
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA molecular electron density theory study of the [3 + 2] cycloaddition reaction of nitrones with ketenes†
Mar
Ríos-Gutiérrez
a,
Andrea
Darù
b,
Tomás
Tejero
b,
Luis R.
Domingo
 *a and
Pedro
Merino
*b
*a and
Pedro
Merino
*b
aDepartment of Organic Chemistry, University of Valencia, Dr. Moliner 50, E-46100 Burjassot, Valencia, Spain. E-mail: domingo@utopia.uv.es; Web: http://www.luisrdomingo.com
bLaboratorio de Síntesis Asimétrica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), Universidad de Zaragoza-CSIC, Zaragoza 50009, Spain. E-mail: pmerino@unizar.es; Web: http://www.pmerino.com
First published on 16th January 2017
Abstract
The [3 + 2] cycloaddition (32CA) reaction between nitrones and ketenes has been studied within the Molecular Electron Density Theory (MEDT) at the Density Functional Theory (DFT) MPWB1K/6-311G(d,p) computational level. Analysis of the conceptual DFT reactivity indices allows the explanation of the reactivity, and the chemo- and regioselectivity experimentally observed. The particular mechanism of this 32CA reaction involving low electrophilic ketenes has been elucidated by using a bonding evolution theory (BET) study. It is determined that this reaction takes place in one kinetic step only but in a non-concerted manner since two stages are clearly identified. Indeed, the formation of the second C–O bond begins when the first O–C bond is already formed. This study has also been applied to predict the reactivity of nitrones with highly electrophilic ketenes. Interestingly, this study predicts a switch to a two-step mechanism due to the higher polar character of this zw-type 32CA reaction. In both cases, BET supports the non-concerted nature of the 32CA reactions between nitrones and ketenes.
1. Introduction
Since the first examples gathered by Irvin in 1938,1 demonstrating that nitrones are capable of undergoing 1,3-additions, the [3 + 2] cycloaddition (32CA) reaction of nitrones with alkenes2 has been widely used as a key step for the synthesis of heterocycles and natural products.3 The ready availability and ease of use of nitrones,4 the tuneability of the reaction by using chiral Lewis acids5 and the high efficiency of this transformation6 combine to make this reaction a powerful method for heterocyclic synthesis.7 Of particular interest are cycloadditions with allenes 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 and heteroallenes such as isocyanates 39 and ketenes 4,10 which present alternative reaction paths leading to different heterocyclic compounds depending on the allenic part of the system in which the cycloaddition takes place (Scheme 1).
8 and heteroallenes such as isocyanates 39 and ketenes 4,10 which present alternative reaction paths leading to different heterocyclic compounds depending on the allenic part of the system in which the cycloaddition takes place (Scheme 1).
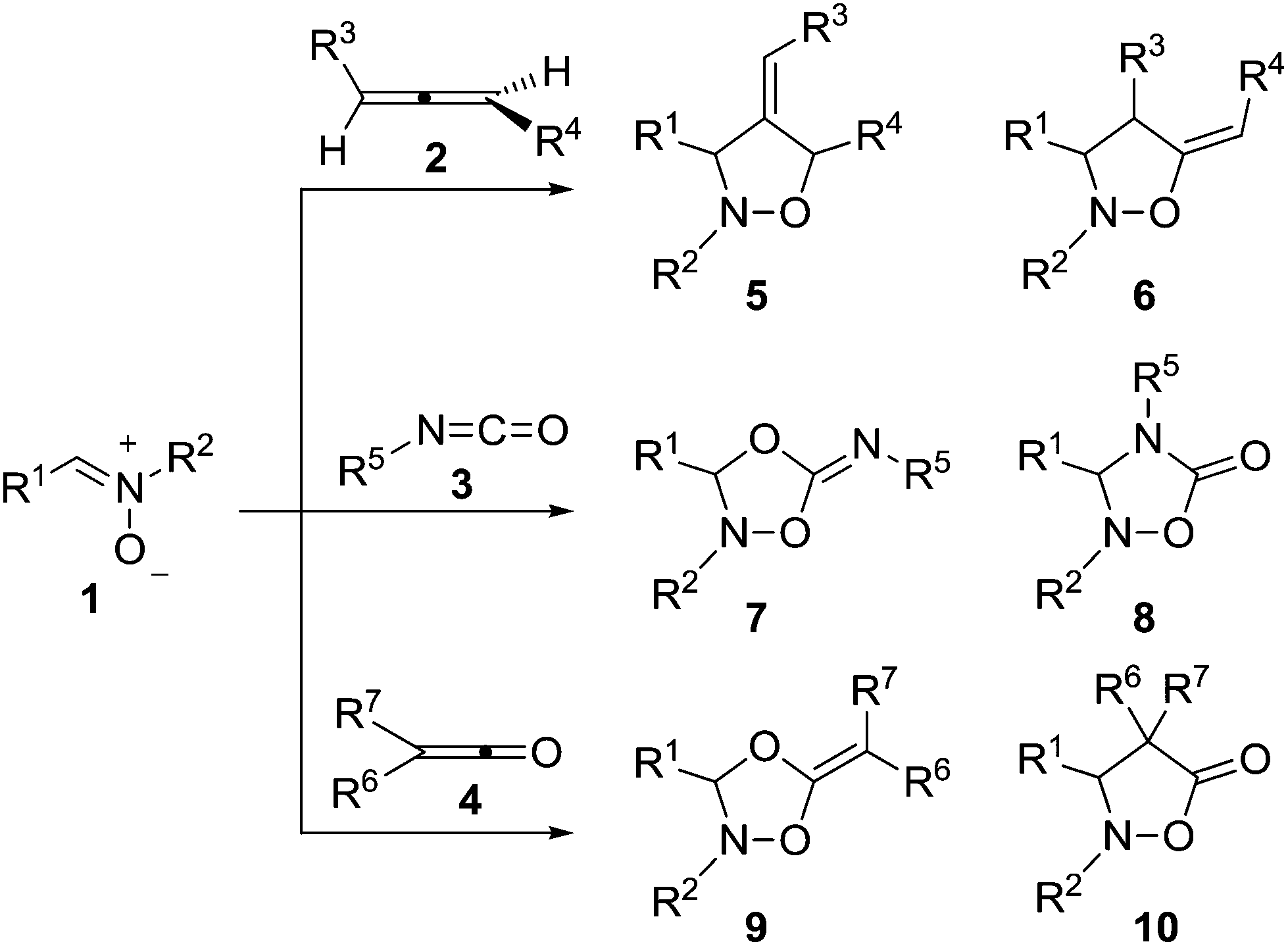 | ||
| Scheme 1 Reactions of nitrones with allenes and heteroallenes. | ||
Despite this synthetic utility, mechanistic studies that allow the interpretation and prediction of the adducts to be obtained with allenes or heteroallenes showing different alternatives like 2–4 are scarce. The reaction of the simplest nitrone 1a (R1, R2 = H) with the simplest allene 2 (R3,R4 = H) was computationally studied and it was determined that the reaction follows a stepwise mechanism.11 Very recently, we observed the same preference for a stepwise mechanism in the reaction with isocyanates.12 Houk and co-coworkers studied the reaction between N-aryl nitrones and alkylarylketenes giving rise to a “pericyclic” cascade with chirality transfer.13 However, their study only referred to a particular process in which it was suggested that the first cycloaddition step between the nitrone and the ketene is a one-step reaction but highly asynchronous.
