
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic investigations of the asymmetric hydrosilylation of ketimines with trichlorosilane reveals a dual activation model and an organocatalyst with enhanced efficiency†
X.
Li
,
A. T.
Reeder
,
F.
Torri
,
H.
Adams
and
S.
Jones
 *
*
Department of Chemistry, University of Sheffield, Dainton Building, Brook Hill, Sheffield, UK S3 7HF. E-mail: simon.jones@sheffield.ac.uk
First published on 22nd February 2017
Abstract
Structural probes used to help elucidate mechanistic information of the organocatalyzed asymmetric ketimine hydrosilylation have revealed a new catalyst with unprecedented catalytic activity, maintaining adequate performance at 0.01 mol% loading. A new ‘dual activation’ model has been proposed that relies on the presence of both a Lewis basic and Brønsted acidic site within the catalyst architecture.
Introduction
The organocatalyzed asymmetric hydrosilylation of ketimines provides an effective method to prepare chiral amines in good yield and enantioselectivity. Since the first chiral Lewis-base derived catalyst was reported in 2001,1 there have been many variants of catalyst prepared and evaluated;2,3 selected examples include formamides,4–7 sulfinimides,8,9 pyridines,10–12 and organophosphorus compounds.13–15 The main focus of research from this group has been using an imidazole derived catalyst that has been shown to function as efficiently as the reported analogous picolinoyl series,16 in addition to being amenable to reductive amination protocols.17 Despite it being more than a decade since the first asymmetric catalysts for this type of transformation were reported, mechanistic data and insights have been sporadic, and only tentative models have been put forward to explain the selectivity.18 The most comprehensive study to date is that put forward by Malkov et al.,19 crucially noting the dependence of adventitious Brønsted acids to ensure the success of these reactions. Given the scarcity of mechanistic detail for these catalytic processes, a more in depth structural analysis of the imidazole catalyst system developed in these laboratories has now been conducted, providing new insights into the mechanism of this process, and has delivered a new catalyst with even higher activity.Results and discussion
The role of the Lewis basic amide in relation to the imidazole ring system was first investigated. A series of N-substituted imidazoles and their corresponding dimethylamides were prepared. 1-Methylimidazole 1 is commercially available, whereas 1-tert-butylimidazole 2 was prepared by condensing a mixture of glyoxal, aqueous formaldehyde, aqueous ammonia and tert-butylamine.20 These were converted to the dimethylamides 3 and 4 by deprotonation with n-BuLi and quenching with dimethylcarbamoyl chloride (Scheme 1), and evaluated in a benchmark HSiCl3 reduction of the N-PMP acetophenone ketimine 5 under a standard set of reaction conditions by removing aliquots and analyzing the conversion by 1H NMR spectroscopy (Fig. 1). N,N-Dimethyl benzamide was included as a benchmark to compare the role of the imidazole nitrogen atom.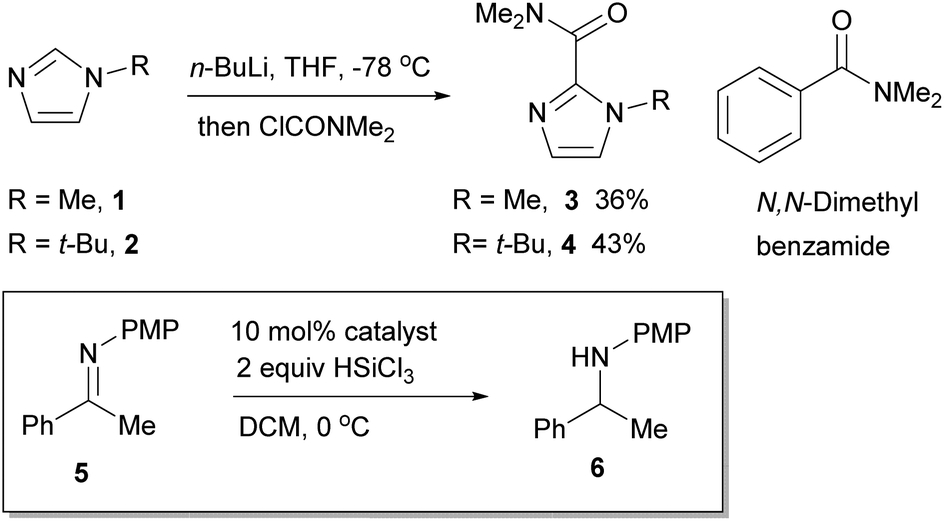 | ||
| Scheme 1 Synthesis and evaluation of imidazole catalysts 1–4 and N,N-dimethyl benzamide. | ||
 | ||
| Fig. 1 Relative reaction rates of imidazole catalysts 1–4 and N,N-dimethyl benzamide. Reactions performed using 10 mol% catalyst, 2 equiv. HSiCl3 in DCM (1 mL mmol−1) at 0 °C. Conversion calculated from comparison of appropriate integrals in 1H NMR spectrum of aliquots removed from reaction. | ||
The data obtained show slightly different reactivities between the N-Me and N-t-Bu catalysts 1 and 2 but this may be due to the N-Me catalyst not being completely soluble in DCM. However, what is noticeable is the significant difference in reaction rate between the two model dimethylamide catalysts; the N-Me catalyst 3 showed a significant increase in relative reaction rate compared to the parent imidazole, whilst the N-t-Bu catalyst 4 reaction rate dropped. This leads to the relative initial rate of the N-Me catalyst being of the order of 4 times faster than that of the N-t-Bu one. By comparison to the imidazole catalysts, N,N-dimethyl benzamide was a very poor catalyst, barely operating above the background reaction rate for these type of processes.
In order to probe this further, both dimethylamide catalysts 3 and 4 were studied using in situ infra-red experiments. The absorption signal of the carbonyl group of N-Me catalyst 3 in DCM initially appeared at 1635 cm−1 and after addition of one equivalent of HSiCl3, a new signal immediately appeared at 1668 cm−1, while the original signal at 1635 cm−1 simultaneously disappeared (Fig. 2). Addition of further equivalents of HSiCl3 led to no additional change other a dilution effect (observed in the slight drop of the signal at 1668 cm−1 after 8 min). This suggests that coordination of HSiCl3 with the amide is rapid and complete for this catalyst. In the case of the N-t-Bu catalyst 4, the original signal of the carbonyl group was at 1645 cm−1 with a smaller new signal appearing at 1671 cm−1 once treated with one equivalent of HSiCl3. Noticeably, in this case significant amounts of the un-coordinated catalyst were still present compared to the N-Me catalyst 3 and this ratio did not change with time, or when a further equivalent of HSiCl3 was added. This suggests that coordination of this catalyst to HSiCl3 is more difficult than in the case of the N-Me catalyst 3, and that steric effects of the imidazole N-alkyl group play an important role in the activity of such catalysts. In complete contrast, N,N-dimethyl benzamide, showed no change in the carbonyl group signal, correlating with its poor reactivity.
 | ||
| Fig. 2 From left to right; in situ IR data of N-Me catalyst 3 treated with HSiCl3 as a function of time; in situ IR data of catalyst N-t-Bu 4 treated with HSiCl3 as a function of time; in situ IR data of N,N-dimethyl benzamide treated with HSiCl3 as a function of time. | ||
For this series of compounds, it seems that the extent of coordination of the HSiCl3 with the carbonyl group is a reasonable measure of catalytic activity. The N-Me and N-t-Bu catalysts are likely to incur different and significant energetic penalties for the interaction of the amide N-methyl groups with the imidazole alkyl substituent, in either case probably ruling out a truly co-planar species 7a (Fig. 3). This probably favours a stable conformation that places the carbonyl group and imidazole ring orthogonal to each other i.e. structure 7b in Fig. 3 (note that these are depicted as reactive conformations, not active catalytic species that might be protonated at the imidazole nitrogen atom, vide infra). The increasing torsional angle then in-turn increases the distance of the silicon and imidazole nitrogen atoms, reducing reactivity, fitting well with observations with the N,N-dimethyl benzamide system, that lacks any cooperative effect of the imidazole nitrogen atom.
 | ||
| Fig. 3 Reactive conformations of catalysts 3 and 4. | ||
Naturally, these catalyst fragments lack the prolinol moiety that might have synergistic effects when considering the whole catalyst system, and any speculation about mechanism for these simple amides might not directly translate to more complex systems. Thus, in order to further probe such interactions, a series of chiral heterocyclic catalysts were investigated containing a prolinol derived backbone that were prepared starting from their parent carboxylic acids. 1-Methylimidazole-4-carboxylic acid 9 is commercially available, and 1-methylpyrrole-2-carboxylic acid 10 was obtained by hydrolysis of the methyl ester 12 with aqueous potassium hydroxide. 1-t-Butylimidazole-4-carboxylic acid 11 was accessed by deprotonation of 1-t-butylimidazole 2 with n-BuLi then quenching with ethyl chloroformate to give the ester 8, followed by basic hydrolysis. These three heterocyclic carboxylic acids 9–11 were transformed into the respective acid chlorides with oxalyl chloride, followed by immediate coupling with α,α-diphenylprolinol 13 giving catalysts 14–16, with previously reported catalyst 17 being prepared by a similar method as previously described (Scheme 2).16
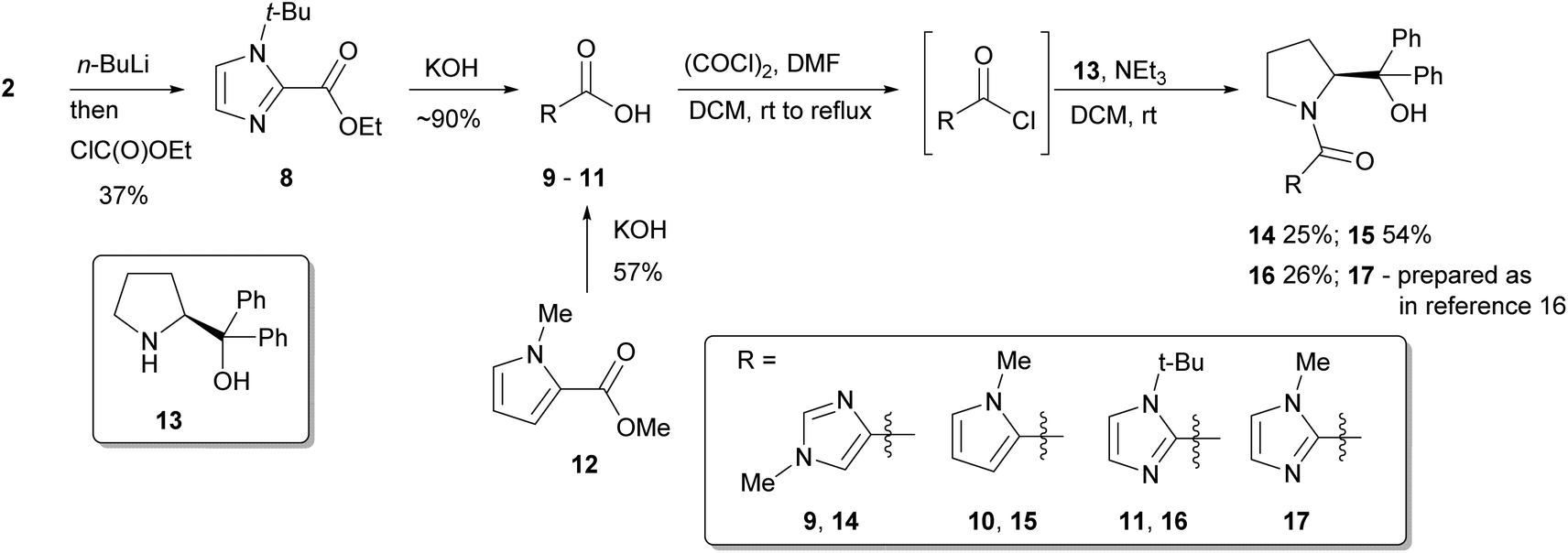 | ||
| Scheme 2 Synthesis of catalysts 14–17. | ||
All of these new catalysts were then evaluated under the standard set of reaction conditions (Scheme 1 and Fig. 4). The isomeric imidazole 14 was active but led to poor selectivity delivering the opposite enantiomer of product amine 6 to that normally observed. Given the torsional freedom that this isomeric system has compared to the parent imidazole catalyst 17, it is likely that an alternative reactive conformation may be accessible in this case, perhaps even a co-planar species analogous to 7a, resulting in a competitive pathway with a different stereochemical bias. The N-Me pyrrole catalyst 15 was essentially inactive which is interesting as it indicates that the amide functionality alone is not sufficient to promote this reaction. Based on the kinetic data for the dimethylamide catalyst 4, perhaps unsurprisingly the N-t-Bu catalyst 16 was essentially inactive, the 15% conversion being typical of the background reaction rate for this process. None of these surpassed the performance of the previously reported catalyst 17. This compares well with the findings obtained with the simple, achiral catalysts 3 and 4, whereby torsional freedom around the imidazole–carbonyl bond to appropriately locate the imidazole nitrogen atom is critical for high catalytic activity.
