
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtecting group free synthesis of glycosyl thiols from reducing sugars in water; application to the production of N-glycan glycoconjugates†
S. R.
Alexander
a,
D.
Lim
 a,
Z.
Amso
bc,
M. A.
Brimble
bc and
A. J.
Fairbanks
*ad
a,
Z.
Amso
bc,
M. A.
Brimble
bc and
A. J.
Fairbanks
*ad
aDepartment of Chemistry, University of Canterbury, Private Bag 4800, Christchurch 8140, New Zealand. E-mail: antony.fairbanks@canterbury.ac.nz
bSchool of Chemical Sciences, The University of Auckland, 23 Symonds St, Auckland 1142, New Zealand
cMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
dBiomolecular Interaction Centre, University of Canterbury, Private Bag 4800, Christchurch 8140, New Zealand
First published on 13th February 2017
Abstract
Glycosyl thiols may be accessed from the corresponding reducing sugars in water without recourse to any sugar projecting groups by way of a DMC mediated reaction with thioacetic acid in the presence of base, and hydrolysis of the anomeric thioacetate. Glycosyl thiols produced by this method may be used to access glycoconjugates, such as glycopeptides by use of the thiol–ene click reaction.
Protein glycosylation is the most diverse form of post-translational modification. Unsurprisingly the roles of the carbohydrate portions of glycoproteins are correspondingly numerous,1 and indeed in many cases the glycan is essential for effective bioactivity.
The synthesis of neo-glycoproteins by the chemical conjugation of carbohydrates to non-glycosylated proteins, typically expressed in bacterial culture, has been an area of significant interest.2 The majority of synthetic approaches that attach carbohydrates in which the innermost monosaccharide pyranose ring remains intact require the pre-synthesis of a functionalised glycoside as the protein modifying agent; a process which usually requires multiple steps and protecting group manipulations.3
In 2009 Shoda and co-workers reported the remarkable application of the dehydrating reagent 2-chloro-1,3-dimethylimidazolinium chloride (DMC, 1)4 for the direct aqueous synthesis of glycosyl oxazolines from un-protected sugars with an N-acetyl glucosamine residue at the reducing terminus.5 Subsequently the synthesis of N-glycan oxazolines in water using DMC has proven to be a cornerstone for the production of wide variety of biologically important glycopeptides and glycoproteins in homogenous form using endo-β-N-acetylglucosaminidase (ENGase) catalysis.6 Key to the success of this process is the greater acidity of the anomeric hydroxyl group,7 which, under the mildly basic reaction conditions, may be selectively de-protonated and so is able to outcompete both solvent water and other sugar hydroxyl-groups as a nucleophile for reaction with DMC. Reaction of the anomeric hydroxyl with DMC activates it to subsequent displacement, for example by the acetamide at position-2 leading to oxazoline. Alternatively, an activated intermediate may be trapped if a good external nucleophile is present. Along these lines Shoda also reported the use of DMC activation of reducing sugars in the presence of a large excess of azide for the direct synthesis of glycosyl azides.8 The development of the related reagent ADMP,9 which is capable of both activating the anomeric hydroxyl group and furnishing a source of azide, allowed the one-pot conjugation of un-protected sugars, including large N-glycan oligosaccharides isolated from natural sources, to other species via click chemistry of glycosyl azides that were made in situ.10 Other selective nucleophilic substitution processes at the anomeric centre are also possible, most notably involving attack of sulfur nucleophiles11 on activated intermediates, and have been applied to the synthesis of pyridyl and aryl thioglycosides,12 and for the linking of peptides to carbohydrates via the side chains of cysteine residues.13
A mechanistic rationalisation of all of these differing reaction outcomes is shown in Scheme 1. Reaction of the mutatorating mixture of un-protected sugars 2β and 2α with DMC 1 initially gives rise to activated intermediates 3β and 3α (Scheme 1), following which a variety of mechanistic scenarios are possible. Direct attack of a nucleophile on the 1,2-cis glycoside 3α could lead to the 1,2-trans substitution product 4; if that nucleophile is solvent water then 2β is produced by hydrolysis.
 | ||
| Scheme 1 Proposed mechanistic pathways for DMC activation of un-protected sugars in water. | ||
Alternatively intramolecular attack of the 6-hydroxyl group could lead to the 1,6-anhydro sugar 5, although Shoda has used the inability of 2-deoxy sugars to produce 1,6-anhydro sugars as evidence that this pathway is in fact not followed.
