
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic esterification under Mitsunobu reaction conditions mediated by flavin and visible light†
M.
März
a,
J.
Chudoba
b,
M.
Kohout
a and
R.
Cibulka
 *a
*a
aDepartment of Organic Chemistry, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague, Czech Republic. E-mail: cibulkar@vscht.cz
bCentral Laboratories, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague, Czech Republic
First published on 3rd February 2017
Abstract
The usefulness of flavin-based aerial photooxidation in esterification under Mitsunobu reaction conditions was demonstrated, providing aerial dialkyl azodicarboxylate recycling/generation from the corresponding dialkyl hydrazine dicarboxylate. Simultaneously, activation of triphenylphosphine (Ph3P) by photoinduced electron transfer from flavin allows azo-reagent-free esterification. An optimized system with 3-methylriboflavin tetraacetate (10%), oxygen (terminal oxidant), visible light (450 nm), Ph3P, and dialkyl hydrazine dicarboxylate (10%) has been shown to provide efficient and stereoselective coupling of various alcohols and acids to esters with retention of configuration.
A systems of triphenylphosphine (Ph3P) or a related phosphine activated by an oxidant allow efficient esterification under mild conditions.1 In the Mitsunobu reaction,2,3 PPh3 is oxidized by a dialkyl azodicarboxylate (usually DEAD [1a] or DIAD [1b]) to form a betain species, which facilitates transformation of an alcohol to a reactive alkoxyphosphonium intermediate (Scheme 1). The latter undergoes SN2 substitution with carboxylate giving an ester with inversion of configuration. There are a few cases in which an acyloxyphosphonium intermediate predominates under Mitsunobu reaction conditions, subsequent SNAc substitution of which gives the product with retention of configuration.4–6
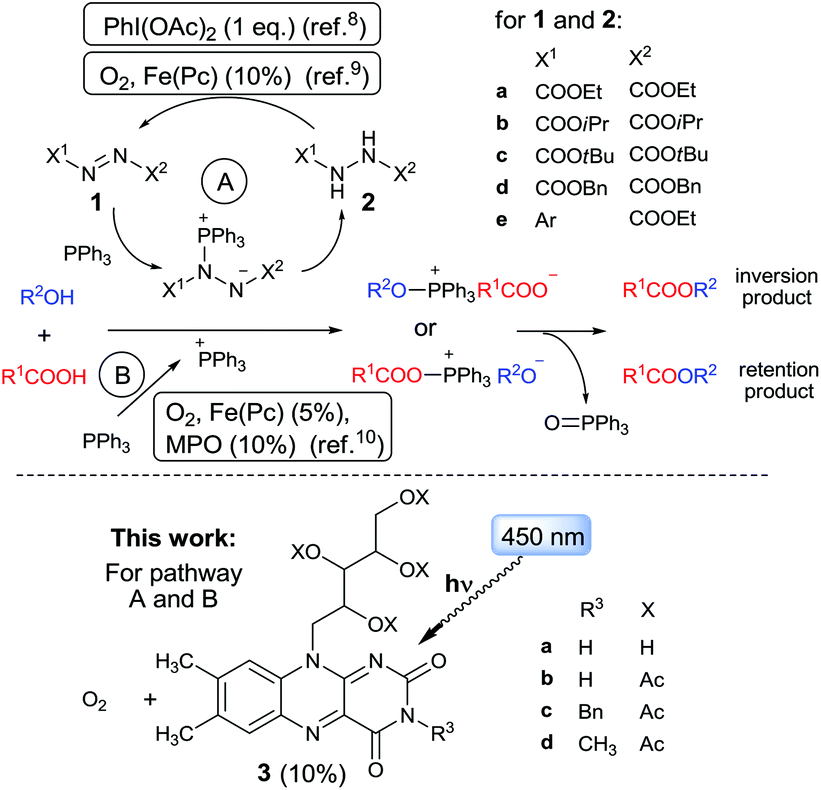 | ||
| Scheme 1 Catalytic esterification mediated by activated Ph3P under Mitsunobu (A) and azo-reagent-free (B) conditions. | ||
The Mitsunobu reaction has become an extremely useful tool in organic synthesis.3b,7 However, its further expansion, in particular towards large-scale applications, is limited by the need for a stoichiometric amount of oxidant, azodicarboxylate 1, which is toxic and unstable and the use of which generates dialkyl hydrazine dicarboxylate 2 as a waste by-product. An attempt to solve this problem led to recent pioneering studies on catalytic Mitsunobu reaction.1b Toy and co-workers applied a stoichiometric amount of the oxidant PhI(OAc)2 to convert the generated hydrazine 2a back to azo compound 1a, allowing only 10 mol% 1a to be used.8 Taniguchi, et al. developed and optimized catalytic Mitsunobu reaction using ethyl N-aryl-azocarboxylates 1e (10 mol%) instead of 1a or 1b, in conjunction with a catalytic amount (10 mol%) of Fe(II)-phthalocyanine (Fe[Pc]), which re-oxidized arylhydrazine, being simultaneously re-generated by air oxygen.9 Notably, Fe[Pc] was not able to recycle original Mitsunobu reagents, DEAD or DIAD, due to high oxidation potential of corresponding hydrazines 2. Another approach is to use a procedure free from azo reagent 1 in which PPh3 is activated by aerial oxidation catalyzed by Fe[Pc]. This esterification occurs via an acylphosphonium intermediate giving an ester with retention of configuration.10 Another problem of the Mitsunobu type reactions is bulk production of phosphine oxide. O'Brien11 and later Aldrich and Buonomo12 showed that phosphine oxide can be reduced in situ with phenylsilane, thus allowing use of the phosphine reagent in a catalytic amount.13
We surmise that, due to their redox character, Mitsunobu-type reactions represent a typical challenge for photoredox catalysis,14 which have undergone rapid development in recent years. Upon excitation, molecules become stronger oxidizing agents compared to their ground-state forms, which considerably extends the possibilities for regeneration of Mitsunobu reagents or alternative activation of phosphines. However, to the best of our knowledge, no application of photoredox catalysis in catalytic Mitsunobu reactions or phosphine-mediated esterifications has hitherto been reported. Herein, we present a photocatalytic system (Scheme 1) based on flavin 3d (a derivative of vitamin B2 [3a]), oxygen, and visible light, which is able to regenerate commercial dialkyl azodicarboxylates 1. The system was found to simultaneously activate Ph3P by photoinduced electron-transfer, thus providing azo-reagent-free esterification.
