
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetraceno[2,1,12,11-opqra]tetracene-extended tetrathiafulvalene – redox-controlled generation of a large PAH core†
Søren Lindbæk
Broman
a,
Cecilie Lindholm
Andersen
a,
Tanguy
Jousselin-Oba
b,
Mads
Mansø
a,
Ole
Hammerich
a,
Michel
Frigoli
*b and
Mogens Brøndsted
Nielsen
 *a
*a
aDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen Ø, Denmark. E-mail: mbn@chem.ku.dk
bUMR CNRS 8180, UVSQ, Institut Lavoisier de Versailles, 45 Avenue des Etats-Unis, 78035 Versailles Cedex, France. E-mail: michel.frigoli@uvsq.fr
First published on 22nd December 2016
Abstract
Two tetraceno[2,1,12,11-opqra]tetracene-extended tetrathia-fulvalenes were prepared and found to undergo reversible conversion into their planar polycyclic aromatic hydrocarbons (PAHs) upon electrochemical oxidation – at potentials probing the best valence bond representations.
Tetrathiafulvalene (TTF) is a redox-active molecule undergoing two reversible one-electron oxidations, generating two aromatic 1,3-dithiolium rings,1 and has for this reason found wide interest in both materials and supramolecular chemistry.2 Its redox properties can be finely tuned by incorporation of a π-extended spacer between the two dithiafulvene (DTF) units.3 By incorporation of a 9,10-anthracenediylidene spacer as in compound 1 (Fig. 1), two-electron oxidation does not only lead to generation of two aromatic dithiolium rings but also to an aromatic anthracene core.4 Similarly, tetracene and pentacene cores have been obtained by two-electron oxidation of the corresponding extended TTFs (ext-TTFs)5 and a pentacene core from a derivative of compound 2 by removal of four electrons.6 The oxidations have significant consequences for the molecular geometries, changing from “butterfly-like” or “saddle-like” shapes to planar acene cores to which the 1,3-dithiolium rings are nearly perpendicular. Synthesis of large polycyclic aromatic hydrocarbons (PAHs) or nanographenes, being small sections of graphene, has in parallel found wide interest due to their electronic properties of relevance for development of organic devices such as light-emitting diodes, field-effect transistors, and photovoltaic devices.7 The combination of PAHs and TTFs should thus have the potential to furnish molecules and materials with new interesting properties and possessing multiple redox states with distinct conformations. For compounds including two DTF units, the dication would consist of the intact PAH π-core in regard to geometry, conjugation pattern and number of Clar's aromatic sextets. Very recently, Giguère and Morin8 reported derivatives of one such super-extended TTF, 3 (ANT-TTF), based on an anthanthrene (ANT) core containing six fused benzene rings. We became interested in expanding the number of rings further and turned our attention to the dibenzo-anthanthrene which is a tetraceno[2,1,12,11-opqra]tetracene scaffold (TT) containing a total of eight fused benzene rings.
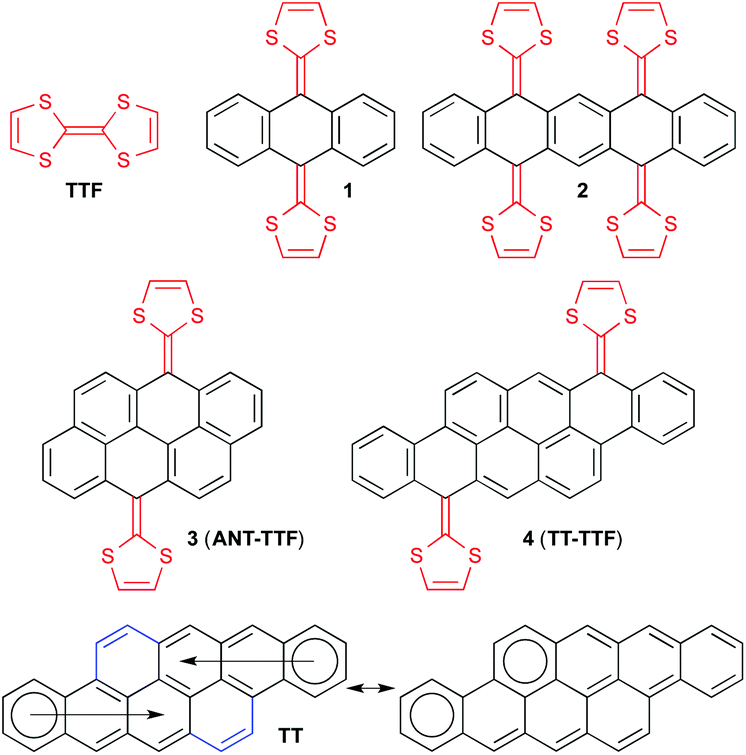 | ||
| Fig. 1 Extended tetrathiafulvalenes (ext-TTFs) and Clar's representation of TT. | ||
This PAH was first reported in 1937 by Vollmann et al.9 It has been shown recently by Frigoli and co-workers10 that TT derivatives functionalized with (triisopropylsilyl)acetylene substituents showed very similar optical and electrochemical properties as compared to those of the 6,13-bis(triisopropylsilylethynyl)pentacene, but with much higher stability. This stability was attributed to the aromaticity of the TT unit, which is comparable to an anthracene along each tetracene (Fig. 1). Indeed, according to the Clar's sextet rule, the TT core is best described as two anthracenes fused at the a face and bridged by two 1,2-vinylidene spacers (Fig. 1). This description was based on simple structural considerations supported by NICS calculations. Here we present the synthesis and properties of ext-TTFs (4, TT-TTF) based on this core (functionalized with substituents to furnish solubility and to prevent aggregation in solution). By comparing the redox properties to those of the previously reported ext-TTFs, we find that the value of the oxidation potential of such molecules is probing the change of aromaticity when proceeding from the ext-TTF to the acene and consequently provides important information on the inherent aromatic character of TT.