A variety of scenarios are open to the 1,2-trans intermediate 3β. In the case of a 2-hydroxy sugar (X = OH), the 2-hydroxyl group of 3β can attack the anomeric centre, leading to the 1,2-anhydro sugar 6 as a transitory reactive intermediate. Reaction of 6 with an external nucleophile leads to the formation of the 1,2-trans substitution product 4, or alternatively a competing process is attack of the 6-hydroxyl group at the anomeric centre, leading to the formation of the corresponding 1,6-anhydro sugar 5. For a variety of carbohydrates 1,6-anhydro sugar 5 is the sole product in the absence of an effective external nucleophile.14 Alternatively, as previously mentioned, in the case of a 2-acetamido sugar (X = NHAc) the 1,2-trans glycoside 3β can undergo neighboring group participation and loss of a proton leading to the stable glycosyl oxazoline 7. Under appropriate (i.e. acidic/Lewis acidic conditions) oxazoline 7 may be opened by an external nucleophile, leading to the formation of the 1,2-trans substitution product 8.
Glycosyl thiols have proven to be extremely useful species for the production of neoglyconjugates, using a variety of strategies to link the thiol to a peptide or protein.15,16 Typically their synthesis has required multistep reaction sequences and protecting group manipulations.17 Although a method for the direct production18 of glycosyl thiols from reducing sugars has been reported,19 difficulties associated with the use of Lawesson's reagent, an apparent inapplicability to 2-acetamido sugars (which comprise all N- and O-glycans), and the necessity to then fully protect and then de-protect the sugars as part of the purification process, indicated that the development of an alternative process would be desirable.
Our attention therefore turned to the potential use of DMC activation to directly convert un-protected reducing sugars to the corresponding glycosyl thiols in water.20
DMC mediated reaction of GlcNAc 9 with a series of S-nucleophiles, including Na2S, NaSH, thiourea and KSAc was unsuccessfully attempted. However, stirring GlcNAc 9 with thioacetic acid (5 equiv.) and triethylamine (10 equiv.) in water at 0 °C and then adding DMC 1 (5 equiv.) surprisingly led to the production of the glycosyl acetate 10 as the sole reaction product in 86% yield (Scheme 2, ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mix of anomers).21 Reasoning that 10 had been formed by reaction of thioacetate with DMC, the order of addition of reagents was changed. Pleasingly, stirring GlcNAc 9 with DMC 1 (5 equiv.) and triethylamine (10 equiv.) in water at 0 °C for 30 min and then adding a large excess of thioacetic acid (15 equiv.) gave the glycosyl thioacetate 11 in an excellent 90% yield as the pure β-anomer. Analysis of reaction progress by 1H NMR indicated that the formation of 11 resulted from opening of the glycosyl oxazoline, formed in situ, by the added thioacetic acid (see ESI Fig. S1†). When identical reaction conditions were applied to glucose 12, 1,6-anhydro glucose 13 was the only observable product.
:1 mix of anomers).21 Reasoning that 10 had been formed by reaction of thioacetate with DMC, the order of addition of reagents was changed. Pleasingly, stirring GlcNAc 9 with DMC 1 (5 equiv.) and triethylamine (10 equiv.) in water at 0 °C for 30 min and then adding a large excess of thioacetic acid (15 equiv.) gave the glycosyl thioacetate 11 in an excellent 90% yield as the pure β-anomer. Analysis of reaction progress by 1H NMR indicated that the formation of 11 resulted from opening of the glycosyl oxazoline, formed in situ, by the added thioacetic acid (see ESI Fig. S1†). When identical reaction conditions were applied to glucose 12, 1,6-anhydro glucose 13 was the only observable product.
