
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilization of peptides against proteolysis through disulfide-bridged conjugation with synthetic aromatics†
Yaqi
Chen‡
a,
Tao
Li‡
a,
Jianguo
Li
bc,
Shiyan
Cheng
a,
Jinghui
Wang
a,
Chandra
Verma
cde,
Yibing
Zhao
a and
Chuanliu
Wu
 *a
*a
aThe MOE Key Laboratory of Spectrochemical Analysis and Instrumentation, State Key Laboratory of Physical Chemistry of Solid Surfaces, Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, P. R. China. E-mail: chlwu@xmu.edu.cn
bSingapore Eye Research Institute, 20 College Road, 169856, Singapore
cBioinformatics Institute (A*STAR), 30 Biopolis Street, Matrix, 138671, Singapore
dNational University of Singapore, Department of Biological Sciences, Singapore
eNanyang Technological University, School of Biological Sciences, Singapore
First published on 1st February 2017
Abstract
Peptides have been promising molecular scaffolds for the development of potential therapeutics with high affinity and specificity to biomacromolecules. However, their inherent proteolytic instability significantly hampers their biological applications. Strategies that can stabilize peptides against proteolytic digestion on the basis of noncovalent interactions—without extensive manipulation of the sequence or use of unnatural residues—are greatly desired. In this work, we developed a general, convenient, and efficient strategy for the stabilization of peptides against proteolysis, which involves noncovalent π–π interactions between aromatic amino acid residues in peptides and synthetic electron-deficient aromatics (NDI), together with the implication of steric hindrance (from the bulky NDI moiety), and the enhancement of peptide α-helicity. This strategy is complementary in concept to the conventional well-established covalent approaches for peptide stabilization, and is thus promising for being utilized, in combination with the latter ones, to circumvent the problem of proteolytic instability of peptides. We envisioned that this study should provide invaluable guidelines to the design and synthesis of organic molecule–peptide hybrids with significantly improved proteolytic resistance, and benefit the development of peptide therapeutics and probes.
Introduction
Peptides have been considered as promising molecular scaffolds for the development of potential therapeutic agents and chemical probes with high affinity and specificity to biomacromolecules.1–3 However, the issue of inherent instability towards proteolytic digestion limits their applications in biological environments, mainly due to the functional diversity and aggressivity of endogenous proteases.1,4 Although many strategies have been developed to improve the proteolytic stability of peptides,1,3,5–7 including cyclization, backbone/side chain modification, and unnatural residue substitution, poor proteolytic stability is still one of the most formidable challenges facing the development of peptide-based therapeutics/materials. In contrast to the conventional strategies generally exploiting covalent modifications,6,8 nature has evolved to use noncovalent interactions such as hydrogen bonding, aromatic interactions, electrostatics, and hydrophobic packing to stabilize the structures of proteins and natural peptides, and prevent them from enzymatic digestion in the body.9,10 These interactions have also been rationally regulated in artificial peptide constructs to enhance their structural/proteolytic stability.11–14 However, the effectiveness of this strategy generally relies on the precise placement of paired (or multiple) key amino-acid residues11,12 or unnatural residues14 in the sequence, which can be awkward to implement, thus severely restricting its applicability. We argue that simple strategies to stabilize peptides from enzymatic digestion on the basis of noncovalent interactions—without extensive manipulation of the sequence or use of unnatural residues—should be extremely valuable and desired, especially considering the possibility of combining orthogonally the two fundamentally different strategies (i.e., covalent and noncovalent) for maximally solving the problem of proteolytic instability of peptides.In this work, we developed novel hybrid constructs to enhance the proteolytic stability of peptides by linking peptides with an electron-deficient aromatic compound, 1,4,5,8-naphthalenediimide (NDI),15,16 through dynamic disulfide bonds (Fig. 1). Hybrid constructs can be conveniently designed and synthesized by arbitrary inclusion of double (or single) cysteine residues in the peptides, and then conjugating with the thiol-bearing NDI (C-NDI-C) through thiol–disulfide exchanges. Using model peptides derived from the known inhibitors of the E3 ubiquitin ligase MDM2,17,18 the contribution of aromatic interactions between peptides (i.e., aromatic ring of residues phenylalanine F, tyrosine Y, or tryptophan W) and NDI to the proteolytic stability of peptides was unambiguously revealed. Peptides incorporated into the hybrid constructs display exceptionally high resistance to proteolysis. The dynamic properties of disulfide bonds allow the incorporated peptides to be released specifically under endogenous redox stimulation.11,15,19–21 This study thus presents a general, convenient and efficient strategy for the stabilization of peptides against proteolysis, which is conceptually orthogonal to the conventional well-established covalent modification-based approaches.
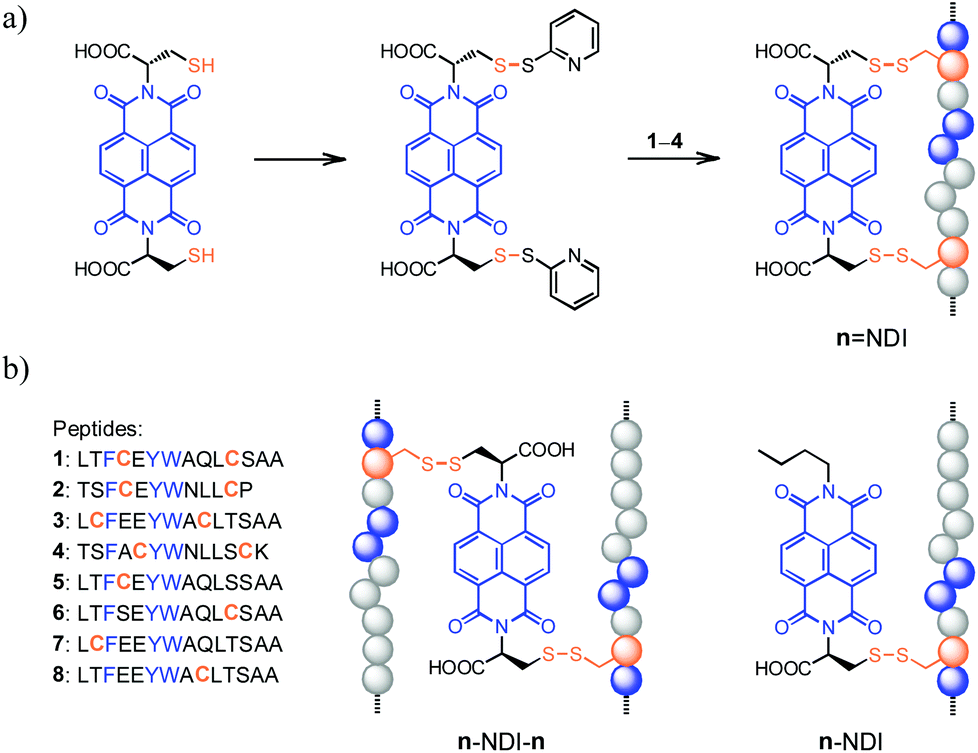 | ||
Fig. 1 (a) General routes for the synthesis of hybrid dimers (n![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NDI, n = 1–4) linked by two disulfide bonds; blue balls represent the random distribution of aromatic amino acid residues. (b) The sequence of peptides (1–8) used in this study and schematic drawings illustrating the structure of hybrid trimers (n–NDI–n, n = 5–8; linked by two disulfide bonds) and dimers (n–NDI, n = 5–8; linked by one disulfide bond), respectively. See Fig. S1–S12† for characterization of products. NDI, n = 1–4) linked by two disulfide bonds; blue balls represent the random distribution of aromatic amino acid residues. (b) The sequence of peptides (1–8) used in this study and schematic drawings illustrating the structure of hybrid trimers (n–NDI–n, n = 5–8; linked by two disulfide bonds) and dimers (n–NDI, n = 5–8; linked by one disulfide bond), respectively. See Fig. S1–S12† for characterization of products. | ||
Experimental section
Reagents
Peptides were purchased from either Sangon Biotech (Shanghai, China) or KE Biochem Co., Ltd (Shanghai, China) with >95% purity. All peptides, except 2, were N-terminally acetylated and C-terminally amidated. Analytical chromatograms and mass spectra were used to confirm the identity and purity of peptides. MCF7 and U251 cells were purchased from CoBioer Biosciences Co., Ltd (Nanjing, China). Dulbecco's Modified Eagle's Medium (DMEM) and 4′,6-diamidino-2-phenylindole (DAPI) were obtained from Thermo Scientific (Beijing, China). Eppendorf tubes (1.5 mL), 6-well chambers, 24-well chambers, 96-well flat-bottomed plates and cell culture dishes were purchased from JET BIOFIL (Guangzhou, China). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) and dithiothreitol (DTT) were purchased from Sigma-Aldrich. Millipore ultrapure water was used throughout the experiments.Synthesis of NDI–peptide conjugates