Several Density Functional Theory14 (DFT) studies carried out within the Molecular Electron Density Theory (MEDT)15 devoted to the study of the reactivity of three-atom-components (TACs) participating in 32CA reactions have allowed the establishment of a useful classification of this class of cycloaddition reactions depending on the electronic structure and behaviour of the TAC into pseudodiradical-type (pr-type, typically an azomethineylide 11),16 carbenoid-type (cb-type, typically a nitrile ylide 12)17 and zwitterionic-type (zw-type, typically a nitrone 1a)16 reactions (Scheme 2). The feasibility of the zw-type 32CA reactions depends on the polar character of the reactions, i.e. the nucleophilic character of the nitrones and the electrophilic character of the ethylene derivatives, or vice versa. In general, nitrones are good nucleophiles that react with electron-deficient alkenes.18
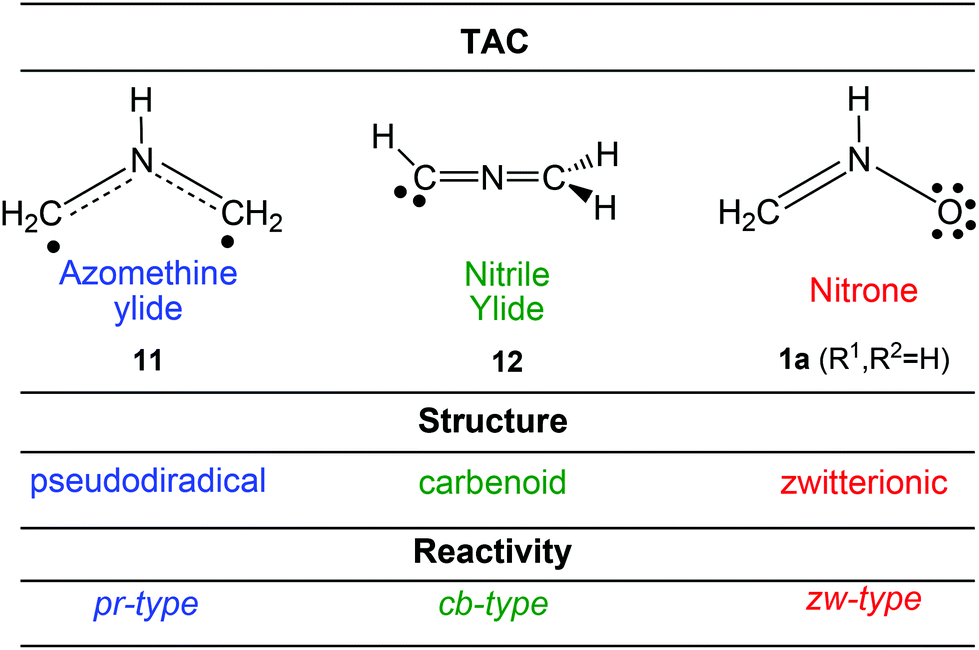 | ||
| Scheme 2 Electronic structure of TACs and the proposed reactivity types in 32CA reactions. | ||
In order to understand the zw-type reactivity16,18 of nitrones towards ketenes, the 32CA reaction of nitrone 1b (R1, R2 = Me) with ketenes 4b and 4c yielding cycloadducts (CAs) 9b,c and 10b,c are herein studied within the MEDT through DFT calculations at the MPWB1K/6-311G(d,p) computational level (see Scheme 3). The chemoselectivity associated with the 32CA reactions of nitrone 1b with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC double bonds of ketenes 4b and 4c is first studied. Then, a Bonding Evolution Theory19 (BET) analysis of the most favourable chemoisomeric channel associated with the 32CA reaction of nitrone 1b with ketene 4b is carried out in order to understand the mechanism of these zw-type reactions. Finally, an ELF comparative analysis of the TSs and intermediate related to the most favourable channel associated with the 32CA reaction of nitrone 1b with ketene 4c is performed.
O and CC double bonds of ketenes 4b and 4c is first studied. Then, a Bonding Evolution Theory19 (BET) analysis of the most favourable chemoisomeric channel associated with the 32CA reaction of nitrone 1b with ketene 4b is carried out in order to understand the mechanism of these zw-type reactions. Finally, an ELF comparative analysis of the TSs and intermediate related to the most favourable channel associated with the 32CA reaction of nitrone 1b with ketene 4c is performed.
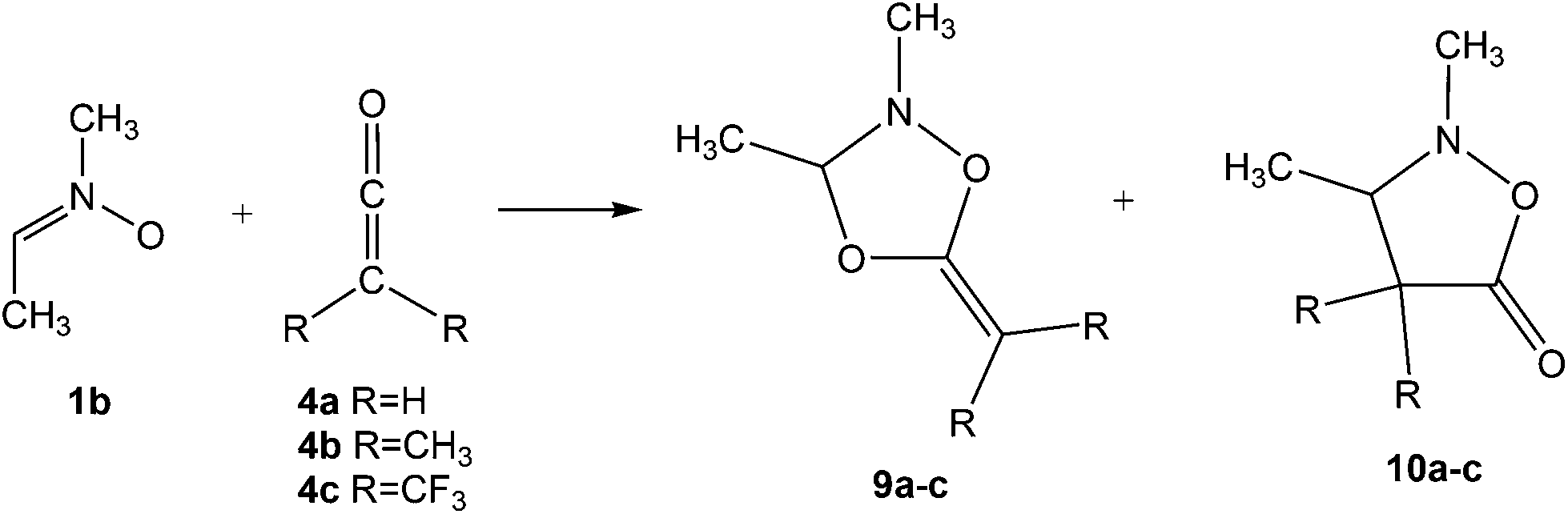 | ||
| Scheme 3 32CA reactions of nitrone 1b with ketenes 4a–c yielding cycloadducts 9a–c and 10a–c. | ||
2. Computational methods
DFT calculations were performed using the MPWB1K functional20 together with the 6-311G(d,p) basis set.21 Optimisations were carried out using the Berny analytical gradient optimization method.22 The stationary points were characterised by frequency computations in order to verify that TSs have one and only one imaginary frequency. The IRC paths23 were traced in order to check the energy profiles connecting each TS to the two associated minima of the proposed mechanism using the second order González–Schlegel integration method.24 The solvent effects of benzene were taken into account by full optimization of the gas phase structures at the MPWB1K/6-311G(d,p) computational level using the polarisable continuum model (PCM) developed by Tomasi's group25 in the framework of the self-consistent reaction field (SCRF).26 The values of enthalpies, entropies and Gibbs free energies in benzene were calculated with the standard statistical thermodynamics at 25 °C and 1 atm.21 The electronic structures of the stationary points were characterised by a natural population analysis (NPA),27 and by the electron localisation function (ELF)28 topological analysis of the electron density. All computations were carried out with the Gaussian 09 suite of programs.29 ELF studies were performed with the TopMod30 program using the corresponding MPWB1K/6-311G(d,p) monodeterminantal wavefunctions. For the BET study, the corresponding reaction channel was followed by performing the topological analysis of the ELF for 862 nuclear configurations along the IRC path. ELF calculations were computed over a grid spacing of 0.1 a.u. for each structure and ELF basin isosurfaces were obtained for an ELF value of 0.75 a.u.Conceptual DFT (CDFT) provides different indices to rationalise and understand chemical structure and reactivity.31 The global electrophilicity index,32ω, is given by the following expression, ω = (μ2/2η), in terms of the electronic chemical potential μ and the chemical hardness η. Both quantities may be approached in terms of the one-electron energies of the frontier molecular orbitals HOMO and LUMO, εH and εL, as μ = (εH + εL)/2 and η = (εL − εH), respectively.33 The global nucleophilicity index,34N, based on the HOMO energies obtained within the Kohn–Sham scheme,35 is defined as N = EHOMO(Nu) − EHOMO(TCE), where tetracyanoethylene (TCE) is the reference because it presents the lowest HOMO energy in a long series of molecules already investigated in the context of polar organic reactions. The electrophilic Pk+ and nucleophilic Pk− Parr functions,36 which allow for the characterisation of the electrophilic and nucleophilic centers of a molecule, were obtained through the analysis of the Mulliken atomic spin density (ASD) of the radical cation of nitrone 1b and the radical anion of ketenes 4a and 4c, by single-point energy calculations from the optimised neutral geometries.