Our original idea to regenerate azodicarboxylates 1 from the corresponding hydrazides 2 led us to flavin photocatalysts.15,16 Upon excitation with blue light (450 nm), flavins become oxidizing agents17 ( for riboflavin tetraacetate (3b)), strong enough to oxidize 2 (a value of
for riboflavin tetraacetate (3b)), strong enough to oxidize 2 (a value of  has been reported for BocNHNHBoc18), but not so strong as to mediate undesired oxidation of alcohols, even when they are activated, e.g. benzyl alcohols, which are often substrates of Mitsunobu reactions. The only exceptions are electron-rich benzyl alcohols such as 4-methoxybenzyl alcohol (
has been reported for BocNHNHBoc18), but not so strong as to mediate undesired oxidation of alcohols, even when they are activated, e.g. benzyl alcohols, which are often substrates of Mitsunobu reactions. The only exceptions are electron-rich benzyl alcohols such as 4-methoxybenzyl alcohol (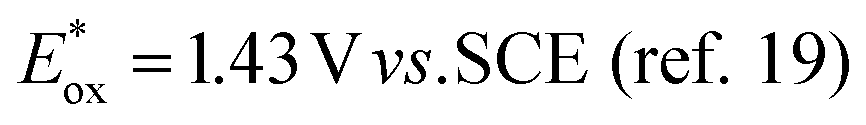 ), which is a traditional substrate for testing flavins in photooxidations.16e,17,20 Importantly, flavins can be converted back to their oxidized forms by oxygen, and can thus be applied in catalytic amounts (cf.Scheme 2).15
), which is a traditional substrate for testing flavins in photooxidations.16e,17,20 Importantly, flavins can be converted back to their oxidized forms by oxygen, and can thus be applied in catalytic amounts (cf.Scheme 2).15
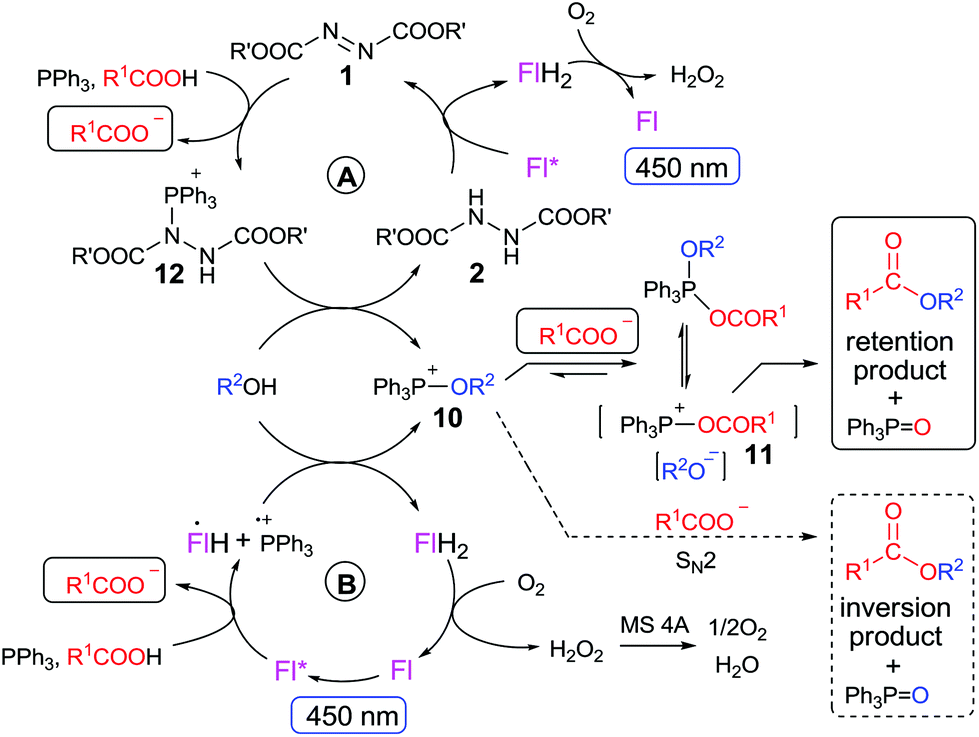 | ||
| Scheme 2 Proposed mechanism of flavin (Fl)-mediated photocatalytic esterification with (A) and without (B) contribution from azodicarboxylate 1. | ||
Preliminary experiments confirmed that 3b oxidizes dialkyl hydrazine-dicarboxylates 2a–d to the corresponding azo compounds 1 when irradiated by light of wavelength 450 nm (see ESI†). The oxidation proceeded remarkably in acetonitrile, and thus we used this solvent in model photocatalytic esterification with 3-nitrobenzoic acid 4a and benzyl alcohol 5a using 10 mol% of 3b, 10 mol% of 1b, and 2 equivalents of PPh3 (Table 1). The reaction was performed at 25 °C under oxygen and irradiation with a blue diode (450 nm), in the presence of molecular sieves (4 Å) to remove hydrogen peroxide formed during flavin regeneration.9c To our delight, we observed formation of ester 6a in moderate conversion after 24 h (entry 1), and a positive result was found even when 2b (10 mol%) was used instead of 1b (entry 2). Only a small amount of aldehyde was formed with 3b, confirming the suitability of flavins in this reaction. Use of stronger oxidants, such as 9-mesityl-10-methylacridinium perchlorate (7; 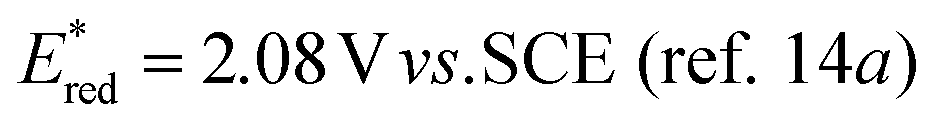 ) or triphenylpyrylium tetrafluoroborate (8;
) or triphenylpyrylium tetrafluoroborate (8; 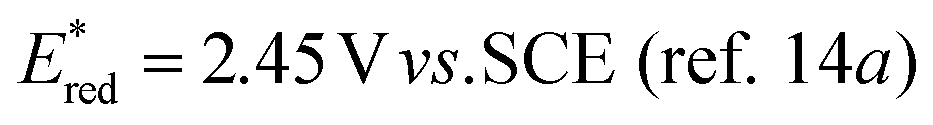 ) preferentially led to benzylic oxidation (entries 3 and 4). Notably, the N–H bond in flavins is sufficiently acidic for 3b to be a substrate of Mitsunobu reaction. We observed formation of N-benzyl derivative 3c, which was still active in esterification, but less than 3b (cf. entries 2 and 5). Thus, N-methylflavin 3d seemed to be a good choice of photocatalyst (entries 6 and 7).