Synthetic procedures for C-NDI-C and C-NDI have been reported in the literature.15,22 The thiol groups of C-NDI-C (or C-NDI) were first activated by 2,2′-dithiodipyridine through a reaction of C-NDI-C with excessive amounts of 2,2′-dithiodipyridine in 100 mM carbonate bicarbonate buffer (pH 9.16). Then, the products (thiol-activated C-NDI-C and C-NDI) were purified using a HPLC. NDI–peptide conjugates were obtained by the thiol–disulfide exchange reactions between cysteine residue-containing peptides and thiol-activated C-NDI-C (or C-NDI) in 100 mM phosphate buffer (pH 7.4) containing ∼40 vol% TFE. The reaction processes were monitored, and the final products were purified using a HPLC (Waters Xbridge C18 column (4.6 mm × 250 mm, 5 μm), 1.0 mL min−1 flow rate, isocratic with 5 vol% acetonitrile (ACN) + 0.1% trifluoroacetic acid (TFA) for 5 min followed by a linear gradient of ACN + 0.1% TFA (5–80 vol%) over 30 min). The obtained NDI–peptide conjugates were identified by MALDI-TOF mass spectrometry with saturated 2,5-dihydroxybenzoic acid solution as the matrix. The products were then lyophilized and stored at −20 °C until use.Analysis of digestion kinetics
All digestion reactions were performed in phosphate buffer (100 mM, pH = 7.4) at room temperature. The stock solutions of peptides (or NDI–peptide conjugates) were prepared in DMSO. The final concentration of peptides (or NDI–peptide conjugates) was 50 μM, containing less than 4% DMSO (from the stock solutions). We have confirmed that 4% DMSO in buffers does not affect the kinetics of peptide digestion. The concentration of protease was rationally changed according to the proteolytic stability of the peptides. Aliquots were taken from the samples at predefined times, and were immediately treated with 10% HPO3 to quench the reactions. The samples were then monitored using a Waters Xbridge C18 column (4.6 mm × 250 mm, 5 μm) on a Shimadzu HPLC system (1.0 mL min−1 flow rate, isocratic with 5 vol% acetonitrile (ACN) + 0.1% trifluoroacetic acid (TFA) for 5 min followed by a linear gradient of ACN + 0.1% TFA (5–80 vol%) over 30 min). The digestion kinetics was calculated by the area of absorption peaks at the 280 nm (free peptides) and 363 nm (NDI–peptide conjugates) signals, respectively. The half-lives of the peptides were analyzed by a pseudo-first-order method using OriginPro 8.5 software (OriginLab Corp.).Circular dichroism (CD) spectroscopy
CD spectra were recorded at room temperature (25 °C) using a 0.1 cm path length cuvette. The spectra were recorded in a wavelength range of 190–260 nm and averaged over 3 scans with a resolution of 1 nm, a bandwidth of 2 nm, and a response time of 8 s. The sensitivity and the speed of the spectrometer were set to 100 mdeg and 50 nm min−1, respectively. The baseline signal (phosphate buffer) was subtracted from each spectrum. All peptides were dissolved in aqueous solution or mixed ACN/phosphate buffer to reach a concentration of 30 μM.Molecular dynamics simulations
Atomistic molecular dynamics simulations were carried out to study the conformational space of peptides 5 and 6 and their NDI-conjugates (5–NDI, 6–NDI, and 6–NDI–6) in explicit solvents. The peptides were modelled using the AMBER99sb force field,23 and the NDI moiety was parameterized based on the AMBER general force field.24 The peptides were solvated using the TIP3P water model and counter ions were added to make the system neutral. Hamiltonian replica exchange molecular dynamics (HREMD) simulations25 were employed to enhance the sampling of the conformational phase space of the peptides. In each HREMD simulation, eight replicas were run in parallel and a Monte Carlo move was carried out to exchange adjacent replicas every 1 ps, resulting in an average acceptance ratio of 20%. Each HREMD simulation run was carried out for 200 ns and the last 100 ns were used for analysis. For all HREMD simulations, a cutoff of 1.0 nm was used for both Lennard-Jones potential and short range electrostatic interactions, while long range electrostatic interactions were computed using PME.26 All the simulations were carried out under the NPT ensemble, with temperature and pressure maintained at 300 K and 1 bar, respectively. Secondary structure evolution was calculated using GROMACS tool do_dssp. Cluster analysis was carried out using the g_cluster and the radius of gyration was calculated using the g_gyrate. All simulations were carried out using GROMACS 4.627 in combination with PLUMED 2.1.28Protein expression and purification
E. coli BL21 (DE3) cells transfected with pSUMO-Mut-MDM2 (residues 17–125) plasmids were purchased from Zoonbio Biotechnology Co., Ltd (Nanjing, China). BL21 (DE3) cells were grown in 1.2 L LB medium containing 50 μg mL−1 kanamycin at 37 °C to OD600 = 0.6–0.8. Then, 0.2 mM isopropyl-β-D-thiogalactopyranoside (IPTG) was added to induce SUMO-MDM2 expression, and the cells were incubated for 12 h at 16 °C. After harvesting by centrifugation, the cell pellets were re-suspended in buffer A (100 mM Tris, 500 mM NaCl, 1 mM PMSF, pH 7.9) and lysed by sonication; then the lysate was clarified by centrifugation at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min. The clarified supernatant was applied to a Ni Sepharose column and washed with buffer B (100 mM Tris, 500 mM NaCl, 200 mM imidazole, pH 7.9); then the protein was purified by gel filtration on a SuperdexTM 75 10/300 GL column (GE) into 1× PBS and stored at −80 °C until use.
000 rpm for 30 min. The clarified supernatant was applied to a Ni Sepharose column and washed with buffer B (100 mM Tris, 500 mM NaCl, 200 mM imidazole, pH 7.9); then the protein was purified by gel filtration on a SuperdexTM 75 10/300 GL column (GE) into 1× PBS and stored at −80 °C until use.
Fluorescence polarization (FP) assays
Fluorescence polarization assays were conducted by using a filter-based microplate reader (Tecan Infinite F200 PRO) with a 485 nm excitation filter, a 535 nm emission filter, and an integration time of 20 μs. The binding affinity of the fluorescently labeled peptide FITC-PMI was measured by titrating 20 nM FITC-PMI with increasing concentrations of SUMO-MDM2 in non-treated black 96-well flat-bottomed plates (Promega), and the data were fitted using Graphpad Prism to a FP direct binding model. Competition fluorescence polarization assays were performed by using FITC-PMI as a probe peptide in 96-well plates. Titrations were carried out with the concentrations of SUMO-MDM2 held constant at 80 nM and the probe peptide at 20 nM, respectively. The competing peptides were then titrated against the complex of FITC-PMI and SUMO-MDM2. Curve fitting was carried out using Prism 6.0 (GraphPad). All experiments were carried out in 1× PBS (pH 7.4), each sample was performed in duplicate wells, and the titrations were conducted in triplicate.Cell culture
MCF7 and U251 cells were maintained in DMEM medium (high glucose) supplemented with 10% FBS and 1% penicillin/streptomycin (penicillin: 10000 U mL−1, streptomycin: 10000 U mL−1) at 37 °C under a humidified atmosphere containing 5% CO2. The cells were passaged at about 80% cell confluency using a 0.25% trypsin solution.