3. Results and discussion
The present MEDT study is organised as follows: in section 3.1, an analysis of the electronic structure of nitrones 1a,b is performed in order to understand the zw-type reactivity of 1b in 32CA reactions. Section 3.2 contains an analysis of the CDFT reactivity indices of the reagents involved in the 32CA reactions of nitrone 1b with ketenes 4b,c. In section 3.3, the potential energy surfaces (PESs) associated with the chemoisomeric channels associated with the 32CA reactions of nitrone 1b with ketenes 4b,c are explored and characterised. Section 3.4 discusses a BET study characterising the bonding changes, as well as the energies related to those changes, along the most favourable reaction channel associated with the 32CA reaction of nitrone 1b with ketene 4b. And finally, in section 3.5, a comparative ELF topological analysis of the TSs and intermediate involved in the most favourable reaction channel associated with the 32CA reaction of nitrone1b with electrophilic ketene 4c is performed.3.1. ELF topological analysis of the electronic structure of nitrones 1a,b
As commented in the Introduction part, the reactivity of TACs can be correlated with their electronic structure.16,17 Thus, an ELF topological analysis of the simplest nitrone 1a and nitrone 1b was first performed in order to characterise the electronic structure of these TACs. The representation and attractor positions of ELF valence basins, as well as ELF electron populations, natural atomic charges and the Lewis structures arising from the ELF topological analysis for nitrones 1a,b are shown in Fig. 1.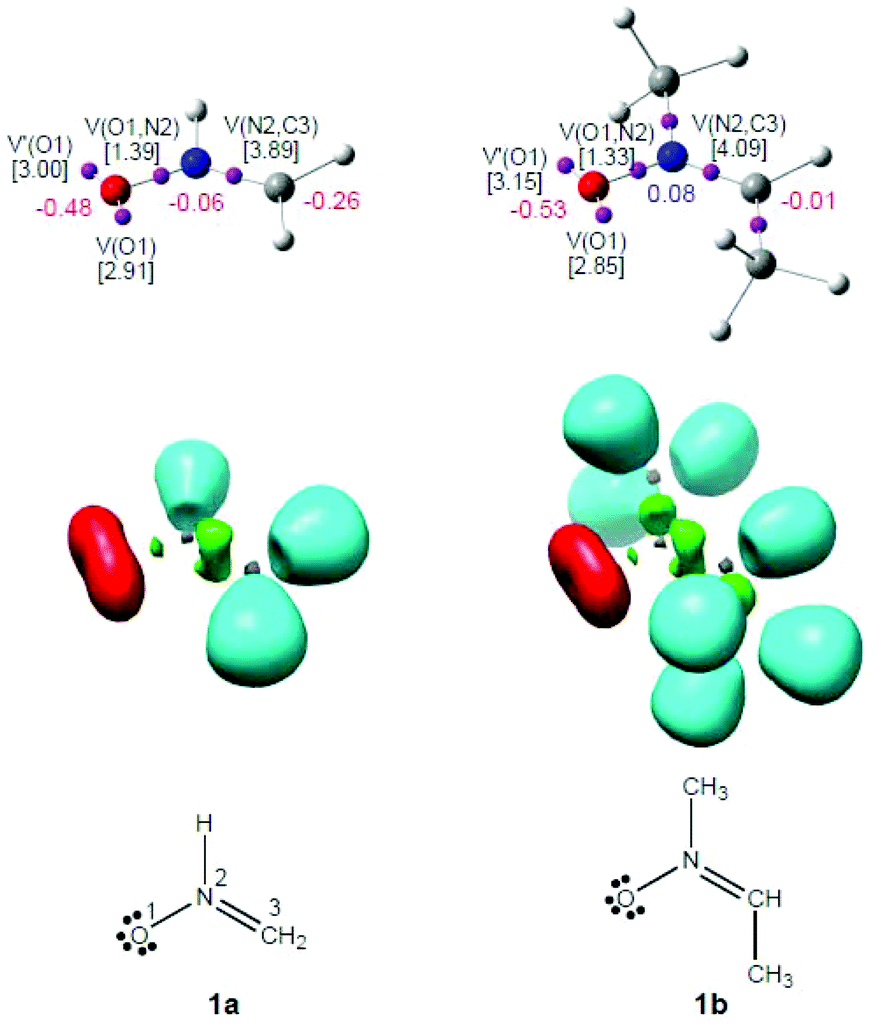 | ||
| Fig. 1 Representation of ELF attractors and valence basin populations, in e, natural atomic charges (negative in red and positive in blue), ELF valence basins and proposed Lewis structures for nitrones 1a,b. | ||
The ELF topology of the simplest nitrone 1a permits the establishment of the Lewis structure of this TAC (see Fig. 1). As can be seen, nitrone 1a presents two V(O1) and V′(O1) monosynaptic basins, integrating a total of 5.91e, and one V(O1,N2) disynaptic basin with a population of 1.39e. This behaviour suggests that the O1–N2 bonding region is strongly polarised towards the O1 oxygen atom. In addition, the presence of one V(N2,C3) disynaptic basin integrating 3.89e indicates that the N2–C3 bonding region has a strong double bond character. Consequently, the ELF topology of the simplest nitrone 1a clearly indicates that this TAC is able to participate only in zw-type 32CA reactions, as it neither presents a pseudodiradical37 nor a carbenoid17 electronic structure that would enable it to participate in pr- or cb-type 32CA reactions.16,17
On the other hand, the ELF topology of nitrone 1b shows a very similar bonding pattern to that found in the simplest nitrone 1a. Indeed, the substitution of the hydrogen atoms bound to the N2 nitrogen and C3 carbon by a methyl group is expected to produce no significant electronic changes. The ELF valence basin populations have varied very slightly (see Fig. 1), although in this case the N2–C3 bonding region is characterised by the presence of two disynaptic basins, V(N2,C3) and V′(N2,C3), integrating a total population of 4.09e. Therefore, according to the ELF topological analysis, nitrone 1b will behave as a zwitterionic TAC participating only in zw-type 32CA reactions such as the simplest nitrone 1a.16
Although the ELF topological analysis of nitrones 1a and 1b allows the establishment of a bonding pattern in these TACs, NPA indicates that neither nitrone has zwitterionic charge distribution. Note that although the O1 oxygen has a high negative charge, −0.48e (1a) and−0.53e (1b), the N2 nitrogen presents practically no charge, −0.06e (1a) and 0.08e (1b). Moreover, the C3 carbon appears negatively charged at nitrone 1a, −0.26e, while at nitrone 1b it presents a null charge, −0.01e.
Thus, although ELF topological analysis provides a bonding pattern concordant with the commonly accepted Lewis structures of nitrones 1a,b, the NPA is completely in disagreement with the representation of their electronic structure as a 1,2-zwitterionic structure. Nevertheless, ELF topological characterisation of the electron density distribution at these nitrones accounts for their zw-type reactivity.
3.2 Analysis of the CDFT reactivity indices of the reagents involved in the 32CA reactions of nitrone 1b with ketenes 4b,c
Studies devoted to polar organic reactions have shown that the analysis of the reactivity indices defined within the CDFT31d,38 is a powerful tool to understand the reactivity in polar cycloadditions. Global DFT indices, namely, the electronic chemical potential, μ, chemical hardness, η, electrophilicity, ω, and nucleophilicity, N, at the ground state of the reagents involved in these 32CA reactions are given in Table 1.| μ | η | ω | N | |
|---|---|---|---|---|
| 4c | −5.26 | 5.62 | 2.47 | 1.05 |
| 4a | −3.76 | 5.57 | 1.27 | 2.58 |
| 1a | −3.43 | 5.55 | 1.06 | 2.92 |
| 4b | −3.23 | 5.08 | 1.02 | 3.35 |
| 1b | −2.95 | 5.42 | 0.80 | 3.46 |
The electronic chemical potential of nitrone 1b, −2.95 eV, is higher than that of ketenes, −3.23 (4b) and −5.26 (4c) eV, indicating that along polar reactions the global electron density transfer (GEDT)39 will flux from the nitrone framework towards the ketene one.
Along a polar reaction, there is an electron density transfer from the nucleophilic to the electrophilic species, which is measured by the GEDT39 value computed at the TS of the reaction; the larger the GEDT at the TS, the more polar the reaction. Note that the GEDT concept comes from the observation that the electron density transfer taking place from the nucleophile to the electrophile along a polar reaction is not a local process, but a global one involving the two interacting frameworks39 and depending on the electrophilic/nucleophilic interactions taking place between them.
The electrophilicity ω and nucleophilicity N indices of the simplest nitrone 1a are 1.06 eV and 2.92 eV, being classified as a moderate electrophile and on the borderline of strong nucleophiles within the electrophilicity40 and nucleophilicity41 scales. Inclusion of the two electron-releasing (ER) methyl groups at the N2 and C3 atoms of the simplest nitrone 1a decreases the electrophilicity ω index of nitrone 1b to 0.80 eV and increases its nucleophilicity N index to 3.46 eV. Consequently, nitrone 1b will behave as a strong nucleophile participating in zw-type 32CA reactions.18
On the other hand, the electrophilicity ω and nucleophilicity N indices of the simplest ketene 4a are 1.27 eV and 2.58 eV, being classified as a moderate electrophile and nucleophile. Inclusion of the two ER methyl groups at the terminal C6 carbon of ketene 4a decreases the electrophilicity ω index of ketene 4b to 1.02 eV and increases its nucleophilicity N index to 2.58 eV. Thus, ketene 4b will behave as a moderate electrophile and a strong nucleophile. Regarding ketene 4c, inclusion of the two electron-withdrawing (EW) trifluoromethyl (CF3) groups at the terminal C6 carbon of ketene 4a notably increases the electrophilicity ω index to 2.47 eV and decreases the nucleophilicity N index to 1.05 eV. Therefore, ketene 4c will behave as a strong electrophile and a marginal nucleophile. This early analysis of the CDFT global reactivity indices suggests that, as expected, the zw-type 32CA reaction of nitrone 1b with the more electrophilic ketene 4c will be more favourable than that with ketene 4b.