) preferentially led to benzylic oxidation (entries 3 and 4). Notably, the N–H bond in flavins is sufficiently acidic for 3b to be a substrate of Mitsunobu reaction. We observed formation of N-benzyl derivative 3c, which was still active in esterification, but less than 3b (cf. entries 2 and 5). Thus, N-methylflavin 3d seemed to be a good choice of photocatalyst (entries 6 and 7).
|
|
||||
|---|---|---|---|---|
| Entry | Catalytic system | Temp [°C] | Conversion after 24 h [%] | |
| Ester | Aldehyde | |||
| a PPh3 added in three portions at the time 0, 4 and 8 hours. b PhSiH3 (2 equiv.) added. | ||||
| 1 | 3b/1b | 25 | 58 | 2 |
| 2 | 3b/2b | 25 | 48 | 2 |
| 3 | 7/2b | 25 | 46 | 31 |
| 4 | 8/2b | 25 | 8 | 20 |
| 5 | 3c/2b | 25 | 41 | 7 |
| 6 | 3d/1b | 25 | 64 | 2 |
| 7 | 3d/2b | 25 | 60 | 2 |
| 8a | 3d/2b | 25 | 79 | Trace |
| 9b | 3d/2b | 25 | 85 | Trace |
| 10 | 3d/2b | 50 | 66 | Trace |
| 11b | 3d/2b | 50 | 92 | Trace |
| 12 | 3d/— | 25 | 17 | 2 |
| 13b | 3d/— | 25 | 26 | — |
| 14 | 3d/— | 50 | 47 | 2 |
| 15b | 3d/— | 50 | 69 | — |
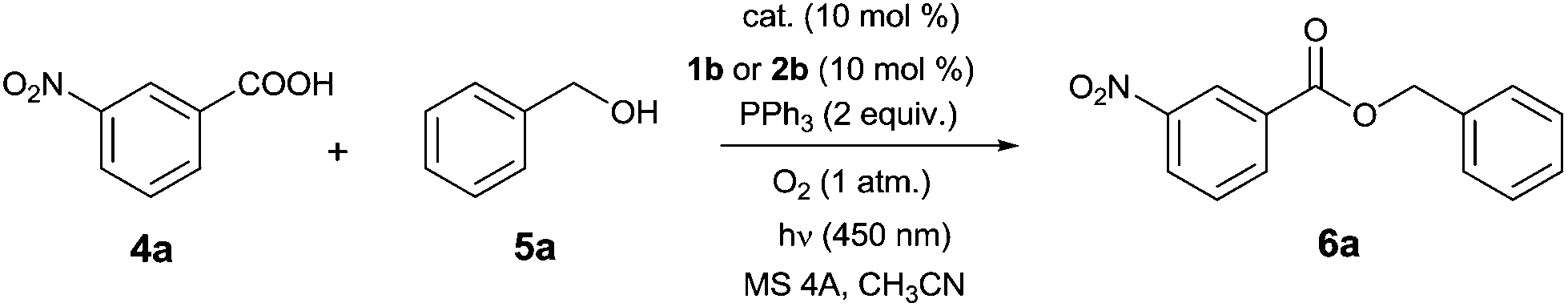
Monitoring the reaction course showed undesired direct oxidation of Ph3P to Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, which retarded ester formation. We assume that excited flavin participates in this oxidation by electron transfer from Ph3P (Eox = 1.06 vs. SCE21) (for quenching experiment, see ESI†). The formed Ph3P˙+ reacts with oxygen to form phosphine oxide, which is known to occur by several mechanisms.22 This side reaction can be suppressed by subsequent addition of PPh3, which increases the yield significantly (entry 8). A similar effect was achieved by adding PhSiH3
O, which retarded ester formation. We assume that excited flavin participates in this oxidation by electron transfer from Ph3P (Eox = 1.06 vs. SCE21) (for quenching experiment, see ESI†). The formed Ph3P˙+ reacts with oxygen to form phosphine oxide, which is known to occur by several mechanisms.22 This side reaction can be suppressed by subsequent addition of PPh3, which increases the yield significantly (entry 8). A similar effect was achieved by adding PhSiH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 for in situ reduction of Ph3PO back to Ph3P (entry 9). Notably, positive effects of elevated temperature on the reaction in both the absence and presence of PhSiH3 were observed, achieving yields of up to 92% (entries 10 and 11). It should be noted that formation of 6a was not observed in blank experiments (see ESI†) in the absence of the flavin photocatalyst, light, Ph3P or molecular sieves.‡ On the other hand, a little 6a was formed with omission of the azo compound or its precursor, irrespective of the presence of PhSiH3 (entries 12 and 13). The amount of ester formed by the azo-free process was increased at elevated temperature (entries 14 and 15).