Cell confocal microscopy
MCF7 cells were seeded at a density of 6 × 104 cells per well into a 24-well chamber and a coverslip was put on the bottom of each well. After incubation for 24 h at 37 °C, the medium was removed and the cells were washed with PBS. 1 mL medium (supplemented with 10% FBS) containing 1.0 μM rhodamine-labeled 6–NDI–6 was then added. After incubation for 24 h at 37 °C, the cells were thoroughly washed three times with PBS. Then, the cell nuclei were stained with DAPI, and the cells were fixed with 4% triformol for 20 min. After the washing step with PBS, the cells were made into smears for imaging. The fluorescence of the cells was detected by a confocal microscopy system (Zeiss Exciter 5) at different detection channels (DAPI channel: λex = 405 nm, λem = 420–480 nm; rhodamine channel: λex = 543 nm, λem = 560–615 nm).Flow cytometry
MCF7 cells were seeded at 2 × 105 cells per well in a 6-well plate. After incubation for 24 h at 37 °C, the medium was removed and the cells were washed with PBS. 2 mL medium (supplemented with 10% FBS) containing 1.0 μM 6–NDI–6 (or 6 and 6–NDI) was then added. After incubation for 24 h at 37 °C, the cells were thoroughly washed three times with PBS. Then, the cells were detached by a treatment with 0.25% trypsin solution. The cells were collected by centrifugation and washed twice with cold PBS. After that, the cells were re-suspended in 1 mL cold PBS. Fluorescent signals in the cells were detected by flow cytometry (BD FACSAria II), and at least 10000 fluorescent events were counted per sample. All experiments were conducted in triplicate.
Cell viability assay
The bioactivity of 6, 6–NDI, 6–NDI–6, and 9–NDI–9 and the cytotoxicity of NDI were evaluated by MTT assays. The cells were seeded in a 96-well plate at an initial cell density of 6000 cells per well (MCF7 cells) or 10000 cells per well (U251 cells) and grown at 37 °C and 5% CO2 for 24 h. The stock solutions of 6, 6–NDI, 6–NDI–6, 9–NDI–9, and NDI were prepared in DMSO, which were then diluted into different concentrations (from 0 to 20 μM) by using DMEM medium (with or without 10% FBS supplemented). The final concentration of DMSO in the medium does not exceed 1% (i.e., no obvious cytotoxicity from DMSO). After the addition of the peptide or NDI–peptide conjugate medium and incubating at 37 °C for 24 h, the viability of the cells was evaluated using MTT according to its manufacturer's protocol. The absorbance was measured at 490 nm using an ELISA reader (PerkinElmer Enspire®). All experiments were conducted in triplicate.
Results and discussion
Two peptides (1 and 2) derived from the inhibitors of MDM2 were designed and synthesized.17,18 Each peptide contains two cysteine residues placed at positions i and i + 7 that were not considered pivotal to the binding activity to MDM2. Hybrid dimers, connected by two disulfide bonds, were then prepared through thiol–disulfide exchange reactions between peptides and the C-NDI-C molecule activated by 2,2′-dithiopyridine (Fig. 1a). To compare the proteolytic stability of the hybrid dimers and the free peptides, the kinetics of degradation by chymotrypsin were monitored by high-performance liquid chromatography (HPLC).11 Free peptides 1 and 2 can be rapidly digested with 0.02 μM chymotrypsin, with a half-life of 36 and 10 min (extracted from the kinetic curves of peptide digestion), respectively (Fig. 2a, S16 and S17†). In contrast, the hybrid dimers are extremely stable under the same conditions, and the degradation can only be observed while the concentration of chymotrypsin was increased to 10 μM (Fig. S18 and S19†), suggesting that the peptides can be significantly stabilized against proteolysis by the disulfide-linked NDI (Fig. 2a). We surmised that the remarkable increase in the proteolytic stability should result from the interplay of disulfide bonds, π–π interactions between the aromatic residues of peptides and aromatic NDI, steric hindrance from NDI, and the enhanced α-helicity arising from the cyclization. Our study further revealed that the enhanced proteolytic stability is tolerant to the variation in positions where the two cysteine residues are placed (peptides 3 and 4; improvement in proteolytic stability observed when the peptides are conjugated to C-NDI-C by disulfide bonds, Fig. 2a and S20–S23†), though the extent of enhancement in proteolytic stability reduced to some extent. Importantly, as all of the hybrid dimers display some degree of α-helicity (10–60%) in aqueous solution (Fig. S24–S27†), the helical wheel representations of peptides 1–4 might indicate the arrangement of aromatic residues (F, Y and W) in peptides relative to their appended NDI (Fig. 2b). Compared to that in the other three hybrid dimers, the F/Y/W face of the α-helix and the disulfide-linked NDI in the dimeric 4NDI is obviously more efficiently separated in structural space (Fig. 2b) and more interestingly, it can indeed be more rapidly cleaved by chymotrypsin than other dimers (Fig. 2a). In addition, both 1NDI and 2NDI exhibit a higher stability compared to 3NDI and 4NDI, a result that is in agreement with the degree of α-helicity (Fig. S24–S27†). These findings thus strongly suggest the important effect of the π–π interactions between the peptides and aromatic NDI and the consequent enhancement of α-helicity on the enhanced proteolytic stability of the peptides.
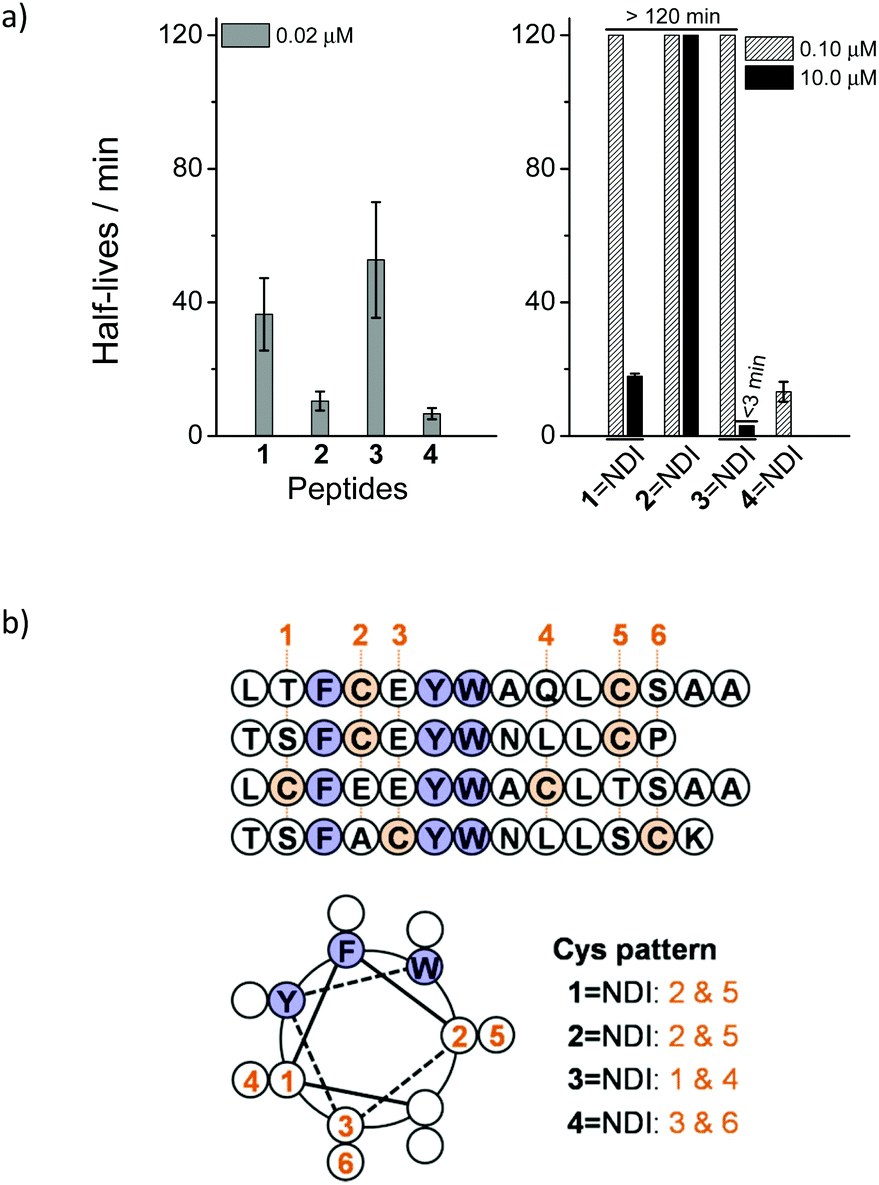 | ||
| Fig. 2 (a) Half-lives of peptide digestions by chymotrypsin in 100 mM phosphate buffer, pH 7.4; to quantitatively compare their proteolytic stability, the concentration of chymotrypsin was increased from 0.02 to 0.1 or 10 μM while the peptides (or hybrids dimers) are too stable to be digested under the lower enzyme concentrations; peptide concentration: 50 μM. (b) Helical wheel representation of peptides 1–4 indicating the relative arrangement of aromatic residues in the peptides relative to the two cysteines or the disulfide-linked NDI. | ||