 | ||
| Scheme 2 Reaction of un-protected sugars with DMC and thioacetic acid in the presence of base. | ||
The competitive formation of the 1,6-anhydro sugar, taken together with the fact that thioacetate out-competes the anomeric hydroxyl group for reaction with DMC meant that the process could not be used for 2-hydroxy sugars. However, although this may at first appear as a limitation, in fact all N- and O-linked glycoprotein oligosaccharides possess either a GlcAc or a GalNAc at the reducing terminus, and therefore this method should be widely applicable for the synthesis of the glycosyl thiols of a wide range of biologically important oligosaccharides. The reaction was therefore extended to other 2-acetamido monosaccharides and a disaccharide terminated in a 2-acetamido sugar (Table 1). For the monosaccharides per-acetylation of the free hydroxyl groups of intermediate glycosyl thioacetate expedited isolation and purification; in the majority of cases Zemplen de-acetylation (NaOMe in MeOH, 0.5 h RT) yielded the completely de-protected glycosl thiols in quantitative yield, except in the case of ManNAc (Table 1, entry 2), when decomposition was observed. In the case of diacetyl chitobiose (Table 1, entry 4) purification by HPLC yielded the glycosyl thioacetate 16a, which was then smoothly converted into the glycosyl thiol 16b. Following glycosyl thiol production, conjugation to other species using the photo-initiated thiol–ene click reaction22,23 was investigated. GlcNAc thiol 11b was used a model substrate, and conjugated to a variety of alkenes in un-buffered aqueous solution using 2,2-dimethoxy-2-phenyl acetophenone (DMPA) as the initiator in a photo-reactor (254 nm) (Table 2). Conjugation to the allylated monosaccharide 18a gave disaccharide mimic 18b, whilst use of the allylated serine derivative 19a correspondingly gave the glycosyl amino acid 19b.
| Entry | Substrate | Thioacetate product/% yield | Thiol product/% yield |
|---|---|---|---|
| a Isolated yields following purification by column chromatography. b Reaction conditions: 0.2 M NaOMe in MeOH, 0.5 h, RT. c Isolated yields following purification by HPLC. d Reaction conditions: 1 M aq. NaOH, 1 h, RT. | |||
| 1 | GlcNAc 9 |

|

|
| 2 | ManNAc |
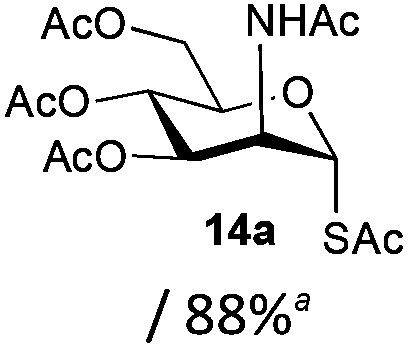
|
Decomposition |