In polar cycloaddition reactions involving the participation of non-symmetric reagents, the most favourable reactive channel is that involving the initial two-center interaction between the most electrophilic center of the electrophile and the most nucleophilic center of the nucleophile. Recently, Domingo et al. proposed the electrophilic Pk+ and nucleophilic Pk− Parr functions36 derived from the changes of spin electron density reached via the GEDT process from the nucleophile to the electrophile as powerful tools in the study of the local reactivity in polar processes. Accordingly, the nucleophilic Pk− Parr functions of nitrone 1b and the electrophilic Pk+ Parr functions of ketenes 4b,c were analysed in order to characterise the most electrophilic and nucleophilic centers of the species involved in these 32CA reactions and, thus, to explain the regio- and chemoselectivity experimentally observed (see Fig. 2).
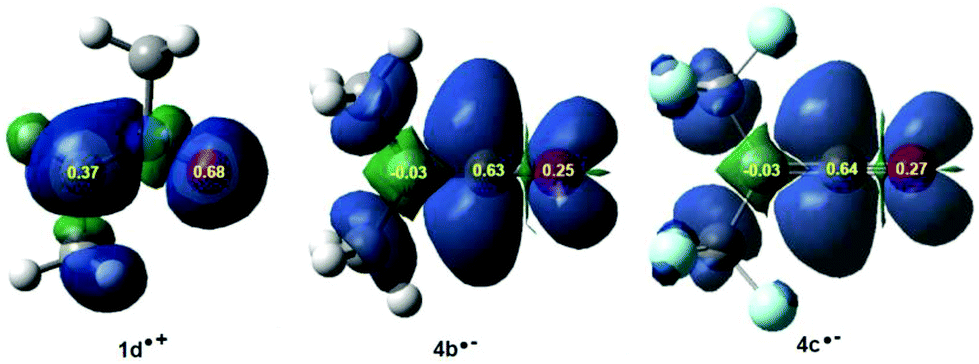 | ||
| Fig. 2 3D representations of the ASD of the radical cation 1b˙+ and the radical anions 4b,c˙−, together with the nucleophilic Pk− Parr functions of nitrone 1b and the electrophilic Pk+ Parr functions of ketenes 4b,c. | ||
Analysis of the nucleophilic Pk− Parr functions of nitrone 1b indicates that the O1 oxygen, Pk− = 0.68, is twice as nucleophilically activated as the C3 carbon, Pk− = 0.37, while the electrophilic Pk+ Parr functions of ketenes 4b,c indicate that the central C5 carbon is the most electrophilic center of these molecules, Pk+ = 0.63 (4b) and 0.64 (4c). Consequently, the most favourable electrophile–nucleophile interaction along the polar zw-type 32CA reactions of nitrone 1b with ketenes 4b,c will take place between the most nucleophilic center of nitrone 1b, the O1 oxygen atom, and the most electrophilic center of ketenes 4b,c, the central C5 carbon, in clear agreement with the regioselectivity experimentally reported first by Taylor42 and further by Houk.13
On the other hand, analysis of the electrophilic Pk+ Parr functions of ketenes 4b,c also indicates that while the ketene O4 oxygen atom presents some electrophilic activation, Pk+ = 0.25 (4b) and 0.27 (4c), the terminal C6 carbon is slightly electrophilically deactivated, Pk+ = −0.03. This means that along the zw-type 32CA reaction of nitrone 1b with ketenes 4b,c, the terminal carbon does not participate in the reaction, and thereby, this zw-type 32CA reaction will present a complete CO chemoselectivity.
3.3. Analysis of the PESs of the 32CA reactions of nitrone 1b with ketenes 4b,c
Due to the non-symmetry of both the reagents, the 32CA reactions between nitrone 1b and ketenes 4b,c can take place through four competitive reaction channels, which are related to the chemoisomeric attacks of nitrone 1b on the CO and CC double bonds of ketenes 4b,c, and with the two regioisomeric approach modes of nitrone 1b towards each one of the two double bonds of ketenes 4b,c. Due to the high regioselectivity predicted by the analysis of the electrophilic Pk+ and nucleophilic Pk− Parr functions, only the two chemoisomeric channels associated with the initial nucleophilic attack of the nitrone O1 oxygen on the central C5 carbon of ketenes 4b,c were studied.
Interestingly, analysis of the PESs associated with the two 32CA reactions indicates that they take place through different mechanisms; the 32CA reaction between nitrone 1b and ketene 4b takes place via a one-step mechanism, while that of nitrone 1b with electrophilic ketene 4c takes place via a two-step mechanism (see Scheme 4). Total and relative energies, in the gas phase and benzene, as well as total and relative enthalpies, entropies and Gibbs free energies in benzene, of the stationary points involved in the 32CA reactions between nitrone 1b and ketenes 4b,c are given in the ESI.†
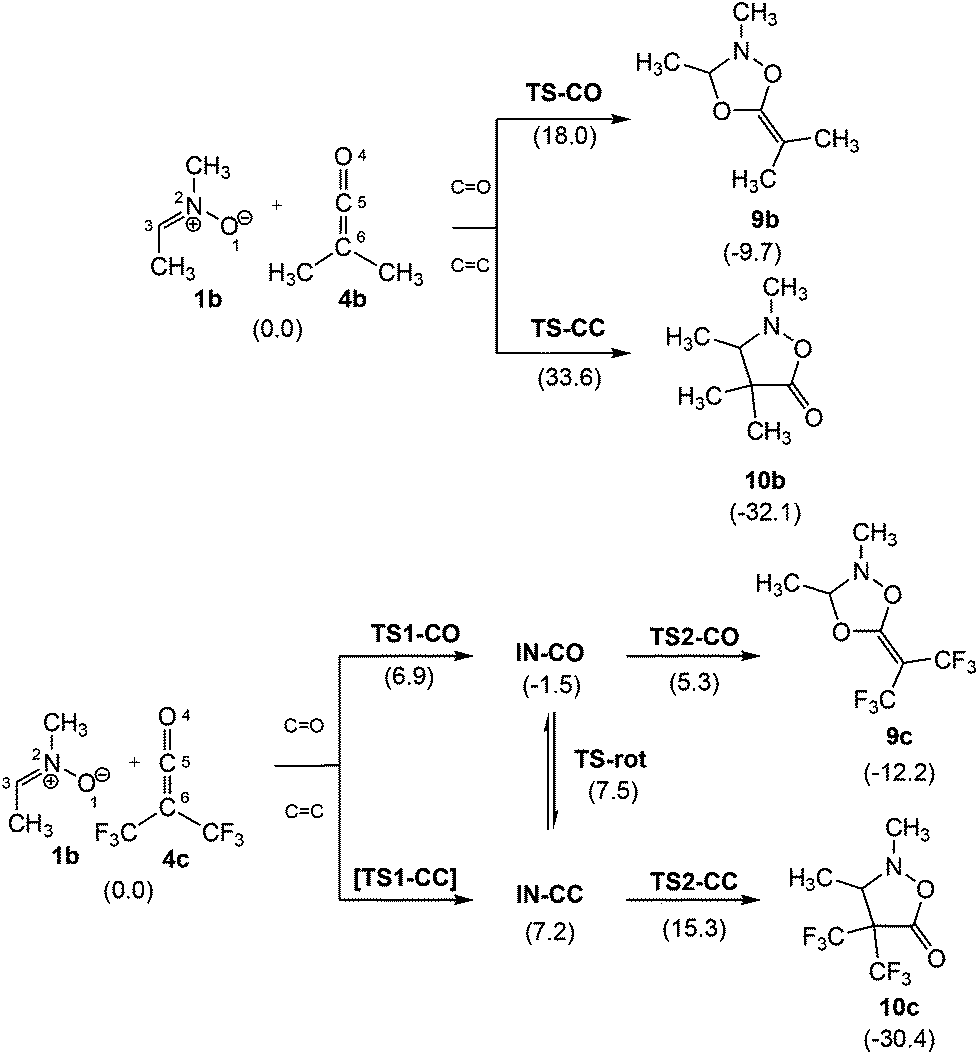 | ||
| Scheme 4 CO and CC chemoisomeric channels associated with the zw-type 32CA reaction between nitrone 1b and ketenes 4b,c. Relative Gibbs free energies, in kcal mol−1, are given in parentheses. | ||
The activation Gibbs free energy of the 32CA reaction between nitrone 1b and ketene 4b presents a low value, 18.8 kcal mol−1, the reaction being exergonic, −9.7 kcal mol−1 (9b) (see relative Gibbs free energies in Scheme 4). Analysis of the relative Gibbs free energies of TS-CO and TS-CC indicates that this 32CA is kinetically completely chemoselective, as TS-CC is 15.6 kcal mol−1 higher in Gibbs free energy than TS-CO. However, the formation of CA 10b is thermodynamically more favourable than 9b. Consequently, while CA 9b is the product of a kinetic control, CA 10b could become the product of a thermodynamic control under thermal equilibrium conditions.