Considering that the introduction of a pair of fixed cysteine residues into peptides can significantly restrict the general applicability of the developed peptide-stabilization strategy, we next examined if peptide–NDI hybrids, linked by a single disulfide bond, can also take advantage of π–π interactions between the aromatic residues and the electron-deficient NDI to increase the peptide proteolytic stability. Four single cysteine-containing peptide analogues derived from peptides 1 and 3 were designed and synthesized. Hybrid trimers comprised of two peptide chains and a C-NDI-C molecule cross-linked by two disulfide bonds were then synthesized through thiol–disulfide exchange reactions (Fig. 1b). In addition, hybrid and single disulfide-linked dimers were also prepared by a reaction of peptides with a single cysteine-bearing NDI molecule (C-NDI) (Fig. 1b). To quantitatively compare the proteolytic stability of the hybrid trimers/dimers to that of the relevant free peptides, the half-lives of the digestion by chymotrypsin were extracted from their kinetic curves of degradation and are given in Table 1 (see Fig. S28–S44† for cleavage kinetics). As hybrid trimers or dimers are, in general, significantly more resistant to proteolysis compared to free peptides, the concentration of protease was increased and a stability “enhancement factor” was calculated by dividing the digestion half-life by the protease concentration.11 Interestingly, a maximum 985-fold and 478-fold increase in stability was observed for the hybrid trimer (6–NDI–6) and dimer (5–NDI), respectively (Table 1). In addition, though only moderate improvement in stability was observed for the 8–NDI dimer (28-fold), their hybrid trimer (8–NDI–8) exhibits a >900-fold increase in stability. In contrast, some hybrid dimers (5–NDI and 7–NDI) are more stable than their hybrid trimers. We further evaluated the proteolytic resistance of the peptide–NDI hybrids to a more aggressive protease, proteinase K,7 which cleaves peptide bonds which are covered but not limited to the predominant cleavage sites of chymotrypsin (i.e., the carboxyl side of both aliphatic and aromatic residues). The most stable trimer, 6–NDI–6, still displays an ∼50-fold increase in stability as compared to the free peptide 6 (Fig. S40 and S41†).
| Peptides | τ 1/2 of peptides in chymotrypsin (min) | EF | ||
|---|---|---|---|---|
| 0.02 μM | 0.10 μM | 10.0 μM | ||
| a Peptide concentration, 50 μM; n.deg., no degradation observed by HPLC within 2 hours; —, not determined; EF, stability enhancement factor relative to the monomer. Data are presented as mean ± s.d. (n = 3). | ||||
| 5 | 6.8 ± 3.6 | — | — | |
| 5–NDI | — | — | 6.5 ± 0.3 | 478 |
| 5–NDI–5 | — | n.deg. | 2.9 ± 0.2 | 213 |
| 6 | 17.0 ± 3.4 | — | — | |
| 6–6 | — | 58.6 ± 5.3 | — | 17 |
| 6–NDI | — | n.deg. | 12.0 ± 1.3 | 353 |
| 6–NDI–6 | — | n.deg. | 33.5 ± 2.2 | 985 |
| 6–Bph–6 | — | — | 9.4 ± 0.4 | 276 |
| 6–Bme–6 | — | 24.0 ± 1.5 | — | 7 |
| 7 | 69.8 ± 15.0 | — | — | |
| 7–NDI | — | — | 8.2 ± 0.6 | 59 |
| 7–NDI–7 | — | — | 1.7 ± 0.4 | 12 |
| 8 | 17.8 | — | — | |
| 8–NDI | — | — | 1.0 | 28 |
| 8–NDI–8 | — | — | 33.2 | 933 |
The homodimers of 6 (6–6, 6–Bme–6 and 6–Bph–6, linked by a disulfide bond, 1,2-bismaleimidoethane and a 4,4′-bis(bromomethyl)biphenyl crosslinker, respectively; Fig. S13–S15†) were also synthesized as controls for comparison. Only a 17- and 7-fold increase in stability for 6–6 and 6–Bme–6 respectively was observed relative to the peptide monomer (against cleavage by chymotrypsin), which is significantly less pronounced than its NDI–hybrid dimer or trimer (Fig. S42 and S43† and Table 1). In contrast, 6–Bph–6 is significantly more tolerant to enzymatic degradation than both 6–6 and 6–Bme–6, but it is still less stable than 6–NDI–6 (Fig. S44† and Table 1). These results have a strong implication of the important role of the aromaticity of the crosslinker in the enhanced proteolytic stability of peptides. In addition, the bulky and electron-deficient aromatic NDI provides better protection to peptides against proteolysis than the smaller Bph linker.
To further elucidate the effect of NDI conjugation on the enhancement of peptide stability, the location of proteolytic cleavage was analyzed by HPLC and mass spectrometry (MS). Theoretically, peptide 6 possesses three cleavage sites at the carboxyl sides of aromatic residues F, Y, and W when against chymotrypsin. However, analysis of digestion fragments indicates an additional cleavage site at the carboxyl side of L. 6 and 6–6 can be digested at every possible cleavage site by chymotrypsin (Fig. 3, S45, and S46†). In contrast, the cleavage sites of 6–NDI are located only at the carboxyl sides of Y and L, suggesting that the other two cleavage sites are protected against attacks by proteolysis (Fig. S47†). 6–NDI–6 which exhibits the highest stability against proteolysis can only be digested by the cleavage of the carboxyl side of F, and other cleavage sites are strictly tolerant to the proteolysis. Moreover, even the partially digested fragments of 6–NDI–6 exhibit a high stability in the digestion buffer (Fig. S48†). These results have unambiguously demonstrated that the proteolytic stability of peptides can indeed be significantly enhanced by covalent conjugation with aromatic NDI molecules, though the extent of enhancement in stability depends strongly on the amino acid sequence, the position of NDI conjugation, and the manner of hybridization (e.g., trimeric or dimeric form).