12 for in situ reduction of Ph3PO back to Ph3P (entry 9). Notably, positive effects of elevated temperature on the reaction in both the absence and presence of PhSiH3 were observed, achieving yields of up to 92% (entries 10 and 11). It should be noted that formation of 6a was not observed in blank experiments (see ESI†) in the absence of the flavin photocatalyst, light, Ph3P or molecular sieves.‡ On the other hand, a little 6a was formed with omission of the azo compound or its precursor, irrespective of the presence of PhSiH3 (entries 12 and 13). The amount of ester formed by the azo-free process was increased at elevated temperature (entries 14 and 15).
Taking into account conversions of ester 6a achieved in the presence and absence of 2b (cf. entries 7 vs. 12, 9 vs. 13, 10 vs. 14, and 11 vs. 15), the initial results gave evidence that: (i) after irradiation, flavin 3d can mediate a Mitsunobu reaction that is catalytic in azo reagent by virtue of its generation/recycling from the corresponding hydrazine; (ii) another mechanistic pathway not requiring the azo component is involved, especially at elevated temperature. Such a “background” reaction has been analogously observed using recycling systems with PhI(OAc)28 and Fe(II)-phthalocyanine.9c
Next, we examined the substrate scope of photocatalytic esterification alternating various alcohols 5 (Table 2) and acids 4 (Table 3) under selected conditions: (i) without PhSiH3 with 10% of 1b at 25 °C, characterized by a major contribution from the azo-compound-mediated reaction (method I, analogous to entry 6 in Table 1), and (ii) with PhSiH3 and 10% of 2b at 50 °C, whereupon the non-azo-reagent-free pathway predominated (method II, analogous to entry 11 in Table 1). In the Tables 2 and 3, the efficacy of both methods is characterized by conversions and preparative yields of esters after 24 h. Conversions of photocatalytic esterifications in the absence of azo reagent 1b or its precursor 2b are given for comparison to estimate the contribution of azo-reagent-free pathway (blank).
| Entry | Product | Methoda | Conv. [%] | Yieldb [%] | Conv. (blank)c [%] |
|---|---|---|---|---|---|
| a Conditions for method I: n(5) = 0.15 mmol, n(4) = 0.18 mmol, n(3d) = 0.015 mmol, n(1b) = 0.015 mmol, n(PPh3) = 0.3 mmol, MS 4 Å (150 mg), 2 ml CH3CN, 455 nm, 25 °C, 24 h; for method II: 2b instead of 1b; additionally 0.3 mmol of PhSiH3; 50 °C. b Preparative yield. c Conversion of esterification in the absence of 1b (method I) or 2b (method II) from 1H NMR data. d 80% after 72 h. e 14% of aldehyde. f 7% of aldehyde. g 18% of aldehyde. h 15% of aldehyde. | |||||
| 1 |

|
I | 64d | 53 | 17 |
| 2 | II | 92 | 82 | 69 | |
| 3 |
|
I | 73 | 65 | 21 |
| 4 | II | Quant. | 85 | 80 | |
| 5 |

|
I | 67 | 60 | 25 |
