
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A stereoselective synthesis of (E)- or (Z)-β-arylvinyl halides via a borylative coupling/halodeborylation protocol†
Jakub
Szyling
ab,
Adrian
Franczyk
a,
Piotr
Pawluć
b,
Bogdan
Marciniec
ab and
Jędrzej
Walkowiak
 *a
*a
aCentre for Advanced Technologies, Adam Mickiewicz University in Poznan, Umultowska 89c, 61-614 Poznan, Poland. E-mail: jedrzejw@amu.edu.pl; Tel: +48 61 829 18 77
bFaculty of Chemistry, Adam Mickiewicz University in Poznan, Umultowska 89b, 61-614 Poznan, Poland
First published on 7th February 2017
Abstract
A new stereoselective method for the synthesis of (E)-β-arylvinyl iodides and (E)- or (Z)-β-arylvinyl bromides from styrenes and vinyl boronates on the basis of a one-pot procedure via borylative coupling/halodeborylation is reported. Depending on the halogenating agent as well as the mode of the halodeborylation reaction, (E) or (Z) isomers are selectively formed.
Introduction
Transition metal catalyzed cross-coupling reactions, especially the Heck, Suzuki, Hiyama, Stille and Ullmann processes have been the most commonly used methods for generation of new C–C bonds.1–3 These transformations predominantly use aryl or alkenyl halides as reagents and therefore the development of new synthetic protocols for their stereoselective synthesis is still of significant importance. β-Arylvinyl halides are vastly useful intermediates (coupling components) in organic chemistry, in the synthesis of unsaturated, conjugated products as well as natural compound analogues and are applied as precursors of vinyl anions or reagents in the Buchwald–Hartwig reaction.4–7Over the past few decades, several routes to alkenyl halides with a specific stereoselectivity have been developed. β-Arylvinyl halides are classically prepared by the halodecarboxylation of cinnamic acid derivatives (Hunsdiecker reaction),8–10 reduction of 1,1-dihalogenoalkenes11,12 or olefination of aromatic aldehydes (Takai olefination).13 Other effective methods leading to (E)-alkenyl halides are based on the catalytic anti-Markovnikov hydrobromination of alkynes, which occurs via the hydrocupration of alkynes, followed by bromination of the alkenyl copper intermediate,14 homologation and further stereoselective elimination of benzyl bromides with dihalomethanes in the presence of a base15 or intramolecular dehydration of bromohydrins using H-β zeolite as a catalyst.16
(Z)-β-Arylvinyl bromides can be synthesized by the Wittig olefination of an aldehyde with bromomethylene triphenylphosphorane,17,18 by the reaction of aromatic aldehydes with α-bromomethyl sulfones (Julia olefination),19 Pd-catalyzed debromination of 1,1-dibromo-1-alkenes by tributyltin hydride20,21 or debrominative decarboxylation of anti-2,3-dibromo-3-arylpropanoic acids using e.g. NaN3.22,23
Hydrometallation of alkynes followed by halogenation is another effective protocol used for the synthesis of (E)- or (Z)-alkenyl halides. Addition of the H–E bond (where E = Si, B, Sn, Zr) to alkynes leads to vinyl derivatives of these elements, which in the next step are stereospecifically substituted by a halogen atom using different halogenating agents.24–30
The most common methods used for the synthesis of alkenyl halides with different stereoselectivities are presented in Scheme 1.
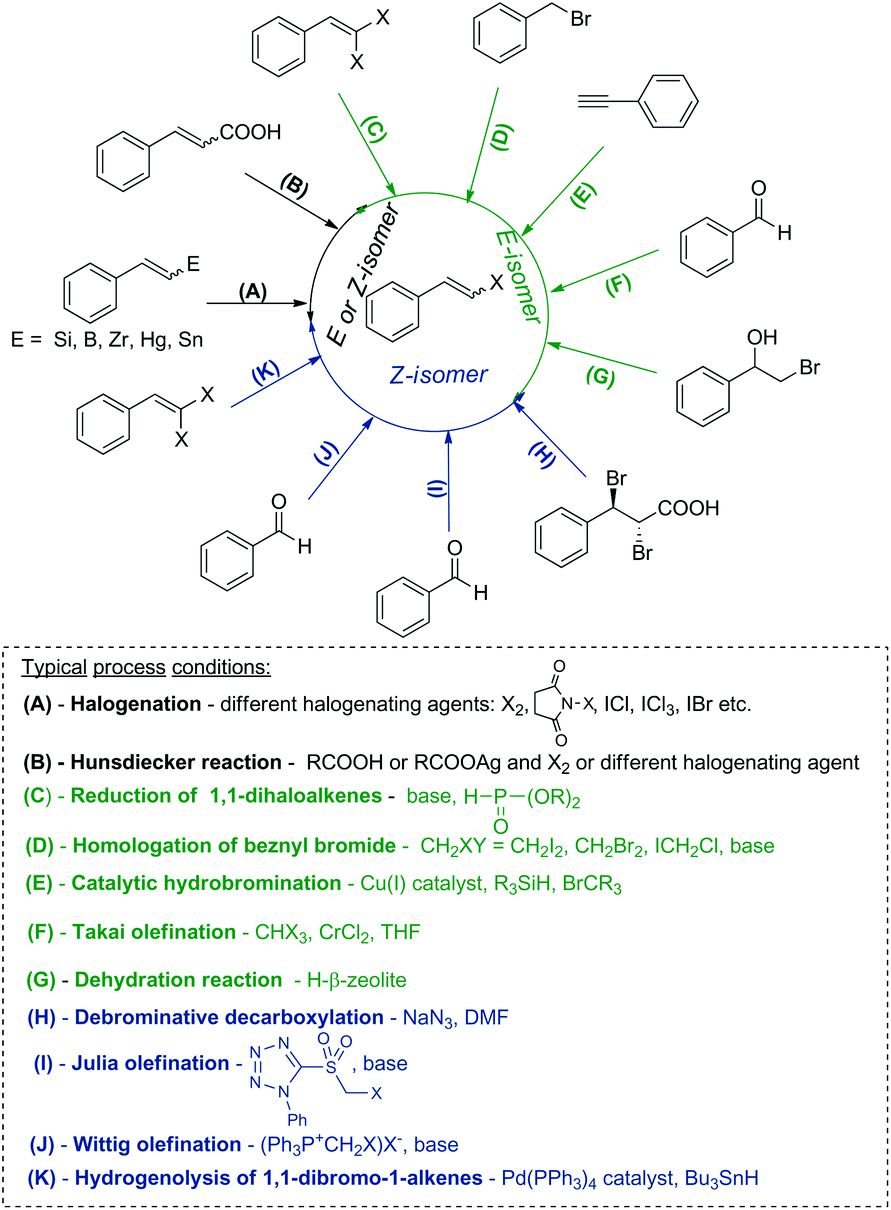 | ||