The dione 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 containing 2,6-dimethylphenyl substituent groups was employed as starting material (Scheme 1). We and others have recently employed the Horner–Wadsworth–Emmons (HWE) reaction to prepare indenofluorene-extended TTFs from a dione and phosphonate esters using sodium hexamethyldisilazide (NaHMDS) as a base,11 and we therefore aimed at using similar conditions here. Phosphonate esters 6a11a and 6b12 with peripheral SEt and SHex groups, respectively, were prepared following known procedures. Dropwise addition of the preformed phosphonate ylide of 6b (prepared at −78 °C by addition of NaHMDS to the phosphonate ester) to 5 only gave trace amount of the mono-coupled intermediate 7b (Fig. 2). Conversely, we found that simply adding NaHMDS to a mixture of 5 and 6b in THF at rt gave the desired super-extended TTF 8b in good yield (40%).13 Although product formation is almost instantaneous upon addition of the base, the reaction does not proceed to completion. Thus, under these conditions, significant amounts of unreacted starting material 5 (51%) as well as a small amount of the mono-coupled intermediate 7b were isolated. The reactivity of the dione 5 in the HWE reaction is generally very low and unreliable. For example we found, to our surprise, that only the SHex-capped phosphonate ester 6b gave good yields whereas for the corresponding SEt derivative 6a only small amounts of the desired product 8a were formed. It is noteworthy that a HWE reaction was previously attempted for the synthesis of a derivative of 3 using a dithiole with no peripheral substituents, but without success.8 It seems that the peripheral substituents play a significant role for the success of this reaction and not only the dione itself.
10 containing 2,6-dimethylphenyl substituent groups was employed as starting material (Scheme 1). We and others have recently employed the Horner–Wadsworth–Emmons (HWE) reaction to prepare indenofluorene-extended TTFs from a dione and phosphonate esters using sodium hexamethyldisilazide (NaHMDS) as a base,11 and we therefore aimed at using similar conditions here. Phosphonate esters 6a11a and 6b12 with peripheral SEt and SHex groups, respectively, were prepared following known procedures. Dropwise addition of the preformed phosphonate ylide of 6b (prepared at −78 °C by addition of NaHMDS to the phosphonate ester) to 5 only gave trace amount of the mono-coupled intermediate 7b (Fig. 2). Conversely, we found that simply adding NaHMDS to a mixture of 5 and 6b in THF at rt gave the desired super-extended TTF 8b in good yield (40%).13 Although product formation is almost instantaneous upon addition of the base, the reaction does not proceed to completion. Thus, under these conditions, significant amounts of unreacted starting material 5 (51%) as well as a small amount of the mono-coupled intermediate 7b were isolated. The reactivity of the dione 5 in the HWE reaction is generally very low and unreliable. For example we found, to our surprise, that only the SHex-capped phosphonate ester 6b gave good yields whereas for the corresponding SEt derivative 6a only small amounts of the desired product 8a were formed. It is noteworthy that a HWE reaction was previously attempted for the synthesis of a derivative of 3 using a dithiole with no peripheral substituents, but without success.8 It seems that the peripheral substituents play a significant role for the success of this reaction and not only the dione itself.
 | ||
| Scheme 1 Synthesis of tetracenotetracene-extended TTFs. | ||
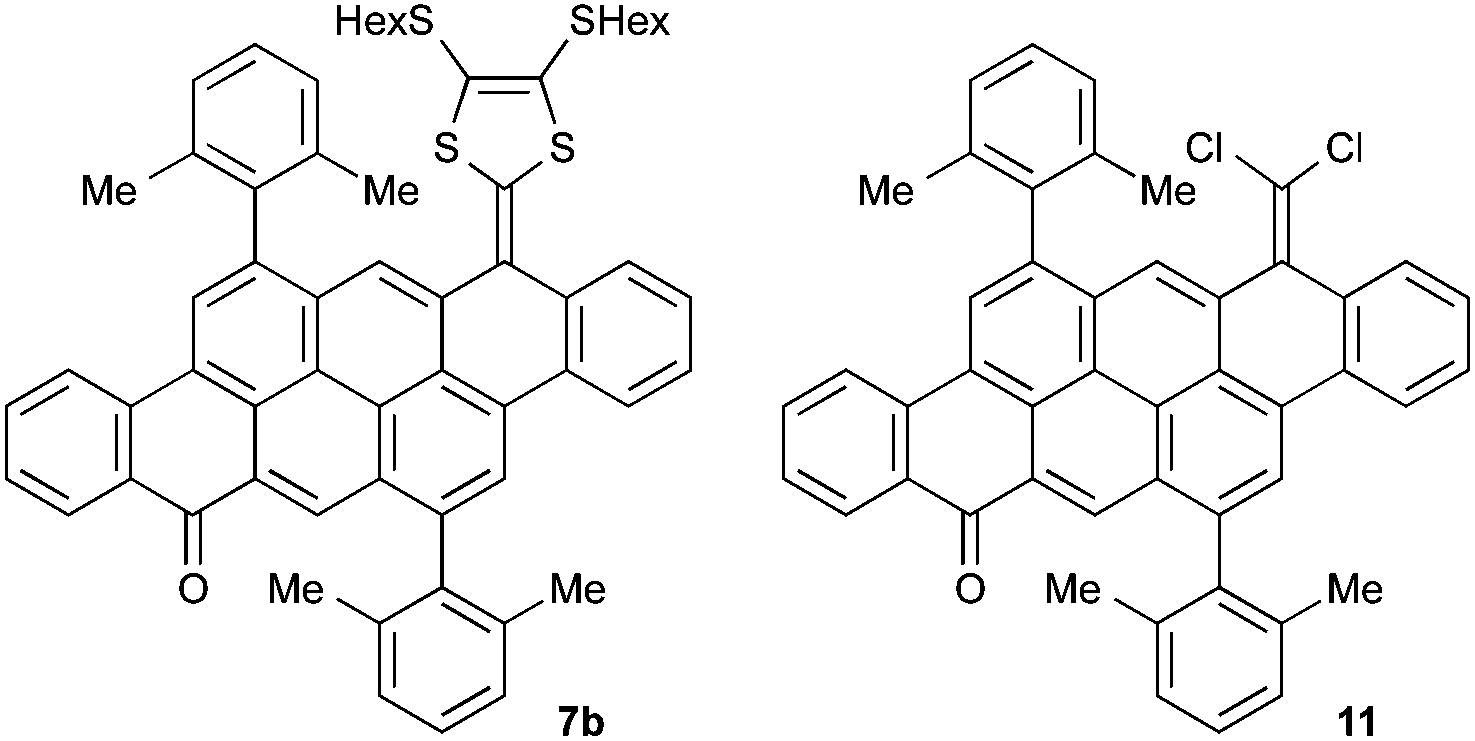 | ||
| Fig. 2 Structures of intermediates 7b and 11. | ||
To further extend the conjugated π-system, we prepared the benzo-fused super-extended TTF 9 following a recent methodology for generating the DTF rings.8 A chloroolefination of 5 gave 10 (72%) along with some of the intermediate 11 (18%). Next, treating 10 with benzene-1,2-dithiole in the presence of Bu4NOH gave 9 (55%). This product suffered from poor solubility, which limited its characterization, but electrochemical characteristics were achieved.
The UV-Vis absorption spectra of 8b in various solvents are shown in Fig. 3. The compound exhibits a longest-wavelength absorption maximum at 571 nm in CH2Cl2 with a remarkably high molar absorptivity of 59.9 × 103 M−1 cm−1. For comparison, the longest-wavelength absorption maximum of the native TT is around 586–589 nm,14 and the disruption of this PAH by the two DTF units is therefore seen to give a small blueshifted absorption. Only minor solvatochromic effects are observed in the studied solvents (CH2Cl2, CHCl3, THF, PhMe, acetone, and MeCN). Thus, only a small blueshift is observed in MeCN15 (λmax = 562 nm; see ESI†). No concentration dependence was observed in the concentration range 2.0 × 10−6–2.2 × 10−5 M in CH2Cl2 as the UV-Vis absorption data followed the Lambert–Beer law when plotting the absorbance at different wavelengths against the concentration (see ESI†). Thus, the aryl substituents efficiently disrupt any aggregation.
 | ||
| Fig. 3 UV-Vis absorption spectra of 8b in various solvents. | ||
To shed light on the possible conformations of the TT-TTF, density functional theory (DFT) calculations (RB3LYP/cc-pVDZ) were performed using Gaussian 09.16 To save computational time, these studies focused on 8a with SEt groups, and the effect of the orientation of the SEt groups was not addressed. Two conformers were obtained (Fig. 4); for the one, the two DTF units are in a syn relationship and the core is U-shaped, while for the other, the two DTF units are in anti relationship and the core is S-shaped. The thermodynamic stabilities of the two conformers are very close, with the syn form being only 1.5 kJ mol−1 more stable. For both conformers, the aryl groups are nearly orthogonal to the core, hence preventing association between molecules in agreement with the spectroscopic studies. For 6,14-bis(2,6-dimethylphenyl)-tetraceno[2,1,12,11-opqra]tetracene, the two aryl groups were also found to be orthogonal to the core (ESI†). Hindered rotation around the TT-aryl single bond makes the two Me groups on each aryl ring of 8b non-identical in the non-planar syn and anti conformations, but they are observed as a singlet in the 1H-NMR spectrum at 300 K (500 MHz; CD2Cl2), which indicates that the DTF units flip fast between up and down positions, interconverting the environment of the Me groups. However, at 270 K the signal is broadened and at 240 K it appears as two singlets (see ESI;† slow exchange situation). Further cooling to 211 K causes each of the two singlets to split into a small and large singlet (ratio of ca. 1:3.5), possibly indicating that the inherently different Me protons on the syn and anti conformations are now distinguishable; thus, each conformer has two different pairs of identical Me groups (pointing up and down).
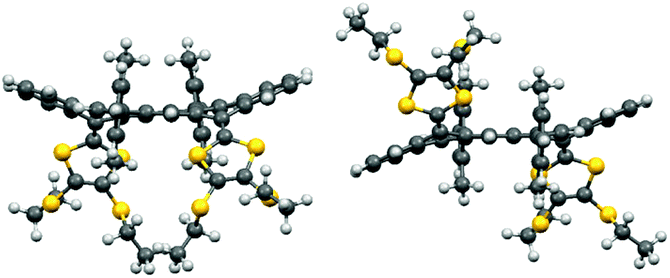 | ||
| Fig. 4 Geometry-optimized structures of 8a-syn (left) and 8a-anti (right). | ||
The redox properties of 8b were studied by cyclic voltammetry in CH2Cl2 (0.1 M Bu4NPF6). The cyclic voltammogram shows three oxidation steps corresponding to the exchange of two, one, and one electron, respectively (Fig. 5). First, a chemically reversible two-electron process is observed at E°′ = 0.008 V (vs. Fc/Fc+) corresponding to the oxidation of the two DTF units to the dithiolium state which is accompanied by the formation of a fully aromatic TT core (Scheme 2). This is in analogy with what has been reported for smaller acenes.5,8,17 DFT calculations supported generation of the planar TT core with bond lengths similar to those calculated for the parent TT (see ESI†). The change in bond length for one of the carbon–carbon bonds of the core upon oxidation is indicated in Scheme 2 (using 8a for calculational convenience). The second reversible and third irreversible oxidation step, observed at E°′ = 0.716 V and Ep = 1.124 V, reflect the one-electron oxidations of the TT core, first to a persistent radical cation and then to a highly reactive dication. The latter undergoes a chemical follow-up reaction, most probably due to nucleophilic attack by residual water, resulting in the formation of a cationic intermediate, the irreversible reduction of which is seen at Ep = 0.25 V during the backward scan. This classical behavior of the aromatic core has earlier been reported for a number of other aromatic hydrocarbons including, e.g., 9,10-disubstituted anthracenes.18 The structurally related compound 9 showed similar behavior (see ESI†) with the two-electron process being observed at E°′ = 0.071 V and electron transfer processes related to the core at E°′ = 0.768 V and Ep = 1.21 V. For a derivative of TT (TTa, see ESI, p. S10†) some non-ideal behavior was observed, but the voltammogram allowed for estimates of the formal potential for the 1st redox couple (E°′ = 0.196 V; formation of TTa˙+), and the peak potential for the 2nd electron transfer process (Ep = 0.869 V; formation and further reaction of TTa2+). Thus, the presence of the positively charged dithiolium rings increases the oxidation potential of the TT core by about 0.5 V.
 | ||
| Fig. 5 Cyclic voltammograms for the oxidation of 8b (1 mM) in CH2Cl2 (0.1 M Bu4NPF6) obtained at three different values of the switch potential. Scan rate 0.1 V s−1. | ||
 | ||
| Scheme 2 Oxidation events of 8b. Ar = 2,6-dimethylphenyl. Calculated lengths of the highlighted bonds are shown in the box (here for 8a instead of 8b). | ||
It is interesting to compare the first two-electron oxidation potentials of 9 to those reported previously4a–c,8 for benzo-fused ext-TTFs (Fig. 6). The oxidation becomes more difficult (larger potential) the longer the resulting acene is. A reverse trend is observed for acenes in which the oxidation potential, the aromaticity, and the stability decrease along with increasing acene length.19 Based on these two observations, two statements can be given. The oxidation potential gives information about the change of aromaticity from the ext-TTF to the acene, which can be seen from the difference in number of aromatic sextets between the two forms. Secondly, the aromaticity of the acene produced upon oxidation can be gauged. The ratio of aromatic sextets between TT-TTF and TT is 4:2; the ratio is 2:1 for any ext-TTF and its corresponding acene (≥3 rings). However, the oxidation potential of the TT-TTF is close to that of the anthracenediylidene-extended TTF (as we expect a slightly lower potential in MeCN than in CH2Cl2), suggesting that TT has an aromaticity similar to anthracene (in agreement with other work10). According to NICS calculations, ANT should have an aromaticity similar to that of naphthalene.20 The ratio of aromatic sextets between ANT-TTF and ANT is 2:2; likewise a ratio of 1:1 is observed between the naphthalenediylidene-extended TTF and naphthalene. The oxidation potential of the smaller ANT-TTF derivative is lower than that of the TT-TTF and indeed comparable to that of the naphthalenediylidene-extended TTF.
 | ||
| Fig. 6 Comparison of the first two-electron oxidation potential (vs. Fc/Fc+). Potentials in italics are estimated values from literature potentials against SCE by subtracting 0.39 V (in MeCN). Benzenoid rings are indicated by Clar structures. | ||
In conclusion, combination of TT and TTF has provided the to-date largest PAH-extended TTF derivative, which was shown to undergo a reversible conversion into the planar PAH core upon oxidation. Oxidation/reduction of exocyclic substituents is thus a convenient way of switching between PAH conformations. It supplements the structural approach where nonhexagonal defects are incorporated into nanographenes, such as 5- or 7-membered rings, causing distortions from planarity.21 Importantly, a comparison of redox potentials of PAH-ext-TTFs shows that the first two-electron oxidation potential is a convenient probe for the aromatic character of the 2-dimensional PAH formed. It is in agreement with an electronic description of TT as two fused anthracenes (with 1,2-vinylidene spacers) and of ANT as two fused naphthalenes.
We acknowledge the Villum Foundation and ANR (ANR-16-CE07-0024) for support. We are grateful to Mr Christian G. Tortzen for running variable temperature NMR experiments.
Notes and references
- M. B. Nielsen and S. P. A. Sauer, Chem. Phys. Lett., 2008, 453, 136 CrossRef CAS.
- (a) Chem. Rev., Special issue on Molecular Conductors, ed. P. Batail, 2004, 104, 4887; ; (b) S. Dolder, S.-X. Liu and S. Decurtins, Chimia, 2006, 60, 256 CrossRef CAS; (c) D. Canevet, M. Sallé, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245 RSC; (d) N. Martín, Chem. Commun., 2013, 49, 7025 RSC.
- (a) J. Roncali, J. Mater. Chem., 1997, 7, 2307 RSC; (b) A. Gorgues, P. Hudhomme and M. Sallé, Chem. Rev., 2004, 104, 5151 CrossRef CAS PubMed; (c) P. Frère and P. J. Skabara, Chem. Soc. Rev., 2005, 34, 69 RSC.
- (a) Y. Yamashita and T. Miyashi, Chem. Lett., 1988, 661 CrossRef CAS; (b) Y. Yamashita, Y. Kobayashi and T. Miyashi, Angew. Chem., Int. Ed. Engl., 1989, 28, 1052 CrossRef; (c) M. R. Bryce, E. Fleckenstein and S. Hünig, J. Chem. Soc., Perkin Trans. 2, 1990, 1777 RSC; (d) A. J. Moore and M. R. Bryce, J. Chem. Soc., Perkin Trans. 1, 1991, 157 RSC; (e) F. G. Brunetti, J. L. López, C. Atienza and N. Martín, J. Mater. Chem., 2012, 22, 4188 RSC.
- N. Martín, L. Sánchez, C. Seoane, E. Ortí, P. M. Viruela and R. Viruela, J. Org. Chem., 1998, 63, 1268 CrossRef.
- E. A. Younes and Y. Zhao, RSC Adv., 2015, 5, 88821 RSC.
- X. Feng, W. Pisula and K. Müllen, Pure Appl. Chem., 2009, 81, 2203 CrossRef CAS.
- J.-B. Giguère and J.-F. Morin, J. Org. Chem., 2015, 80, 6767 CrossRef PubMed.
- H. Vollmann, H. Becker, M. Corell and H. Streeck, Justus Liebigs Ann. Chem., 1937, 531, 1 CrossRef CAS.
- K. Sbargoud, M. Mamada, Y. Takeda, S. Tokito, A. Yassar, J. Marrot and M. Frigoli, submitted.
- (a) M. A. Christensen, C. R. Parker, T. J. Sørensen, S. de Graaf, T. J. Morsing, T. Brock-Nannestad, J. Bendix, M. M. Haley, P. Rapta, A. Danilov, S. Kubatkin, O. Hammerich and M. B. Nielsen, J. Mater. Chem. C, 2014, 2, 10428 RSC; (b) M. Mansø, M. Koole, M. Mulder, I. J. Olavarria-Contreras, C. L. Andersen, M. Jevric, S. L. Broman, A. Kadziola, O. Hammerich, H. S. J. van der Zant and M. B. Nielsen, J. Org. Chem., 2016, 81, 8406 CrossRef PubMed.
- For details, see ESI.†.
- For a related procedure where BuLi was added directly to a mixture of carbonyl derivative and phosphonate, see: P. Frère, K. Boubekeur, M. Jubault, P. Batail and A. Gorgues, Eur. J. Org. Chem., 2001, 3741 CrossRef.
- (a) E. Clar, Chem. Ber., 1943, 76, 328 CrossRef; (b) I. Bergman, Trans. Faraday Soc., 1956, 52, 829 RSC; (c) J. Sepiol, R. Kolos, A. Renn and U. P. Wild, Chem. Phys. Lett., 2002, 355, 71 CrossRef CAS.
- The solubility of 8b is extremely low in MeCN; the absorption spectrum is included in ESI.†.
- M. J. Frisch, et al., Gaussian 09, EM64L-G09RevB.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed. Full reference in ESI.†.
- (a) R. Glass, in Organic Electrochemistry, ed. O. Hammerich and B. Speiser, CRC Press, Boca Raton, 5th edn, 2015, ch. 27, pp. 1035–1102 Search PubMed; (b) N. E. Gruhn, N. A. Macías-Ruvalcaba and D. H. Evans, Langmuir, 2006, 22, 10683 CrossRef CAS PubMed.
- O. Hammerich and V. D. Parker, J. Am. Chem. Soc., 1974, 96, 4289 CrossRef CAS.
- (a) M. S. Deleuze, L. Claes, E. S. Kryachko and J.-P. François, J. Chem. Phys., 2003, 119, 3106 CrossRef CAS; (b) G. Portella, J. Poater, J. M. Bofill, P. Alemany and M. J. Sola, J. Org. Chem., 2005, 70, 2509 CrossRef CAS PubMed.
- G. Portella, J. Poater and M. Solà, J. Phys. Org. Chem., 2005, 18, 785 CrossRef CAS.
- K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols, electrochemical, spectroscopic and calculational data, and NMR spectra. See DOI: 10.1039/c6ob02666d |
| This journal is © The Royal Society of Chemistry 2017 |