| 6 | II | Quant. | 79 | 80 | |
| 7 |
|
I | 64 | 54 | 60 |
| 8 | II | 82 | 72 | 75 | |
| 9 |
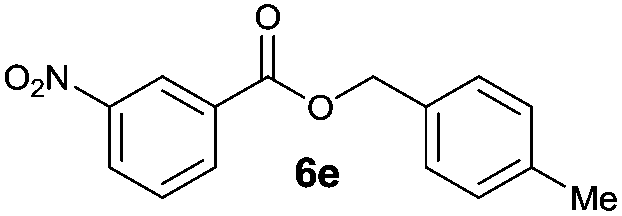
|
I | 62e | 54 | 28 |
| 10 | II | 76f | 65 | 40 | |
| 11 |
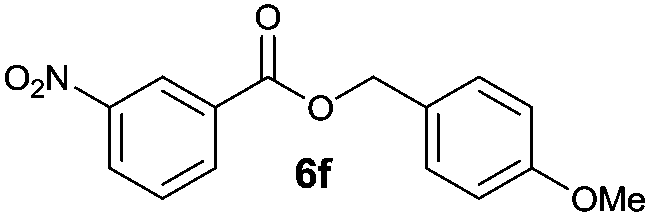
|
I | 50g | 39 | 39 |
| 12 | II | 85h | 71 | 68 | |
| 13 |
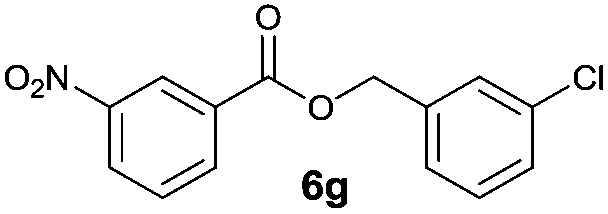
|
I | 52 | 46 | 46 |
| 14 | II | 78 | 60 | 59 | |
| 15 |
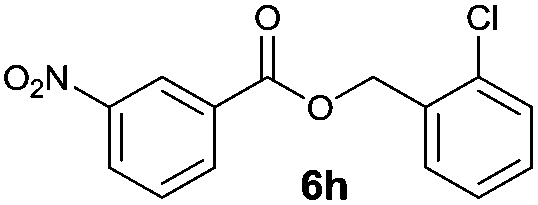
|
I | 53 | 44 | 43 |
| 16 | II | 78 | 71 | 68 | |
| 17 |
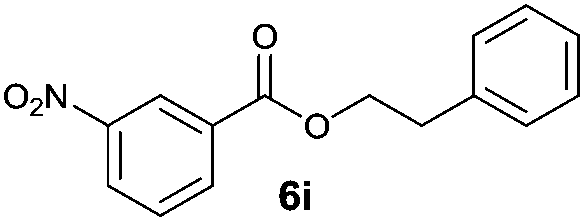
|
I | 18 | n.d. | 10 |
| 18 | II | Quant. | 80 | 20 | |
| 19 |
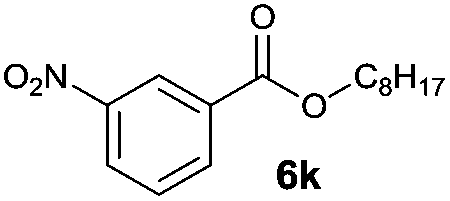
|
I | 51 | 39 | 22 |
| 20 | II | 72 | 61 | 63 | |
| Entry | Product | Methoda | Conv. [%] | Yieldb [%] | Conv. (blank)c [%] |
|---|---|---|---|---|---|
| a Conditions for method I: n(5) = 0.15 mmol, n(4) = 0.18 mmol, n(3d) = 0.015 mmol, n(1b) = 0.015 mmol, n(PPh3) = 0.3 mmol, MS 4 Å (150 mg), 2 ml CH3CN, 455 nm, 25 °C, 24 h; for method II: 2b instead of 1b; additionally 0.3 mmol of PhSiH3; 50 °C. b Preparative yield. c Conversion of esterification in the absence of 1b (method I) or 2b (method II) from 1H NMR data. d 72 h. | |||||
| 1 |
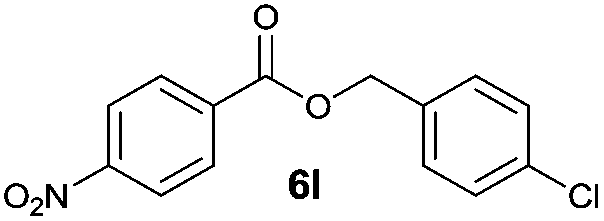
|
I | 30 | 19 | 27 |
| 2 | II | Quant. | 79 | 80 | |
| 3 |

|
I | 5 | n.d. | n.d. |
| 4 | II | 58 | 43 | 5 | |
| 5 |
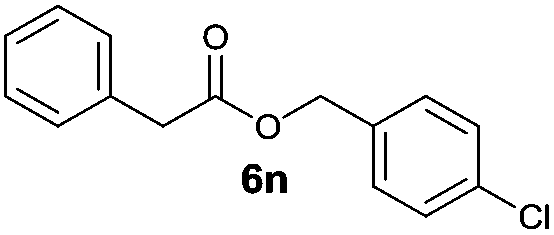
|
I | 68 | 45 | 12 |
| 6 | II | Quant. | 75 | 60 | |
| 7 |

|
Id | 33 | 25 | 7 |
| 8 | IId | 40 | 32 | 16 | |
| 9 |
|
I | 44 | 32 | 31 |
| 10 | II | 65 | 58 | 44 | |
| 11 |
|
I | 8 | n.d. | n.d. |
| 12 | II | 16 | n.d. | 11 | |
| 13 |
|
I | 6 | n.d. | n.d. |
| 14 | II | 66 | 57 | 20 | |
Method I (Table 2, odd entries) provided moderate to good conversions and yields of esters 6a–h by esterification of 3-nitrobenzoic acid (4a) with substituted benzyl alcohols regardless of the character and position of the substituent.
Octyl ester 6k (representative of esters with aliphatic alcohols) was also obtained in good yield by method I while ester 6i with 2-phenylethanol (5i) was formed in poor conversion only (entries 17 and 19). On the other hand, at elevated temperature (method II), high conversions and good to high yields of esters 6a–k were achieved with all alcohols investigated (Table 2, even entries). As could be expected, significant amount of benzaldehyde (15–28%) was formed during esterification of 4-methyl- (5e) and 4-methoxybenzyl (5f) alcohols because of their low oxidation potentials (entries 9–12).19 Both methods proved to be effective for secondary alcohols, albeit requiring longer reaction times (Table 4).
| Entry | Alcohol | Product | Methoda | Conv.b [%] | Yieldc [%] | erd |
|---|---|---|---|---|---|---|
|
a For conditions, see Table 2; reaction time: 72 h.