The PES associated with the 32CA reaction between nitrone 1b and electrophilic ketene 4c is more complex. In order to understand the reaction mechanism, the Gibbs free energy profiles of the two competitive channels are depicted in Fig. 3. Analysis of these energy profiles allows one to reach some appealing conclusions: (i) the nucleophilic attack of nitrone 1b to ketene 4cviaTS1-CO has an activation Gibbs free energy of 6.9 kcal mol−1; (ii) the activation Gibbs free energy associated with the ring closure from IN-COviaTS2-CO is 6.8 kcal mol−1, and 8.1 kcal mol−1 from IN-CCviaTS2-CC; (iii) this reaction is completely chemoselective, as TS2-CO is 10.5 kcal mol−1 lower in Gibbs free energy than TS2-CC; (iv) formation of CAs 9c and 10c is exergonic by 12.2 and 30.4 kcal mol−1, respectively; (v) interestingly, IN-CO and IN-CC are two conformers being interconvertible by a O1–C5 single bond rotation implying only 0.3 kcal mol−1. Consequently, these intermediates connect both chemoisomeric channels. Thus, under thermodynamic control, CA 9c could be converted into the more thermodynamically stable 10c with an activation energy of 27.5 kcal mol−1; and finally, (vi) a comparison between the energy profiles of the 32CA reactions of nitrone 1b with ketenes 4b,c allows one to reach two appealing conclusions: (a) the electrophilic activation of the ketene changes the molecular mechanism and activation energies, but it does not change the thermodynamics of the reaction; and (b) in both reactions, the ring closure is the step controlling the chemoselectivity.
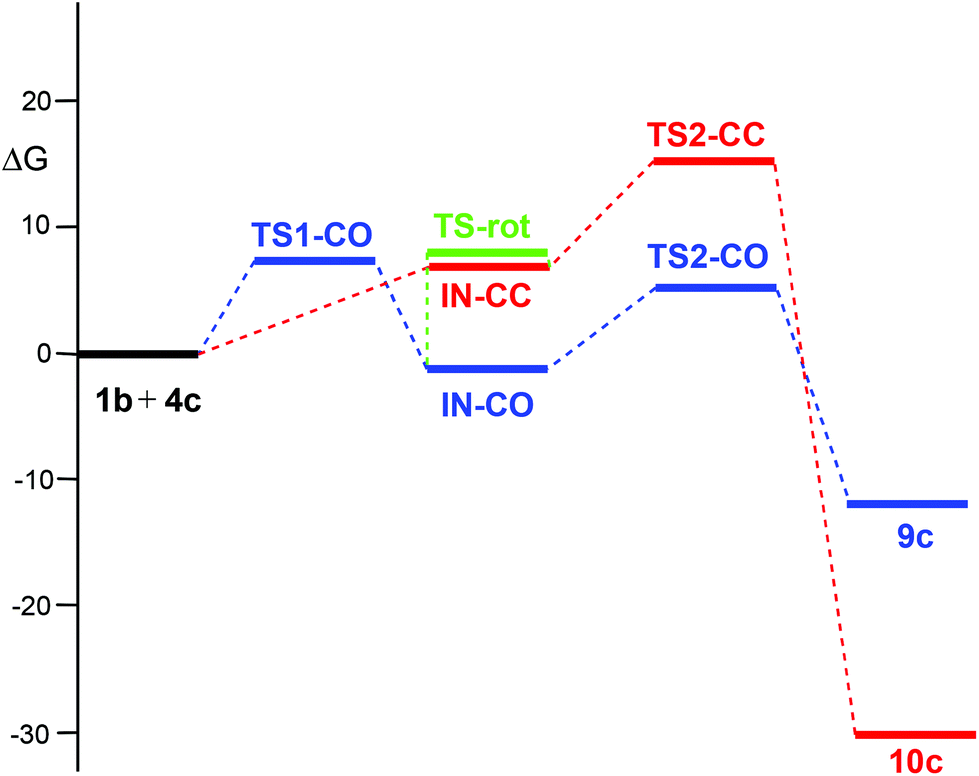 | ||
| Fig. 3 MPWB1K/6-311G(d,p) Gibbs free energy profile (ΔG, kcal mol−1), in benzene, of the 32CA reaction between nitrone 1b and electrophilic ketene 4c. | ||
The geometries of the TSs and intermediates involved in the 32CA reactions of nitrone 1b with ketenes 4b,c are given in Fig. 4. At the TSs associated with the 32CA reaction of nitrone 1b with ketene 4b, the distances between the two O1 and C5, and the two C3 and O4/C6 interacting atoms are: 1.560 Å and 2.219 Å at TS-CO and 1.604 Å and 2.546 Å at TS-CC, respectively. The short distance between the O1 and C5 atoms at both TSs indicates that the O1–C5 single bond is already formed.43 Consequently, these TSs can be associated with the formation of the second C3–O4 or C3–C6 single bonds.
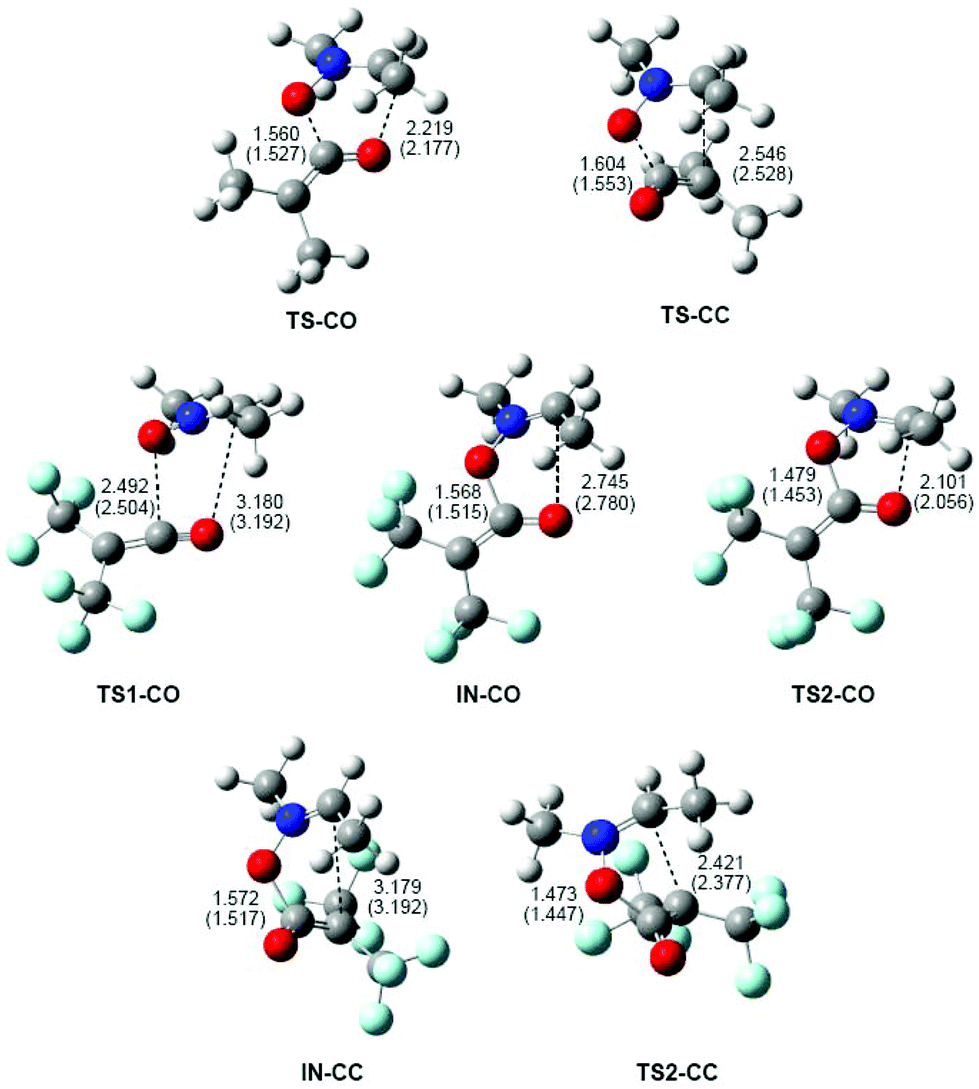 | ||
| Fig. 4 Geometries of the TSs and intermediates involved in the 32CA reaction of nitrone 1b with ketenes 4b,c. Lengths are given in Angstroms. Values in parentheses are associated with the lengths in benzene. | ||
At TS1-CO, related to the nucleophilic attack of the nitrone O1 oxygen atom on the C5 carbon of ketene 4c, the distances between the O1 and C5, and the C3 and O4 atoms are 2.492 and 3.180 Å. These distances are coherent with an early-transition state, in clear agreement with its null activation energy. At intermediates IN-CO and IN-CC, the lengths of the O1–C5 single bonds are 1.568 and 1.572 Å, respectively, while the distances between the C3 and O4/C6 atoms are 2.745 and 3.179 Å. Finally, at the TSs associated with the ring closure along the two reaction paths, the lengths of the formed O1–C5 single bonds are 1.479 Å (TS2-CO) and 1.473 Å (TS2-CC), while the distances between the C3 and O4/C6 atoms are 2.101 (TS2-CO) and 2.421 (TS2-CC) Å, respectively.