| 3 | GalNAc |
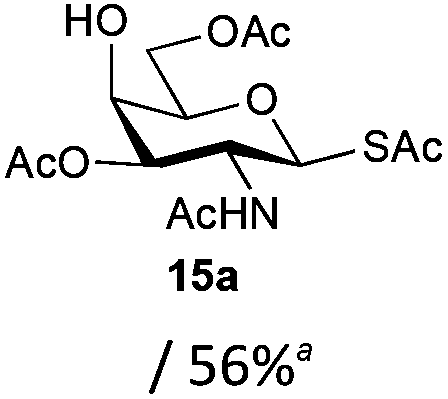
|

|
| 4 | Diacetyl-chitobiose |

|
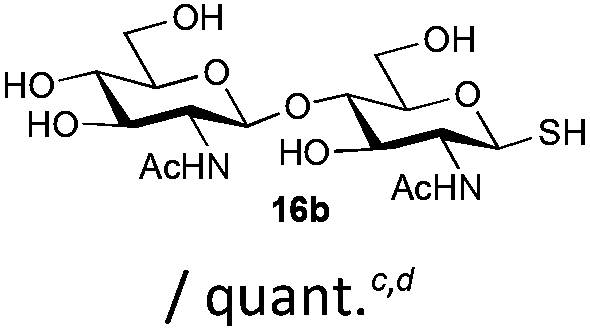
|



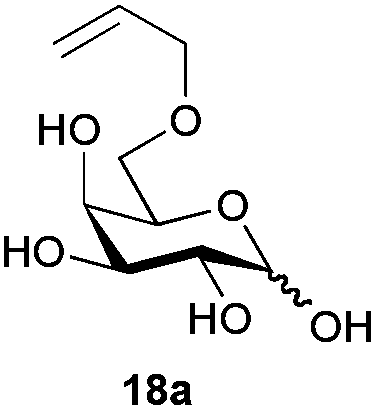
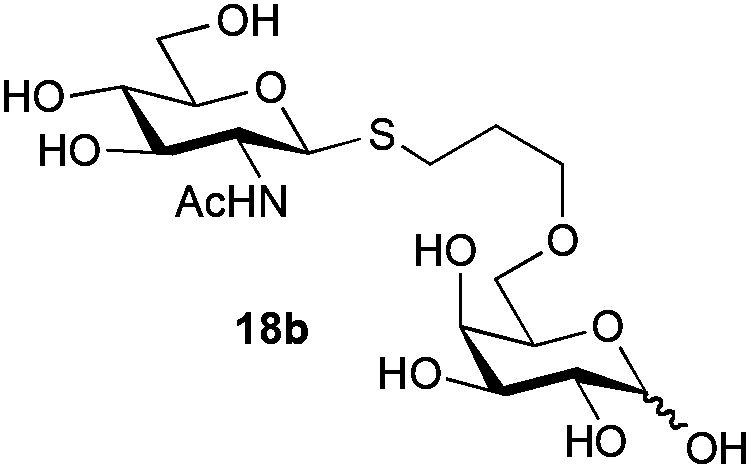
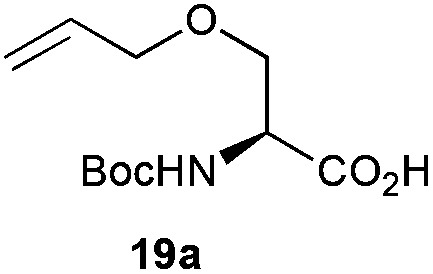

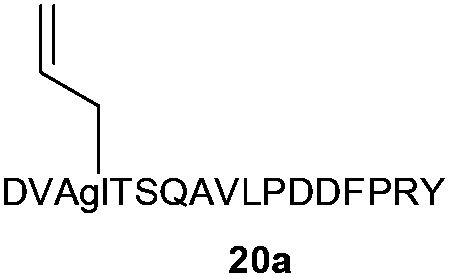

Next, the 16-mer peptide 20a, which corresponds to residues 1–16 of the rat pancreatic hormone preptin24 in which the natural serine at position-3 has been replaced by an allyl glycine residue (Agl), was produced by using microwave-assisted 9H-fluoren-9-ylmethoxycarbonyl-solid phase peptide synthesis (Fmoc-SPPS), as described previously for the synthesis of structurally-similar peptides,25 as a model substrate to investigate the effectiveness of the thiol–ene click conjugation method with more extended peptide structures. Conjugation of GlcNAc thiol 11b to peptide 20a was essentially quantitative as assessed by HPLC (Table 2, entry 4).
As a demonstration of the utility of the approach to access glycosyl thiols from complex oligosaccharides isolated from natural sources, the complex bi-antennary glycan 21, isolated from egg yolks as previously described,26 was subjected to the DMC/AcSH-mediated processes, and gave the decasaccharide thioacetate 22 in 60% yield (Scheme 3). De-acetylation using aqueous sodium hydroxide and immediate conjugation to peptide 20a was then achieved using the same photochemical free radical process, and yielded the glycopeptide 23 in essentially quantitative conversion as assessed by HPLC.
 | ||
| Scheme 3 Conversion of a complex bi-antennary N-glycan into its glycosyl thiol and conjugation to a 16-mer peptide. | ||
In summary the application of DMC and AcSH, added in the correct order, allows the conversion of un-protected 2-acetamido terminated reducing sugars to the corresponding glycosyl thioacetates, including complex N-glycans derived from natural sources. Facile thioacetate removal gives glycosyl thiols which may be used directly for conjugation with alkenes, including complex peptide substrates. Application of this methodology to proteins comprised of non-natural amino acids bearing alkenes should allow the production of homogeneous neo-glycoproteins bearing complex oligosaccharides without recourse to any sugar protecting groups.