b Conversion of esterification in the presence/in the absence of 1b (method I) or 2b (method II) from 1H NMR data.
c Preparative yields.
d From HPLC, see ESI.
e er 98:2 was found in the absence of 2b.
|
||||||
| 1 |
|
|
I | 46/7 | 30 | 98:2 |
| 2 | II | 76/61e | 65 | 99:1 |
||
| 3 |
|
|
I | 44/7 | 25 | 99:1 |
| 4 | II | 70/61 | 59 | 99:1 |
||
| 5 |

|

|
I | 48/44 | 39 | 99:1 |
| 6 | II | 58/49 | 50 | 98:2 |
||
| 7 |
|
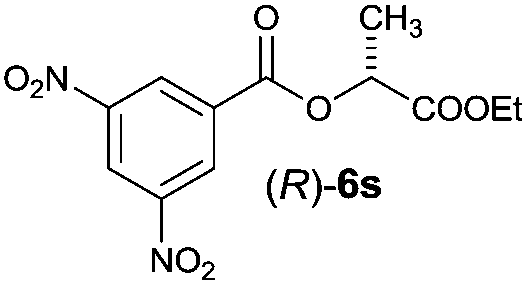
|
I | 45/44 | 38 | 97:3 |
| 8 | II | 60/36 | 48 | 98:2 |
||
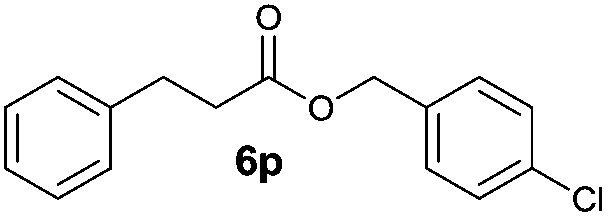
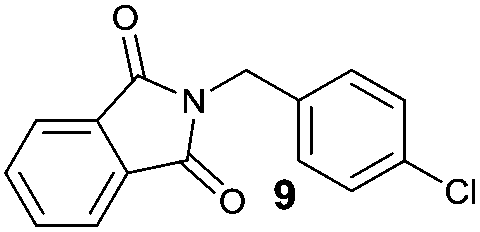
Besides 4a, other aromatic acids (4-nitrobenzoic [4b] and benzoic acid [4c]) as well as phenylalkanoic acids (phenylacetic [4d] and 3-phenylpropanoic acid [4f]) gave good to high yields of esters 6l–n and 6p with 4-chlorobenzyl alcohol (5b) by method II (Table 3, even entries). For these acids, method I seemed to be less efficient giving lower yields of 6n and 6p (entries 5 and 9) and almost no ester 6m (entry 3). Interestingly, both methods afforded ester 6o with sterically hindered α,α-disubstituted acid 4e albeit in low yields and after prolonged reaction time (entries 7 and 8). Low efficacy of our protocols was shown for esterification of hexanoic acid (4g) as a representative of non-substituted aliphatic acids (entries 11 and 12). Method II was proved to be usefull also for phthalimide as a representative of N-nucleophile (entry 14).
Notably, a significant contribution of azodicarboxylate 1b or its precursor 2b was observed with most substrates (Tables 2 and 3, cf. conversions of methods I or II with azo-free pathway [blank]). This contribution became even more pronounced for esterification of some benzylic esters, e.g.6a–6c and 6e (Table 2, entries 1, 3, 5 and 9), provided by method I or for some esterifications by method II, e.g. towards 6i (Table 2, entry 18), 6m and 9 (Table 3, entries 4 and 14). On the other hand, in some cases, especially for method II, azo-free reaction pathway predominates as indicated by high conversions of blank. The conversions of esters are indeed high under optimal conditions (method II) but not quantitative in many cases. The reason is probably bleaching of the flavin photocatalyst which was observed during esterifications. Similar photodecomposition of flavins was reported to occur also during other photocatalytic processes.15,16,20
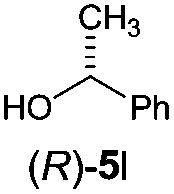
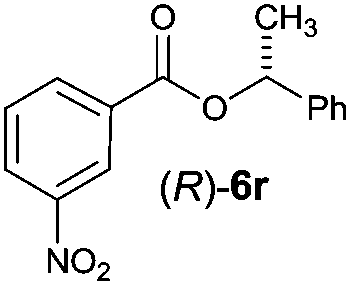
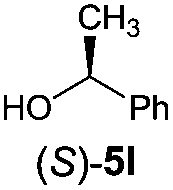
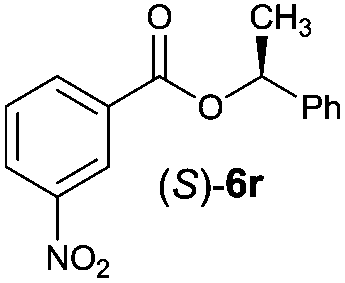
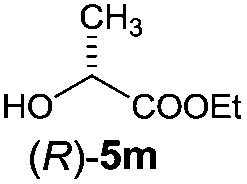
Irrespective of the method, highly stereoselective production of esters (Table 4) with retention of configuration was observed for two types of secondary alcohols, 1-phenylethanol (5l, entries 1–4) and ethyl lactate (5m, entries 5–8), indicating that our photocatalytic esterification occurs not through alkoxyphosphonium 10 but through acylphosphonium species 11 (see Scheme 2). This intermediate must also predominate in the photocatalytic “Mitsunobu reaction pathway” occurring through betain intermediate 12 (Scheme 2A), as indicated by the very high stereoselectivities observed with 5l at 25 °C (method I), i.e. at the conditions where a significant contribution of the catalytic amount of 1b was detected (entries 1 and 3, cf. conversions in the presence and the absence of 1b). Most probably, our photocatalytic esterification just met the conditions favoring acyloxyphosphonium species 11 in the delicate acyloxy/alkoxyphosphonium equilibrium.5 Notably, the inversion product predominated (er = 85:15§) in standard (stoichiometric) Mitsunobu esterification of 5l, but it is not formed exclusively indicating not only SN2 mechanism is involved.