Inclusion of benzene in the optimisations does not significantly modify the geometries of the TSs (see Fig. 4). The changes in distances in the gas phase and in benzene are found between 0.05 and 0.01 Å. In general, the distances between the O1 and C5, and the C3 and O4/C6 atoms are shorter in benzene as a consequence of the solvent stabilisation of the corresponding species. Only at TS1-CO and intermediates, are the distances between the non-bound atoms slightly increased.
Thorough studies have made it possible to establish good correlations between the polar character of the reactions and their feasibility.44 In order to evaluate the electronic nature of the 32CA reaction of nitrone 1b with ketenes 4b,c, the GEDT was analysed. The GEDT of a reaction is computed by the sum of the natural atomic charges of the atoms belonging to each framework at the corresponding TSs; the sign indicates the direction of the electron density flux in such a manner that positive values mean a flux from the considered framework to the other one. Reactions with GEDT values near 0.0e correspond to non-polar processes, whereas values higher than 0.2e correspond to polar processes. Thus, at TS-CO and TS-CC associated with the 32CA reaction nitrone 1b with ketene 4b, the GEDT that takes place from the nitrone to the ketene framework is 0.21e and 0.33e, respectively. These values indicate that these TSs have some polar character. At the TSs and intermediates associated with the 32CA reaction nitrone 1b with electrophilic ketene 4c, the GEDT that takes place from the nitrone to the ketene framework is 0.05e at TS1-CO, 0.45e at IN-CO, 0.44e at IN-CC, 0.41e at TS2-CO and 0.37e at TS2-CC. From these GEDT values some appealing conclusions can be drawn: (i) the negligible GEDT found at TS1-CO is a consequence of the earlier character of this TSs; (ii) a high GEDT is found at the corresponding intermediates, ca.0.45e emphasising the zwitterionic character of these species; (iii) a decrease of the GEDT is found at the TSs associated with the ring closure step as a consequence of a retrodonation process; (iv) the GEDT along the two chemoisomeric channels presents similar values, in spite of the energy differences. This behaviour supports the global character of the electron density transfer from the nucleophilic to the electrophilic framework; and (v) the GEDT at TS2-CO and TS2-CC presents higher values than that at TS-CO and TS-CC as a consequence of the higher electrophilic character of ketene 4c, ω = 2.47 eV, than ketene 4b, ω = 1.02 eV.
Analysis of the GEDT along the two competitive reaction paths associated with these reactions is in agreement with the low computed activation energies and confirms the zw-type character of these 32CA reactions, in which the feasibility of the reaction depends on the electrophilic character of the ketene.
3.4. BET characterisation of the molecular mechanism of the zw-type 32CA reaction between nitrone 1b and ketene 4b
When trying to achieve a better understanding of bonding changes in organic chemical reactions, the so-called BET19 has proved to be a very useful methodological tool.45 BET applies Thom's catastrophe theory (CT) concepts46 to the topological analysis of the gradient field of the ELF28 along the reaction coordinate. Several theoretical studies have shown that the topological analysis of the ELF offers a suitable framework for the study of the changes of electron density. This methodological approach is used as a valuable tool to understand the bonding changes along the reaction path and, consequently, to establish the nature of the electronic rearrangement associated with a given molecular mechanism.47Recently, a BET study of the bonding changes along the zw-type 32CA reactions of nitrone 1a with ED acrolein 13 was carried out in order to understand the O–C and C–C bond formation processes and to determine the molecular mechanism of these zw-type 32CA processes.43 On the other hand, a recent BET study of the molecular mechanism of the ketene–imine Staudinger reaction48 permitted one to characterise the participation of the C–C double bond of the ketene in the second step of this reaction. Herein, in order to understand the participation of the O–C double bond of ketenes, a BET study of the most favourable reaction channel associated with the 32CA reaction between nitrone 1b and ketene 4b is performed with the aim of characterising the molecular mechanism of 32CA reactions involving ketenes. The complete BET study is reported in the ESI.† The equivalence between the topological characterisation of the different phases and the associated chemical process is given in Table 2.
| Phases | d(O1–C5) | d(C3–O4) | ΔE | GEDT | Topological characterisation | Chemical process | |
|---|---|---|---|---|---|---|---|
| a | I–IV | 3.04 ≥ d > 1.79 | 3.26 ≥ d > 2.39 | 8.6 | ≤0.34 | Depopulation of the V(O4,C5) disynaptic basin | Depopulation of the O4–C5 bonding region |
| b | V | 1.64 ≥ d > 1.61 | 2.39 ≥ d > 2.33 | 9.0(0.4) | 0.34 | Formation of the V(O1–C5) disynaptic basin | Formation of the O1–C5 single bond |
| c | VI | 1.61 ≥ d > 1.46 | 2.33 ≥ d > 1.91 | 4.5(−4.5) | ≤0.34 | Depopulation of the V(N2,C3) disynaptic basin and the formation of the V(N2) monosynaptic basin | Rupture of the N2–C3 double bond and the formation of the N2 nitrogen lone pair |
| d | VII | 1.46 ≥ d > 1.44 | 1.91 ≥ d > 1.84 | 1.7(−3.1) | ≤0.24 | Formation of the non-bonding V(C3) monosynaptic basin | Formation of the C3 pseudoradical center |
| e | VIII | 1.44 ≥ d ≥ 1.37 | 1.84 ≥ d ≥ 1.42 | −22.7(−24.1) | ≤0.21 | Formation of the V(C3–O4) disynaptic basin | Formation of the C3–O4 single bond |
Some appealing conclusions can be drawn from this BET study: (i) the IRC associated with the 32CA reaction of nitrone 1b with ketene 4b is divided in eight differentiated phases. A behaviour that clearly indicates that the bonding changes along this one-step mechanism are non-concerted; (ii) formation of the first O1–C5 single bond takes place at a C–O distance of 1.64 Å, by the donation of some electron density of the O1 oxygen lone pairs of the nitrone to the C5 carbon atom of the ketene moiety. Note that the O1 oxygen is the most nucleophilic center of nitrone 1b and the C5 carbon corresponds to the most electrophilic center of ketene 4b (see the chemical process taking place in phase V in Table 2, and the V(O1,C5) disynaptic basin in P4 in Fig. 5); (iii) formation of this bond demands the asymmetric depopulation of the O4–C5 bonding region of ketene 4b. The large GEDT, 0.34e, taking place along this zw-type 32CA reaction favours these bonding changes according to the electronic behaviour anticipated by the Parr functions, which is in agreement with the chemoselectivity experimentally observed; (iv) formation of the second C3–O4 single bond takes place at a C–O distance of 1.84 Å by the donation of some of the electron density of the O4 oxygen lone pairs to the C3 pseudoradical center of the nitrone framework (see the chemical process taking place in phase VIII in Table 2, and the V(C3,O4) disynaptic basin in P7 in Fig. 5). This carbon participates with a residual electron density of 0.03e in the formation of the C3–O4 single bond; (v) the reaction follows a two-stage one-step mechanism49 in which the formation of the second C3–O4 one begins when the first O1–C5 single bond is practically already formed. This fact also emphasises that the bonding changes in this one-step reaction are non-concerted processes; (vi) the activation energy associated with this 32CA reaction, 9.3 kcal mol−1, can be mainly associated with the depopulation of the O4–C5 and N2–C3 bonding regions towards the O4 oxygen and the N2 nitrogen, respectively, which is demanded before the donation of the electron density of the O4 oxygen to the C3 carbon; (vii) the present BET study allows the establishment of the molecular mechanism of the zw-type 32CA reactions between nitrones and ketenes as a [2n,2n] mechanism,50i.e. only two non-bonding electrons of the oxygen lone pairs of nitrone 1b and two non-bonding electrons of the oxygen lone pairs of ketene 4b are mainly involved in the formation of the two C–O single bonds in CA 9b.