Acknowledgements
The authors thank the University of Canterbury (PhD Scholarship to SRA) and the Biomolecular Interaction Centre for financial support, and Dr Marie Fitchett for technical assistance.Notes and references
- A. Varki, Glycobiology, 1993, 3, 97 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- For some examples see: (a) V. P. Kamath, P. Diedrich and O. Hindsgaul, Glycoconjugate J., 1996, 13, 315 CrossRef CAS; (b) S. I. van Kasteren, H. B. Kramer, D. P. Gamblin and B. G. Davis, Nat. Protoc., 2007, 2, 3185–3194 CrossRef CAS PubMed; (c) D. P. Gamblin, P. Garnier, S. Ward, N. J. Oldham, A. J. Fairbanks and B. G. Davis, Org. Biomol. Chem., 2003, 1, 3642 RSC.
- (a) T. Isobe and T. J. Ichikawa, J. Org. Chem., 1999, 64, 6984 CrossRef CAS; (b) T. Isobe and T. J. Ichikawa, Org. Chem., 1999, 64, 6989 CrossRef CAS.
- M. Noguchi, T. Tanaka, H. Gyakushi, A. Kobayashi and S.-i. Shoda, J. Org. Chem., 2009, 74, 2210 CrossRef CAS PubMed.
- (a) A. J. Fairbanks, Pure Appl. Chem., 2013, 85, 1847 CrossRef CAS; (b) L.-X. Wang and M. N. Amin, Chem. Biol., 2014, 21, 51 CrossRef CAS PubMed; (c) P. Priyanka, T. B. Parsons, A. Miller, F. M. Platt and A. J. Fairbanks, Angew. Chem., Int. Ed., 2016, 55, 5058 CrossRef CAS PubMed.
- The pKa of the anomeric hydroxyl group of un-protected sugars in dilute aqueous solution ranges from 12.1–12.5. See: S. Feng, C. Bagia and G. J. Mpourmpakis, Phys. Chem. A, 2013, 117, 5211 CrossRef CAS PubMed; J. J. Christensen, J. H. Rytting and R. M. Izatt, J. Chem. Soc. B, 1970, 1646 RSC.
- T. Tanaka, H. Nagai, M. Noguchi, A. Kobayashi and S.-i. Shoda, Chem. Commun., 2009, 3378 RSC.
- (a) M. Kitamura, N. Tashiro, Y. Takamoto and T. Okauchi, Chem. Lett., 2010, 39, 732 CrossRef CAS; (b) M. Kitamura, S. Kato, M. Yano, N. Tashiro, Y. Shiratake, M. Sando and T. Okauchi, Org. Biomol. Chem., 2014, 12, 4397 RSC.
- D. Lim, M. A. Brimble, R. Kowalczyk, A. J. A. Watson and A. J. Fairbanks, Angew. Chem., Int. Ed., 2014, 53, 11907 CrossRef CAS PubMed.
- S. R. Alexander and A. J. Fairbanks, Org. Biomol. Chem., 2016, 14, 6679 CAS.
- (a) N. Yoshida, M. Noguchi, T. Tanaka, T. Matsumoto, N. Aida, M. Ishihara, A. Kobayashi and S.-i. Shoda, Chem. – Asian J., 2011, 6, 1876 CrossRef CAS PubMed; (b) T. Tanaka, T. Matsumoto, M. Noguchi, A. Kobayashi and S.-I. Shoda, Chem. Lett., 2009, 38, 458 CrossRef CAS.
- A. Novoa, S. Barluenga, C. Serba and N. Winssinger, Chem. Commun., 2013, 49, 7608 RSC.
- T. Tanaka, W. C. Huang, M. Noguchi, A. Kobayashi and S.-i. Shoda, Tetrahedron Lett., 2009, 50, 2154 CrossRef CAS.