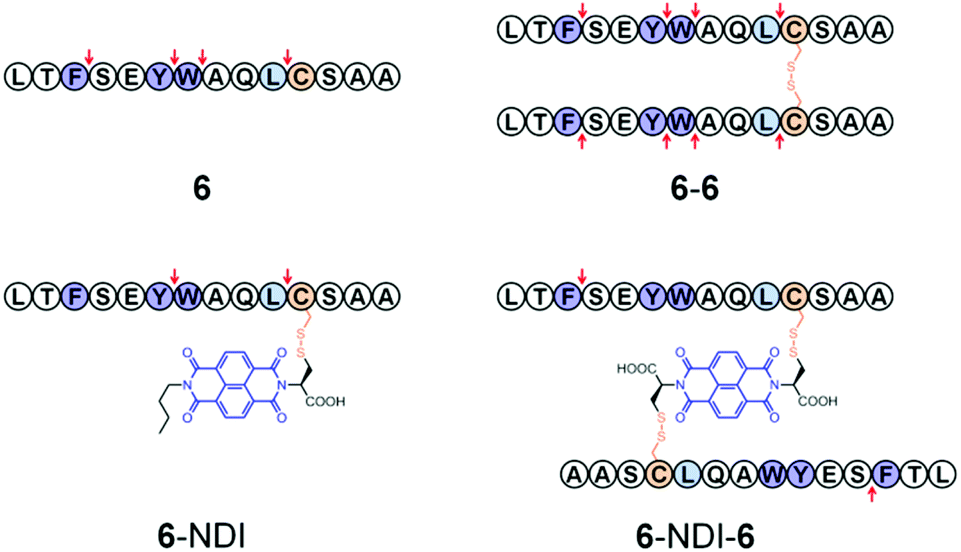 | ||
| Fig. 3 Diagram indicating the chymotrypsin cleavage sites (↓) within peptides. | ||
NDI is an electron-deficient aromatic compound,29 which displays a strong propensity of stacking with electron-rich aromatic residues (e.g., F, Y and W) through π–π interactions.10,30 Thus, the nonspecific π–π interactions between the NDI moiety and the three aromatic residues (one or several of them) in peptides 5–8 very likely contribute to the enhanced proteolytic stability. Noncovalent π–π stacking interactions are solvent-sensitive, which are thus difficult to be probed by NMR techniques, because an organic cosolvent has to be used to solubilize the peptide–NDI hybrids for NMR measurements. Accordingly, to further understand how the interactions between NDI and the peptide chain affect the proteolytic (or structural) stability of peptides, Hamiltonian replica exchange molecular dynamics (HREMD) simulations25,28 were carried out to examine the conformational spaces of two hybrid dimers (i.e., 5–NDI and 6–NDI) and one hybrid trimer (i.e., 6–NDI–6). All hybrid peptides displayed enhanced propensities of α-helical conformations with respect to their parent peptides, as shown in the evolution of secondary structures of the peptides (Fig. S49†), consistent with the results of circular dichroism (CD) spectral characterization (Fig. S51 and S52†). The last 100 ns of each trajectory were then clustered based on root mean square deviation (RMSD) and the representative conformations of the hybrid peptides are shown in Fig. 4. For the hybrid dimers 5–NDI and 6–NDI, the representative conformations revealed that the NDI moiety stacks with one or more aromatic residues, forming an aromatic cluster. For the hybrid peptide trimer 6–NDI–6, the NDI moiety was found to stack with two aromatic residues (i.e., Trp), forming a sandwich structure (Fig. 4c) and resulting in a remarkable enhancement of α-helicity. This stacking interaction is also supported by the observation of extremely drastic quenching of fluorescence of the Trp in 6–NDI–6 (Fig. S53†). As the preferable cleavage site for protease contains aromatic residues,11 the stacking of the NDI moiety with aromatic residues provides steric hindrance to the hydrolysis reaction between the peptides and proteases, which would reduce essentially the accessibility or susceptibility of the cleavage sites of the peptides to the proteases. Moreover, the aromatic cluster was further stabilized by the hydrogen bonds between the NDI moiety and the polar groups of the peptide, which results in more compact conformations and reduced flexibility of the hybrid peptides, as evidenced by the smaller values and narrower distributions of the radius of gyration of the hybrid peptides (Fig. S50†). The parent peptides without the NDI moiety are more flexible than the hybrid peptides, leaving their backbone readily susceptible to proteases. These results clearly confirm the important role of the π–π interactions between NDI and aromatic residues in the relatively rigid structures of the hybrids (relative to the flexible free peptides), which in turn leads to enhanced proteolytic resistance.
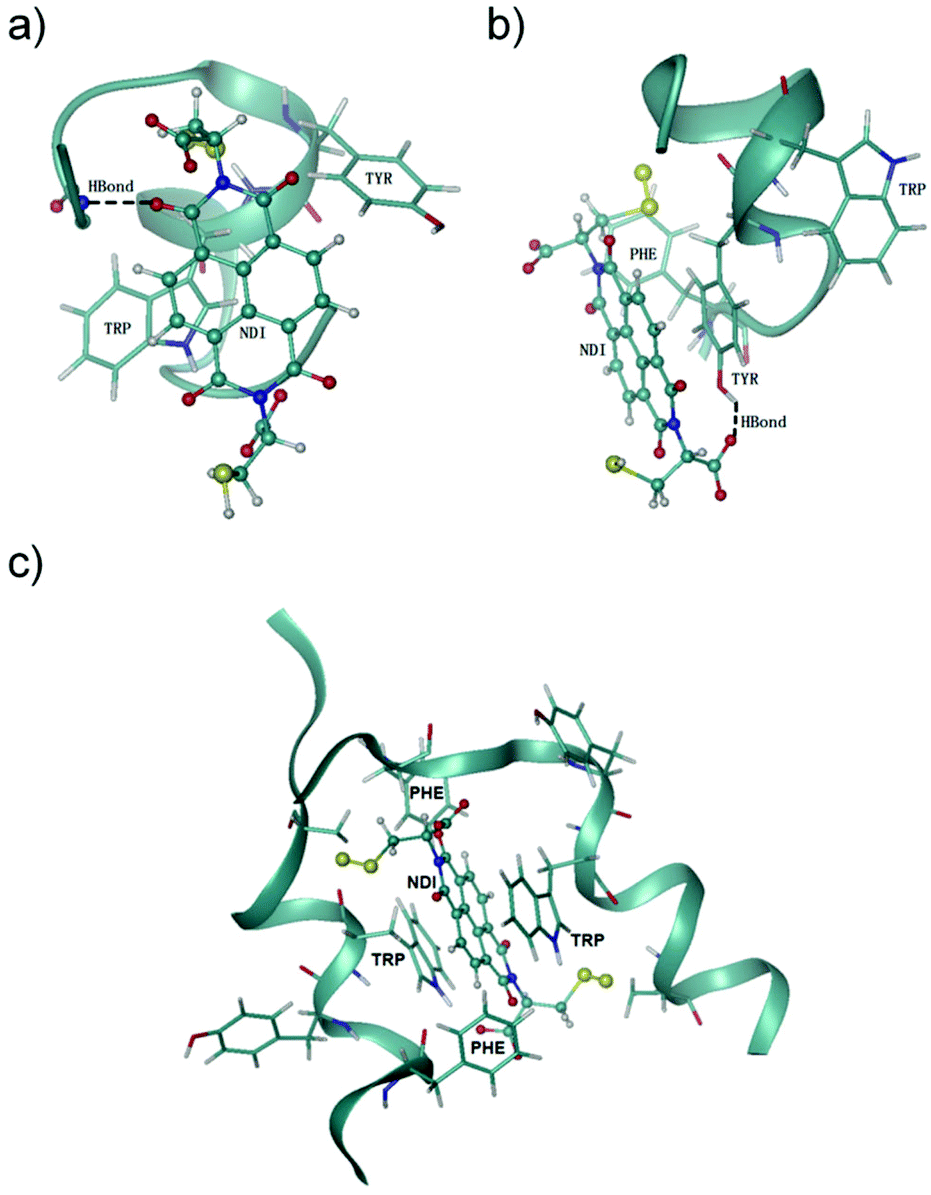 | ||
| Fig. 4 Representative conformations of NDI-peptide conjugates; (a) 5–NDI, (b) 6–NDI, and (c) 6–NDI–6. NDI is represented as a ball and stick while aromatic residues are represented as sticks; dashed black lines denote hydrogen bonds. | ||
Disulfide bonds are redox-responsive,20,31,32 which allows for reversible modulation of the conformation and structural/proteolytic stability of the disulfide-linked peptide–NDI hybrids according to the local redox-environments they reside in. This dynamic feature is important for the practical application of peptide–NDI hybrids, especially considering that the conjugation of a bulky NDI moiety to peptides might disturb their bioactive binding to target proteins. Thus, the hybrid trimer or dimer platforms presented here can be exploited as prodrug formulations that would be stable and highly resistant to proteolysis in oxidizing environments (e.g., in blood circulation, the proximal digestive tract, and the endocytic compartments), but can ultimately release the active monomeric peptides under highly reducing conditions (e.g., in the cytosol) (Fig. 5a).11,15,19,32 As expected, the representative trimer 6–NDI–6 and dimer 5–NDI are both reducible under reducing conditions mimicking the cytosols (Fig. S54 and S55†). The released monomeric peptides (5 and 6) both display relative high affinities to MDM2 (Ki: 24.8 ± 2.0 and 28.1 ± 6.8 nM, respectively) (Fig. 5b), which are comparable to the binding affinities of the parent peptides reported in the literature,17,18 indicating that the introduction of a cysteine residue into the peptide does not obviously reduce its protein-binding activity.