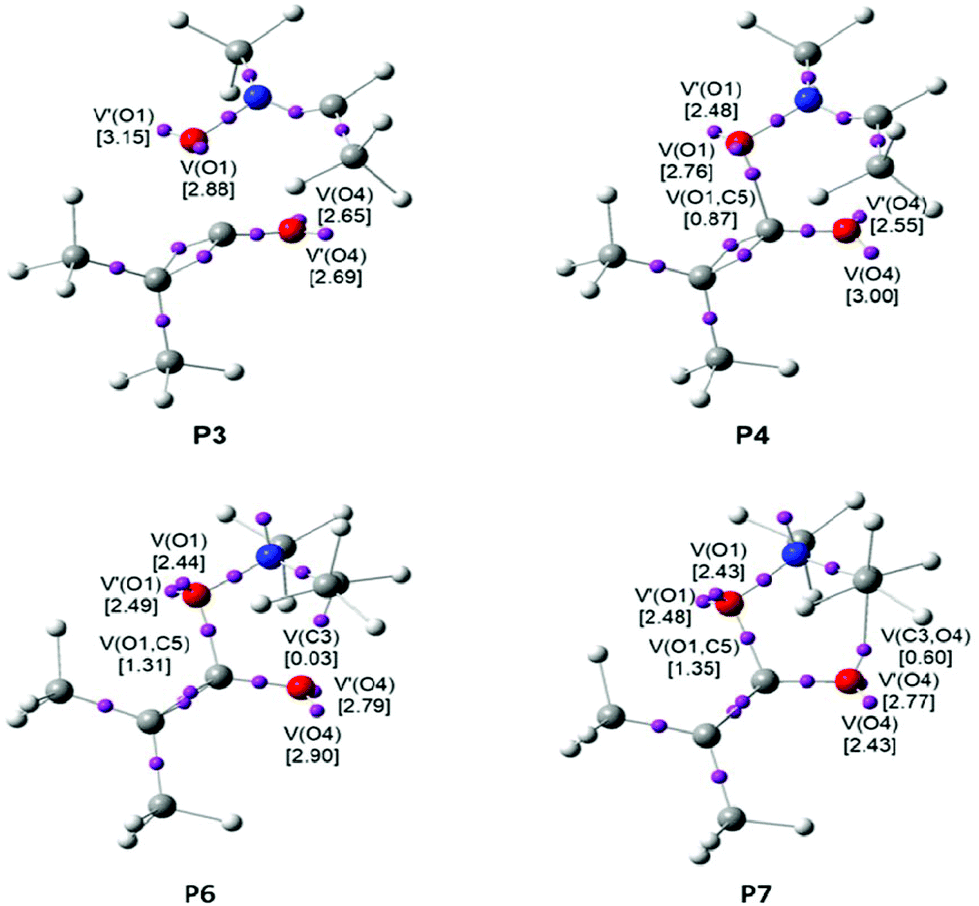 | ||
| Fig. 5 ELF attractor positions for the points of the IRC defining phases IV, V, VII and VIII involved in the formation of the O1–C5 and C3–O4 single bonds along the most favourable reactive channel associated with the zw-type 32CA reaction between nitrone 1b and ketene 4b. The electron populations, in e, are given in brackets. | ||
3.5. ELF topological analysis of the stationary points involved in the most favourable reactive channel associated with the 32CA reaction of nitrone 1b with electrophilic ketene 4c
In order to understand how the EW substitution in the ketene can modify the molecular mechanism of the zw-type 32CA reaction of nitrones with electrophilic ketenes, the bonding patterns of the stationary points involved in the most favourable reactive channel associated with the 32CA reaction of nitrone 1b with electrophilic ketene 4c were characterised by ELF topological analysis. ELF topological analysis is reported in the ESI.†Some appealing conclusions can be obtained from this ELF topological analysis: (i) at TS1-CO, no bonding change with respect to MC-c is observed. The bonding pattern of TS1-CO resembles that associated with the structures located at the end of phase I of the reaction between nitrone 1b and ketene 4b, emphasising its earlier character; (ii) as expected, at IN-CO, while the formation of the O1–C5 single bond is very advanced, the formation of the C3–O4 single bond has not yet begun; (iii) the bonding pattern of IN-CO resembles that of point P5 of the reaction between nitrone 1b and ketene 4b. The main difference between IN-CO and P5 is the C3–O4 distance: 2.745 at IN-CO and 2.329 Å at P5. The short C3–O4 distance at P5 justifies that this species is not a stationary point in the PES of the 32CA reaction between nitrone 1b and ketene 4b; (iv) the bonding pattern of TS2-CO resembles that of TS-CO. The only difference is that TS2-CO is slightly more advanced than TS-CO; and (v) the GEDT at IN-CO and TS2-CO is larger than that at P5 and TS-CO as a consequence of the higher electrophilic character of ketene 4c than ketene 4b. The higher polar character of the zw-type 32CA reaction between nitrone 1b and electrophilic ketene 4c permits the stabilisation of the zwitterionic intermediate, thus changing the molecular mechanism from a one-step mechanism to a two-step one, but the bonding changes in both 32CA reactions are essentially the same. This similar bonding pattern along the two 32CA reactions makes it possible to establish the non-concerted nature of the two-stage one-step mechanism.49
4. Conclusions
A comprehensive MEDT study of the reaction between nitrones and ketenes has been carried out using DFT methods at the MPWB1K/6-311G(d,p) computational level. ELF analysis of nitrones confirmed their electronic structure as TACs participating in zw-type 32CA reactions although NPA clearly shows that the common representation as 1,2-zwitterions is incorrect. The analysis of CDFT reactivity indices of nitrones and ketenes correctly predicts the experimentally observed complete CO regio- and chemoselectivities. A detailed analysis of the PES corresponding to the reaction between nitrones and non-electrophilically activated ketenes indicates that the reaction follows a two-stage one-step mechanism in which the formation of the second bond takes place once the first one is almost completely formed. No significant changes are observed by the inclusion of solvent effects. Analysis of the thermodynamic data indicates that while the initial 32CA reaction of the nitrone towards the ketene C–O double bond is kinetically regio- and chemoselectively controlled, the products resulting from the addition of the ketene C–C double bond can be obtained under thermal equilibrium conditions, in clear agreement with the experimental outcomes.
A complete BET study confirms the non-concerted nature of the reaction identifying the mechanism as a [2n,2n] process, i.e. only two non-bonding electrons of the oxygen lone pairs of the nitrone and two non-bonding electrons of the oxygen lone pairs of the ketene are mainly involved in the formation of the two C–O single bonds. Finally, an ELF topological analysis of the electron density distribution of the stationary points involved in the most favourable reactive channel associated with the 32CA reaction involving electrophilic ketenes predicts a switch of mechanism from two-stage one-step to two-step. Despite the change of mechanism, the bonding pattern is the same for the two reactions, thus the present computational study confirms the non-concerted nature of the cycloaddition between nitrones and ketenes.
Acknowledgements
This research was supported by the Spanish MINECO (FEDER CTQ2016-78669-P and CTQ2016-76155-R), and the Government of Aragón (Grupos Consolidados, E.10). M. R.-G. thanks MINECO for a pre-doctoral contract co-financed by the European Social Fund (BES-2014-068258). The authors thankfully acknowledge the resources from the supercomputers “Memento” and “Cierzo”, technical expertise and assistance provided by BIFI-ZCAM (Universidad de Zaragoza, Spain).References
- L. Irvin, Chem. Rev., 1938, 23, 193–285 CrossRef.
- P. N. Confalone and E. M. Huie, Org. React., 1988, 36, 1–174 CrossRef CAS.
- J. N. Martin and R. C. F. Jones, in Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry toward Heterocycles and Natural Products, ed. A. Padwa and W. H. Pearson, Wiley, Chichester, United Kingdom, 2002, vol. 59, pp. 1–81 Search PubMed.
- (a) P. Merino, in Science of Synthesis, ed. D. Bellus and A. Padwa, George Thieme, Stuttgart, 2004, vol. 27, pp. 511–580 Search PubMed; (b) P. Merino, in Science of Synthesis, ed. E. Schaumann, George Thieme, Stuttgart, 2011, vol. 2010/4, pp. 325–403 Search PubMed.
- (a) S. Kanemasa, Synlett, 2002, 1371–1387 CrossRef CAS; (b) S. Kanemasa, in Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry toward Heterocycles and Natural Products, ed. A. Padwa and W. H. Pearson, Wiley, Chichester, United Kingdom, 2002, vol. 59, pp. 755–815 Search PubMed; (c) D. A. Evans, F. Kleinbeck and M. Rüping, in Asymmetric Synthesis - The Essentials, ed. M. Christmann and S. Bräse, Wiley-VCH, Weinheim, 2006, pp. 72–77 Search PubMed.
- K. Rück-Braun, T. H. E. Freysoldt and F. Wierschem, Chem. Soc. Rev., 2005, 34, 507–516 RSC.
- (a) J. Revuelta, S. Cicchi, A. Goti and A. Brandi, Synthesis, 2007, 485–504 CAS; (b) A. Brandi, F. Cardona, S. Cicchi, F. M. Cordero and A. Goti, Chem. – Eur. J., 2009, 15, 7808–7821 CrossRef CAS PubMed; (c) M. Frederickson, Tetrahedron, 1997, 53, 403–425 CrossRef CAS; (d) K. V. Gothelf, in Cycloaddition Reactions in Organic Synthesis, ed. S. Kobayashi and K. A. Jorgensen, Wiley-VCH, Inc., New York, 2002, pp. 211–247 Search PubMed.
- (a) J. Malinina, T. Q. Tran, A. V. Stepakov, V. V. Gurzhiy, G. L. Starova, R. R. Kostikov and A. P. Molchanov, Tetrahedron Lett., 2014, 55, 3663–3666 CrossRef CAS; (b) T. Kawai, K.-H. Kodama, T. Ooi and T. Kusumi, Tetrahedron Lett., 2004, 45, 4097–4099 CrossRef CAS; (c) A. Padwa, D. N. Kline, K. F. Koehler, M. Matzinger and M. K. Venkatramanan, J. Org. Chem., 1987, 52, 3909–3917 CrossRef CAS.