- (a) N. Floyd, B. Vijayakrishnan, J. R. Koeppe and B. G. Davis, Angew. Chem., Int. Ed., 2009, 48, 7798 CrossRef CAS PubMed; (b) D. P. Gamblin, P. Garnier, S. I. van Kasteren, N. J. Oldham, A. J. Fairbanks and B. G. Davis, Angew. Chem., Int. Ed., 2004, 43, 828 CrossRef CAS PubMed.
- (a) Y. Zhu and W. A. van der Donk, Org. Lett., 2001, 3, 1189 CrossRef CAS PubMed; (b) M. D. Gieselman, Y. Zhu, H. Zhou, D. Galonic and W. A. van der Donk, ChemBioChem, 2002, 3, 709 CrossRef CAS PubMed; (c) D. P. Galonić, W. A. van der Donk and D. Y. Gin, Chem. – Eur. J., 2003, 9, 5997 CrossRef PubMed; (d) D. P. Galonić, N. D. Ide, W. A. van der Donk and D. Y. Gin, J. Am. Chem. Soc., 2005, 127, 7359 CrossRef PubMed; (e) J. Wang, S. M. Schiller and P. G. Schultz, Angew. Chem., Int. Ed., 2007, 46, 6849 CrossRef CAS PubMed; (f) G. J. L. Bernardes, J. M. Chalker, J. C. Errey and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 5052 CrossRef CAS PubMed.
- For some recent indirect methods see: (a) J. M. MacDougall, X. D. Zhang, W. E. Polgar, T. V. Khroyan, L. Toll and J. R. Cashman, J. Med. Chem., 2004, 47, 5809 CrossRef CAS PubMed; (b) B. D. Johnston and B. M. Pinto, J. Org. Chem., 2000, 65, 4607 CrossRef CAS PubMed; (c) O. B. Wallace and D. M. Springer, Tetrahedron Lett., 1998, 39, 2693 CrossRef CAS; (d) F. M. Ibatulin, K. A. Shabalin, J. V. Jänis and A. G. Shavva, Tetrahedron Lett., 1998, 39, 7961 Search PubMed.
- For a single example of a non-stereoselective method requiring the use of HF and H2S see: J. Defaye, A. Gadelle and C. Pedersen, Carbohydr. Res., 1991, 217, 51 CrossRef CAS PubMed.
- G. J. L. Bernardes, D. P. Gamblin and B. G. Davis, Angew. Chem., Int. Ed., 2006, 45, 4007 CrossRef CAS PubMed.
- During the submission of this manuscript a similar method for the production of glycosyl thiols using DMC was reported by Rademann and co-workers. See: S. Köhling, M. P. Exner, S. Nojoumi, J. Schiller, N. Budisa and J. Rademann, Angew. Chem., Int. Ed., 2016, 55, 15510 CrossRef PubMed.
- D. Lim and A. J. Fairbanks, Chem. Sci., 2017, 8 10.1039/C6SC04667C , in press.
- A. Dondoni, A. Massi, P. Nanni and A. Roda, Chem. – Eur. J., 2009, 15, 11444 CrossRef CAS PubMed.
- For some recent reviews see: (a) A. Dondoni and A. Marra, Chem. Soc. Rev., 2012, 41, 573 RSC; (b) L. McSweeney, F. Dénès and E. M. Scanlan, Eur. J. Org. Chem., 2016, 2080 CrossRef CAS.
- C. M. Buchanan, A. R. Phillips and G. J. Cooper, Biochem. J., 2001, 360, 431 CrossRef CAS PubMed.
- See ESI Scheme S1.† For details of the synthesis of related peptides see: Z. Amso, R. Kowalczyk, M. Watson, Y.-E. Park, K. E. Callon, D. S. Musson, J. Cornish and M. A. Brimble, Org. Biomol. Chem., 2016, 14, 9225 CAS.
- M. Umekawa, T. Higashiyama, Y. Koga, T. Tanaka, M. Noguchi, A. Kobayashi, S.-i. Shoda, W. Huang, L.-X. Wang, H. Ashida and K. Yamamoto, Biochim. Biophys. Acta, 2010, 1800, 1203 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, including full compound characterisation and spectra. See DOI: 10.1039/c7ob00112f |
| This journal is © The Royal Society of Chemistry 2017 |