Based on literature data and our own experimental results, we propose a mechanism for the pathway not involving the azo compound or its hydrazine precursor (Scheme 2B). Excited flavin Fl* oxidizes Ph3P to Ph3P˙+, as it is evident from the redox potentials (see above) and confirmed by the observation of efficient emission quenching (KS = 22 L mol−1; for the Stern–Volmer plot, see ESI†). The flavin radical anion (pKa of conjugated acid is 8.4, ref. 23) is immediately protonated by a carboxylic acid to form radical FlH˙. Ph3P˙+ then reacts with an alcohol to form (after subsequent oxidation and deprotonation) an alkoxyphosphonium species 10, which is in equilibrium with the corresponding acyloxyphosphonium species 11. Nevertheless, 11 may also be formed directly from Ph3P˙+ and carboxylate.¶ Finally, 11 undergoes substitution with alkoxide to form ester and the reduced flavin FlH2 is re-oxidized by oxygen in a dark procedure.17,20a,24 Hydrogen peroxide, formed as a by-product, is decomposed by molecular sieves.9c
Quantum yields of ester 6a production by methods I and II was found to be 0.04 and 0.07, respectively, thus supporting closed catalytic cycle and indicating that an open radical propagation mechanism is not involved. We also not observed corresponding anhydride when monitoring the reaction mixture with 4b and 5b by 1H NMR. Nevertheless presence of this alternative by-product/intermediate3b,5 in low concentration cannot be excluded.
Conclusions
In conclusion, we have shown for the first time that the original Mitsunobu reagents, DIAD (1b) and DEAD (1a), can be regenerated from the corresponding hydrazines by photocatalytic and/or organocatalytic system,25 thus allowing esterifications that are catalytic in 1. The method is based on visible light and readily available riboflavin derivative 3d, which is used in a catalytic amount, being recycled with molecular oxygen, an inexpensive and green terminal oxidant. The only drawback is side photooxidation of Ph3P to Ph3PO under aerial conditions, which can be eliminated by in situ back-reduction. We have observed a completely new esterification pathway being involved in our photocatalytic esterification that occurs without any contribution from the azo compound 1 or hydrazine 2. It proceeds through Ph3P˙+ generated by photoinduced electron transfer to flavin. To the best of our knowledge, no similar synthetic application of photochemically generated Ph3P˙+ has hitherto been reported. As there is still room for improvement, optimization of both concepts is currently being pursued in our laboratories.
Acknowledgements
This project was supported by the Czech Science Foundation (Grant No. 16-09436S). R. C. thanks the German National Science Foundation (GRK 1626 “Chemical Photocatalysis”).Notes and references
- (a) A. Voituriez and A. N. Saleh, Tetrahedron Lett., 2016, 57, 4443 CrossRef CAS; (b) J. An, R. M. Denton, T. H. Lambert and E. D. Nacsa, Org. Biomol. Chem., 2014, 12, 2993 RSC; (c) J. Otera and J. Nishikido, in Esterification, Wiley-VCH Verlag GmbH & Co. KGaA, 2010 Search PubMed.
- O. Mitsunobu and Y. Masaaki, Bull. Chem. Soc. Jpn., 1967, 40, 2380 CrossRef CAS.
- (a) S. Fletcher, Org. Chem. Front., 2015, 2, 739 RSC; (b) K. C. K. Swamy, N. N. B. Kumar, E. Balaraman and K. V. P. P. Kumar, Chem. Rev., 2009, 109, 2551 CrossRef CAS PubMed; (c) T. Y. S. But and P. H. Toy, Chem. – Asian J., 2007, 2, 1340 CrossRef CAS PubMed.
- (a) J. McNulty, A. Capretta, V. Laritchev, J. Dyck and A. J. Robertson, Angew. Chem., Int. Ed., 2003, 42, 4051 CrossRef CAS PubMed; (b) G. Wang, J.-R. Ella-Menye, M. St. Martin, H. Yang and K. Williams, Org. Lett., 2008, 10, 4203 CrossRef CAS PubMed; (c) A. B. Hughes and M. M. Sleebs, J. Org. Chem., 2005, 70, 3079 CrossRef CAS PubMed; (d) A. B. Smith, I. G. Safonov and R. M. Corbett, J. Am. Chem. Soc., 2002, 124, 11102 CrossRef CAS PubMed; (e) C. Ahn and P. DeShong, J. Org. Chem., 2002, 67, 1754 CrossRef CAS PubMed.
- S. Schenk, J. Weston and E. Anders, J. Am. Chem. Soc., 2005, 127, 12566 CrossRef CAS PubMed.
- T. Mukaiyama, Angew. Chem., Int. Ed. Engl., 1976, 15, 94 CrossRef.
- A. J. Reynolds and M. Kassiou, Curr. Org. Chem., 2009, 13, 1610 CrossRef CAS.
- T. Y. S. But and P. H. Toy, J. Am. Chem. Soc., 2006, 128, 9636 CrossRef CAS PubMed.
