cienc., 1979, 51, 643–645 CAS.
- C. A. N. Catalan, J. E. Thompson, W. C. M. C. Kokke and C. Djerassi, Tetrahedron, 1985, 41, 1073–1084 CrossRef CAS.
- P. De Luca, M. De Rosa, L. Minale and G. Sodano, J. Chem. Soc., Perkin Trans. 1, 1972, 2132–2135 RSC.
- N. S. Lira, R. L. Monte-Neto, J. G. B. Marchi, A. C. S. Lins, J. F. Tavares, M. S. Silva, C. S. Dias, J. M. Barbosa-Filho, C. F. Santos, E. V. L. Cunha, U. S. Pinheiro and R. Braz-Filho, Quim. Nova, 2012, 35, 2189–2193 CrossRef CAS.
- B. Dinda, S. Majumder, S. Arima, N. Sato and Y. Harigaya, J. Nat. Med., 2008, 62, 447–451 CrossRef CAS PubMed.
- E. A. Santalova, V. A. Denisenko, V. P. Glazunov, A. I. Kalinovskii, S. D. Anastyuk and V. A. Stonik, Russ. Chem. Bull., 2011, 60, 570–580 CrossRef CAS.
- E. A. Santalova, V. A. Denisenko and P. S. Dmitrenok, Chem. Nat. Compd., 2013, 49, 75–78 CrossRef CAS.
- R. J. Andersen and D. J. Faulkner, Tetrahedron Lett., 1973, 14, 1175–1178 CrossRef.
- Y. M. Goo, Arch. Pharmacal Res., 1985, 8, 21–30 CrossRef CAS.
- R. Xynas and R. J. Capon, Aust. J. Chem., 1989, 42, 1427–1433 CrossRef CAS.
- M. H. Kossuga, S. P. Lira, A. M. Nascimento, M. T. P. Gambardella, R. G. S. Berlinck, Y. R. Torres, G. G. F. Nascimento, E. F. Pimenta, M. Silva, O. H. Thiemann, G. Oliva, A. G. Tempone, M. S. C. Melhem, A. O. Souza, F. C. S. Galetti, C. L. Silva, B. Cavalcanti, C. O. Pessoa, M. O. Moraes, E. Hajdu, S. Peixinho and R. M. Rocha, Quim. Nova, 2007, 30, 1194–1202 CrossRef CAS.
- X.-P. Huang, Z.-W. Deng, R. W. M. van Soest and W.-H. Lin, J. Asian Nat. Prod. Res., 2008, 10, 253–258 Search PubMed.
- L. A. Shaala, S. I. Khalifa, M. K. Mesbah, R. W. M. van Soest and D. T. A. Youssef, Nat. Prod. Commun., 2008, 3, 219–222 CAS.
- K. H. Shaker, H. Zinecker, M. A. Ghani, J. F. Imhoff and B. Schneider, Chem. Biodiversity, 2010, 7, 2880–2887 CAS.
- J.-j. Zhao and H.-g. Liu, Cent. South Pharm., 2013, 11, 185–186 CAS.
- X.-P. Huang, Z.-W. Deng, R. W. M. van Soest and W.-H. Lin, J. Asian Nat. Prod. Res., 2008, 10, 239–242 CrossRef CAS PubMed.
- A. A. Salim, Z. G. Khalil and R. J. Capon, Tetrahedron, 2012, 68, 9802–9807 CrossRef CAS.
- F. D. Mora, D. K. Jones, P. V. Desai, A. Patny, M. A. Avery, D. R. Feller, T. Smillie, Y.-D. Zhou and D. G. Nagle, J. Nat. Prod., 2006, 69, 547–552 CrossRef CAS PubMed.
- B. A. Gorshkov, I. A. Gorshkova, T. N. Makarieva and V. A. Stonik, Toxicon, 1982, 20, 1092–1094 CrossRef CAS PubMed.
- Bruker, APEX2 Software Suite for Crystallographic Programs, Bruker AXS Inc., Madison, WI, USA, 2009 Search PubMed.
- Bruker, SMART and SAINT. Area Detector Control and Integration Software. Version5.1, Bruker Analytical X-ray Instruments Inc., Madison, WI, USA, 1996 Search PubMed.
- G. M. Sheldrick, SADABS. Program for Absorption Correction. Version 2.10, University of Göttingen, Germany, 1996 Search PubMed.
- M. C. Burla, M. Camalli, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2003, 36, 1103–1103 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565–565 CrossRef CAS.
- Y. Corbett, L. Herrera, J. Gonzalez, L. Cubilla, T. L. Capson, P. D. Coley, T. A. Kursar, L. I. Romero and E. Ortega-Barria, Am. J. Trop. Med. Hyg., 2004, 70, 119–124 CAS.
- W. Trager and J. Jensen, Science, 1976, 193, 673–675 CAS.
- W. Trager and J. B. Jensen, J. Parasitol., 2005, 91, 484–486 CrossRef PubMed.
- M. C. Pangborn and R. J. Anderson, J. Am. Chem. Soc., 1936, 58, 10–14 CrossRef CAS.
- R. Jain, S. Nagpal, S. Jain and S. C. Jain, J. Med. Aromat. Plant Sci., 2004, 26, 48–50 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S13; Fig. S1–S22 (copies of the 1H, 13C and 2D NMR spectra of compounds 6, 7 and 9a); details of the antimalarial assay. CCDC 1526876. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ob00191f |
| This journal is © The Royal Society of Chemistry 2017 |