- (a) D.-L. Mo, W. H. Pecak, M. Zhao, D. J. Wink and L. L. Anderson, Org. Lett., 2014, 16, 3696–3699 CrossRef CAS PubMed; (b) J. Holt and A. Fiksdahl, J. Heterocycl. Chem., 2007, 44, 375–379 CrossRef CAS.
- (a) N. Celebi-Oelcuem, Y.-h. Lam, E. Richmond, K. B. Ling, A. D. Smith and K. N. Houk, Angew. Chem., Int. Ed., 2011, 50, 11478–11482 CrossRef CAS PubMed; (b) E. Richmond, N. Duguet, A. M. Z. Slawin, T. Lebl and A. D. Smith, Org. Lett., 2012, 14, 2762–2765 CrossRef CAS PubMed; (c) A. R. Evans, M. Hafiz and G. A. Taylor, J. Chem. Soc., Perkin Trans. 1, 1984, 1241–1245 RSC.
- K. Kavitha and P. Venuvanalingam, J. Chem. Soc., Perkin Trans. 2, 2002, 2130–2139 RSC.
- A. Darù, D. Roca-López, T. Tejero and P. Merino, J. Org. Chem., 2016, 81, 673–680 CrossRef PubMed.
- E. Richmond, K. B. Ling, N. Duguet, L. B. Manton, N. Çelebi-Ölçüm, Y.-H. Lam, S. Alsancak, A. M. Z. Slawin, K. N. Houk and A. D. Smith, Org. Biomol. Chem., 2015, 13, 1807–1817 CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- L. R. Domingo, Molecules, 2016, 21, 1319 CrossRef PubMed.
- L. R. Domingo and S. R. Emamian, Tetrahedron, 2014, 70, 1267–1273 CrossRef CAS.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, Tetrahedron, 2016, 72, 1524–1532 CrossRef CAS.
- L. R. Domingo, M. J. Aurell and P. Pérez, Tetrahedron, 2014, 70, 4519–4525 CrossRef CAS.
- X. Krokidis, S. Noury and B. Silvi, J. Phys. Chem. A, 1997, 101, 7277–7282 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 6908–6918 CrossRef CAS.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- (a) H. B. Schlegel, J. Comput. Chem., 1982, 3, 214–218 CrossRef CAS; (b) Modern Electronic Structure Theory, ed. H. B. Schlegel and D. R. Yarkony, World Scientific Publishing, Singapore, 1994 Search PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- (a) C. González and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5525 CrossRef; (b) C. González and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853–5860 CrossRef.
- (a) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS; (b) B. Y. Simkin and I. Sheikhet, Quantum Chemical and Statistical Theory of Solutions– Computational Approach, Ellis Horwood, London, 1995 Search PubMed.
- (a) E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS; (b) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS; (c) V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404–417 CrossRef CAS.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- S. Noury, X. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597–604 CrossRef CAS.
- (a) R. G. Parr and W. Yang, Annu. Rev. Phys. Chem., 1995, 46, 701–728 CrossRef CAS PubMed; (b) H. Chermette, J. Comput. Chem., 1999, 20, 129–154 CrossRef CAS; (c) F. De Proft and P. Geerlings, Chem. Rev., 2001, 101, 1451–1464 CrossRef CAS PubMed; (d) P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1873 CrossRef CAS PubMed; (e) P. W. Ayers, J. S. M. Anderson and L. J. Bartolotti, Int. J. Quantum Chem., 2005, 101, 520–534 CrossRef CAS; (f) J. L. Gázquez, J. Mex. Chem. Soc., 2008, 52, 3–10 Search PubMed; (g) R. F. Nalewajski, J. Korchowiec and A. Michalak, Density Functional Theory IV, Topics in Current Chemistry, Springer, Berlin, Heidelberg, 1996, vol. 183, p. 25 Search PubMed; (h) P. Geerlings, S. Fias, Z. Boisdenghien and F. De Proft, Chem. Soc. Rev., 2014, 43, 4989–5008 RSC.
- R. G. Parr, L. von Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- (a) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS; (b) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- (a) L. R. Domingo, E. Chamorro and P. Pérez, J. Org. Chem., 2008, 73, 4615–4624 CrossRef CAS PubMed; (b) L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2011, 9, 7168–7175 RSC.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, 1133–1138 CrossRef.
- L. R. Domingo, P. Pérez and J. A. Sáez, RSC Adv., 2013, 3, 1486–1494 RSC.
- L. R. Domingo, E. Chamorro and P. Pérez, Lett. Org. Chem., 2010, 7, 432–439 CrossRef CAS.
- L. R. Domingo, M. Ríos-Gutérrez and P. Pérez, Molecules, 2016, 21, 748 CrossRef PubMed.
- L. R. Domingo, RSC Adv., 2014, 4, 32415–32428 RSC.
- L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, Tetrahedron, 2002, 58, 4417–4423 CrossRef CAS.
- P. Jaramillo, L. R. Domingo, E. Chamorro and P. Pérez, J. Mol. Struct. (THEOCHEM), 2008, 865, 68–72 CrossRef CAS.
- (a) R. Pratt, D. P. Stokes and G. A. Taylor, J. Chem. Soc., Perkin Trans. 1, 1975, 498–503 RSC; (b) A. F. Gettins, D. P. Stokes and G. A. Taylor, J. Chem. Soc., Perkin Trans. 1, 1977, 1849–1855 RSC; (c) M. Hafiz and G. A. Taylor, J. Chem. Soc., Perkin Trans. 1, 1980, 1700–1705 RSC; (d) A. R. Evans, M. Hafiz and G. A. Taylor, J. Chem. Soc., Perkin Trans. 1, 1984, 1241–1245 RSC; (e) C. P. Falshaw, M. Hafiz and G. A. Taylor, J. Chem. Soc., Perkin Trans. 1, 1985, 1837–1843 RSC.
- M. Ríos-Gutiérrez, P. Pérez and L. R. Domingo, RSC Adv., 2015, 5, 58464–58477 RSC.
- L. R. Domingo and J. A. Sáez, Org. Biomol. Chem., 2009, 7, 3576–3583 CAS.
- (a) S. Berski, J. Andrés, B. Silvi and L. R. Domingo, J. Phys. Chem. A, 2003, 107, 6014–6024 CrossRef CAS; (b) V. Polo, J. Andrés, S. Berski, L. R. Domingo and B. Silvi, J. Phys. Chem. A, 2008, 112, 7128–7136 CrossRef CAS PubMed; (c) J. Andrés, P. Gonzáez-Navarrete and V. Safont, Int. J. Quantum Chem., 2014, 114, 1239–1252 CrossRef.
- (a) R. Thom, Structural Stability and Morphogenesis: An Outline of a General Theory of Models, Inc., Reading, MA, 1976 Search PubMed; (b) A. E. R. Woodcock and T. Poston, A Geometrical Study of Elementary Catastrophes, Spinger-Verlag, Berlin, 1974 Search PubMed; (c) R. Gilmore, Catastrophe Theory for Scientists and Engineers, Dover, New York, 1981 Search PubMed.
- (a) V. Polo, J. Andrés, R. Castillo, S. Berski and B. Silvi, Chem. – Eur. J., 2004, 10, 5165–5172 CrossRef CAS PubMed; (b) J. C. Santos, J. Andrés, A. Aizman, P. Fuentealba and V. Polo, J. Phys. Chem. A, 2005, 109, 3687–3693 CrossRef CAS PubMed; (c) S. Berski, J. Andrés, B. Silvi and L. R. Domingo, J. Phys. Chem. A, 2006, 110, 13939–13947 CrossRef CAS PubMed; (d) V. Polo and J. Andrés, J. Chem. Theory Comput., 2007, 3, 816–823 CrossRef CAS PubMed; (e) S. Berski and Z. Ciunik, Mol. Phys., 2015, 113, 765–781 CrossRef CAS.
- L. R. Domingo, M. Ríos-Gutiérrez and J. A. Sáez, RSC Adv., 2015, 5, 37119–37129 RSC.
- L. R. Domingo, J. A. Saéz, R. J. Zaragozá and M. Arnó, J. Org. Chem., 2008, 73, 8791–8799 CrossRef CAS PubMed.
- M. Ríos-Gutiérrez, P. Pérez and L. R. Domingo, RSC Adv., 2015, 5, 84797–84809 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: BET characterisation of the molecular mechanism of the zw-type 32CA reaction between nitrone 1b and ketene 4b. ELF topological analysis of the stationary points involved in the most favourable reactive channel associated with the 32CA reaction of nitrone 1b with electrophilic ketene 4c. Tables with the MPWB1K/6-311G(d,p) total and relative electronic energies, in the gas phase and in benzene, and enthalpies, entropies and Gibbs free energies, and the relative ones, computed at room temperature and 1 atm in benzene, for the stationary points involved in the 32CA reactions between nitrone 1b and ketenes 4b,c. See DOI: 10.1039/c6ob02768g |
| This journal is © The Royal Society of Chemistry 2017 |