| Scheme 1 Methods used for the synthesis of β-arylvinyl halides with different stereoselectivities. | ||
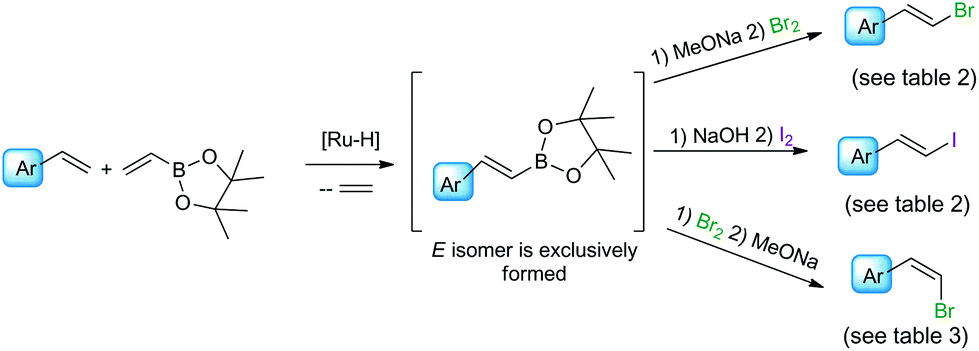
The range of commercially available substituted styrenes is much wider than that of phenylacetylene analogues and therefore the methods for their application in the synthesis of metalloid-substituted compounds and further halodemetallation to the corresponding β-arylvinyl halides are important from the synthetic point of view (see Scheme 2). Terminal phenylacetylenes are often more expensive than their vinyl analogues, their preparation needs several steps and due to their tendency for dimerization and polymerization, they are less attractive in the synthesis. The formation of (E)-arylvinyl iodides and (Z)-arylvinyl bromides from styrenes in cross-metathesis with vinyl or 1-propenyl boronates, followed by the halogenation of alkenyl boronate intermediates was reported by Grubbs and co-workers.31 β-Arylvinyl bromides were synthesized in a one-pot two-step procedure, while β-arylvinyl iodides were obtained only from isolated boronate intermediates. The cross-metathesis of 4-methoxystyrene with (E)-1,2-dichloroethene led to (Z)-4-methoxystyryl chloride as a main product.32
 | ||
| Scheme 2 The application of styrenes as reagents in the synthesis of β-arylvinyl halides. | ||
(E)-Vinyl iodides can be also prepared by a two-step procedure: oxidative cleavage of terminal olefins with ozone or OsO4/NaIO4 and Takai iodoolefination.33,34
In 2009, our group reported an efficient method for the preparation of (E)-β-arylvinyl halides (bromides and iodides) in a one-pot procedure on the basis of a sequential silylative coupling reaction of vinylsilanes with styrenes and N-halosuccinimide-mediated halodesilylation (see Scheme 2).35 Developed in our group, the silylative coupling reaction of olefins with vinylsilanes catalyzed by complexes with the Ru–H or Ru–Si bonds yields silyl-substituted alkenes with the simultaneous evolution of ethylene. The reaction occurs via activation of the C–Si bond in vinylsilane and C–H in olefin. Depending on the activation of different carbon–hydrogen bonds in olefin, (E), (Z) or gem isomers can be obtained. This mode of reactivity has also been extended to other vinyl metalloids: vinyl boronates and vinyl germanes.36 Vinyl boronates give exclusively (E)-boryl-substituted ethenes in the reaction with styrenes and other olefins (see Scheme 3).37
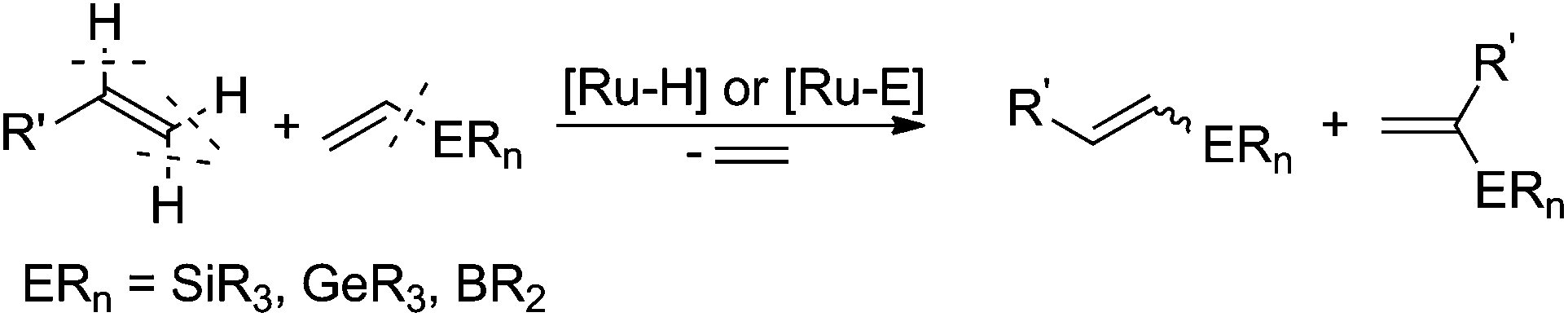 | ||
| Scheme 3 Trans-metallation of olefins with vinyl metalloids. | ||
The presented coupling reactions lead, in an easy and simple way, to metalloid-functionalized olefins, which can be used in many organic transformations. Our group has made a significant contribution to the development of one-pot procedures based on vinylsilanes in the preparation of organic compounds with various functional groups.38–42
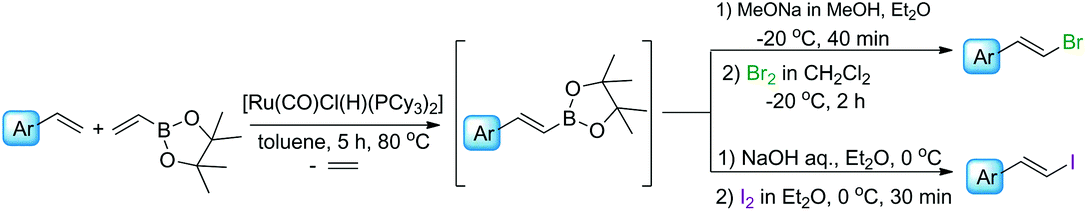
Herein, we would like to report our investigation on the application of vinyl boronates as reagents for the synthesis of β-arylvinyl halides with (E) or (Z) geometry and propose new one-pot procedures for the preparation of such compounds via sequential borylative coupling and electrophilic halodeborylation reactions. This method constitutes an alternative for the synthesis of alkenyl halides according to the metathesis/halogenation or hydroboration/halogenation protocols, from easily accessible styrenes.