- (a) D. Hirose, M. Gazvoda, J. Kosmrlj and T. Taniguchi, Chem. Sci., 2016, 7, 5148 RSC; (b) T. Hashimoto, D. Hirose and T. Taniguchi, Adv. Synth. Catal., 2015, 357, 3346 CrossRef CAS; (c) D. Hirose, T. Taniguchi and H. Ishibashi, Angew. Chem., Int. Ed., 2013, 52, 4613 CrossRef CAS PubMed.
- T. Taniguchi, D. Hirose and H. Ishibashi, ACS Catal., 2011, 1, 1469 CrossRef CAS.
- C. J. O'Brien, PCT Int. Appl, WO2010/118042A2.2010, 2010 Search PubMed.
- J. A. Buonomo and C. C. Aldrich, Angew. Chem., Int. Ed., 2015, 54, 13041 CrossRef CAS PubMed.
- Very recently, idea on system catalytic both from the point of view of azo-compound and phosphine based on the above mentioned recycling agents was introduced (ref. 12) but realization is still subject of discussion; see: D. Hirose, M. Gazvoda, J. Kosmrlj and T. Taniguchi, Org. Lett., 2016, 18, 4036 CrossRef CAS PubMed.
- (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (b) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 985 CrossRef CAS PubMed; (c) S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059 RSC; (d) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97 RSC.
- S. Kümmel, R. Cibulka and B. König, in Chemical Photocatalysis, ed. B. König, de Gruyter, Berlin, 2013, p. 45 Search PubMed.
- (a) T. Neveselý, E. Svobodová, J. Chudoba, M. Sikorski and R. Cibulka, Adv. Synth. Catal., 2016, 358, 1654 CrossRef; (b) B. Mühldorf and R. Wolf, Angew. Chem., Int. Ed., 2016, 55, 427 CrossRef PubMed; (c) J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 1040 CrossRef CAS PubMed; (d) T. Hartman and R. Cibulka, Org. Lett., 2016, 18, 3710 CrossRef CAS PubMed; (e) B. Mühldorf and R. Wolf, Chem Commun., 2015, 51, 8425–8428 RSC.
- S. Fukuzumi, K. Yasui, T. Suenobu, K. Ohkubo, M. Fujitsuka and O. Ito, J. Phys. Chem. A, 2001, 105, 10501 CrossRef CAS.
- G. Jürmann, O. Tšubrik, K. Tammeveski and U. Mäeorg, J. Chem. Res., 2005, 661 CrossRef.
- M. Yasuda, M. Nakai, Y. Kawahito and T. Shiragami, Bull. Chem. Soc. Jpn., 2003, 76, 601 CrossRef CAS.
- (a) J. Daďová, S. Kümmel, C. Feldmeier, J. Cibulková, R. Pažout, J. Maixner, R. M. Gschwind, B. König and R. Cibulka, Chem. – Eur. J., 2013, 19, 1066 CrossRef PubMed; (b) U. Megerle, R. Lechner, B. Konig and E. Riedle, Photochem. Photobiol. Sci., 2010, 9, 1400 RSC; (c) J. Svoboda, H. Schmaderer and B. König, Chem. – Eur. J., 2008, 14, 1854 CrossRef CAS PubMed.
- S. Fukuzumi, K. Shimoosako, T. Suenobu and Y. Watanabe, J. Am. Chem. Soc., 2003, 125, 9074 CrossRef CAS PubMed.
- (a) S. Yasui, S. Kobayashi and M. Mishima, J. Phys. Org. Chem., 2016, 29, 443 CrossRef CAS; (b) S. Tojo, S. Yasui, M. Fujitsuka and T. Majima, J. Org. Chem., 2006, 71, 8227 CrossRef CAS PubMed; (c) O. Kei, N. Takashi and F. Shunichi, Bull. Chem. Soc. Jpn., 2006, 79, 1489 CrossRef; (d) S. Yasui, S. Tojo and T. Majima, J. Org. Chem., 2005, 70, 1276 CrossRef CAS PubMed.
- A. Ehrenberg, F. Müller and P. Hemmerich, Eur. J. Biochem., 1967, 2, 286 CrossRef CAS PubMed.
- C. Feldmeier, H. Bartling, K. Magerl and R. M. Gschwind, Angew. Chem., Int. Ed., 2015, 54, 1347 CrossRef CAS PubMed.
- Oxidation of di-tert-butyl hydrazinedicarboxylate to azo compound with CuI/O2 system was reported during preparation of the manuscript: D. Jung, M. H. Kim and J. Kim, Org. Lett., 2016, 18, 6300–6303 CrossRef CAS PubMed.
- (a) H. Maeda, T. Koide, T. Maki and H. Ohmori, Chem. Pharm. Bull., 1995, 43, 1076 CrossRef CAS; (b) H. Ohmori, M. Maeda, M. Kikuoka, T. Maki and M. Masui, Tetrahedron, 1991, 47, 767 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and copies of NMR spectra and HPLC chromatograms. See DOI: 10.1039/c6ob02770a |
| ‡ Notably only trace of ester was formed in stoichiometric Mitsunobu reaction in the presence of hydrogen peroxide (1 equiv.) in acetonitrile while 90% of ester was formed after addition of MS 4A thus demonstrating H2O2-quenching of Mitsunobu reaction. Hydrogen peroxide was not detected by iodometry after photocatalytic esterifications in the presence of MS 4A. |
| § This value was not affected by either light or the flavin (see ESI†). |
| ¶ Notably analogous formation of alkoxyphosphonium and acyloxyphosphonium species was observed when Ph3P˙+ was generated electrochemically, see ref. 26. |
| This journal is © The Royal Society of Chemistry 2017 |