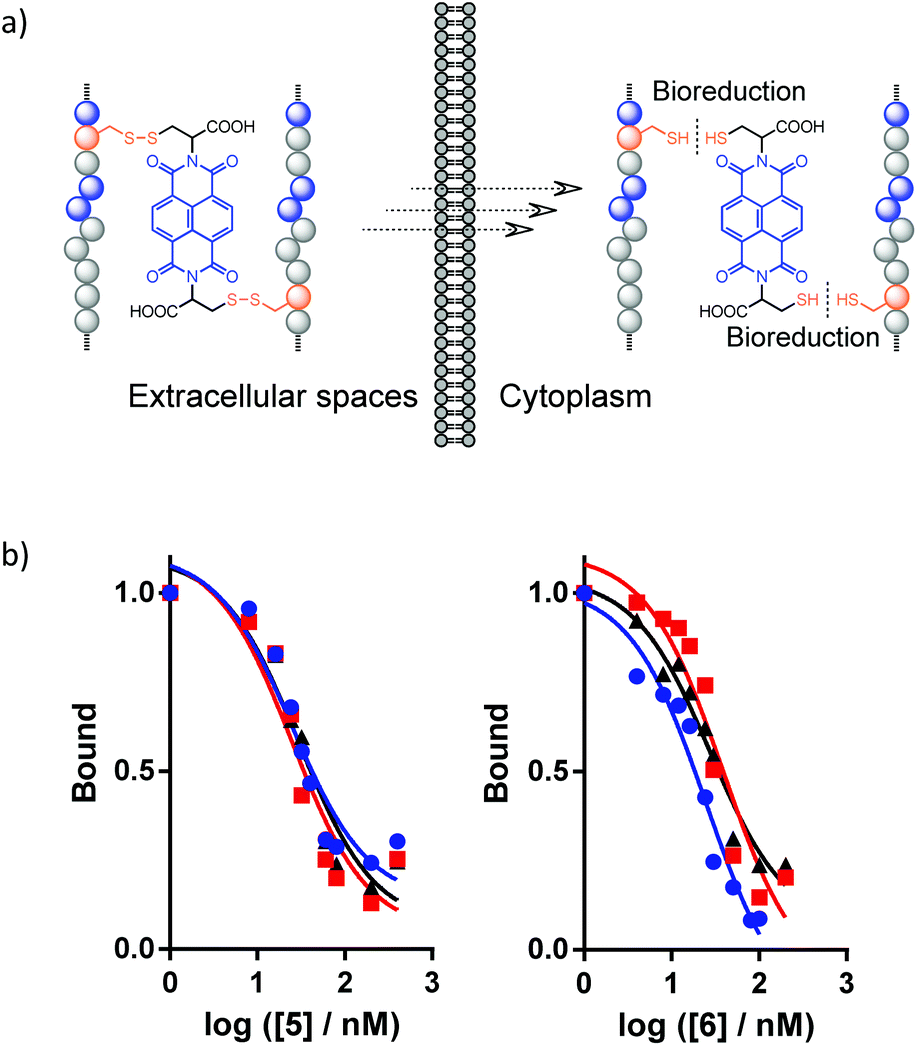 | ||
| Fig. 5 (a) Diagram illustrating the cellular uptake of peptide–NDI hybrids and their bioreduction in the cytoplasm. (b) Fluorescence polarization competition assays determine the binding interaction of 5 (left) and 6 (right) with MDM2; curves were obtained from three independent measurements. | ||
The above promising results suggest that peptide–NDI hybrids (e.g., 6–NDI–6) could be potential candidates for modulating intracellular targets or interactomes. Fluorophore-labeled 6–NDI–6 was then prepared to investigate first whether the hybrid trimer can enter into cells efficiently. MCF7 breast cancer cells, a cell line expressing wild-type p53, were used for the study.18 As shown in Fig. 6a, we observed strong intracellular red fluorescence from the rhodamine-labeled 6 after 24 h incubation. The cellular uptake of 6–NDI–6 was evaluated further by flow cytometry and compared with the free peptide 6 and the dimer 6–NDI (Fig. 6b). The uptake of 6–NDI–6 by MCF7 cells was as efficient as that of 6 and 6–NDI, though the hybrid trimer had nearly doubled in size compared to the monomer or dimer. We surmised that the stabilized secondary structure and enhanced hydrophobicity provided by the NDI moiety would, at least to some extent, facilitate the cellular uptake of the hybrid trimer. In addition, it was demonstrated that 6–NDI–6 was indeed completely reduced within the cells due to the highly reducing conditions of the intracellular environments, because no fluorescence recovery was observed when treating the cells (pre-incubated with 6–NDI–6) with a strong and cell-penetrating reducing agent dithiothreitol (Fig. S56;† note: the fluorescence of rhodamine labeled on the peptide can be significantly quenched by the central NDI). The bioactivity of 6–NDI–6 towards MCF7 cells was then assessed using MTT assays, as the inhibition of MDM2 and/or MDMX by 6 would lead to p53 activation and subsequent cell apoptosis.18,33 Upon incubation of 6–NDI–6 at concentrations from 0.01 to 20 μM with cells for 24 h, we observed concentration-dependent apoptosis (Fig. 6c). To confirm that the bioactivity of 6–NDI–6 is closely associated with p53 activation, U251 human glioma cells, a p53 mutant cancer cell line, were used for comparison. The U251 cells were largely resistant to the treatment of 6–NDI–6 (Fig. 6c). Besides, a deactivated random peptide (9: LWTSYAEQAFLCSA), derived from 6, was designed, and its NDI–hybrid trimer (9–NDI–9) displays a negligible bioactivity towards MCF7 cells (Fig. 6c). Based on these results, it is expected that the released and bioactive 6 (from the bioreduction of 6–NDI–6), through its modulation of the MDM2/MDMX-p53 interactions, should take responsibility for the apoptosis, though the detailed mechanism for apoptosis remains to be explored. The monomer 6 displays a weaker cell-killing capability at equivalent concentrations compared to 6–NDI–6 and 6–NDI (Fig. 6c; though their cellular uptake efficiencies are comparable), suggesting likely that the aromatic NDI would protect the disulfide-linked peptides from proteolysis during cellular uptake (i.e., before the bioreduction in the cytoplasm). In addition, we found that 6–NDI–6 exhibits moderate bioactivity even under more stringent physiological conditions (i.e., in the presence of 10% serum) (Fig. S57†).