Results and discussion
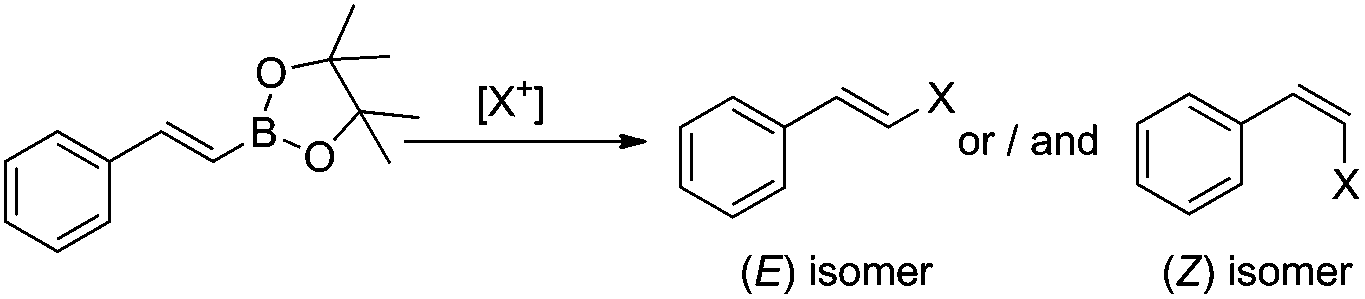
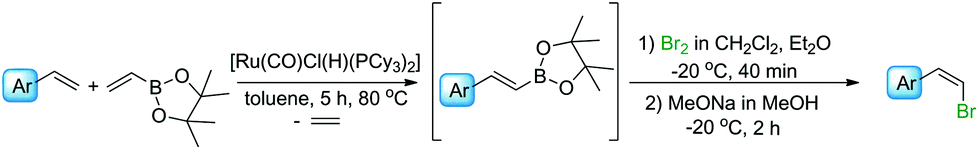
We have previously found that a borylative coupling of a series of substituted styrenes with vinyl boronates occurs with high regio- and stereoselectivity with the use of [Ru(CO)Cl(H)(PCy3)2] or [Ru(CO)Cl(H)(PPh3)3].37 The first catalyst shows a much higher activity. (E)-2-Aryl-1-borylethenes were exclusively formed with quantitative yields, while no by-product of vinyl boronate homocoupling under the applied reaction conditions was observed.37 This is an advantage when compared to the metathesis of analogous reagents carried out in the presence of a Grubbs catalyst (1st and 2nd generation), in which the homocoupling products of olefin and vinyl boronate were also detected.31 The selectivity of metathesis was slightly lower compared to borylative coupling reaction and some amount of the (Z) isomer was formed. It means that the mixture of products obtained in the metathesis process after halodeborylation includes both isomers at different molar ratios.For this reason, in our studies we decided to use borylative coupling – for the synthesis of (E)-2-aryl-1-borylethenes – as the first step of a sequential method for the preparation of β-arylvinyl halides (Scheme 4).
 | ||
| Scheme 4 A new synthetic protocol to (E)- or (Z)-β-arylvinyl halides via trans-metallation/halodemetallation reactions. | ||
Commercially available, 4,4,5,5-tetramethyl-2-vinyl-1,3-dioxaborolane was used as a reagent for the borylative coupling of a group of styrenes, which had not been previously tested in this process. We found that only 1.1 molar excess of styrenes or even their equimolar amounts are enough to prevent vinyl boronate homocoupling, when 1 mol% of [Ru(CO)Cl(H)(PCy3)2] catalyst was used. Applying these conditions, the complete conversion of the reagents was observed after 5 h. The process was carried out in toluene at 80 °C, under an inert atmosphere. The formation of desired products, as well as the consumption of substrates, was monitored using GC and GC-MS analyses. 1H NMR analysis showed that only the (E) isomer was formed in this catalytic transformation.
Three representatives of the obtained boryl-substituted ethenes were isolated to confirm the exact geometry of their structures and purity (see the ESI†). The rest of them were prepared in situ and were directly used in the halodeborylation process.
The excellent regio- and stereoselectivity of (E)-2-aryl-1-borylethene synthesis via borylative coupling is essential for the following halogenation process and for obtaining targeted β-arylvinyl halides with strictly defined configuration.
In the next step of the studies, the halodeborylation reaction (iododeborylation and bromodeborylation) was optimized using (E)-1-(4′,4′,5′,5′-tetramethyl)—1′,3′,2′-dioxaborolanyl-2-phenylethene and different halogenating agents: molecular I2, Br2 as well as NIS and NBS, applying the appropriate reaction conditions for both types of reagents (Table 1). Halodeborylations with molecular iodine or bromine were carried out in diethyl ether at 0 °C or −20 °C respectively, according to the procedure described by Brown's and co-workers (Table 1, entries 1, 2 and 4, 5).43 We observed that depending on the order of addition of the base and halogen for the bromination process, the (E) or (Z) isomer was preferentially formed. When a base was added before Br2 – the product was (E)-β-bromostyrene (Table 1, entry 4), while the opposite order of reagents led to (Z)-β-bromostyrene (Table 1, entry 5).
|
|
|||
|---|---|---|---|
| Entry | [X+] | Yieldg [%] | Selectivity E/Zh |
| a 1. Et2O, 3 M aq. NaOH, 0 °C. 2; I2 in Et2O, 30 min, 0 °C. b 1. I2 in Et2O, 30 min, 0 °C; 2. Et2O, 3 M aq. NaOH, 0 °C. c 1.5 eq. of NIS, acetonitrile (0.1 M), rt. d 1. Et2O, 3 M MeONa in MeOH, 40 min, −20 °C; 2. Br2 in CH2Cl2, 2 h, −20 °C. e 1. Et2O, Br2 in CH2Cl2, −20 °C; 2. 3 M MeONa in MeOH, 2 h, −20 °C. f 1.5 eq. of NBS, acetonitrile (0.1 M), rt. g Reaction yields were calculated on the basis of GC analysis. h GC-MS and 1H NMR were used for selectivity determination. | |||
| 1a | I2 | 100 | 1/0 |
| 2b | I2 | 97 | 1/0 |
| 3c | NIS | 94 | 1/0 |
| 4d | Br2 | 100 | 1/0 |
| 5e | Br2 | 99 | 1/20 |
| 6f | NBS | 92 | 9/1 |
On the other hand, the iododeborylation of (E)-1-(4′,4′,5′,5′-tetramethyl)—1′,3′,2′-dioxaborolanyl-2-phenylethene irrespective of the order of addition of reagents, leads to (E)-β-iodostyrene. A slightly better yield of the reaction was obtained when the base was added before iodine (Table 1, entries 1 and 2).
We also observed that the rate of addition of halogen into the reaction mixture was essential for the process selectivity. When the reagent was added rapidly, the dihalogenated product was formed as well. A slight excess of halogen relative to the boronate reagent should be used during the entire process to prevent this side reaction. The temperature control is also important to obtain the desired monohalogenated product exclusively, and should be strictly maintained during the halogenation process.
Applying NIS or NBS as halogenating agents, β-arylvinyl halides were synthesized with the retention of configuration and with slightly lower yield compared to Brown's method (Table 1, entries 3 and 6).
The positive results on borylative coupling reaction as well as halodeborylation (bromodeborylation and iododeborylation), especially regarding the high efficiency and selectivity of both processes, encouraged us to check whether β-arylvinyl halides can be synthesized in one-pot protocol, without isolation and purification of intermediates – boryl-substituted ethenes.