 | ||
| Fig. 4 Evaluation of prolinol catalysts 14–17 according to Scheme 1. | ||
The previously reported development of the imidazole catalyst 17 clearly demonstrated some key requirements in order to obtain both high yields and selectivity.16 Most critical for obtaining high selectivities was use of a gem-diaryl group. This most likely relays stereochemical information from the adjacent proline stereogenic center to create a chiral environment that facilitates the required facial differentiation of the planar ketimine.
In order to investigate this further, the gem-dimethyl catalyst 21 was prepared by coupling (S)-α,α-dimethylprolinol 19 (obtained from reaction of ester 18 with an excess of MeMgI, followed by hydrolysis) with ethyl N-methyl imidazole-1-carboxylate 20 and evaluated in the benchmark reduction of the N-PMP-ketimine of acetophenone 5 (Fig. 5 and Scheme 1). Noticeably, although the conversion to product was approximately the same as other catalysts previously prepared, the enantioselectivity was lower than that of the gem-diaryl catalyst 17, and similar to the catalyst 22 without the gem-dimethyl group. Thus, the role of this component may not simply be steric, an effect that has been previously documented for gem-diaryl groups.21
 | ||
| Fig. 5 Synthesis of catalyst 21 and comparison of activity with previous catalysts in the reduction of N-PMP ketimine 5 as shown in Scheme 1. | ||
In the final stage of catalyst evaluation, the role of the hydroxyl group was examined. Matsumura's work had previously shown that the deoxy-picoline derivative 24 catalyzed the asymmetric hydrosilylation of N-phenyl ketimine 25 but led to poor selectivity compared to the parent catalyst 23, inferring the need for a hydrogen bond to effectively coordinate with the ketimine substrate, leading to the commonly accepted model shown.12 In order to compare this effect in the imidazole series, removal of the hydrogen bond acceptor was achieved by methylation of catalyst 17 with sodium hydride and methyl iodide giving the O-Me catalyst 26 in 56% yield (Scheme 3). For comparison purposes, Matsumura's picolinic acid derived catalyst 23 was prepared from commercially available 2-picolinic acid by treating with oxalyl chloride, then reaction with α,α-diphenylprolinol 13. This was also O-methylated in analogous fashion to give catalyst 27 in 83% yield. These catalysts were then evaluated in the benchmark reduction of the N-PMP ketimine 5, each being challenged at low catalyst loadings to better determine efficacy (Table 1). Commercially available Sigamide® 28 was also included to provide a frame of reference.
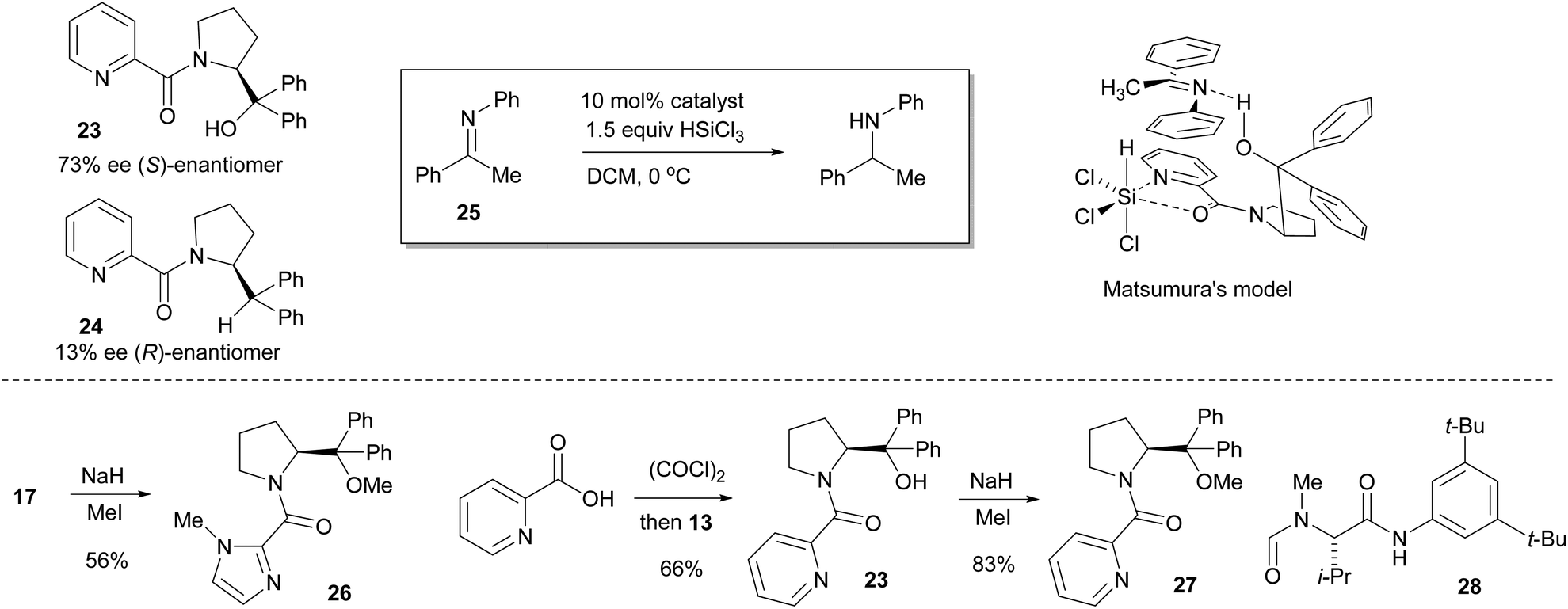 | ||
| Scheme 3 Matsumura's picolinoyl catalysts 23 & 24 and selectivity model, synthesis of O-methyl catalysts 26 & 27, and structure of Sigamide® 28. | ||
| Entry | Catalyst | Catalyst loading (mol%) | Conversionb (%) | eec (%) | ACEd |
|---|---|---|---|---|---|
| a Reactions conducted with N-PMP-ketimine 5 (1 mmol), HSiCl3 (2 mmol) at the specified catalyst loading in DCM (0.5 mL) at 0 °C for 4 h, followed by aqueous workup. b Conversion calculated by comparison of the value of the integrals for the product to residual ketimine and ketone (from hydrolysis). c Determined by chiral phase HPLC analysis. d See ref. 22. e Not determined. | |||||
| 1 | 17 | 1 | 100 | 90 | 56 |
| 2 | 0.1 | 88 | 87 | 481 | |
| 3 | 0.01 | 60 | 80 | 3018 | |
| 4 | 26 | 1 | 100 | 91 | 58 |
| 5 | 0.1 | 100 | 89 | 538 | |
| 6 | 0.01 | 96 | 84 | 4881 | |
| 7 | 23 | 1 | 100 | 79 | 50 |
| 8 | 27 | 1 | 100 | 56 | 34 |
| 9 | 0.1 | 95 | 62 | 359 | |
| 10 | 0.01 | 25 | NDe | ||
| 11 | 28 | 1 | 60 | 80 | 31 |
Imidazole derived catalyst 17 performed as expected, its reactivity dropping off slightly with reduced loading, but still acting in a reasonably stereoselective manner (Table 1, entries 1–3). However, completely unexpectedly, methylated imidazole catalyst 26 showed superior activity and selectivity at even as low as 0.01 mol% loading (entries 4–6). Calculation and comparison of the Asymmetric Catalytic Efficiency (ACE)22 factor at each of the catalyst loadings highlights the exceptional activity of methoxy catalyst 26 at low catalyst loading (Table 1, compare entries 3 and 6). Given the accepted hypothesis of the importance of the hydrogen bond, this is a remarkable and unprecedented result. Interestingly, a broadly similar trend was observed with the picoline catalysts 23 and 27 although not at the same performance level as the imidazole ones (entries 7–10). Under the conditions used, Sigamide® 28 did not perform as well as the other catalysts (entry 11).
The enhanced catalytic activity of catalyst 26 was exemplified with a small representative selection of ketimines 29–33, with conversion and enantioselectivities of the product amines 34–38 comparable to those obtained at higher loading using catalyst 17 (Scheme 4 and Table 2). Given the structural similarity of these catalysts it is not surprising that the stereodifferentiating elements result in comparable selectivities and further exemplification and optimisation of reaction parameters was not carried out.
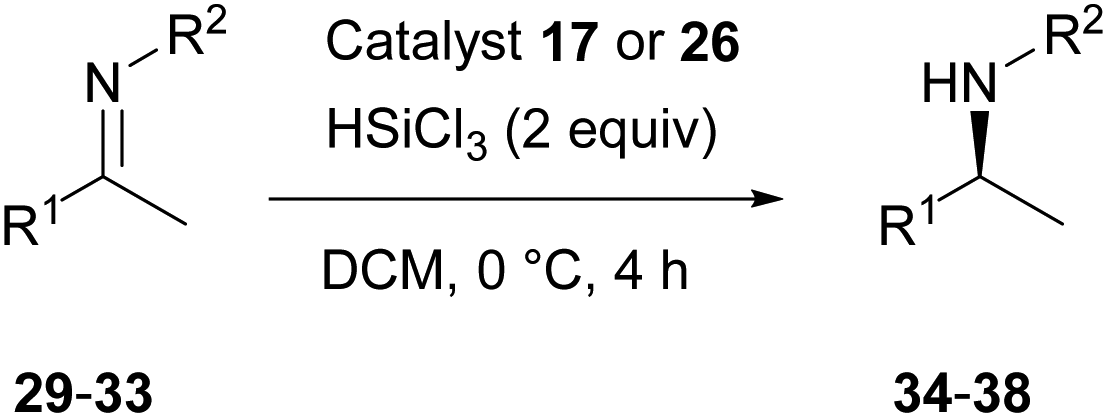 | ||
| Scheme 4 Substrate scope with catalysts 17 and 26. | ||
| Entry | Catalyst (loading per mol%) | R1 | R2 | Imine | Amine | Conversionb (%) | eec (%) |
|---|---|---|---|---|---|---|---|
| a Reactions conducted with ketimine (1 mmol), HSiCl3 (2 mmol) at the specified catalyst loading in DCM (0.5 mL) at 0 °C for 4 h, followed by aqueous workup. b Conversion calculated by comparison of the value of the integrals for the product to residual ketimine and ketone (from hydrolysis). c Determined by chiral phase HPLC analysis. d Literature values, see ref. 16. e Using reductive amination protocol, see ref. 17. | |||||||
| 1 | 26 (0.1) | Ph | 4-FC6H4 | 29 | 34 | 95 | 86 |
| 2 | 17 (1) | Ph | 4-FC6H4 | 29 | 34 | 96 | 87 |
| 3 | 26 (0.1) | 4-MeOC6H4 | 4-MeOC6H4 | 30 | 35 | 97 | 84 |
| 4 | 17 (1)d | 4-MeOC6H4 | 4-MeOC6H4 | 30 | 35 | 81 | 85 |
| 5 | 26 (0.1) | 4-NO2C6H4 | 4-MeOC6H4 | 31 | 36 | 98 | 84 |
| 6 | 17 (1)d | 4-NO2C6H4 | 4-MeOC6H4 | 31 | 36 | 85 | 86 |
| 7 | 26 (0.1) | 2-Thienyl | 4-MeOC6H4 | 32 | 37 | 77 | 80 |
| 8 | 17 (1)e | 2-Thienyl | 4-MeOC6H4 | 32 | 37 | 61 | 79 |
| 9 | 26 (0.1) | β-Naphthyl | 4-MeOC6H4 | 33 | 38 | 99 | 92 |
| 10 | 17 (1)d | β-Naphthyl | 4-MeOC6H4 | 33 | 38 | 86 | 86 |
The remarkable and unexpected catalyst activity, presumably due to the absence of the hydrogen bond, warranted further investigation. The difference in hydroxy and methoxy catalysts 17 and 26 is first evident from their 1H NMR spectra. Hydroxy catalyst 17 has a very broad and almost uninterpretable spectrum in CDCl3 at room temperature and is best recorded in DMSO at 100 °C in order to obtain the best resolution signals, presumably due to the presence of an internal hydrogen bond, although this is not evident in the single crystal X-ray crystal structure (see ESI†). In contrast, methoxy catalyst 26 provides a much better resolved spectrum at room temperature in CDCl3, existing as two rotameric forms in a 4.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.
:1 ratio.
In an attempt to elucidate the mechanism further, the relationship of product ee and catalyst ee was examined using the N-PMP-ketimine substrate 5 and hydroxy catalyst 17. Plotting this gave a linear relationship (Fig. 6), indicating that one molecule of catalyst was involved in formation of the catalytically active species. Furthermore, the ee of the product remained constant over the duration of the experiment.
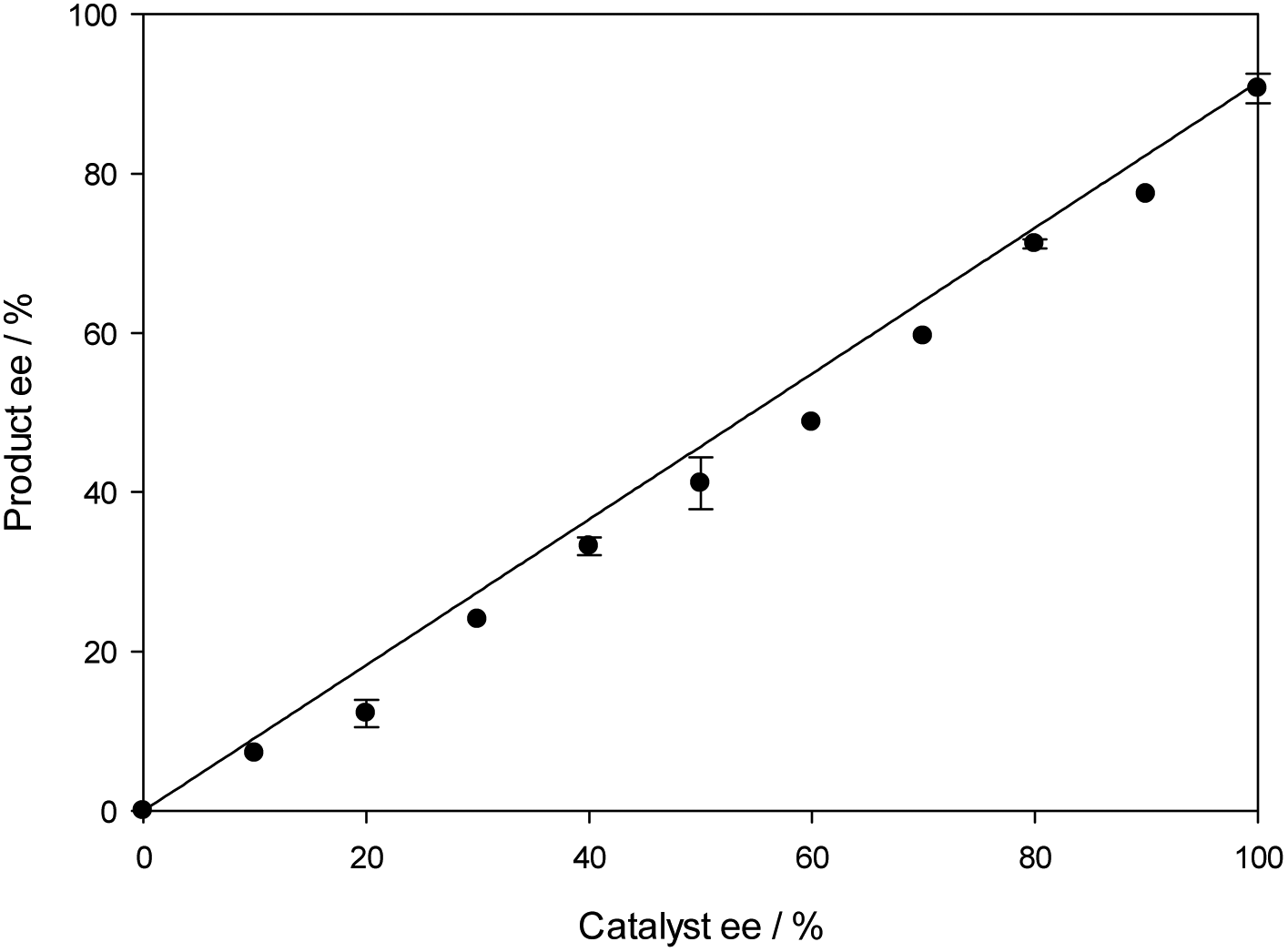 | ||
| Fig. 6 Plot of product 5 ee vs. catalyst ee 17. | ||
Since the critical unusual feature that was observed in this catalyst screen related to the hydroxyl group, further efforts were made to examine the chemistry around this centre by 1H NMR and 29Si NMR experiments. The reduction of N-PMP ketimine 5 was performed with HSiCl3 (2 equiv.) in CDCl3 with 1 mol% imidazole catalyst 17.
After 4 h, 1H NMR and 29Si NMR spectroscopy indicated that the main species resulting from the reduction were N-SiCl2H amine 39 (δH 5.55 ppm and δSi −9.7 ppm) and silicon tetrachloride (δSi −18.9 ppm) (Fig. 7). The identity of the former species was confirmed by independent synthesis by addition of trichlorosilane to a sample of N-PMP-α-methylbenzylamine 6, and the latter by comparison with a reference sample. It is noticeable that the silicon tetrachloride results from the reaction and is not present in the HSiCl3.
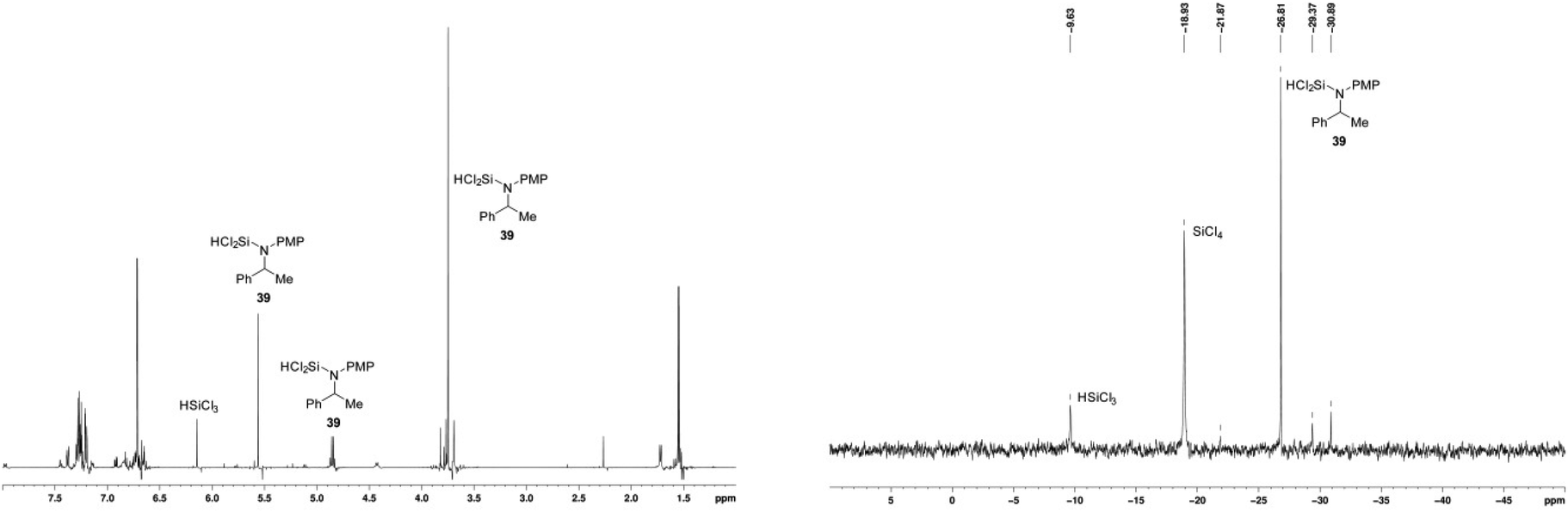 | ||
| Fig. 7 In situ 1H and 29Si NMR spectra of reaction products as depicted in Scheme 1 with catalyst 17. | ||
When hydroxy catalyst 17 was treated with an excess of trichlorosilane, new signals were observed in the 1H NMR (δH 4.99 ppm) and 29Si NMR (δSi −33.2 ppm) spectra. These signals are consistent with an oxosilane species formed by silylating the hydroxyl group of catalyst 17, confirmed by comparison of the 1H and 29Si chemical shifts of the related EtOSiCl2H in the literature, including JSi–H coupling of 29Si satellite signals in the 1H NMR spectrum (Fig. 8).23 Importantly, the 29Si NMR spectrum only provided evidence for a tetra-coordinate silicon atom. The 1H NMR spectrum also shows a significant downfield chemical shift of the methine proton adjacent to the amide nitrogen atom that might result as a combination of the loss of the internal hydrogen bond and from complexation of the carbonyl group to the silane. The spectrum also has considerably better resolved signals than that obtained for the parent catalyst at room temperature, implying fewer equilibrating rotameric and/or hydrogen bonded species. In retrospect, this is what one would expect from the natural reactivity of a chlorosilane with an alcohol to generate a silyl ether and HCl. Given the formation of the latter, it is also very likely that the imidazole of the catalyst will be protonated. Importantly, this preformed catalyst – trichlorosilane complex 40 demonstrated the same catalytic activity in the standard asymmetric hydrosilylation reaction, and returned unchanged catalyst after work-up. In contrast, no new signals were observed in the 29Si NMR spectrum of the methoxy catalyst 26 when treated with trichlorosilane, which was corroborated by the presence of only a very small signal at δH 5.27 ppm in the 1H NMR spectrum where evidence of a Si–H signal might be observed (Fig. 9). However, significant chemical shift changes were observed in the imidazole and methine protons, in addition to those signals corresponding to the pyrrolidine ring. As in the case of the hydroxyl-catalyst 17, these changes may have resulted from protonation of the imidazole, in this case from adventitious HCl usually found in HSiCl3.
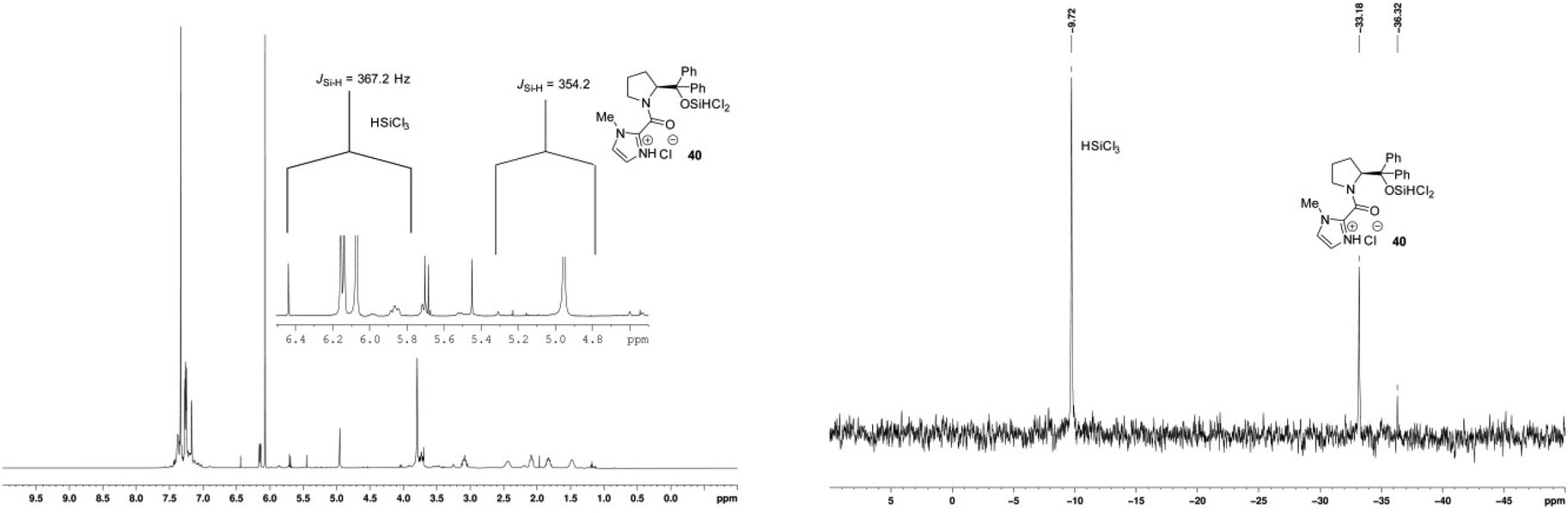 | ||
| Fig. 8 1H and 29Si NMR spectra of catalyst 17 and excess HSiCl3. | ||
 | ||
| Fig. 9 1H NMR spectrum of catalyst 26 before and after addition of 4 equiv. HSiCl3. | ||
The presence of HCl and protonation of the catalysts appeared to be a key factor in their activity, and related studies have also shown that additives such as water (to form HCl in situ), acetic acid, and benzoic acid can improve the catalytic activity.6,7,9,24,25 As previously indicated, HCl is most likely also present in commercial HSiCl3, and to confirm the effect of this, a series of reactions were performed under diluted conditions in order to slow them down sufficiently to observe positive or negative effects of the effects of proton sponge, HCl and chloride ions (in the form of n-BuN4Cl). None of these additives catalysed the reaction in the absence of the catalysts, and reactions conducted with the hydroxy 17 and methoxy 26 catalysts showed that the presence of HCl was essential for reactivity (Table 3, entries 2 and 7).
| Entry | Catalyst (loading per mol%) | Additive (equiv.) | Conversionb/% |
|---|---|---|---|
| a Reactions conducted with N-PMP-ketimine 5 (1 mmol), HSiCl3 (2 mmol) with the specified additive and catalyst loading in DCM (5 mL) at 0 °C for 1 h, followed by aqueous workup. b Conversion calculated by comparison of the value of the integrals for the product to residual ketimine and ketone (from hydrolysis). | |||
| 1 | 17 (0.1) | None | 57 |
| 2 | 17 (0.1) | Proton sponge (1) | 0 |
| 3 | 17 (0.1) | n-Bu4NCl (0.1) | 61 |
| 4 | 17 (0.1) | HCl (0.005) | 60 |
| 5 | 17 (0.1) | HCl (0.05) | 58 |
| 6 | 26 (0.01) | None | 13 |
| 7 | 26 (0.01) | Proton sponge (1) | 0 |
| 8 | 26 (0.01) | n-Bu4NCl (0.1) | 15 |
| 9 | 26 (0.01) | HCl (0.005) | 15 |
| 10 | 26 (0.01) | HCl (0.05) | 16 |
As conducted with catalysts 3 and 4, investigation of the reactive catalyst species was conducted by in situ infra-red analysis (Fig. 10 and 11). Upon addition of 1 equiv. of HSiCl3, the hydroxy catalyst 17 shows a rapid and complete change in the stretching frequency of the carbonyl group, whilst catalyst 26 shows a rapid but incomplete change, suggesting an equilibrium process. This is very similar to the behaviour with the simpler catalysts 3 and 4, however the reactivity is inverted, with catalyst 26 showing incomplete complexation being more active. Since no direct reaction with HSiCl3 is possible here as corroborated by 29Si NMR data, the changes in the 1H NMR and IR data indicate a different resting state for the catalyst, perhaps with the imidazole being protonated by the small quantities of HCl present in the HSiCl3. Catalyst 17 reacts stoichiometrically with HSiCl3, in doing so generating HCl to completely protonate the imidazole.
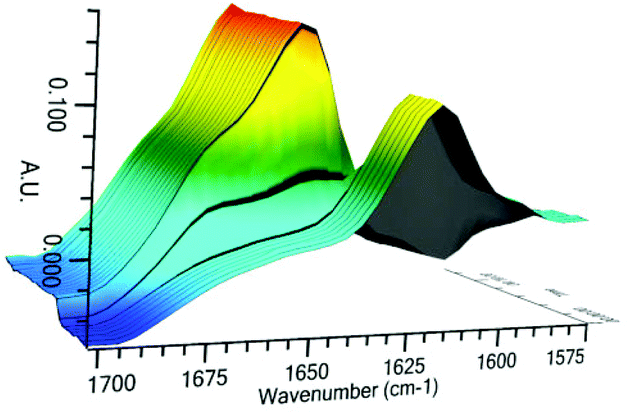 | ||
| Fig. 10 In situ IR data of catalyst 17 treated with HSiCl3 as a function of time. | ||
 | ||
| Fig. 11 In situ IR data of catalyst 26 treated with HSiCl3 as a function of time. | ||
Based on these results, a new ‘dual activation’ mechanistic model is proposed that involves a Lewis basic site to activate the HSiCl3 and an additional Brønsted base capable of reacting with HCl generated or present in the reaction (Fig. 12). Reaction of the hydroxy catalyst 17 with HSiCl3 provides a covalently linked siloxane 40 where the imidazole is protonated by HCl, either as a by-product from reaction with the alcohol, or from adventitious HCl in the HSiCl3. This is the resting state for the catalyst that is set up for forming a key hydrogen bond with the ketimine substrate.
 | ||
| Fig. 12 Proposed dual activation mechanism for catalyst 17. | ||
Coordination of the silane with the Lewis basic amide inferred from the in situ infra-red studies must involve twisting of the amide bond to negate steric interactions, leading to a reactive hexa-coordinate species 41. The ketimine substrate is now activated by a hydrogen bond to the protonated imidazole, and placed in the correct orientation to facilitate hydride delivery from the activated siloxane. Importantly, the N-PMP-amine product 6 then undergoes reaction with more trichlorosilane to generate the observed N-SiHCl2 product 39, whilst the inactive catalyst 42 turns-over by protonation and reaction with more trichlorosilane, generating silicon tetrachloride. This accounts for the observation that the reaction slows considerably when using less than 2 equivalents of HSiCl3.
This model essentially inverts that proposed by Matsumura (Scheme 3), with the crucial hydrogen bond now being located at the imidazole nitrogen atom. However, the deoxy catalyst 23 may still react via the previously accepted model, especially as there is some similarity between the sense and magnitude of the selectivity observed with catalyst 14 (Fig. 4).
The process with methoxy catalyst 26 is more complex since no evidence could be obtained for a species whereby HSiCl3 is bound to the catalyst. Clearly there is a change in the 1H NMR and IR data on addition of HSiCl3 that may be due to reaction with HCl (Fig. 13). Naturally this means that no Si–O bond needs to be broken after reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond and therefore the catalyst can turn-over more quickly, perhaps explaining the enhanced catalytic activity. A completely different mechanism is unlikely due to the similar levels of enantioselectivity that catalysts 17 and 26 provide.
N bond and therefore the catalyst can turn-over more quickly, perhaps explaining the enhanced catalytic activity. A completely different mechanism is unlikely due to the similar levels of enantioselectivity that catalysts 17 and 26 provide.
 | ||
| Fig. 13 Postulated resting state 43 and intermediate in dual activation mechanism with methoxy catalyst 26. | ||
Such a dual activation model is obviously transferable to the Matsumura and related picoline systems, the difference in reactivity probably now coming from the different torsional freedom and basicity provided by the 2-pyridyl moiety. The model previously proposed for related N-formamide catalysts such as Sigamide® 28 also contains key elements of this dual activation mechanism, with the Brønsted acid now being the relatively acidic aniline NH proton (Fig. 14).
 | ||
| Fig. 14 Previously reported model for Sigamide 28 illustrating dual activation. | ||
Conclusion
In summary, a dual-activation mechanism is proposed for the organocatalyzed asymmetric hydrosilylation of ketimines. This model fits the available mechanistic and characterization data and in doing so has provided an example of a new catalyst system displaying catalytic activity at unprecedented low levels of catalyst loading. Computational studies on these systems may reveal more details to support these studies. This work should provide valuable insights for the future design and mechanistic rationale for new catalysts in this area.Experimental section
General experimental methods
All solvents were obtained from a dry solvent system and glassware was flame dried and cooled under vacuum before use. All dry reactions were carried out under a nitrogen atmosphere. TLC was carried out using aluminium TLC sheets (silica gel 60 F254), and visualised using a UV lamp or by dipping in KMnO4 solution then exposure to heat. Flash column chromatography was carried out with silica gel 40–63 μ 60 Å. 1H, 13C NMR and 29Si spectra were measured using CDCl3 as solvent unless otherwise stated. 13C NMR spectra were recorded using the JMOD method. Specific rotations were measured at 589 nm (Na D-Line) at 20 °C unless otherwise stated, and are given in 10−1 deg cm2 g−1. All chemicals were used as received without further purification. Previously reported methods were used to prepare the following compounds: N-PMP-ketimine 5, (S)-α,α-diphenylprolinol 13, N-Me-catalyst 17, (S)-N-ethoxycarbonylproline methyl ester 18, ethyl N-methyl imidazole-1-carboxylate 20, 4-methoxy-N-[1-(4-nitrophenyl)ethylidene]benzenamine 31, 4-methoxy-N-[1-(2-naphthalenyl)ethylidene]benzenamine 33;16 4-fluoro-N-(1-phenylethylidene)benzenamine 29, 4-methoxy-N-[1-(4-methoxyphenyl)ethylidene]benzenamine 30;26N-4-methoxy-N-[1-(2-thienyl)ethylidene]benzenamine 32.191-t-Butylimidazole 2
The title compound was prepared using a modified literature procedure.20 A 250 mL three-necked flask containing distilled water (50 mL) was equipped with two dropping funnels and a condenser. One dropping funnel was filled with a mixture of 40% aqueous glyoxal (11.5 mL, 0.1 mol) and 37% aqueous formaldehyde (8.10 g, 0.1 mol), the other with t-butylamine (10.6 mL, 0.1 mol) and 35% aqueous ammonia (4.86 g, 0.1 mol). The water was heated until boiling, and then both solutions were added simultaneously at the same addition rate. The resulting mixture turned brown and was stirred for 30 min at reflux after complete addition, and then cooled to rt. The mixture was extracted with DCM (3 × 150 mL), the combined organic phases were washed with brine (50 mL) and dried over MgSO4. The desiccant was filtered off and the filtrate was evaporated. The brown residue was purified by vacuum distillation to give the product 2 as a colourless oil (4.80 g, 39%); δH (400 MHz, CDCl3): 1.55 [9H, s, (CH3)3C], 7.03 (1H, s, ArCH), 7.04 (1H, s, ArCH), 7.60 (1H, s, ArCH); δC (100 MHz, CDCl3): 30.6 [C(CH3)3], 54.7 [C(CH3)3], 116.3 (ArCH), 129.1 (ArCH), 134.3 (ArCH); Data were in accordance with the literature.1-Methyl-N,N-dimethyl-1H-imidazole-2-carboxamide 327
N-Methylimidazole 1 (1.64 g, 20 mmol) was dissolved in THF (20 mL) and cooled to −78 °C. n-BuLi (2.5 M in hexanes, 10 mL, 25 mmol) was added dropwise by syringe. The resulting mixture was stirred at −78 °C for 2 h. This solution was added dropwise to a solution of dimethylcarbamoyl chloride (2.99 g, 28 mmol) in diethyl ether (25 mL) at −78 °C and stirred for 1 h at the same temperature. The reaction mixture was then warmed to rt and quenched with water (50 mL). The organic solvent was evaporated and the aqueous was extracted with DCM (3 × 30 mL) and dried over MgSO4. The desiccant was filtered off and the filtrate was evaporated to give the crude residue, which was purified by chromatography on silica gel [50%–100% ethyl acetate in petroleum ether (40–60)] to yield the product 3 as a colourless oil (1.10 g, 36%); δH (400 MHz, CDCl3): 3.12 (3H, s, CH3), 3.41 (3H, s, CH3), 3.89 (3H, s, CH3), 6.94 (1H, d, J 1.0, ArCH), 7.05 (1H, d, J 1.0, ArCH); δC (100 MHz, CDCl3): 35.0 (CH3), 35.7 (CH3), 39.3 (CH3), 123.6 (ArCH), 127.3 (ArCH), 140.1 (ArC), 160.9 (CO); Data was in accordance with the literature.1-t-Butyl-N,N-dimethyl-1H-imidazole-2-carboxamide 4
1-t-Butylimidazole 2 (2.48 g, 20 mmol) was dissolved in THF (20 mL) and cooled to −78 °C. n-BuLi (2.0 M in hexanes, 12.5 mL, 25 mmol) was added dropwise by syringe. The resulting mixture was stirred at −78 °C for 2 h. This solution was added dropwise to a solution of dimethylcarbamoyl chloride (2.99 g, 28 mmol) in diethyl ether (20 mL) at −78 °C and stirred for 1 h at the same temperature. The reaction mixture was then warmed to rt and quenched with water (50 mL). The organic solvent was evaporated and the aqueous was extracted with DCM (3 × 100 mL) and dried over MgSO4. The desiccant was filtered off and the filtrate was evaporated to give the crude residue. Pure product 4 was obtained as a white solid after recrystallization from ethyl acetate (1.69 g, 43%); m.p. 146–148 °C; νmax (thin film): 3157 (w), 2931 (w), 1638 (s), 1524 (w); δH (400 MHz, CDCl3): 1.60 [9H, s, (CH3)3C], 2.96 (3H, s, CH3), 3.09 (3H, s, CH3), 6.97 (1H, d, J 1.2, ArCH), 7.05 (1H, d, J 1.2, ArCH); δC (100 MHz, CDCl3): 30.4 (CH3), 35.0 (CH3), 38.6 (CH3), 57.0 [(CH3)2C], 117.9 (ArCH), 126.8 (ArCH), 140.3 (ArC), 164.5 (CO); HRMS (ES+ TOF): found 196.1442 ([M + H]+); C10H18N3O requires 196.1450.Kinetic analysis of the reduction of ketimine 5 to amine 6
N-PMP-ketimine 5 (0.225 g, 1.0 mmol) and catalyst (mol% as specified) were dissolved in DCM (1 mL) at 0 °C. HSiCl3 (0.2 mL, 2.0 mmoL) was added and the reaction left to stir for the specified time. Work-up, either after the reaction was complete, or by removal of samples during the reaction, was accomplished by dilution with DCM (20 mL per mL aliquot), quenching with aqueous NaOH (1 M, approximately 20 mL per mL aliquot), and separation of the organic layer. The aqueous layer was further extracted with DCM (2 × 20 mL per mL aliquot), the combined organic layers dried over MgSO4, filtered and evaporated. Conversion was calculated from comparison of the integrals corresponding to ketimine starting material(s) and ketone (from hydrolysis of the ketimine) and that of the product amine 6. δH (400 MHz, CDCl3): 1.52 (3H, d, J 7.1, CH3), 3.70 (3H, s, CH3), 4.43 (1H, q, J 7.1, CH), 6.49 [2H, (AX)2, ArCH], 6.72 [2H, (AX)2, ArCH], 7.21–7.40 (5H, m, ArCH).Ethyl 1-t-butyl-1H-imidazole-2-carboxylate 8
1-t-Butylimidazole 2 (4.34 g, 35 mmol) was dissolved in THF (30 mL) and cooled to −78 °C. n-BuLi (2 M in hexanes, 20 mL, 40 mmol) was added dropwise by syringe. The resulting mixture was stirred at −78 °C for 2 h, and this solution added to a solution of ethyl chloroformate (7.56 g, 70 mmol) in THF (20 mL) by cannula at −78 °C, followed by stirring for 1 h at the same temperature. The reaction mixture was then warmed to rt and quenched with saturated aqueous ammonium chloride (50 mL). The resulting mixture was extracted with ethyl acetate (4 × 100 mL), and the combined organic layers were washed with brine (100 mL) and dried over MgSO4. The desiccant was filtered off and the filtrate was evaporated. The residue was purified by chromatography on silica gel [(20%–30%) ethyl acetate in petroleum ether (40–60)] to yield the product 8 as a colourless oil (2.50 g, 37%); νmax (thin film) 2980 (m), 1721 (s), 1403 (m); δH (400 MHz, CDCl3): 1.41 (3H, t, J 7.1, CH3), 1.50 [9H, s, (CH3)3], 4.38 (2H, q, J 7.1, CH2), 7.06 (1H, s, ArCH), 7.23 (1H, s, ArCH); δC (100 MHz, CDCl3): 14.2 (CH3), 30.1 [(CH3)3], 58.7 [(CH3)3C], 61.6 (CH2), 122.2 (ArCH), 127.8 (ArCH), 137.4 (ArC), 160.0 (CO); HRMS (ES+ TOF): found 197.1299 ([M + H]+); C10H17N2O2 requires 197.1290.1-Methyl-2-pyrrolecarboxylic acid 10
Aqueous KOH (2.5 M, 20 mL, 50 mmol) was added to a solution of methyl 1-methylpyrrole-2-carboxylate 12 (2.1 g, 6.6 mmol) dissolved in methanol (20 mL) at rt and the resulting reaction mixture was stirred overnight. Methanol was evaporated and the remaining solution was carefully acidified to pH 2 with hydrochloric acid (6 M). The white precipitate was collected by filtration and washed with water. The precipitate was dissolved in DCM (50 mL) and dried over MgSO4. The desiccant was filtered off and the filtrate was evaporated to give the product 10 as a white solid (1.1 g, 57%); m.p. 136–138 °C (lit.28 135–136 °C); δH (400 MHz, CDCl3): 3.96 (3H, s, CH3), 6.18 (1H, dd, J 4.0, 2.1, ArCH), 6.87 (1H, t, J 2.1, ArCH), 7.13 (1H, dd, J 4.0, 2.1, ArCH), 12.20 (1H, br s, OH); δC (63 MHz, CDCl3): 37.0 (CH3), 108.4 (ArCH), 120.1 (ArCH), 121.8 (ArC), 130.8 (ArCH), 166.6 (CO). NMR data was not previously reported in the literature.1-t-Butyl-1H-imidazole-2-carboxylic acid 11
Aqueous KOH (2.5 M, 20 mL, 50 mmol) was added to a solution of ethyl 1-t-butyl-1H-imidazole-2-carboxylate 8 (1.29 g, 6.6 mmol) in methanol (20 mL) at rt. The reaction mixture was stirred overnight, the methanol was evaporated and the mixture carefully acidified to pH 2 with concd. HCl. The resulting mixture was evaporated to give a white solid that was treated with methanol (30 mL) and filtered. The filtrate was evaporated to give the crude product 11 as a white solid (0.70 g) that was contaminated with a small amount of the t-butyl imidazole compound formed by decarboxylation (∼10%) but was used in the next step without further purification; δH (400 MHz, MeOD): 1.86 [9H, s, (CH3)3C], 7.71 (1H, d, J 1.8, ArCH), 8.02 (1H, d, J 1.8, ArCH); δC (100 MHz, MeOD): 28.3 [(CH3)3], 63.9 [(CH3)3C], 119.1 (ArCH), 124.3 (ArCH), 134.8 (ArC), 154.8 (CO); HRMS (ES+ TOF): found 169.0978 [M + H]+; C8H13N2O2 requires 169.0977; Data for the N-t-butylimidazole contaminant; δH (400 MHz, MeOD): 1.72 [9H, s, (CH3)3], 7.63 (1H, s, ArCH), 7.91 (1H, s, ArCH), 9.10 (1H, s, ArCH).(S)-[2-(Hydroxydiphenylmethyl)pyrrolidin-1-yl](1-methyl-1H-imidazol-4-yl)methanone 14
Oxalyl chloride (0.18 mL, 2.1 mmol) was added to a mixture of 1-methyl-1H-imidazole-4-carboxylic acid 9 (132 mg, 1.05 mmol) in DCM (5 mL) at rt. One drop of DMF was added and the reaction mixture was stirred at room temperature for 2 h. Excess oxalyl chloride and DCM were removed by evaporation under reduced pressure. The residue was dissolved in DCM (5 mL) and added to a solution of amino alcohol 13 (253 mg, 1.0 mmol) and triethylamine (0.2 mL, 1.4 mmol) in DCM (5 mL). The resulting mixture was stirred at rt for 2 h and then quenched with aqueous saturated NaHCO3 (15 mL). The aqueous phase was extracted with DCM (3 × 15 mL), and the combined organic extracts washed with brine (15 mL) and dried over MgSO4. The desiccant was filtered off and the filtrate was evaporated. The residue was purified by chromatography on silica gel (diethyl ether to flush off excess amino alcohol then 2% methanol in ethyl acetate) to yield the product 14 as a white solid (91 mg, 25%); m.p. 68–70 °C; [α]20D −116.3 (c 0.94, CHCl3); νmax (thin film) 2924 (s), 2854 (m), 1587 (m), 1556 (m), 1446 (m), 1434 (m); δH (500 MHz, 100 °C, DMSO-d6): 1.30–1.39 (1H, m, CHH), 1.51–1.60 (1H, m, CHH), 1.88–1.94 (1H, m, CHH), 1.99–2.07 (1H, m, CHH), 3.28–3.34 (1H, m, CHH), 3.63 (3H, s, CH3), 4.08–4.13 (1H, m, CHH), 5.67 (1H, br s, CH), 6.63 (1H, s, ArCH), 7.15–7.20 (3H, m, ArCH), 7.22–7.25 (1H, m, ArCH), 7.28–7.36 (5H, m, ArCH), 7.43–7.45 (2H, m, ArCH), 7.50 (1H, s, ArCH); δC (126 MHz, 100 °C, DMSO-d6): 22.4 (CH2), 27.6 (CH2), 32.4 (CH3), 48.3 (CH2), 64.3 (CH), 80.8 (C), 125.1 (ArCH), 125.7 (ArCH), 126.0 (ArCH), 126.5 (2 × ArCH), 126.7 (2 × ArCH), 126.8 (2 × ArCH), 127.1 (2 × ArCH), 136.4 (ArCH), 136.9 (ArC), 144.6 (ArC), 146.1 (ArC), 163.6 (CO); HRMS (ES+ TOF): found 362.1876 [M + H]+; C22H24N3O2 requires 362.1869.(S)-[2-(Hydroxydiphenylmethyl)pyrrolidin-1-yl](1-methyl-1H-pyrrol-2-yl)methanone 15
Oxalyl chloride (0.18 mL, 2.1 mmol) was added to a solution of 1-methyl-2-pyrrolecarboxylic acid 10 (131 mg, 1.05 mmol) in DCM (5 mL) at room temperature. One drop of DMF was added and then the reaction mixture was stirred at rt for 2 h. Excess oxalyl chloride and DCM were removed by evaporation under reduced pressure. The residue was dissolved in DCM (5 mL) and added to a solution of amino alcohol 13 (253 mg, 1 mmol) and triethylamine (0.2 mL, 1.4 mmol) in DCM (5 mL). The resulting mixture was stirred at rt for 2 h and then quenched with aqueous saturated NaHCO3 (15 mL). The aqueous phase was extracted with DCM (3 × 15 mL), and the combined organic extracts washed with brine (15 mL) and dried over MgSO4. The desiccant was filtered off, the filtrate evaporated, and the residue purified by chromatography on silica gel [15% ethyl acetate in petroleum ether (40–60)] to yield the product 15 as a white solid (195 mg, 54%); m.p. 184–186 °C; [α]20D −83.7 (c 0.86, CHCl3); (Found: C, 76.28; H, 6.64; N, 7.69. C23H24N2O2 requires C, 76.64; H, 6.71; N, 7.77%); νmax (thin film) 3246 (w), 2958 (w), 1588 (s), 1435 (s); δH (400 MHz, CDCl3): 1.42 (1H, pent d, J 7.5, 4.1, CHH), 1.61 (1H, d pent, J 12.4, 8.8, CHH), 1.94–2.03 (1H, m, CHH), 2.18 (1H, dtd, J 12.4, 8.8, 4.1, CHH), 2.92 (1H, td, J 10.0, 7.3, CHH), 3.76–3.79 (1H, m, CH), 3.81 (3H, s, CH3), 5.50 (1H, dd, J 8.4, 7.4, CH), 6.08 (1H, dd, J 3.9, 2.0, ArCH), 6.23 (1H, dd, J 3.9, 2.0, ArCH), 6.70 (1H, app t, J 2.0, ArCH), 6.75 (1H, br s, OH), 7.27–7.39 (6H, m, ArCH), 7.48–7.50 (2H, m, ArCH), 7.55–7.58 (2H, m, ArCH); δC (100 MHz, CDCl3): 24.5 (CH2), 29.3 (CH2), 36.3 (CH3), 51.6 (CH2), 66.8 (CH), 82.7 (C), 107.1 (ArCH), 114.6 (ArCH), 125.5 (ArC), 127.1 (ArCH), 127.2 (ArCH), 127.3 (ArCH), 127.4 (2 × ArCH), 127.9 (2 × ArCH), 128.0 (2 × ArCH), 128.2 (2 × ArCH), 143.1 (ArC), 145.7 (ArC), 165.5 (CO); HRMS (ES+ TOF): found 361.1913 [M + H]+; C23H25N2O2 requires 361.1916.(S)-(1-t-Butyl-1H-imidazol-2-yl)[2-(hydroxydiphenylmethyl)pyrrolidin-1-yl]methanone 16
Oxalyl chloride (0.18 mL, 2.1 mmol) was added to the crude mixture of 1-t-butyl-1H-imidazole-2-carboxylic acid 11 (0.133 mg, 1.05 mmol) in DCM (5 mL) at room temperature. One drop of DMF was added and then the reaction mixture was stirred at rt for 2 h. The excess oxalyl chloride and DCM were removed by evaporation under reduced pressure. The residue was dissolved in DCM (5 mL) and this added to a solution of amino alcohol 13 (253 mg, 1 mmol) and triethylamine (0.2 mL, 1.4 mmol) in DCM (5 mL). The resulting mixture was stirred at rt for 2 h and then quenched with aqueous saturated NaHCO3 (15 mL). The aqueous phase was extracted with DCM (3 × 15 mL), and the combined organic extracts washed with brine (15 mL) and dried over MgSO4. Filtration and evaporation was followed by purification by chromatography on silica gel [diethyl ether to flush off excess amino alcohol, then 50% ethyl acetate in petroleum ether (40–60)] yielding the product 16 as a white solid (110 mg, 26%); m.p. 157–159 °C; [α]20D −153.7 (c 0.8, CHCl3); νmax (thin film) 2979 (w), 1618 (s), 1449 (s); δH (500 MHz, 100 °C, DMSO-d6): 1.40 (1H, br s, CHH), 1.45 [9H, s, (CH3)3], 1.53–1.62 (1H, m, CHH), 1.93–1.99 (1H, m, CHH), 2.01–2.08 (1H, m, CHH), 3.19–3.24 (1H, m, CHH), 3.46 (1H, br s, CHH), 5.41 (1H, dd, J 8.3, 3.3, CH), 6.22 (1H, br s, ArCH), 6.86 (1H, s, ArCH), 7.13–7.16 (1H, m, ArCH), 7.20–7.25 (4H, m, ArCH), 7.29–7.32 (2H, m, ArCH), 7.42 (2H, d, J 7.5, ArCH), 7.48 (2H, d, J 7.7, ArCH); δC (126 MHz, 100 °C, DMSO-d6): 22.2 (CH2), 27.6 (CH2), 29.5 [(CH3)3C], 48.6 [(CH3)3C], 56.4 (CH2), 64.5 (CH), 80.4 (C), 118.4 (ArCH), 125.4 (ArCH), 125.8 (ArCH), 126.1 (ArCH), 126.6 (2 × ArCH), 126.7 (2 × ArCH), 126.9 (ArCH), 140.4 (ArC), 144.9 (ArC), 145.8 (ArC), 163.7 (CO); HRMS (ES+ TOF): found 404.2348 [M + H]+; C25H30N3O2 requires 404.2338.General procedure A for the reduction of ketimines with catalysts
Ketimine (1.0 mmol) and catalyst (mol% as specified) were dissolved in DCM (1 mL) at 0 °C. HSiCl3 (0.2 mL, 2.0 mmoL) was added and the reaction left to stir for the specified time. The reaction was quenched with aqueous HCl (1 M, 2 mL – care, gas evolution), followed by addition of DCM (20 mL) and aqueous NaOH (1 M, 20 mL), and stirred until the precipitate dissolved. The organic layer was separated and the aqueous layer was further extracted with DCM (2 × 20 mL). The combined organic layers were dried over MgSO4, filtered and evaporated. Purification and analysis is detailed in individual amine products.Catalyst evaluation using (S)-N-(4-methoxyphenyl)-α-methyl-benzenemethanamine 6
Prepared by general procedure A, using 4-methoxy-N-(1-phenylethylidene)-benzenamine 5 (225 mg, 1 mmol) and specified catalyst, gave the title compound 6. Conversion to product was determined by comparison of the integrals of signals in the 1H NMR spectrum, whilst the enantiomeric excess was determined by chiral phase HPLC analysis; δH (400 MHz, CDCl3): 1.52 (3H, d, J 7.1, CH3), 3.70 (3H, s, CH3), 4.43 (1H, q, J 7.1, CH), 6.49 [2H, (AX)2, ArCH], 6.72 [2H, (AX)2, ArCH], 7.21–7.40 (5H, m, ArCH); Phenomenex Lux 3 μm cellulose-1, hexane/i-PrOH = 98/2, flow rate = 1.0 mL min−1, 254 nm, (R) enantiomer tR = 16.5 min, (S) enantiomer tR = 19.3 min.(S)-α,α-Dimethyl-2-pyrrolinol 1929
Mg (1.92 g, 80 mmol), iodine (one granule) and diethyl ether (20 mL) were added to a dry three-necked flask. A solution of MeI (4.98 g, 80 mmol) in diethyl ether (80 mL) was added dropwise (initiated by heat after about 5–10 mL addition). The mixture was stirred at rt for 2 h and then cooled to 0 °C. A solution of (S)-N-ethoxycarbonylproline methyl ester 18 (4.02 g, 20 mmol) in diethyl ether (20 mL) was added dropwise. The resulting mixture was stirred at rt overnight, cooled to 0 °C and quenched with saturated aqueous ammonium chloride (50 mL). The organic layer was separated and the aqueous layer extracted with diethyl ether (2 × 50 mL). The combined organic extracts were evaporated to afford a yellow oil (3.36 g). This residue was treated with KOH (9.3 g, 0.166 mol), water (60 mL) and ethanol (40 mL), and heated at reflux overnight. The mixture was cooled to rt and the ethanol removed under reduced pressure. The resulting solution was extracted with DCM (2 × 50 mL), the combined extracts were washed with brine (30 mL) and dried over MgSO4. After filtration, the filtrate was evaporated under reduced pressure to afford the title compound 19 as a brown, low melting solid (1.87 g, 72%); [α]20D −28.0 (c 2, MeOH, lit. −32.3,29c 1.84, MeOH); δH (400 MHz, CDCl3): 1.14 (3H, s, CH3), 1.18 (3H, s, CH3), 1.60–1.82 (4H, m, 2 × CH2), 2.84 (2H, br s, NH & OH), 2.90–3.05 (3H, m, CH2 & CH); δC (100 MHz, CDCl3): 25.2 (CH3), 26.1 (CH2), 26.3 (CH2), 28.6 (CH3), 47.0 (CH2), 67.0 (CH), 70.3 (C); Data was in accordance with the literature.(S)-[2-(2-Hydroxypropan-2-yl)pyrrolidin-1-yl](1-methyl-1H-imidazol-2-yl)methanone 21
(S)-α,α-Dimethyl-2-pyrrolinol 19 (1.29 g, 10 mmol) was dissolved in toluene (10 mL) and NaH (60% dispersion in mineral oil, 600 mg, 15 mmol) was added at rt. After stirring for 30 min, ethyl 1-methylimidazole-2-carboxylate 20 (1.69 g, 11 mmol) was added. The resulting reaction mixture was slowly warmed to 70 °C and stirred for 40 h. The reaction was cooled to rt, quenched by addition of water (20 mL) and extracted with ethyl acetate (3 × 30 mL). The combined organic phases were dried over MgSO4. After filtration, the filtrate was evaporated and purified by chromatography on silica gel [50%–100% ethyl acetate in petroleum ether (40–60)] to yield the product 21 (0.896 g, 39%) as a white solid; m.p. 84–86 °C; [α]20D −124 (c 1.1, CHCl3); (Found: C, 60.70; H, 7.98; N, 17.70. C12H19N3O2 requires C, 60.74; H, 8.07, N, 17.71%); νmax (thin film) 3353 (br m), 2974 (m), 1607 (s), 1455 (s); δH (500 MHz, DMSO-d6, 100 °C): 1.07 [6H, s, 2 × CH3], 1.65–1.70 (1H, m, CHH), 1.89–1.95 (3H, m, CH2 & CHH), 3.49–3.54 (1H, m, CHH), 3.79 (3H, s, CH3), 3.97–4.03 (1H, m, CHH), 4.42–4.62 (1H, br m, OH), 4.71 (1H, br s, CH), 6.95 (1H, s, ArCH), 7.19 (1H, s, ArCH); δC (126 MHz, DMSO-d6, 100 °C): 23.4 (br, CH2), 25.6 (CH2), 25.7 (CH3), 26.9 (CH3), 33.8 (CH3), 48.7 (br CH2), 65.2 (CH), 72.4 (C), 123.5 (ArCH), 126.1 (ArCH), 140.4 (ArC), 160.4 (CO); HRMS (ES+ TOF): found 238.1556 [M + H]+; C12H20N3O2 requires 238.1550.(S)-[2-(Hydroxydiphenylmethyl)pyrrolidin-1-yl](pyridin-2-yl)methanone 23
Oxalyl chloride (1.32 mL, 15 mmol) was added dropwise to a solution of picolinic acid (615 mg, 5 mmol) in DCM (15 mL). The resulting mixture was stirred at rt for 4 h, followed by evaporation to give a black solid. DCM (20 mL) was added and the suspension transferred into a solution of (S)-α,α-diphenylprolinol 13 (1.02 g, 4 mmol) in toluene (20 mL) and the resulting mixture stirred at rt overnight. The reaction was quenched by aqueous ammonium chloride (20 mL) and extracted with ethyl acetate (3 × 20 mL). The combined organic extracts were dried over Na2SO4, filtered, evaporated under reduced pressure, and the residue purified by chromatography on silica gel [20%–100% ethyl acetate in petroleum ether (40–60)] to yield the product 23 as white solid (950 mg, 66%); m.p. 129–131 °C (not reported in literature); [α]20D −118.7 (c 0.8, CHCl3, not reported in literature); νmax (thin film) 3058 (w), 2989 (w), 1608 (s), 1565 (s); δH (500 MHz, DMSO-d6, 100 °C): 1.63–1.80 (2H, m, CH2), 1.92–1.98 (1H, m, CHH), 2.05–2.12 (1H, m, CHH), 3.47–3.52 (1H, m, CHH), 3.65 (1H, br s, CHH), 5.72 (1H, br s, OH), 5.85 (1H, br s, CH), 7.06–7.26 (7H, m, ArCH), 7.32–7.35 (3H, m, ArCH), 7.48 (2H, d, J 7.6, ArCH), 7.69 (1H, br s, ArCH), 8.48 (1H, d, J 3.9, ArCH); δC (126 MHz, d6-DMSO, 100 °C): 22.7 (br, CH2), 27.4 (CH2), 48.6 (br, CH2), 63.9 (CH), 81.0 (C), 122.8 (ArCH), 123.7 (ArCH), 125.8 (2 × ArCH), 126.1 (2 × ArCH), 126.5 (2 × ArCH), 126.7 (2 × ArCH), 127.0 (2 × ArCH), 136.1 (ArCH), 145.0 (ArC), 145.4 (ArC), 146.9 (br, ArC), 154.0 (ArCH), 166.9 (br, CO); HRMS (ES+ TOF): found 359.1769 [M + H]+; C23H23N2O2 requires 359.1760.(S)-[2-(Methoxydiphenylmethyl)pyrrolidin-1-yl](1-methyl-1H-imidazol-2-yl)methanone 26
NaH (60% dispersion in mineral oil, 44 mg, 1.1 mmol) was added to a solution of hydroxy catalyst 17 (361 mg, 1 mmol) in THF (10 mL). After stirring for 20 min at rt, MeI (156 mg, 1.1 mmol) was added dropwise at the same temperature and then stirred overnight. The resulting mixture was quenched with saturated aqueous ammonium chloride (10 mL). The organic phase was separated and the aqueous phase was extracted with DCM (3 × 10 mL). The combined organic extracts were dried over MgSO4. After filtration and evaporation, the residue was purified by chromatography on silica gel [20%–50% ethyl acetate in petroleum ether (40–60)] to yield the product 26 as a white foam as a 4.6:1 mixture of two rotamers A and B (0.21 g, 56%); m.p. 93–95 °C; [α]20D −68 (c 0.5, CHCl3); νmax (thin film) 2927 (w), 1624 (s), 1524 (m), 1463 (s); δH (400 MHz, CDCl3, rotamer A): 1.03–1.16 (1H, m, CHH), 1.57–1.68 (1H, m, CHH), 1.98–2.05 (1H, m, CHH), 2.25–2.48 (2H, m, CH2), 2.38 (3H, s, CH3), 3.54–3.61 (1H, m, CH), 3.79 (3H, s, CH3), 6.31 (1H, d, J 8.9, CH), 6.79 (1H, d, J 0.9, ArCH), 6.95 (1H, d, J 0.9, ArCH), 7.26–7.36 (7H, m, ArCH), 7.46–7.52 (1H, m, ArCH); δH (400 MHz, CDCl3, rotamer B): 1.03–1.16 (1H, m, CHH), 1.45–1.53 (1H, m, CHH), 1.98–2.05 (1H, m, CHH), 2.95–3.02 (1H, m, CHH), 3.00 (3H, s, CH3), 3.70–3.76 (1H, m, CHH), 3.86 (3H, s, CH3), 5.72 (1H, dd, J 8.7, 3.1, CH), 6.89 (1H, s, ArCH), 7.07 (1H, s, ArCH), 7.26–7.36 (8H, m, ArCH); δC (100 MHz, CDCl3, rotamer A): 21.8 (CH2), 27.6 (CH2), 34.2 (CH3), 47.4 (CH2), 51.8 (CH3), 63.5 (CH), 86.8 (C), 122.4 (ArCH), 126.6 (ArCH), 127.6 (3 × ArCH), 127.7 (3 × ArCH), 129.4 (2 × ArCH), 130.0 (2 × ArCH), 138.8 (ArC), 139.7 (ArC), 142.6 (ArC), 161.2 (CO); δC (100 MHz, CDCl3, rotamer B): 23.9 (CH2), 26.6 (CH2), 29.7 (CH2), 35.4 (CH3), 50.7 (CH2), 52.5 (CH3), 60.4 (CH), 86.7 (C), 123.9 (ArCH), 127.4 (2 × ArCH), 127.5 (2 × ArCH), 127.8 (3 × ArCH), 129.6 (3 × ArCH), 129.9 (ArCH), 140.1 (ArC), 140.9 (ArC), 141.2 (ArC), 161.2 (CO); HRMS (ES+ TOF): found 376.2023 [M + H]+; C23H26N3O2 requires 376.2025.
(S)-[2-(Methoxydiphenylmethyl)pyrrolidin-1-yl](pyridin-2-yl)methanone 27
NaH (60% dispersion in mineral oil, 80 mg, 2 mmol) was added to a solution of alcohol 23 (358 mg, 1 mmol) in THF (8 mL). After stirring for 15 min at rt, MeI (284 mg, 2 mmol) was added dropwise at rt and left to stir overnight. The resulting mixture was quenched with saturated aqueous ammonium chloride (20 mL). The organic phase was separated and the aqueous phase was extracted with ethyl acetate (3 × 15 mL). The combined organic extracts were dried over Na2SO4, filtered, evaporated, and the resulting residue purified by chromatography on silica gel [10%–100% ethyl acetate in petroleum ether (40–60)] to yield the product 27 as a yellow solid as a 2:1 mixture of rotamers A and B (308 mg, 83%); m.p. 104–106 °C; [α]20D −154 (c 1.0, CHCl3); νmax (thin film) 3056 (m), 2961 (m), 1626 (s), 1587 (s), 1556 (s); δH (400 MHz, CDCl3, rotamer A): 1.04–1.16 (1H, m, CHH), 1.59–1.70 (1H, m, CHH), 1.94–2.01 (1H, m, CHH), 2.31 (3H, s, CH3), 2.37–2.45 (2H, m, CH2), 3.65–3.72 (1H, m, CH), 6.20 (1H, dd, J 9.2, 1.6, CH), 7.25–7.39 (9H, m, ArCH), 7.52 (1H, d, J 9.1, ArCH), 7.60 (1H, d, J 6.7, ArCH), 7.59–7.78 (2H, m, ArCH), 8.54 (1H, d, J 4.8, ArCH); δH (400 MHz, CDCl3, rotamer B): 1.04–1.16 (1H, m, CH), 1.44–1.55 (1H, m, CH), 2.02–2.11 (1H, m, CH), 2.13–2.23 (1H, m, CH), 2.71–2.78 (1H, m, CH), 3.07 (3H, s, CH3), 3.15–3.21 (1H, m, CH), 5.80 (1H, dd, J 9.1, 3.2, CH), 7.25–7.39 (11H, m, ArCH), 7.59–7.78 (2H, m, ArCH), 8.65 (1H, d, J 4.6, ArCH); δC (100 MHz, CDCl3, rotamer A): 21.6 (CH2), 27.8 (CH2), 47.7 (CH2), 50.7 (CH), 64.0 (CH3), 87.0 (C), 123.7 (ArCH), 124.3 (ArCH), 127.3 (2 × ArCH), 127.6 (ArCH), 127.7 (2 × ArCH), 127.8 (ArCH), 127.9 (ArCH), 129.6 (ArCH), 129.8 (ArCH), 129.9 (ArCH), 130.0 (ArCH), 138.4 (ArC), 138.6 (ArC), 146.9 (ArCH), 156.6 (ArC), 168.5 (CO); δC (100 MHz, CDCl3, rotamer B): 24.0 (CH2), 26.9 (CH2), 50.6 (CH2), 52.3 (CH), 59.8 (CH3), 86.6 (C), 124.5 (ArCH), 127.3 (ArCH), 127.6 (ArCH), 127.7 (2 × ArCH), 127.8 (ArCH), 127.9 (ArCH), 129.6 (ArCH), 129.8 (ArCH), 129.9 (ArCH), 130.0 (ArCH), 136.4 (2 × ArCH), 140.5 (ArC), 141.9 (ArC), 148.7 (ArCH), 155.0 (ArC), 169.1 (CO); HRMS (ES+ TOF): found 373.1917 [M + H]+; C24H25N2O2 requires 373.1916.
(S)-N-[1-Phenylethyl]-4-fluoroaniline 3426
Obtained using general procedure A with ketimine 29, employing catalyst 17 (1 mol%, 96% conversion, 87% ee) or catalyst 26 (0.1 mol%, 95% conversion, 86% ee) as a light yellow oil; [α]20D +23.8 (c 1.05, CHCl3, 87% ee, lit.26 −16.8, c 1.0, CHCl3, 84% ee for R-enantiomer); δH (400 MHz, CDCl3): 1.52 (3H, d, J 6.6, CH3), 4.03 (1H, br s, NH), 4.45 (1H, q, J 6.6, CH), 6.43–6.47 (2H, m, ArCH), 6.79–6.84 (2H, m, ArCH), 7.23–7.28 (1H, m, ArCH), 7.32–7.38 (4H, m, ArCH); δC (101 MHz, CDCl3): 25.1 (CH3), 54.1 (CH), 114.1 (d, JC–F 6.9, ArCH), 115.5 (d, JC–F 22.2, ArCH), 125.9 (ArCH), 127.0 (ArCH), 128.8 (ArCH), 143.7 (ArC), 145.1 (ArC), 155.5 (d, JC–F 234.6, ArC); HPLC (Kromasil 3-Cellucoat OD, hexane/i-PrOH = 99/1, flow rate = 1.0 mL min−1, 254 nm): (R) enantiomer tR = 6.9 min, (S) enantiomer tR = 7.8 min.(S)-N-[1-(4′-Methoxyphenyl)ethyl]-4-methoxyaniline 3516
Obtained using general procedure A with ketimine 30, employing catalyst 26 (0.1 mol%, 97% conversion, 84% ee) as a white solid; m.p. 93–95 °C (not reported in literature); [α]20D −14.5 (c 0.96, CHCl3, 81% ee, lit. −15.5,16c 1.1, CHCl3, 85% ee); δH (250 MHz, CDCl3): 1.49 (3H, d, J 6.6, CH3), 3.72 (3H, s, CH3), 3.76 (1H, br s, NH), 3.80 (3H, s, CH3), 4.39 (1H, q, J 6.6, CH), 6.49 [2H, (AX)2, ArCH], 6.71 [2H, (AX)2, ArCH], 6.87 [2H, (AX)2, ArCH], 7.29 [2H, (AX)2, ArCH]; δC (63 MHz, CDCl3): 25.1 (CH3), 53.7 (CH3), 55.3, (CH3), 55.8 (CH3), 114.0 (2 × ArCH), 114.6 (2 × ArCH), 114.8 (2 × ArCH), 127.0 (2 × ArCH), 137.6 (ArC), 141.7 (ArC), 151.9 (ArC), 158.5 (ArC); HPLC (Phenomenex Lux 3 μm Cellulose-1, hexane/i-PrOH = 90/10, flow rate = 1.0 mL min−1, 254 nm): (R) enantiomer tR = 9.1 min, (S) enantiomer tR = 10.0 min.(S)-N-[1-(4′-Nitrophenyl)ethyl]-4-methoxyaniline 3616
Obtained using general procedure A with ketimine 31, employing catalyst 26 (0.1 mol%, 98% conversion, 84% ee) as a brown oil; [α]20D −30.8 (c 1.0, CHCl3, 84% ee, lit. −25.9,16c 0.54, CHCl3, 86% ee); δH (400 MHz, CDCl3): 1.55 (3H, d, J 6.8, CH3), 3.72 (3H, s, CH3), 3.93 (1H, br s, NH), 4.53 (1H, q, J 6.8, CH), 6.43 (2H, d, J 8.2, ArCH), 6.72 (2H, d, J 8.2, ArCH), 7.57 (2H, d, J 8.8, ArCH), 8.20 (2H, d, J 8.8, ArCH); δC (101 MHz, CDCl3): 25.0 (CH3), 54.0 (CH3), 55.7 (CH), 114.5 (2 × ArCH), 114.8 (2 × ArCH), 124.1 (2 × ArCH), 126.8 (2 × ArCH), 140.8 (ArC), 147.0 (ArC), 152.3 (ArC), 153.6 (ArC); HPLC (Phenomenex Lux 3 μm Cellulose-1, hexane/i-PrOH = 90/10, flow rate = 1.0 mL min−1, 254 nm): (R) enantiomer tR = 28.3 min, (S) enantiomer tR = 31.8 min.(S)-N-[1-(Thiophen-2-yl)ethyl]-4-methoxyaniline 3717
Obtained using general procedure A with ketimine 32, employing catalyst 26 (0.1 mol%, 77% conversion, 80% ee) as a yellow oil; [α]20D −8.0 (c 1.0, CHCl3, 79% ee, lit.17 −8.0, c 1.0, CHCl3, 79% ee); δH (250 MHz, CDCl3): 1.63 (3H, d, J 6.6, CH3), 3.75 (3H, s, CH3), 4.76 (1H, q, J 6.6, CH), 6.62 [2H, (AX)2, ArCH], 6.75 [2H, (AX)2, ArCH], 6.94–6.98 (2H, m, ArCH), 7.18 (1H, dd, J 4.7, 1.6, ArCH); δC (63 MHz, CDCl3): 24.8 (CH3), 50.6 (CH3), 55.8 (CH), 114.9 (2 × ArCH), 115.2 (2 × ArCH), 123.0 (ArCH), 123.6 (ArCH), 126.7 (ArCH), 141.2 (ArC), 150.6 (ArC), 152.5 (ArC); HPLC (Phenomenex Lux 3 μm Cellulose-1, hexane/i-PrOH = 98/2, flow rate = 1.0 mL min−1, 254 nm): (R) enantiomer tR = 15.1 min, (S) enantiomer tR = 17.3 min.(S)-N-[1-(Naphthalen-2-yl)ethyl]-4-methoxyaniline 3816
Obtained using general procedure A with ketimine 33, employing catalyst 26 (0.1 mol%, 99% conversion, 92% ee) as a light yellow solid; m.p. 96–98 °C (lit.16 95–96 °C); [α]20D −24.0 (c 1.0, CHCl3, 85% ee, lit.16 −26.0, c 1.0, CHCl3, 86% ee); δH (250 MHz, CDCl3): 1.59 (3H, d, J 6.9, CH3), 3.70 (3H, s, CH3), 3.90 (1H, br s, NH), 4.59 (1H, q, J 6.9, CH), 6.53 [2H, (AX)2, ArCH], 6.71 [2H, (AX)2, ArCH], 7.42–7.55 (3H, m, ArCH), 7.80–7.86 (4H, m, ArCH); δC (63 MHz, CDCl3): 25.2 (CH3), 54.6 (CH3), 55.8 (CH), 114.7 (ArCH), 114.8 (ArCH), 124.4 (ArCH), 124.5 (ArCH), 125.5 (ArCH), 126.0 (ArCH), 127.7 (ArCH), 127.9 (ArCH), 128.5 (ArCH), 132.8 (ArC), 133.7 (ArC), 141.7 (ArC), 143.1 (ArC), 152.0 (ArC); HPLC (Phenomenex Lux 3 μm Cellulose-1, hexane/i-PrOH = 90/10, flow rate = 1.0 mL min−1, 254 nm): (R) enantiomer tR = 10.1 min, (S) enantiomer tR = 12.0 min.Acknowledgements
We thank The University of Sheffield (XL and FT) and EPSRC (EP/K007955/1 – ATR) for funding.Notes and references
- F. Iwasaki, O. Onomura, K. Mishima, T. Kanematsu, T. Maki and Y. Matsumura, Tetrahedron Lett., 2001, 42, 2525–2527 CrossRef CAS.
- S. Guizzetti and M. Benaglia, Eur. J. Org. Chem., 2010, 5529–5541 CrossRef CAS.
- S. Jones and C. J. A. Warner, Org. Biomol. Chem., 2012, 10, 2189–2200 CAS.
- A. V. Malkov, A. Mariani, K. N. MacDougall and P. Kočovský, Pavel, Org. Lett., 2004, 6, 2253–2256 CrossRef CAS PubMed.
- Z. Wang, X. Ye, S. Wei, P. Wu, A. Zhang and J. Sun, Org. Lett., 2006, 8, 999–1001 CrossRef CAS PubMed.
- C. Baudequin, D. Chaturvedi and S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 2623–2629 CrossRef CAS.
- Y.-C. Xiao, C. Wang, Y. Yao, J. Sun and Y.-C. Chen, Angew. Chem., Int. Ed., 2011, 50, 10661–10664 CrossRef CAS PubMed.
- D. Pei, Z. Wang, S. Wei, Y. Zhang and J. Sun, Org. Lett., 2006, 8, 5913–5915 CrossRef CAS PubMed.
- D. Pei, Y. Zhang, S. Wei, M. Wang and J. Sun, Adv. Synth. Catal., 2008, 350, 619–623 CrossRef CAS.
- S. Guizzetti, M. Benaglia, F. Cozzi and R. Annunziata, Tetrahedron, 2009, 65, 6354–6363 CrossRef CAS.
- H. Zheng, J. Deng, W. Lin and X. Zhang, Tetrahedron Lett., 2007, 48, 7934–7937 CrossRef CAS.
- O. Onomura, Y. Kouchi, F. Iwasaki and Y. Matsumura, Tetrahedron Lett., 2006, 47, 3751–3754 CrossRef CAS.
- C. J. A. Warner, A. T. Reeder and S. Jones, Tetrahedron: Asymmetry, 2016, 27, 136–141 CrossRef CAS.
- M. Bonsignore, M. Benaglia, R. Annunziata and G. Celentano, Synlett, 2011, 1085–1088 CAS.
- M. Sugiura, M. Kumahara and M. Nakajima, Chem. Commun., 2009, 3585–3587 RSC.
- F.-M. Gautier, S. Jones and S. J. Martin, Org. Biomol. Chem., 2009, 7, 229–231 CAS.
- F.-M. Gautier, S. Jones, X. Li and S. J. Martin, Org. Biomol. Chem., 2011, 9, 7860–7868 CAS.
- Z. Zhang, P. Rooshenas, H. Hausmann and P. Schreiner, Synthesis, 2009, 1531–1544 CAS.
- A. V. Malkov, K. Vranková, S. Stončius and P. Kočovský, J. Org. Chem., 2009, 74, 5839–5849 CrossRef CAS PubMed.
- R. E. Cowley, R. P. Bontchev, E. N. Duesler and J. M. Smith, Inorg. Chem., 2006, 45, 9771–9779 CrossRef CAS PubMed.
- M. Braun, Angew. Chem., Int. Ed., 2012, 51, 2550–2562 CrossRef CAS PubMed.
- S. El-Fayyoumy, M. H. Todd and C. J. Richards, Beilstein J. Org. Chem., 2009, 5, 67 Search PubMed.
- M. Hagemann, A. Mix, R. J. F. Berger, T. Pape and N. W. Mitzel, Inorg. Chem., 2008, 47, 10554–10564 CrossRef CAS PubMed.
- A. V. Malkov, S. Stončius, K. Vrankova, M. Arndt and P. Kočovský, Chem. – Eur. J., 2008, 14, 8082–8085 CrossRef CAS PubMed.
- S. Jones and X. Li, Tetrahedron, 2012, 68, 5522–5532 CrossRef CAS.
- T. Imamoto, N. Iwadate and K. Yoshida, Org. Lett., 2006, 8, 2289–2292 CrossRef CAS PubMed.
- R. I. Ngochindo, J. Chem. Soc., Perkin Trans. 1, 1990, 1645–1648 RSC.
- P. A. Shirley, D. A. Gross and B. H. Roussel, J. Org. Chem., 1955, 20, 225–231 CrossRef.
- K. Nakayama and W. J. Thompson, J. Am. Chem. Soc., 1990, 112, 6936–6942 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H & 13C NMR spectra, chromatographs and crystal structure of catalyst 17. CCDC 1517809. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob02537d |
| This journal is © The Royal Society of Chemistry 2017 |