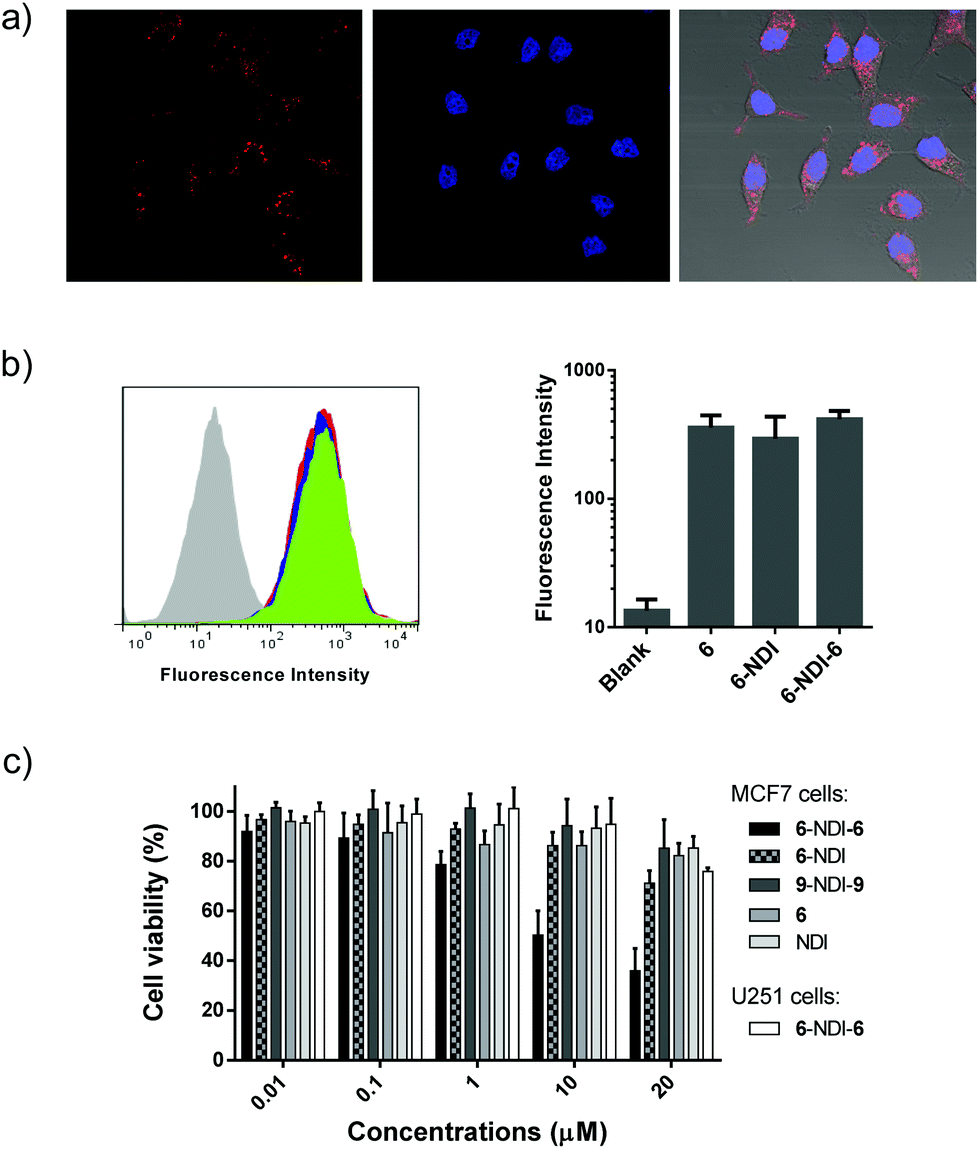 | ||
| Fig. 6 (a) Representative confocal fluorescence images of MCF7 cells exposed to 1.0 μM of 6–NDI–6; DAPI (4′,6-diamidino-2-phenylindole) was used to track the cell nuclei; from left to right: rhodamine channels for peptides, DAPI channels for nuclear staining, and an overlay of bright field and fluorescence images. (b) Flow cytometric analysis of MCF7 cells incubated with 1.0 μM of 6–NDI–6 (green), 6–NDI (blue), and 6 (red), respectively. Mean fluorescence intensity per cell was shown in the right panel (mean ± s.d., n = 3), indicating a comparable cellular uptake efficiency for the three peptides/hybrids. (c) Cell viability determined using MTT assays (MCF7 and U251 cells). The cells were incubated with various concentrations of 6–NDI–6 (or 6, NDI, 6–NDI and 9–NDI–9) in serum-free medium for 24 h. The results are expressed as mean ± s.d. (n = 3). | ||
Conclusions
In summary, this study developed a general, convenient, and efficient strategy for the stabilization of peptides against proteolysis, which involves noncovalent π–π interactions between aromatic amino acid residues in peptides and synthetic electron-deficient aromatics, as well as the implication of steric hindrance and the enhancement of peptide α-helicity. This strategy is complementary in concept to the conventional well-established covalent approaches for peptide stabilization, which is thus promising for being utilized in combination with the latter ones in the future to efficiently solve the problem of proteolytic instability of peptides. A dynamic disulfide bond is considered as an ideal linkage to link (covalently and reversibly) the bioactive peptides and the synthetic aromatics, because it allows the proteolytic stability and bioactivity of the peptides to be manipulated in demand based on the local redox microenvironments. For example, disulfide bonds are usually stable under oxidizing conditions such as the blood circulation, extracellular spaces, and endocytic organelles; therefore, the hybrids can retain their integrity before transporting into their destination where the environments might be highly reducing (e.g., the cytoplasm) and the bioactive peptides can be released from the hybrid entities (i.e., like the activation of prodrugs). In this work, the proposed concept has been well characterized using an electron-deficient NDI and model peptides derived from the inhibitors of MDM2. Peptide–NDI hybrids display exceptionally high resistance to proteolysis, and extensive manipulation of the primary sequence is not required, except that aromatic amino acid residues have to be present. We envisioned that this study should provide invaluable guidelines to the design and synthesis of organic molecule–peptide hybrids with significantly improved proteolytic resistance, and benefit the development of peptide therapeutics and probes.Acknowledgements
We would like to acknowledge the financial support of the National Basic Research Program of China (Grant 2014CB932004), the financial support of the National Natural Science Foundation of China (grants 21375110 and 21475109) and the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (Grant 21521004). We also thank National Super Computing Center for providing computational resources.Notes and references
- P. Vlieghe, V. Lisowski, J. Martinez and M. Khrestchatisky, Drug Discovery Today, 2010, 15, 40–56 CrossRef CAS PubMed.
- V. M. Ahrens, K. Bellmann-Sickert and A. G. Beck-Sickinger, Future Med. Chem., 2012, 4, 1567–1586 CrossRef CAS PubMed; L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef PubMed; C. T. T. Wong, D. K. Rowlands, C. H. Wong, T. W. C. Lo, G. K. T. Nguyen, H. Y. Li and J. P. Tam, Angew. Chem., Int. Ed., 2012, 51, 5620–5624 CrossRef PubMed; R. J. Clark, J. Jensen, S. T. Nevin, B. P. Callaghan, D. J. Adams and D. J. Craik, Angew. Chem., Int. Ed., 2010, 49, 6545–6548 CrossRef PubMed; C. Li, M. Pazgier, M. Liu, W. Y. Lu and W. Y. Lu, Angew. Chem., Int. Ed., 2009, 48, 8712–8715 CrossRef PubMed; K. B. Akondi, M. Muttenthaler, S. Dutertre, Q. Kaas, D. J. Craik, R. J. Lewis and P. F. Alewood, Chem. Rev., 2014, 114, 5815–5847 CrossRef PubMed; M. Góngora-Benítez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2014, 114, 901–926 CrossRef PubMed.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- N. Nischan, A. Chakrabarti, R. A. Serwa, P. H. M. Bovee-Geurts, R. Brock and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2013, 52, 11920–11924 CrossRef CAS PubMed; I. R. Ruttekolk, J. J. Witsenburg, H. Glauner, P. H. M. Bovee-Geurts, E. S. Ferro, W. P. R. Verdurmen and R. Brock, Mol. Pharmaceutics, 2012, 9, 1077–1086 Search PubMed; E. Reits, A. Griekspoor, J. Neijssen, T. Groothuis, K. Jalink, P. van Veelen, H. Janssen, J. Calafat, J. W. Drijfhout and J. Neefjes, Immunity, 2003, 18, 97–108 CrossRef PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed; A. Patgiri, A. L. Jochim and P. S. Arora, Acc. Chem. Res., 2008, 41, 1289–1300 CrossRef PubMed; T. Sawada and S. H. Gellman, J. Am. Chem. Soc., 2011, 133, 7336–7339 CrossRef PubMed; S. Y. Chen, D. Bertoldo, A. Angelini, F. Pojer and C. Heinis, Angew. Chem., Int. Ed., 2014, 53, 1602–1606 CrossRef PubMed; A. P. Blum, J. K. Kammeyer, J. Yin, D. T. Crystal, A. M. Rush, M. K. Gilson and N. C. Gianneschi, J. Am. Chem. Soc., 2014, 136, 15422–15437 CrossRef PubMed; E. Hamed, T. Xu and S. Keten, Biomacromolecules, 2013, 14, 4053–4060 CrossRef PubMed; R. S. Harrison, N. E. Shepherd, H. N. Hoang, G. Ruiz-Gomez, T. A. Hill, R. W. Driver, V. S. Desai, P. R. Young, G. Abbenante and D. P. Fairlie, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11686–11691 CrossRef PubMed; L. Mendive-Tapia, S. Preciado, J. Garcia, R. Ramon, N. Kielland, F. Albericio and R. Lavilla, Nat. Commun., 2015, 6 Search PubMed; Y. H. Lau, P. de Andrade, S.-T. Quah, M. Rossmann, L. Laraia, N. Sköld, T. J. Sum, P. J. E. Rowling, T. L. Joseph, C. Verma, M. Hyvönen, L. S. Itzhaki, A. R. Venkitaraman, C. J. Brown, D. P. Lane and D. R. Spring, Chem. Sci., 2014, 5, 1804–1809 RSC.