In a typical procedure, the olefin, 4,4,5,5-tetramethyl-2-vinyl-1,3,2-dioxaborolane, and 1 mol% of [Ru(CO)Cl(H)(PCy3)2] were dissolved in toluene (0.5 M), placed in a Schlenk's vessel fitted with a plug valve and heated up to 80 °C, under an inert atmosphere. After the total conversion of reagents, which was monitored by GC and GC-MS analyses, the reaction mixture was cooled down to −20 °C or 0 °C, diethyl ether was added and then depending on the protocol, bromine/iodine or the base was added firstly. (E)-β-Arylvinyl bromides and iodides were synthesized by the addition of a base – 3 M solution of sodium methoxide in methanol or sodium hydroxide in water, respectively – to the reaction mixture, followed by the dropwise injection of halogen dissolved in dichloromethane. The iodination was quenched with aqueous saturated solution of Na2S2O3 and the products were separated using flash chromatography. A mixture of hexane with ethyl acetate at the ratio of 9/1 was used as an eluent. Pure (E)-β-arylvinyl bromides and (E)-β-arylvinyl iodides (Table 2, compounds 1–8(a and b)) were obtained with high isolated yields (58–90%).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry |
|
X2 | Yield [%] | Product | Selectivity (E)/(Z) [%] | Isolated yield [%] | |
Borylative coupling procedure: [Ru–H]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[ViB]:[olefin] = 10−2:1:1.1, toluene (0.5 M), 80 °C, 5 h. Bromodeborylation procedure: 1. MeONa (3 M in MeOH), Et2O, 40 min, −20 °C; 2. [Borylated styrene]:[Br2] = 1:1.6, Br2 (1 M in CH2Cl2), −20 °C, 2 h. Iododeborylation procedure: 1. NaOH (3 M aq. solution), Et2O, 0 °C; 2. [Borylated styrene]:[I2] = 1:1.7, I2 (1 M in Et2O), 30 min, 0 °C. Reaction yields were calculated on the basis of GC analysis. GC-MS and 1H NMR were used for selectivity determination. :[ViB]:[olefin] = 10−2:1:1.1, toluene (0.5 M), 80 °C, 5 h. Bromodeborylation procedure: 1. MeONa (3 M in MeOH), Et2O, 40 min, −20 °C; 2. [Borylated styrene]:[Br2] = 1:1.6, Br2 (1 M in CH2Cl2), −20 °C, 2 h. Iododeborylation procedure: 1. NaOH (3 M aq. solution), Et2O, 0 °C; 2. [Borylated styrene]:[I2] = 1:1.7, I2 (1 M in Et2O), 30 min, 0 °C. Reaction yields were calculated on the basis of GC analysis. GC-MS and 1H NMR were used for selectivity determination. |
|||||||
| 1 |
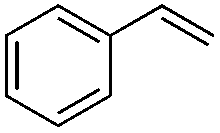
|
Br2 | 100 |
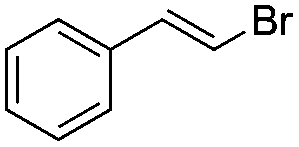
|
(1a) | 1:0 |
87 |
| 2 | I2 | 100 |
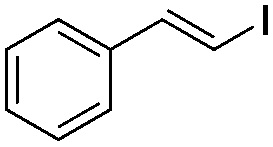
|
(1b) | 1:0 |
82 | |
| 3 |
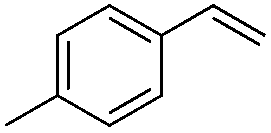
|
Br2 | 98 |
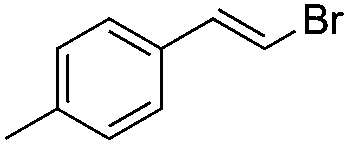
|
(2a) | 15:1 |
83 |
| 4 | I2 | 100 |
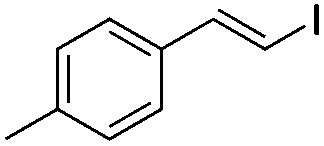
|
(2b) | 1:0 |
84 | |
| 5 |
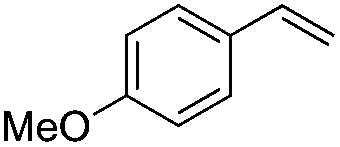
|
Br2 | 100 |
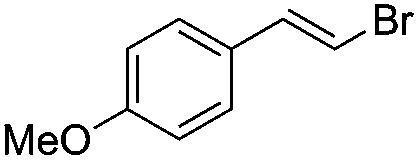
|
(3a) | 20:1 |
76 |
| 6 | I2 | 100 |
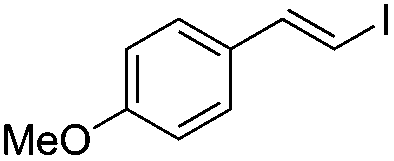
|
(3b) | 1:0 |
77 | |
| 9 |
|
Br2 | 100 |
|
(4a) | 9:1 |
89 |
| 10 | I2 | 100 |

|
(4b) | 1:0 |
79 | |
| 11 |
|
Br2 | 92 |
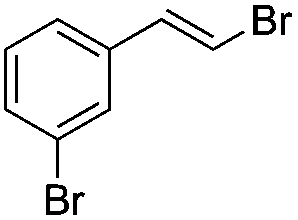
|
(5a) | 1:0 |
86 |
| 12 | I2 | 98 |
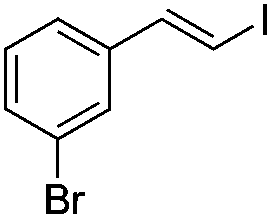
|
(5b) | 1:0 |
81 | |
| 13 |
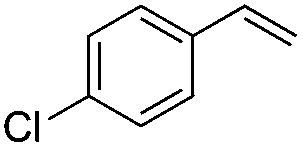
|
Br2 | 99 |
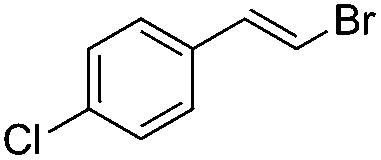
|
(6a) | 1:0 |
71 |
| 14 | I2 | 100 |
|
(6b) | 1:0 |
63 | |
| 15 |
|
Br2 | 94 |
|
(7a) | 20:1 |
67 |
| 16 | I2 | 100 |
|
(7b) | 1:0 |
63 | |
| 17 |
|
I2 | 90 |
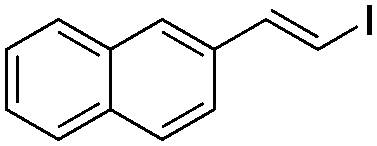
|
(8b) | 1:0 |
58 |
In the case of formation of (Z)-β-arylvinyl bromides, the procedure differs from that described above. Bromine was added to the reactor before the base (Table 3). The time of reaction and the whole isolation procedure were the same as that for the synthesis of (E) isomers. The isolated yields of the (Z)-β-bromostyrenes were similar to those of (E) isomers and varied between 61 and 89%.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry |
|
Yield [%] | Product | Selectivity (E)/(Z) [%] | Isolated yield [%] | |
| Borylative coupling procedure: [Ru–H]:[ViB]:[olefin] = 10−2:1:1.1, toluene (0.5 M), 80 °C, 5 h. Bromodeborylation: 1. [Borylated styrene]:[Br2] = 1:1.6, Br2 (1 M in CH2Cl2), 0 °C, 40 min; 2. MeONa (3 M in MeOH), Et2O, −20 °C, 2 h. Reaction yields were calculated on the basis of GC analysis. GC-MS and 1H NMR were used for selectivity determination. |
||||||
| 1 |
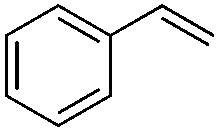
|
98 |
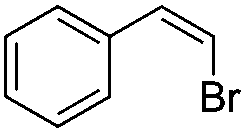
|
(9) | 0/1 | 84 |
| 2 |
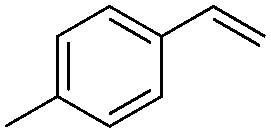
|
93 |
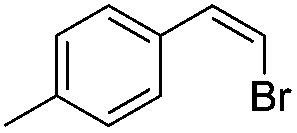
|
(10) | 0/1 | 61 |
| 3 |
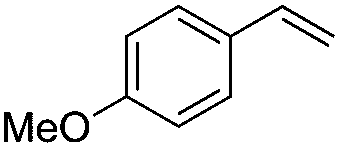
|
100 |
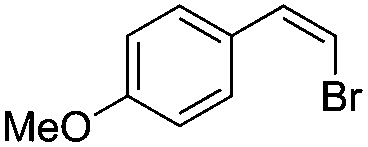
|
(11) | 1/8 | 89 |
| 4 |
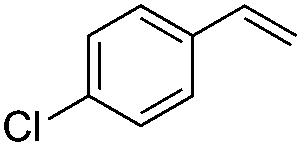
|
96 |
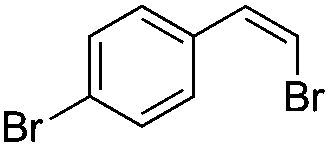
|
(12) | 1/8 | 82 |
| 5 |
|
98 |
|
(13) | 1/8 | 84 |

It is worth emphasizing that all presented products can be prepared from commercially available reagents, without their preliminary purification. Due to the sensitivity of [Ru(CO)Cl(H)(PCy3)2] to air, it is recommended to carry out the borylative coupling reaction under an argon atmosphere in order to obtain total conversion of the reagents before deactivation of the catalyst. However, the halogenation processes were carried out in an air atmosphere. The most important factors affecting the effectiveness of the halodeborylation processes were temperature and the rate of halogen added to the reaction mixture.
Under optimal conditions, applying one-pot reaction sequences, a broad range of (E)-β-arylvinyl iodides and bromides as well (Z)-β-arylvinyl bromides, with miscellaneous substituents attached to the phenyl ring (such as –Me, –Ph, –OMe, –Br, –Cl) in different positions to the vinyl group, were successfully synthesized.
It should be noted that all presented compounds are interesting building blocks for organic synthesis, and can be modified by a wide spectrum of coupling reactions (Suzuki, Sonogashira, Hiyama etc.).
Conclusions
We have devised a one-pot protocol for the stereoselective synthesis of (E)-β-arylvinyl iodides, (E)-β-arylvinyl bromides and (Z)-β-arylvinyl bromides on the basis of highly selective catalytic borylative coupling/halodeborylation reactions. Commercially available substituted styrenes, much cheaper than the analogous terminal alkynes, were used as reagents in the protocols proposed. Not only the accessibility of substrates but also the higher selectivity of borylative coupling reaction vs. metathesis reaction is the reason why this method in an attractive alternative route for the synthesis of desired compounds from styrenes. Moreover, this methodology, compared to the protocol based on silylative coupling/halodesilylation reactions, offers a possibility for the synthesis of (Z)-β-arylvinyl bromides, which cannot be obtained by the previously described Si-mediated reactions.Experimental
Synthetic protocols
Synthesis of (E)-β-arylvinyl bromides
:1) as the eluent. The purity of the product was confirmed by GC-MS and 1H NMR.
:1) as the eluent. The purity of all obtained products were confirmed by GC-MS and 1H NMR and the results were in agreement with the literature.15,16,35
(E)-β-Bromostyrene (1a)35. Isolated yield: 87%; 1H NMR (300 MHz, CDCl3) δ = 6.66 (d, 1H, JH–H = 14.0 Hz, HC
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 7.01 (d, 1H, JH–H = 14.0 Hz, HC), 7.12–7.29 (m, 5H, Ar) ppm; 13C NMR (75 MHz, CDCl3) δ = 106.7 (CH–Br), 126.2, 128.4, 128.9, 136.0 (Ar), 137.3 (CH–Ph) ppm; MS m/z (rel. int., %): 184((M + 2)+, 47), 182(M+, 48), 103(100), 77(60), 51(33).
), 7.01 (d, 1H, JH–H = 14.0 Hz, HC), 7.12–7.29 (m, 5H, Ar) ppm; 13C NMR (75 MHz, CDCl3) δ = 106.7 (CH–Br), 126.2, 128.4, 128.9, 136.0 (Ar), 137.3 (CH–Ph) ppm; MS m/z (rel. int., %): 184((M + 2)+, 47), 182(M+, 48), 103(100), 77(60), 51(33).
(E)-β-Bromo(4-methylstyrene) (2a)15. Isolated yield: 83%; 1H NMR (300 MHz, CDCl3) δ = 2.37 (s, 3H, CH3), 6.74 (d, 1H, JH–H = 14.0 Hz, HC
), 7.11 (d, 1H, JH–H = 14.0 Hz, HC), 7.16–7.18 (d, 2H, Ar), 7.22–7.24 (d, 2H, Ar) ppm; 13C NMR (75 MHz, CDCl3) δ = 21.7 (CH3), 105.9 (CH–Br), 126.5, 129.9, 133.5, 137.4 (Ar), 138.9 (CH–Ar) ppm; MS m/z (rel. int., %): 198((M + 2)+, 57), 196(M+, 58), 117(100), 91(37), 63(20), 57(18) 51(12).
(E)-β-Bromo(4-methoxystyrene) (3a)15. Isolated yield: 76%; 1H NMR (300 MHz, CDCl3) δ = 3.72 (s, 3H, OCH3), 6.52 (d, 1H, JH–H = 14.0 Hz, HC
), 6.76 (d, 2H, JH–H = 8.7 Hz, Ar), 6.95 (d, 1H, JH–H = 14.0 Hz, HC), 7.14 (d, 2H, JH–H = 8.7 Hz, Ar) ppm; 13C NMR (75 MHz, CDCl3) δ = 55.3 (OCH3), 104.0 (CH–Br), 114.2, 127.4, 128.8 (Ar), 136.6 (CH–Ar), 159.7 (Ar) ppm; MS m/z (rel. int., %): 214((M + 2)+, 97), 212(M+, 100), 199(38) 197(40), 169(18), 171(17), 133(47), 118(27), 90(67), 77(16), 63(36).
(E)-β-Bromo(4-bromostyrene) (4a)35. Isolated yield: 89%; 1H NMR (300 MHz, CDCl3) δ = 6.67 (d, 1H, JH–H = 14.0 Hz, HC
), 6.93 (d, 1H, JH–H = 14.0 Hz, HC) 7.04 (d, 2H, Ar), 7.34 (d, 2H, Ar) ppm; 13C NMR (75 MHz, CDCl3) δ = 107.4 (CH–Br), 122.2, 127.6, 132.0, 134.8 (Ar), 136.1 (CH–Ar) ppm; MS m/z (rel. int., %): 264((M + 2)+, 31), 262(M+, 62), 181(33), 102(100), 75(31), 51(30).
(E)-β-Bromo(3-bromostyrene) (5a)16. Isolated yield: 86%; 1H NMR (300 MHz, CDCl3) δ = 6.81 (d, 1H, JH–H = 14.0 Hz, HC
), 7.05 (d, 1H, JH–H = 14.0 Hz, HC) 7.17–7.25 (m, 2H, Ar), 7.41–7.47 (m, 2H, Ar) ppm; 13C NMR (75 MHz, CDCl3) δ = 108.2 (CH–Br), 122.9, 124.7, 129.0, 130.3, 131.2, 135.8 (Ar), 137.9 (CH–Ar) ppm; MS m/z (rel. int., %): 264((M + 2)+, 28), 262(M+, 57), 181(24), 102(100), 75(26), 51(26).
(E)-β-Bromo(4-chlorostyrene) (6a)35. Isolated yield: 71%; 1H NMR (300 MHz, CDCl3) δ = 6.78 (d, 1H, JH–H = 14.0 Hz, HC
), 7.08 (d, 1H, JH–H = 14.0 Hz, HC) 7.23–7.33 (m, 4H, Ar) ppm; 13C NMR (75 MHz, CDCl3) δ = 107.0 (CH–Br), 126.8, 128.9, 133.8, 134.1 (Ar), 135.6 (CH–Ar) ppm; MS m/z (rel. int., %): 220((M + 4)+, 36), 218((M + 2)+, 72), 216(M+, 34), 137(100), 102(68), 75(28).
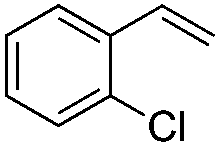
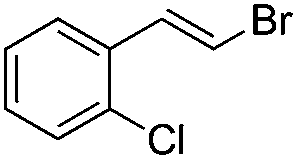
(E)-β-Bromo(2-chlorostyrene) (7a)16. Isolated yield: 67%; 1H NMR (300 MHz, CDCl3) δ = 6.72 (d, 1H, JH–H = 14.0 Hz, HC
), 7.09–7.33 (m, 4H, Ar), 7.39 (d, 1H, JH–H = 14.0 Hz, HC) ppm; 13C NMR (75 MHz, CDCl3) δ = 109.2 (CH–Br), 126.9, 127.0, 129.4, 129.9, 132.5, 133.8 (Ar), 134.1 (CH–Ar) ppm; MS m/z (rel. int. %): 218((M + 2)+, 26), 216(M+, 21), 137(100), 101(53), 75(33), 51(19).
Synthesis of (E)-β-arylvinyl iodides
:1). The purity of the product was confirmed by GC-MS and 1H NMR.
:1). The purity of the product was confirmed by GC-MS and 1H NMR.
:1) as the eluent. The purity of the product was confirmed by GC-MS and 1H NMR and the results were in agreement with the literature.15,35,46
(E)-β-Iodostyrene (1b)35. Isolated yield: 82%; 1H NMR (300 MHz, CDCl3) δ = 6.83 (d, 1H, JH–H = 14.9 Hz, HC
), 7.28–7.36 (m, 5H, Ar), 7.44 (d, 1H, JH–H = 14.9 Hz, HC) ppm; 13C NMR (75 MHz, CDCl3) δ = 66.8 (CH–I), 126.0, 128.4, 128.7, 137.7 (Ar), 145.0 (CH–Ar) ppm; MS m/z (rel. int., %): 230(M+, 100), 127(11), 103(83), 77(49).
(E)-β-Iodo(4-methylstyrene) (2b)35. Isolated yield: 84%; 1H NMR (300 MHz, CDCl3) δ = 2.25 (s, 3H, CH3), 6.65 (d, 1H, JH–H = 14.9 Hz, HC
), 6.97–7.17 (m, 4H, Ar), 7.30 (d, 1H, JH–H = 14.9 Hz, HC) ppm; 13C NMR (75 MHz, CDCl3) δ = 21.3 (CH3), 75.4 (CH–Br), 125.9, 129.4, 135.1, 138.4 (Ar), 144.9 (CH–Ar) ppm; MS m/z (rel. int., %): 244(M+, 100), 127(22), 115(78), 102(24).
(E)-β-Iodo(4-methoxystyrene) (3b)35. Isolated yield: 77%; 1H NMR (300 MHz, CDCl3) δ = 3.85 (s, 3H, OCH3), 6.67 (d, 1H, JH–H = 14.9 Hz, HC
), 6.87–6.89 (d, 2H, Ar), 7.26–7.30 (d, 2H, Ar) 7.40 (d, 1H, JH–H = 14.9 Hz, HC) ppm; 13C NMR (75 MHz, CDCl3) δ = 55.5 (OCH3), 73.6 (CH–I), 114.1, 127.8, 130.7 (Ar), 144.3 (CH–Ar), 159.8 (Ar) ppm; MS m/z (rel. int., %): 260(M+, 100), 133(47), 127(21), 118(20), 103(13), 89(28), 77(23), 63(25), 51(14).
(E)-β-Iodo(4-bromostyrene) (4b)35. Isolated yield: 79%; 1H NMR (300 MHz, CDCl3) δ = 6.70 (d, 1H, JH–H = 14.9 Hz, HC
), 6.99 (d, 2H, JH–H = 8.4 Hz, Ar), 7.20 (d, 1H, JH–H = 14.9 Hz, HC), 7.29 (d, 2H, JH–H = 8.4 Hz, Ar); 13C NMR (75 MHz, CDCl3) δ = 77.6 (CH–I), 122.4, 127.5, 131.9, 136.5 (Ar), 143.8 (CH–Ar) ppm; MS m/z (rel. int., %): 310((M + 2)+, 19), 308(M+, 18), 183(16), 181(17), 127(12), 102(100), 75(40), 51(27).
(E)-β-Iodo(3-bromostyrene) (5b)15. Isolated yield: 81%; 1H NMR (300 MHz, CDCl3) δ = 6.75 (d, 1H, JH–H = 14.9 Hz, HC
), 7.00–7.10 (m, 2H, Ar) 7.20 (d, 1H, JH–H = 14.9 Hz, HC) 7.26–7.34 (m, 2H, Ar), ppm; 13C NMR (75 MHz, CDCl3) δ = 79.0 (CH–I), 123.0, 124.7, 128.9, 130.3, 131.5, 139.5 (Ar), 143.4 (CH–Ar) ppm; MS m/z (rel. int., %): 310((M + 2)+, 40), 308(M+, 39), 183(16), 181(17), 127(6), 102(100), 75(21), 51(12).
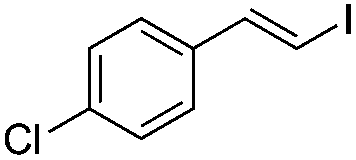
(E)-β-Iodo(4-chlorostyrene) (6b)35. Isolated yield: 63%; 1H NMR (300 MHz, CDCl3) δ = 6.86 (d, 1H, JH–H = 14.9 Hz, HC
), 7.22–7.25 (d, 2H, Ar) 7.30–7.33 (d, 2H, Ar), 7.40 (d, 1H, JH–H = 14.9 Hz) ppm; 13C NMR (75 MHz, CDCl3) δ = 77.6 (CH–I), 127.2, 128.9, 134.2, 136.1 (Ar), 143.7 (CH–Ar) ppm; MS m/z (rel. int., %): 264(M+, 100), 137(83), 127(24), 102(93), 75(23).
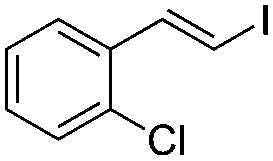
(E)-β-Iodo(2-chlorostyrene) (7b)46. Isolated yield: 63%; 1H NMR (300 MHz, CDCl3) δ = 6.78 (d, 1H, JH–H = 14.9 Hz, HC
), 7.05–7.35 (m, 4H, Ar) 7.69 (d, 1H, JH–H = 14.9 Hz, HC) ppm; 13C NMR (75 MHz, CDCl3) δ = 79.9 (CH–I), 126.9, 127.1, 129.4, 129.9, 132.2, 135.8 (Ar), 141.4 (CH–Ar) ppm; MS m/z (rel. int., %): 264(M+, 37), 137(100), 127(25), 101(71), 75(50), 51(18).
(E)-1-Iodo-2-naphtylethene (8b)15. Isolated yield: 58%; 1H NMR (300 MHz, CDCl3) δ = 6.87 (d, 1H, JH–H = 14.6 Hz, HC
), 7.40–7.65 (m, 4H, napht.), 7.79–7.94 (m, 2H, napht.), 8.03–8.12 (m, 1H, napht.), 8.18 (d, JH–H = 14.6 Hz, HC), ppm; 13C NMR (75 MHz, CDCl3) δ = 79.4 (CH–I), 123.7, 124.2, 125.6, 126.1, 126.5, 128.6, 128.8, 130.2, 133.5, 135.6 (Naph), 142.9 (CH–Naph) ppm; MS m/z (rel. int., %): 280(M+, 29), 153(100), 126(14).
Synthesis of (Z)-β-arylvinyl bromides
:1). The purity of the product was confirmed by GC-MS and 1H NMR.
:1) as the eluent. The purity of the product was confirmed by GC-MS and 1H NMR and the results were in agreement with the literature.19,23,47,48
(Z)-β-Bromostyrene (9)48. Isolated yield: 84%; 1H NMR (300 MHz, CDCl3) δ = 5.27 (d, 1H, JH–H = 7.4 Hz, HC
), 5.89 (d, 1H, JH–H = 7.4 Hz, HC), 7.23–7.37 (m, 5H, Ar) ppm; 13C NMR (75 MHz, CDCl3) δ = 105.8 (CH–Br), 128.0, 128.1, 128.9 131.9 (Ph), 134.9 (CH–Ph) ppm; MS m/z (rel. int., %): 184((M + 2)+, 46), 182(M+, 47) 103(100), 77(58), 51(32).
(Z)-β-Bromo(4-methylstyrene) (10)48. Isolated yield: 61%; 1H NMR (300 MHz, CDCl3) δ = 2.39 (3H, s, CH3), 5.40 (1H, d, JH–H = 7.4 Hz), 6.01 (1H, d, 1H JH–H = 7,4 Hz), 7.16–7.31 (d, 2H, Ar), 7.37–7.40 (2H, d, Ar) ppm; 13C NMR (75 MHz, CDCl3) δ = 21.6 (CH3), 105.3 (
CH–Br), 128.5, 128.9, 131.7 (Ar), 137.9 (CH–Ar) ppm; MS m/z (rel. int., %): 198((M + 2)+, 61), 196(M+, 62) 117(100), 91(41), 63(17), 57(13) 51(13).
(Z)-β-Bromo(4-methoxystyrene) (11)19,48. Isolated yield: 89%; 1H NMR (300 MHz, CDCl3) δ = 3.84 (s, 3H), 5.40 (d, 1H, JH–H = 7.3 Hz), 6.01 (d, 1H, JH–H = 7.3 Hz), 6.90–6.95 (m, 2H, Ar), 7.36–7.44 (m, 2H, Ar) ppm; 13C NMR (75 MHz, CDCl3) δ = 55.0 (CH3), 104.2 (
CH–Br), 112.9, 127.3, 130.3 (Ar), 132.6 (CH–Ar), 159.9 (Ar) ppm; MS m/z (rel. int., %): 214((M + 2)+, 97), 212(M+, 100), 197(42), 169(18), 133(48), 118(28), 90(70), 77(18), 63(41).
(Z)-β-Bromo(4-chlorostyrene) (12)48. Isolated yield: 82%; 1H NMR (300 MHz, CDCl3) – isolated yield: 82%; δ = 5.42 (d, 1H, JH–H = 7.4 Hz, HC
), 6.02 (d, 1H, JH–H = 7.4 Hz, HC), 7.38–7.43 (d, 2H, Ar), 7.47–7.52 (d, 2H, Ar) ppm; 13C NMR (75 MHz, CDCl3) δ = 107.4 (CH–Br), 128.1, 128.9, 131.0, 133.1 (Ar), 133.9 (CH–Ar) ppm; MS m/z (rel. int., %): 220((M + 4)+, 19), 218((M + 2)+, 78), 216(M+, 60), 137(100), 102(74), 75(43).
(Z)-β-Bromo(4-bromostyrene) (13)23,47. Isolated yield: 84%; 1H NMR (300 MHz, CDCl3) δ = 6.52 (d, 1H, JH–H = 7.9 Hz, HC
), 7.03 (d, 1H, JH–H = 7.9 Hz, HC), 7.21–7.41 (d, 2H, Ar), 7.47–7.61 (d, 2H, Ar) ppm; 13C NMR (75 MHz, CDCl3) δ = 108.1 (CH–Br), 122.3, 127.5, 131.1, 131.2, 131.7, 136.9 (CH–Ar) ppm; MS m/z (rel. int., %): 264((M + 2)+, 29), 262(M+, 63), 181(35), 102(100), 75(31), 51(27).
Acknowledgements
This work was supported by the National Centre for Research and Development (Poland) (grant no. LIDER/26/p527/L-5/13/NCBR/2014) and as part of grants financed by the National Science Centre (Poland) Opus 2011/03/B/ST5/01034 and Maestro UMO-2011/02/A/ST5/00472. The authors would like to thank Katarzyna Mielcarek for her contribution to this research as part of her master's thesis.Notes and references
- Metal-catalyzed Cross-coupling Reactions, F. Diederich and P. J. Stang, ed, 2008 Search PubMed.
- H. Doucet, Eur. J. Org. Chem., 2008, 2013–2030 CrossRef CAS.
- Q. Liao, Y. Wang, L. Zhang and C. Xi, J. Org. Chem., 2009, 74, 6371–6373 CrossRef CAS PubMed.
- M. S. Kabir, M. L. Van Linn, A. Monte and J. M. Cook, Org. Lett., 2008, 10, 3363–3366 CrossRef CAS PubMed.
- F. Besselievre, S. Piguel, F. Mahuteau-Betzer and D. S. Grierson, Org. Lett., 2008, 10, 4029–4032 CrossRef CAS PubMed.
- F. A. Davis, G. S. Lal and J. Wei, Tetrahedron Lett., 1988, 29, 4269–4272 CrossRef CAS.
- L. Jiang, G. E. Job, A. Klapars and S. L. Buchwald, Org. Lett., 2003, 5, 3667–3669 CrossRef CAS PubMed.
- J. Sinha, S. Layek, G. C. Mandal and M. Bhattacharjee, Chem. Commun., 2001, 1916–1917 RSC.
- C. Kuang, Q. Yang, H. Senboku and M. Tokuda, Synthesis, 2005, 1319–1325 CrossRef CAS.
- J. P. Das and S. Roy, J. Org. Chem., 2002, 67, 7861–7864 CrossRef CAS PubMed.
- S. Abbas, C. J. Hayes and S. Worden, Tetrahedron Lett., 2000, 41, 3215–3219 CrossRef CAS.
- H. Horibe, K. Kondo, H. Okuno and T. Aoyama, Synthesis, 2004, 986–988 CAS.
- K. Takai, T. Ichiguchi and S. Hikasa, Synlett, 1999, 1268–1270 CrossRef CAS.
- M. R. Uehling, R. P. Rucker and G. Lalic, J. Am. Chem. Soc., 2014, 136, 8799–8803 CrossRef CAS PubMed.
- J. A. Bull, J. J. Mousseau and A. B. Charette, Org. Lett., 2008, 10, 5485–5488 CrossRef CAS PubMed.
- V. Pappula, R. R. Donthiri, C. M. Darapaneni and A. Subbarayappa, Tetrahedron Lett., 2014, 55, 1793–1795 CrossRef CAS.
- X. P. Zhang and M. Schlosser, Tetrahedron Lett., 1993, 34, 1925–1928 CrossRef CAS.
- G. Stork and K. Zhao, Tetrahedron Lett., 1989, 30, 2173–2174 CrossRef CAS.
- M.-E. Lebrun, P. Le Marquand and C. Berthelette, J. Org. Chem., 2006, 71, 2009–2013 CrossRef CAS PubMed.
- J. I. Uenishi, R. Kawahama, O. Yonemitsu and J. Tsuji, J. Org. Chem., 1998, 63, 8965–8975 CrossRef CAS.
- J. I. Uenishi, R. Kawahama, Y. Shiga, O. Yonemitsu and J. Tsuji, Tetrahedron Lett., 1996, 37, 6759–6762 CrossRef CAS.
- W. Xv, W. Zhang, F. Zhang and C. Kuang, J. Chem. Res., 2014, 38, 115–117 CrossRef CAS.
- C. Kuang, Q. Yang, H. Senboku and M. Tokuda, Tetrahedron, 2005, 61, 4043–4052 CrossRef CAS.
- R. B. Miller and G. McGarvey, J. Org. Chem., 1978, 43, 4424–4431 CrossRef CAS.
- H. C. Brown, T. Hamaoka, N. Ravindran, C. Subrahmanyam, V. Somayaji and N. G. Bhat, J. Org. Chem., 1989, 54, 6075–6079 CrossRef CAS.
- H. C. Brown, T. Hamaoka and N. Ravindran, J. Am. Chem. Soc., 1973, 95, 5786–5788 CrossRef CAS.
- N. A. Petasis and I. A. Zavialov, Tetrahedron Lett., 1996, 37, 567–570 CrossRef CAS.
- A. Hamajima and M. Isobe, Angew. Chem., Int. Ed., 2009, 48, 2941–2945 CrossRef CAS PubMed.
- M.-F. Zou and M.-Z. Deng, J. Org. Chem., 1996, 61, 1857–1858 CrossRef CAS PubMed.
- Z. Huang and E.-I. Negishi, Org. Lett., 2006, 8, 3675–3678 CrossRef CAS PubMed.
- C. Morrill and R. H. Grubbs, J. Org. Chem., 2003, 68, 6031–6034 CrossRef CAS PubMed.
- V. Sashuk, C. Samojlowicz, A. Szadkowska and K. Grela, Chem. Commun., 2008, 2468–2470 RSC.
- C. A. Celatka and J. S. Panek, Tetrahedron Lett., 2002, 43, 7043–7046 CrossRef CAS.
- H. J. Kim, R. Pongdee, Q. Wu, L. Hong and H.-W. Liu, J. Am. Chem. Soc., 2007, 129, 14582–14584 CrossRef CAS PubMed.
- P. Pawluc, G. Hreczycho, J. Szudkowska, M. Kubicki and B. Marciniec, Org. Lett., 2009, 11, 3390–3393 CrossRef CAS PubMed.
- B. Marciniec, Acc. Chem. Res., 2007, 40, 943–952 CrossRef CAS PubMed.
- B. Marciniec, M. Jankowska and C. Pietraszuk, Chem. Commun., 2005, 663–665 RSC.
- P. Pawluc, W. Prukala and B. Marciniec, Eur. J. Org. Chem., 2010, 219–229 CrossRef CAS.
- P. Pawluc, J. Szudkowska, G. Hreczycho and B. Marciniec, J. Org. Chem., 2011, 76, 6438–6441 CrossRef CAS PubMed.
- P. Pawluc, A. Franczyk, J. Walkowiak, G. Hreczycho, M. Kubicki and B. Marciniec, Tetrahedron, 2012, 68, 3545–3551 CrossRef CAS.
- P. Pawluc, A. Franczyk, J. Walkowiak, G. Hreczycho, M. Kubicki and B. Marciniec, Org. Lett., 2011, 13, 1976–1979 CrossRef CAS PubMed.
- P. Pawluc, G. Hreczycho, J. Walkowiak and B. Marciniec, Synlett, 2007, 2061–2064 CAS.
- H. C. Brown, T. Hamaoka and N. Ravindran, J. Am. Chem. Soc., 1973, 95, 6456–6457 CrossRef CAS.
- V. S. Rawat and B. Sreedhar, Synlett, 2014, 1132–1136 Search PubMed.
- C. Feng, H. Wang, L. Xu and P. Li, Org. Biomol. Chem., 2015, 13, 7136–7139 CAS.
- J. J. Mousseau, J. A. Bull, C. L. Ladd, A. Fortier, D. S. Roman and A. B. Charette, J. Org. Chem., 2011, 76, 8243–8261 CrossRef CAS PubMed.
- S. H. Kim, H.-X. Wei, S. Willis and G. Li, Synth. Commun., 1999, 29, 4179–4185 CrossRef CAS.
- C. Kuang, H. Senboku and M. Tokuda, Tetrahedron Lett., 2001, 42, 3893–3896 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and 1H NMR spectra of representative examples of obtained halogenated compounds. See DOI: 10.1039/c7ob00054e |
| This journal is © The Royal Society of Chemistry 2017 |