- A. D. de Araujo, H. N. Hoang, W. M. Kok, F. Diness, P. Gupta, T. A. Hill, R. W. Driver, D. A. Price, S. Liras and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 6965–6969 CrossRef CAS PubMed.
- J. W. Checco, E. F. Lee, M. Evangelista, N. J. Sleebs, K. Rogers, A. Pettikiriarachchi, N. J. Kershaw, G. A. Eddinger, D. G. Belair, J. L. Wilson, C. H. Eller, R. T. Raines, W. L. Murphy, B. J. Smith, S. H. Gellman and W. D. Fairlie, J. Am. Chem. Soc., 2015, 137, 11365–11375 CrossRef CAS PubMed.
- A. M. Spokoyny, Y. K. Zou, J. J. Ling, H. T. Yu, Y. S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed; H. Jo, N. Meinhardt, Y. B. Wu, S. Kulkarni, X. Z. Hu, K. E. Low, P. L. Davies, W. F. DeGrado and D. C. Greenbaum, J. Am. Chem. Soc., 2012, 134, 17704–17713 CrossRef PubMed; C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef; S. P. Brown and A. B. Smith III, J. Am. Chem. Soc., 2015, 137, 4034–4037 CrossRef PubMed; C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef PubMed; A. Muppidi, Z. Wang, X. Li, J. Chen and Q. Lin, Chem. Commun., 2011, 47, 9396–9398 RSC; S. Sim, Y. Kim, T. Kim, S. Lim and M. Lee, J. Am. Chem. Soc., 2012, 134, 20270–20272 CrossRef PubMed; N. Assem, D. J. Ferreira, D. W. Wolan and P. E. Dawson, Angew. Chem., Int. Ed., 2015, 54, 8665–8668 CrossRef PubMed; S. Samanta, C. Qin, A. J. Lough and G. A. Woolley, Angew. Chem., Int. Ed., 2012, 51, 6452–6455 CrossRef PubMed; A. Glas, D. Bier, G. Hahne, C. Rademacher, C. Ottmann and T. N. Grossmann, Angew. Chem., Int. Ed., 2014, 53, 2489–2493 CrossRef PubMed.
- A. C. Conibear, K. J. Rosengren, N. L. Daly, S. T. Henriques and D. J. Craik, J. Biol. Chem., 2013, 288, 10830–10840 CrossRef CAS PubMed; M. L. Colgrave and D. J. Craik, Biochemistry, 2004, 43, 5965–5975 CrossRef PubMed; A. J. Wommack, J. J. Ziarek, J. Tomaras, H. R. Chileveru, Y. F. Zhang, G. Wagner and E. M. Nolan, J. Am. Chem. Soc., 2014, 136, 13494–13497 CrossRef PubMed; C. K. L. Wang, R. J. Clark, P. J. Harvey, K. J. Rosengren, M. Cemazar and D. J. Craik, Biochemistry, 2011, 50, 4077–4086 CrossRef PubMed.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- Y. Q. Chen, C. Q. Yang, T. Li, M. Zhang, Y. Liu, M. A. Gauthier, Y. B. Zhao and C. L. Wu, Biomacromolecules, 2015, 16, 2347–2355 CrossRef CAS PubMed.
- S. M. Butterfield, P. R. Patel and M. L. Waters, J. Am. Chem. Soc., 2002, 124, 9751–9755 CrossRef CAS PubMed.
- H. Zheng and J. Gao, Angew. Chem., Int. Ed., 2010, 49, 8635–8639 CrossRef CAS PubMed.
- M. G. Woll, E. B. Hadley, S. Mecozzi and S. H. Gellman, J. Am. Chem. Soc., 2006, 128, 15932–15933 CrossRef CAS PubMed.
- L. X. Zhai, J. J. Liang, X. Q. Guo, Y. B. Zhao and C. L. Wu, Chem. – Eur. J., 2014, 20, 17507–17514 CrossRef CAS PubMed.
- M. S. Cubberley and B. L. Iverson, J. Am. Chem. Soc., 2001, 123, 7560–7563 CrossRef CAS PubMed; H. Y. Au-Yeung, G. D. Pantos and J. K. M. Sanders, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10466–10470 CrossRef PubMed.
- M. Pazgiera, M. Liu, G. Z. Zou, W. R. Yuan, C. Q. Li, C. Li, J. Li, J. Monbo, D. Zella, S. G. Tarasov and W. Lu, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4665–4670 CrossRef PubMed.
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K. H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. L. Cai, P. Berry, E. Feyfant, X. G. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3445–E3454 CrossRef CAS PubMed.
- S. Hyun, S. Lee, S. Kim, S. Jang, J. Yu and Y. Lee, Biomacromolecules, 2014, 15, 3746–3752 CrossRef CAS PubMed; S. Jang, S. Hyun, S. Kim, S. Lee, I. S. Lee, M. Baba, Y. Lee and J. Yu, Angew. Chem., Int. Ed., 2014, 53, 10086–10089 CrossRef PubMed.
- C. L. Wu, S. Wang, L. Brulisauer, J. C. Leroux and M. A. Gauthier, Biomacromolecules, 2013, 14, 2383–2388 CrossRef CAS PubMed; G. Gasparini, E. K. Bang, G. Molinard, D. V. Tulumello, S. Ward, S. O. Kelley, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069–6074 CrossRef PubMed.
- L. Brulisauer, N. Kathriner, M. Prenrecaj, M. A. Gauthier and J. C. Leroux, Angew. Chem., Int. Ed., 2012, 51, 12454–12458 CrossRef PubMed.
- K. Tambara, N. Ponnuswamy, G. Hennrich and G. D. Pantos, J. Org. Chem., 2011, 76, 3338–3347 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- P. Liu, B. Kim, R. A. Friesner and B. J. Berne, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13749–13754 CrossRef CAS PubMed.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- G. Bussi, Mol. Phys., 2014, 112, 379–384 CrossRef CAS.
- F. B. L. Cougnon, N. Ponnuswamy, N. A. Jenkins, G. D. Pantos and J. K. M. Sanders, J. Am. Chem. Soc., 2012, 134, 19129–19135 CrossRef CAS PubMed.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed; F. H. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180–2198 CrossRef PubMed; Y. N. Zhong, W. J. Yang, H. L. Sun, R. Cheng, F. H. Meng, C. Deng and Z. Y. Zhong, Biomacromolecules, 2013, 14, 3723–3730 CrossRef PubMed.
- C. L. Wu, C. Belenda, J. C. Leroux and M. A. Gauthier, Chem. – Eur. J., 2011, 17, 10064–10070 CrossRef CAS PubMed.
- F. Bernal, M. Wade, M. Godes, T. N. Davis, D. G. Whitehead, A. L. Kung, G. M. Wahl and L. D. Walensky, Cancer Cell, 2010, 18, 411–422 CrossRef CAS PubMed; J. A. Kritzer, R. Zutshi, M. Cheah, F. A. Ran, R. Webman, T. M. Wongjirad and A. Schepartz, ChemBioChem, 2006, 7, 29–31 CrossRef PubMed; C. J. Brown, S. T. Quah, J. Jong, A. M. Goh, P. C. Chiam, K. H. Khoo, M. L. Choong, M. A. Lee, L. Yurlova, K. Zolghadr, T. L. Joseph, C. S. Verma and D. P. Lane, ACS Chem. Biol., 2013, 8, 506–512 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S57; characterization of NDI–peptide conjugates; enzymatic digestion kinetic analysis; HPLC and MS analysis of cleavage sites; data on HREMD simulation. See DOI: 10.1039/c6ob02786e |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |