
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the nature of the electronic effect of multiple hydroxyl groups in the 6-membered ring – the effects are additive but steric hindrance plays a role too†
Christian Marcus
Pedersen
 * and
Mikael
Bols
*
* and
Mikael
Bols
*
Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen Ø, Denmark. E-mail: cmp@chem.ku.dk; bols@chem.ku.dk
First published on 13th January 2017
Abstract
Research during the last two decades has shown a remarkable directional component of the substituent effects of hydroxy groups, which has a profound effect on the properties of hydroxylated compounds such as carbohydrates. While the epimerisation of a single hydroxyl function is well studied the consequence of multiple epimerisations is more speculative. In this work the effect of three epimerisations was investigated. To this end epimeric 2-phenyl iminoxylitols that have a phenyl group as a conformational anchor and thus hydroxyl groups in the axial or equatorial position, respectively, were synthesized and their pKa and conformation were studied. The results show that the large difference in the electronic effect between the axial and equatorial hydroxyls is partially cancelled by counteracting steric hindrance from 1,3-diaxial interactions. Hydrogen bonding does not appear to play any role in the electronic influence of the hydroxyl groups.
Introduction
The profound influence of substituents on the reactivity and properties of organic compounds has been recognized for many years.1 The effect of substituents on the basicity of simple amines was nicely compiled and discussed more than half a century ago2 where it was recognized that the hydroxyl was a particularly influential substituent, but was also fickle with respect to its base decreasing effect which could vary profoundly.2a This was presumed to be due to hydrogen bonding. It was however not until recently that the highly directional and systematic aspect of this substituent effect was recognized and the importance of stereochemistry and conformation was revealed.3 From the synthesis of diastereomeric pairs of hydroxylated piperidines we realized that 1-azafagomine and its galacto isomer have different pKa values, which reflect the different stability of the ammonium ions. It was found that the equatorial hydroxyl groups on the piperidine ring decreased the basicity much more than the corresponding axial stereoisomers.3a,4 The substituent effects were also studied for other functional groups and found to be so consistent that the pKa can be calculated using the formula 10.7 − ∑σs for piperidines or 7.3 − ∑σs for hexahydropyridazines, where σs is the substituent contribution of the axial or equatorial substituent. The difference in pKa values can be attributed to several effects, but charge–dipole interactions are in common for all and provide a satisfactory explanation for the observations: each C–O bond constitutes a dipole while each ammonium group has a positive charge (mainly dispersed on the surrounding atoms) – the closer the negative ends of the dipoles are to the positive charge the more favourable the structure. With this in mind it is not surprising that the stereoelectronic effect is coupled with a conformational effect, which is dependent on the protonation stage of the ring nitrogen, i.e. upon protonation it becomes even more favourable to have electro-negative substituents axially oriented as they effectively are less electron withdrawing than when equatorial due to the charge–dipole interaction. The fact that these stereoelectronic effects are strong enough to introduce conformational changes has been observed in a number of cases5e.g. the hexahydropyridazine derivative (Fig. 1), which shifts conformation from 1 to 2 depending on pH6 and also in bicyclic sulfonium salts like 3, which resides in an all axial conformation, which suffers from sterically unfavourable 1,3-diaxial interactions.7 Generally, as the C–X dipole increase, with C–F as the extreme,8 the conformational change placing the dipole in an axial orientation becomes increasingly important for the stabilization of the piperidinium ion.9 In many cases the difference in stability between the two conformations is small and mixtures of conformations are present. The ratio of such mixtures influences the overall pKa value and can be determined directly by NMR from the intermediate J-couplings measured at room temperature10 or even from the pKa itself.6 The pKa values for the two conformational extremes 1 and 2 (Fig. 1) can be calculated using the formula mentioned above giving pKa = 5.5 and 6.8, respectively, whereas the measured value is 6.2 as a result of both conformers being present. The connection between the substituent effects in nitrogen heterocycles, mainly piperidines, and other heterocycles has been thoroughly studied over the last decade,11 and it is well accepted that the stereoelectronic effects observed in piperidines are linearly correlated with the anomeric reactivity in e.g. carbohydrates.3b,12 As an example it has been revealed that there are linear correlations between the rate in glycoside hydrolysis or glycosylation reactions and the acidity of piperidinium salts, with analogous substituent patterns.12a The generality of these effects is such that they with great success have been used to predict the behaviour and reactivity of carbohydrates such as anhydrosugars13 and axial rich glycosyl donors14 as well as hydroxymethyl conformers.15 | ||
| Fig. 1 At the top the calculated pKa values of two conformational extremes of an azasugar (top) and the measured value. Below an all axial bicyclic sulfonium salt illustrating the preference of axial hydroxyl groups in rings with positively charged heteroatoms. | ||
The pKa-calculations of piperidinium derivatives assume that substituent effects are additive, which for a significant number of reference compounds hold true with a relatively small margin of error. However extreme differences in the basicity of conformers are predicted by this method and such differences have so far not been confirmed experimentally due to the unavailability of appropriate reference compounds. The purpose of this work was to investigate whether such extreme differences in base strength would be observed in diastereoisomers. For this purpose we targeted the two epimers of 5-C-phenyliminoxylitol, 6 and 7, and determined their pKa values (Fig. 2). As model compounds 6 and 7 are ideally suited because the very bulky phenyl group in the 5-position (carbohydrate numbering) acts as a conformational anchor fixing the conformation so that the three hydroxyl groups will either be all equatorial or all axial, i.e. giving the two extremes. The differences in pKa values are therefore large and calculated to be 2.0 pKa units, with 7 being the most basic amine (Fig. 2).
 | ||
| Fig. 2 The two target model compounds and the calculation of the difference between their pKa values. | ||
Results and discussion
The two target piperidines have either a D-gluco or L-ido stereochemistry if numbered from the CH2 group using carbohydrate numbering. In view of this, diacetone glucose is an obvious starting material as the stereogenic centers at C2, C3 and C4 can be maintained unchanged while phenyl and nitrogen are installed at C5. Using this approach aldehyde 8 was prepared in 3 steps from glucose acetonide16 and a Grignard addition using phenyl magnesium bromide was carried out to give an approx. 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the epimers (9 and 13), with L-ido (13) being the major product.17 Since both were required, a two-step conversion of 13 involving PDC oxidation and NaBH4 reduction provided the D-gluco derivative 9 as the main product (approx. 10:1 over 13). The L-ido isomer 13 was readily tosylated followed by nucleophilic substitution using NaN3 in DMF to give the fully protected azide 14. Removal of the isopropylidene group and hydrogenation and benzyl hydrogenolysis over Pd/C afforded the 5-C-phenyl-D-gluco-1,5-iminopentitol 6. The same approach could however not be applied for the epimeric alcohol 9 as the tosylation did not proceed even under more forcing conditions. This is probably due to steric hindrance. To circumvent this, the 3-O-benzyl group was removed by hydrogenolysis and a cyclic sulfite 11 was prepared using thionyl chloride. Treating this with NaN3 in DMF gave the desired azide 12. The acidic removal of the isopropylidene protective group followed by hydrogenolysis using Pd/C and triethylsilane (TESH) gave the target compound L-2-phenyl-iminoxylitol 7. The hydrogenation/hydrogenolysis reactions were generally sluggish and attempts to use more forcing conditions mainly gave the pyridine derivative 15. Changing the catalyst to Pearlman's catalyst (Pd(OH)2) or Raney® Ni did not improve the reaction.
:1 ratio of the epimers (9 and 13), with L-ido (13) being the major product.17 Since both were required, a two-step conversion of 13 involving PDC oxidation and NaBH4 reduction provided the D-gluco derivative 9 as the main product (approx. 10:1 over 13). The L-ido isomer 13 was readily tosylated followed by nucleophilic substitution using NaN3 in DMF to give the fully protected azide 14. Removal of the isopropylidene group and hydrogenation and benzyl hydrogenolysis over Pd/C afforded the 5-C-phenyl-D-gluco-1,5-iminopentitol 6. The same approach could however not be applied for the epimeric alcohol 9 as the tosylation did not proceed even under more forcing conditions. This is probably due to steric hindrance. To circumvent this, the 3-O-benzyl group was removed by hydrogenolysis and a cyclic sulfite 11 was prepared using thionyl chloride. Treating this with NaN3 in DMF gave the desired azide 12. The acidic removal of the isopropylidene protective group followed by hydrogenolysis using Pd/C and triethylsilane (TESH) gave the target compound L-2-phenyl-iminoxylitol 7. The hydrogenation/hydrogenolysis reactions were generally sluggish and attempts to use more forcing conditions mainly gave the pyridine derivative 15. Changing the catalyst to Pearlman's catalyst (Pd(OH)2) or Raney® Ni did not improve the reaction.
In order to study the influence of internal hydrogen bonding four additional derivatives were synthesized. The H-bond donating capability of the 3-OH group was investigated by preparing the 3-O-methyl derivatives 18 and 20 (Scheme 2). The D-gluco epimer 18 was synthesized by 3-O-methylation of diacetone glucose followed by selective removal of the 5,6-O-isopropylidene group, periodate cleavage and addition of phenylmagnesium bromide to obtain alcohol 16 predominantly as the L-ido isomer. Tosylation and subsequent SN2 substitution using sodium azide gave azide 17, which was treated with AcOH (75%) to successfully remove the isopropylidene group from where hydrogenolysis to 18 was attempted. Neither hydrogenolysis with H2(g) in the presence of Pd/C, Pearlman's catalyst (Pd(OH)2) or RANEY® Ni gave satisfactory results nor did a two-step procedure of the Staudinger (Ph3P) reaction and NaBH4 reduction give satisfactory yield. However, changing the hydrogen source in the catalytic reduction solved the problem and hence using triethylsilane (TESH) in combination with Pd/C gave the 3-O-methyl 2-phenyl-L-iminoxylitol 18 in reproducible good yields. In order to prepare 20 it was necessary to start with the late intermediate 12 (Scheme 1) where the azide was already present. Compound 12 was methylated with MeI/NaH, deprotected with acetic acid and subjected to reductive amination according to the protocol above (Pd/C, TESH) to give 20 (Scheme 2).
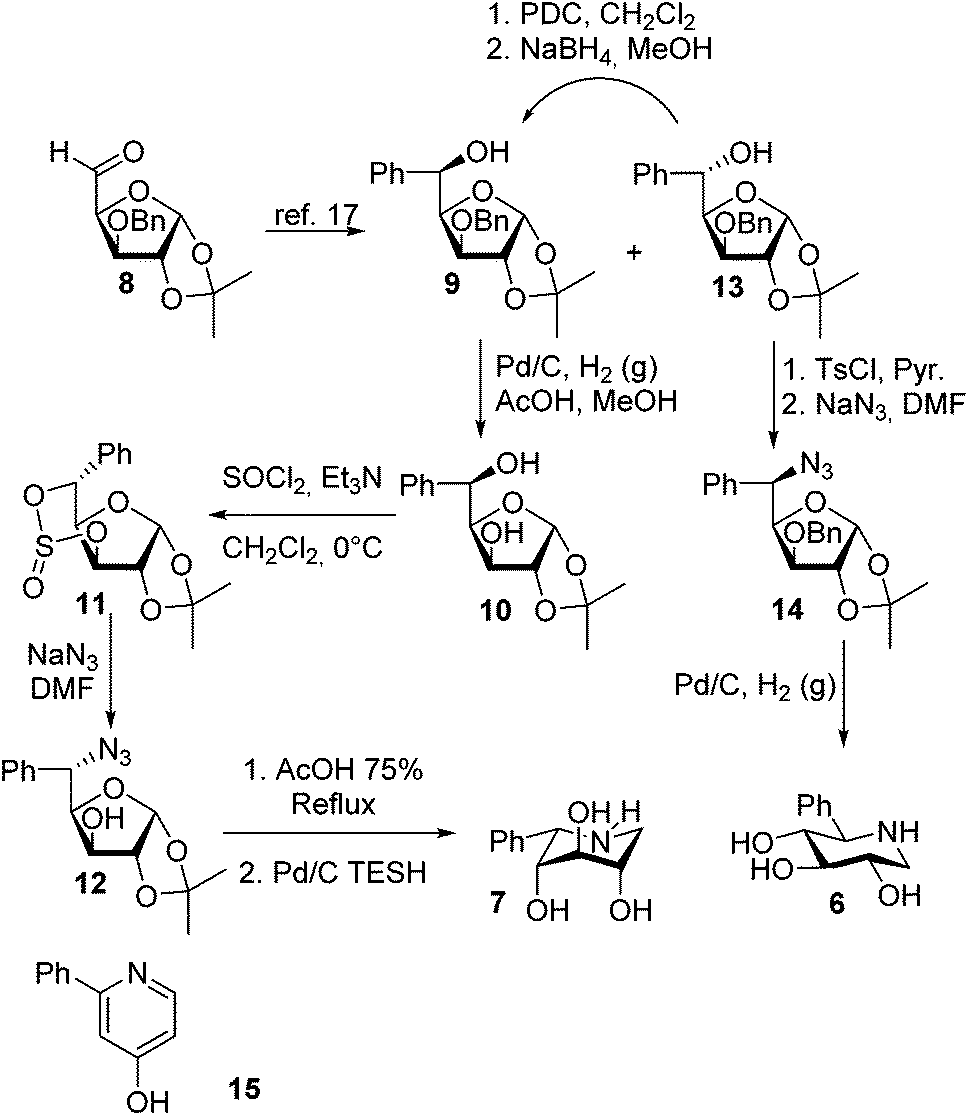 | ||
| Scheme 1 Synthesis of the two model compounds 6 and 7. Pyridine 15 is a dominant side product in the final reductive amination. | ||
 | ||
| Scheme 2 Synthesis of the 3-O-methylated derivatives. | ||
The D-gluco epimer 23 and the L-ido epimer 27 (Scheme 3) having 2- and 4-hydroxyls protected with methyl groups were prepared starting from the late stage intermediates 14 and 12 (Scheme 1). Compound 12 had to be benzylated to the 3-O benzyl derivative 24 (Scheme 3). Then both epimers followed the same reaction sequence of removal of isopropylidene with acetic acid followed by reductive amination, which could be performed using Pd/C and TESH without any loss of benzyl ether. The obtained piperidines 21 and 25 were Boc-protected, methylated and the benzyl groups removed using Pearlman's catalyst and hydrogen. The treatment of the obtained piperidines 22 and 26 with TFA gave the desired compounds 23 and 27 as their corresponding piperidinium trifluoroacetates (Scheme 3).
 | ||
| Scheme 3 Synthesis of the two 2,4-di-O-methylated derivatives. | ||
Conformational studies
With the 6 compounds (6, 7, 18, 20, 23 and 27) prepared, their respective conformations were studied using NMR. Since the conformation can be strongly dependent on the pH, as mentioned in the Introduction, the compounds were studied in both their amine and ammonium forms by adding either NaOD or DCl to the NMR sample. As Table 1 shows there is practically no conformational change upon protonation and hence the role of the 2-phenyl group as a conformational anchor that keeps the conformation fixed in one chair form is confirmed. In the D-gluco series (6, 18 and 23) the compounds, regardless of pH, show large trans vicinal couplings in the range 9–12 Hz for J23, J34 and J45 showing that these compounds are in the 4C1 chair conformation (Table 1). The compounds in the L-ido series (7, 20 and 27) display very small couplings (broad singlets – bs) for J23, J34 and J45, which is consistent with a 1C4 chair conformation (Table 1) and inconsistent with a 4C1 chair or B1,4 boat conformation. In both cases this is as anticipated.| Cmp. | δ H-1ab (J1a1b) | δ H-2 (J1ab2) | δ H-3 (J23) | δ H-4 (J34) | δ H-5 (J45) |
|---|---|---|---|---|---|
| m is multiplet, s is singlet and bs is broad singlet. | |||||
| 6 | 3.58, 3.15 (12.4) | 3.99 (5.1,11.6) | 3.68 (9.3) | 4.02 (9.3) | 4.23 (10.8) |
| 6 (D+) | 3.07 (12.4) | 3.90 (11.6, 0) | 3.58 (9.3) | 3.93 (9.3) | 4.15 (10.8) |
| 18 | 3.14, 2.61 (12.1) | 3.72 (5.3, 11.1) | 3.21 (9.1) | 3.64 (9.1) | 3.50 (10.0) |
| 18 (D+) | 3.58, 3.17 12.2 | 4.05 (5.2, 12.2) | 3.46 (9.3) | 4.09 (9.3) | 4.25 (11.0) |
| 23 | 2.41, 2.47 (12.5) | 3.4 (bm) | 3.56 (∼8.5) | 3.34 (9.1) | 3.53 (9.3) |
| 23 (D+) | 3.82, 3.08 (12.5) | 3.73 (5.2, 11.2) | 3.79 (9.2) | 3.83 (9.2) | 4.27 (10.3) |
| 7 | 3.19, 3.07 (14.5) | 3.77 (bs) | 4.06 (bs) | 3.87 (bs) | 4.20 (bs) |
| 7 (D+) | 3.67, 3.53 (13.7) | 4.15 (bs) | 4.19 (bs) | 4.11 (bs) | 4.71 (bs) |
| 20 | 3.07, 3.03 (14.3) | 3.87 (m) | 3.89 (bs) | 3.65 (bs) | 4.03 (bs) |
| 20 (D+) | 3.59, 3.53 (13.7) | 4.30 (bs) | 3.79 (<3) | 4.25 (<3) | 4.62 (bs) |
| 27 | 3.09, 3.04 (14.4) | 3.33 (m, 3.6) | 4.22 (bs) | 3.46 (m) | 4.22 (bs) |
| 27 (D+) | 3.16, 3.20 (m) | 4.26 (m) | 3.41 (bs) | 3.53 (s) | 4.32 |
Titrations
The pKa values of the 6 compounds (6, 7, 18, 20, 23 and 27) were determined using potentiometric titration. At least two titrations were conducted per compound and the average results are presented in Table 2 with the uncertainty being within 0.1 units. From the results we see some general and interesting trends. Firstly, the difference in the pKa value between the epimeric pairs, 6/7, 18/20 or 23/27, is 1.1 to 1.3 pH units, respectively, with the all axial epimer being more basic. While this qualitatively is the anticipated result it is considerably less than is predicted from calculations as these predict a difference of approximately 2 pKa units. Secondly O-methylation of the 3-OH has no significant influence on the pKa value as seen from the essential identical values obtained from the pairs 6/18 and 7/20. Thirdly the 2,4-di-O-methyl derivatives are found consistently less basic than the hydroxylated counterpart as seen from the 0.4 unit lower pKa value compared to 6 and the 0.6 unit lower pKa value of 27 compared to 7.| Compound | Structure (protonated) | pKa |
|---|---|---|
| 6 |

|
6.1 |
| 18 |

|
6.1 |
| 23 |

|
5.7 |
| 7 |
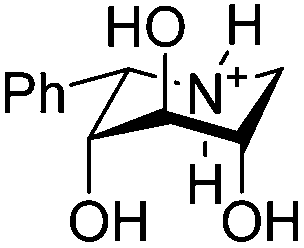
|
7.4 |
| 20 |

|
7.3 |
| 27 |

|
6.8 |
Discussion
The pKa values of the phenylpiperidines in the 4C1 conformation (6, 18 and 23) are 6.1, 6.1 and 5.7 and are very much in line with the values found in similar compounds or are at least readily explained. A comparison of the pKa value of 6.1 for compound 6 with that of 1-deoxynojirimycin (28) of 6.7 (Fig. 3)3a reveals that the value is reasonable since the only difference between these molecules is that the hydroxymethyl group in 28 has been replaced with a phenyl group. This should decrease the basicity since the phenyl group is considerably more electron withdrawing as reflected by its higher inductive substituent constant (0.94 versus 0.66),18 and this is seen as the α-phenyl group reduces the base strength by 1.319 whereas hydroxymethyl only reduces it by 0.7.3a The influence on the pKa of O-methylation in compounds 18 and 23 is also in line with what was previously found with 1-deoxynojirimycin derivatives where the tetra-O-methyl derivative 2911b (Fig. 3) was less basic (pKa 6.0) than 28 (pKa 6.7). The reason for this reduced basicity is the slightly higher electron withdrawing power of O-alkyl over OH, i.e. when 4-OH groups are replaced with OMe pKa drops to 0.7. However a single methylation on the most remote hydroxyl group has little influence as 18 that has the same pKa as 6. But a larger base reducing effect of 0.4 is seen when the closer 2- and 4-OH-groups are methylated (23). Altogether this shows that 6, 18 and 23 behave as we would expect from earlier findings: the pKa is mainly a result of the inductive and electrostatic effects of the phenyl group and the 3 equatorial OH or OMe groups, with OMe being slightly more electron withdrawing compared to an OH group.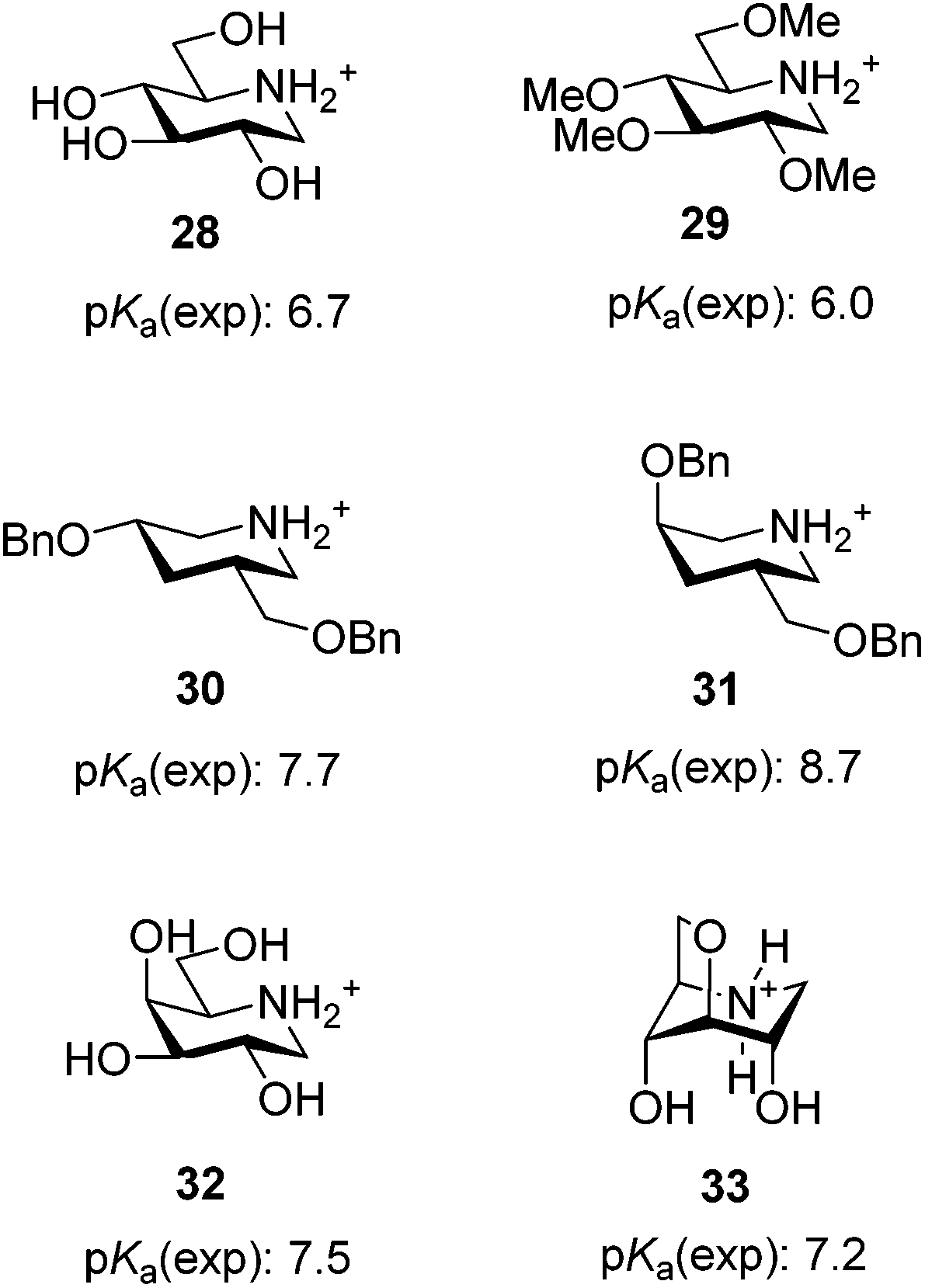 | ||
| Fig. 3 Experimental pKa values from the literature. | ||
However the pKa values of the phenylpiperidines in the 1C4 conformation (7, 20 and 27) are significantly smaller than would be anticipated from earlier findings. If we as above set the base reducing influence of the α-phenyl group to 1.3,19 we find that the pKa of 7 should be 8.2 (pKa = 10.7–1.3–0.5–0.5–0.2) if only inductive and electrostatic effects played a role and not the observed 7.4 (Table 2). The fact that the basicity is remarkably lower is also seen from many literature examples some of which are shown in Fig. 3. Epimerisation of a single benzyloxy group from the equatorial to axial position in piperidines 30 and 31 increases the pKa by 1.0,11b which is almost the same as the difference between the pairs 23 and 27 despite the fact that there are two methoxy groups and one hydroxyl group changing from equatorial to axial. Also galactodeoxynojirimycin 32 (Fig. 3) has a pKa of 7.5 and is thus slightly more basic than 7 despite the fact that it has only 1 and not 3 axial OH groups. Thus the all axial derivatives 7, 20 and 27 are clearly much less basic than anticipated from previous data except for one example: the 3,6-anhydro derivative 33 (Fig. 3) was previously prepared and its pKa value was determined to be 7.2,11b which is also an extraordinary low value. This extraordinary, according to electronic effects, low basicity that compounds 7, 20, 27 and 33 display must be due to the second important factor that influences the base strength of amines – steric hindrance.2a These compounds differ from the other piperidines in that they all have two axial non-hydrogen substituents β to the nitrogen. Therefore the protonation of the nitrogen will lead to two 1,3-diaxial steric interactions between a hydrogen and a hydroxyl or methoxy group, which obviously is unfavourable enough to reduce the pKa of these compounds with 0.8 to 1.0 units (Fig. 4). The fact that the influence of the 1,3-diaxial interactions from hydrogen is high is clearly seen in literature examples: while tert-butylcyclohexane (34) is well known to have the large tert-butyl group, equatorial cis-5-tert-butyl-2-methyl-1,3-dioxane (35) has the tert-butyl group predominantly at the axial position simply due to the lack of 1,3-diaxial interactions with hydrogen (Fig. 4).20 The effect is seen directly on the pKa in base pairs 36/37 and 38/39 (Fig. 4).21 The compounds having the ethylene bridge β to the amine (37 and 39) are 0.87 and 0.75, respectively, less basic than their analogues having the ethylene bridge α (36 and 38). As the electronic effects from the carbon substituents are negligible2a the lower base strength must be due to the steric hindrance caused by the 1,3-diaxial interaction between the β-bridge and ammonium hydrogen. Therefore 7, 20 and 27 do not become as basic as one would expect based on electronic effects because they are more difficult to protonate due to hindrance. The base lowering effect from the steric hindrance of the two axial hydroxyl groups is about 0.8, which is very similar to the pKa difference in base pairs 36/37 and 38/39. This similarity is reasonable as the methylenes in 37 and 39 should be comparable in size to OH and less bulky than methyl.
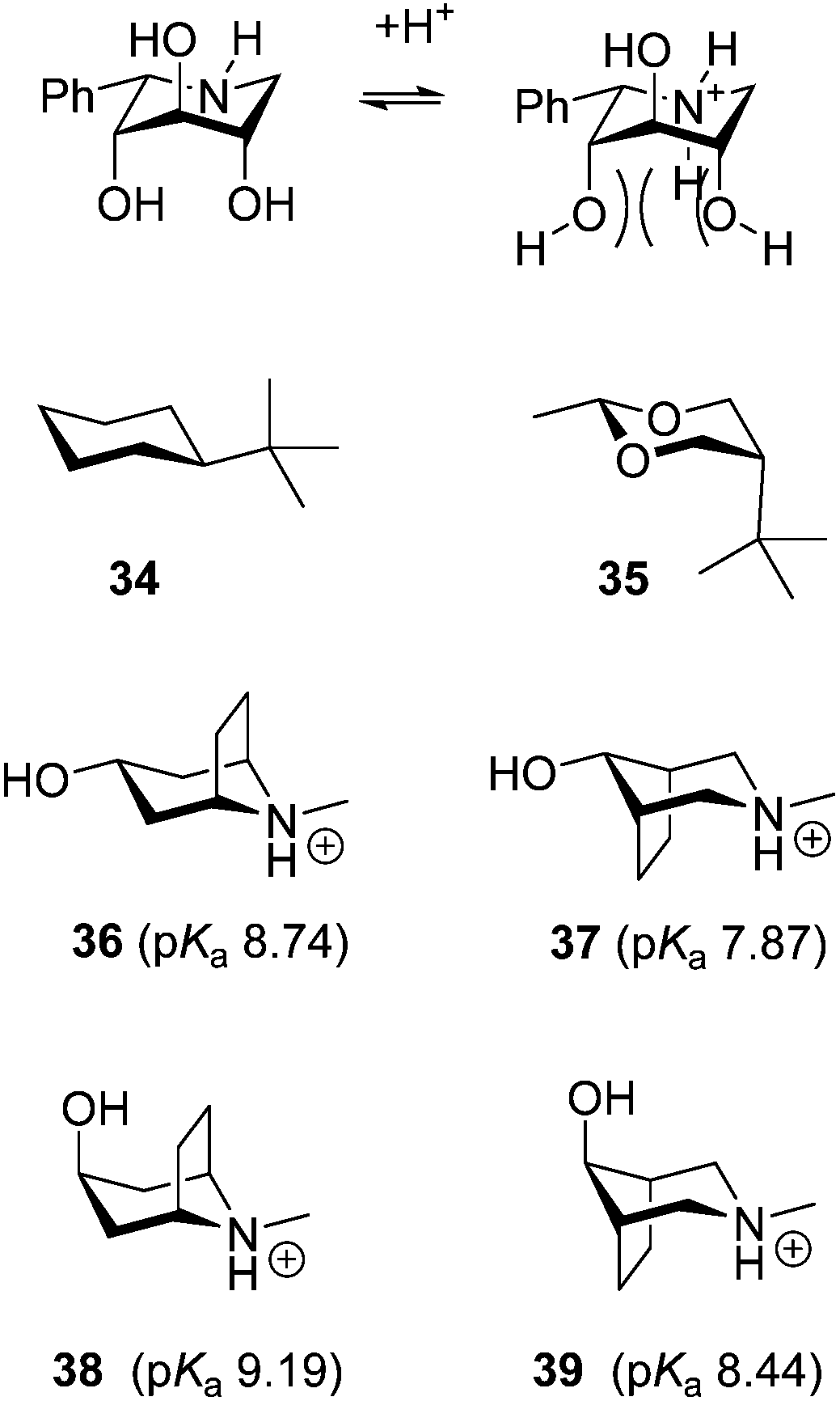 | ||
| Fig. 4 Steric interactions upon protonation of 7 and the preferred conformations of tert-butyl derivatives which show the profound effect of 1,3-diaxial steric interactions with hydrogen. The steric hindrance versus protonation of the amine is the pKa of the base pairs 36/37 and 38/39. | ||
Conclusions
This work has shown that an all-axial 2,3,4-trihydroxypiperidine has a significantly lower base strength than can be accounted for by the electronic effect. The corresponding all equatorial trihydroxypiperidine, on the other hand, has the pKa one would expect based on the electronic effect. The reason for the low pKa of the axial isomer is the two unfavourable 1,3-diaxial interactions that is present when the amine is protonated, which makes it more difficult to protonate. Thus the electronic effects are additive but pKa is reduced by the counteracting steric effect. Partial O-methylation has no significant effect other than reducing the base strength slightly due to the somewhat higher electron-withdrawing effect of OMe compared to OH.Experimental
5-Azido-5-deoxy-1,2-O-isopropylidene-5-C-phenyl-α-L-ido-pentofuranose (12)
Diol 10 (1.66 g) was dissolved in CH2Cl2 (50 mL) together with Et3N (3.24 mL, 23.3 mmol, 4 eq.) and SOCl2 (840 μL, 11.6 mmol, 2 eq.) was added slowly. The reaction was followed by TLC (ethyl acetate:petroleum ether 1:4, product Rf = 0.5) and quenched with water upon completion. The water phase was extracted with CH2Cl2 and the combined organic phases were washed with NH3Cl (aq.) and brine, followed by drying (MgSO4) and concentration in vacuo. Flash chromatography yielded 11 (1.77 g, 91%), which was a ratio of 45:55 stereoisomers at sulphur. [1H NMR (500 MHz, CDCl3): δ 7.49–7.35 (m, 5H, Ar), 6.15 (d, J = 3.6 Hz, 1H, H1), 6.10 (d, J = 3.7 Hz, 1H, H1′), 5.52 (d, J = 7.4 Hz, 1H, H5), 5.16 (d, J = 3.7 Hz, 1H, H3′), 4.98 (d, J = 7.2 Hz, 1H, H5′), 4.81 (2 d, J ∼ 3.6 Hz, 2H, H2, H2′), 4.74 (dd, J = 7.3, 3.8 Hz, 1H, H4′), 4.70 (dd, J = 7.5, 3.8 Hz, 1H, H4), 4.64 (d, J = 3.8 Hz, 1H, H3), 1.46, 1.45, 1.35, 1.34 (4 s, 12 H, Me). 13C NMR (126 MHz, CDCl3): δ 136.2, 136.1, 129.0, 129.0, 128.8, 128.8, 126.5, 126.3 (8 Ar C), 113.1 (CMe2), 113.1 (C′Me2), 106.4 (C1), 106.0 (C1′), 83.3, 83.0, 83.0, 82.3 (4 C, C2, C2′, C4, C4′), 78.7 (C3), 77.0 (C5′), 73.5 (C3′), 71.9 (C5), 27.1, 27.0, 26.5 (4 Me).]
The cyclic sulfite 11 (1.76 g, 5.64 mmol) was dissolved in DMF (25 mL) and NaN3 (2.5 g, 0.038 mmol, 6.8 eq.) was added. The reaction mixture was heated to 105 °C and left overnight. TLC (ethyl acetate–petroleum ether 1:3) showed full conversion (product Rf = 0.25) and the reaction was allowed to cool to room temperature, whereafter it was diluted with water and extracted with ethyl acetate. The organic phases were collected, washed with water and brine followed by drying over MgSO4 and concentration in vacuo. Flash chromatography (ethyl acetate–petroleum ether 1:3) yielded azide 12 (1.51 g, 92%).
1H NMR (500 MHz, CDCl3) δ 7.45–7.32 (m, 5H, Ar), 6.01 (d, J = 3.7 Hz, 1H, H1), 4.79 (d, J = 8.8 Hz, 1H, H5), 4.44 (d, J = 3.7 Hz, 1H, H2), 4.35 (dd, J = 8.7, 2.7 Hz, 1H, H4), 3.77 (bs, 1H, H3), 2.21 (s, 1H, 3-OH), 1.50 (s, 3H, Me), 1.29 (s, 3H, Me).
13C NMR (126 MHz, CDCl3): δ 135.9, 129.0, 129.0, 127.8 (4C, Ar), 112.1 (CMe2), 105.1 (C1), 85.1 (C2), 83.2 (C4), 74.7 (C3), 64.7 (C5), 26.8 (Me), 26.2 (Me).
HRMS calculated C14H17N3O4Na = 314.1117 found: 314.1112.
1,5-Dideoxy-1,5-imino-5-phenyl-L-ido-pentitol (7)
Azide 12 (0.319 g, 1.10 mmol) was dissolved in 30 mL THF and 2 mL HCl (2 M) and refluxed for 20 min. The reaction mixture was neutralized, concentrated and purified by flash chromatography (EA:PE 1:2 to 1:0; Rf = 0.5 in EA) to give the desired hemiacetal intermediate (230 mg; 84%). A portion of this (0.183 g, 0.73 mmol) was then dissolved in EA–MeOH (1:1; 4 mL) and Pd/C (10% Pd ca. 10 mg) was added under a nitrogen atmosphere, which was subsequently exchanged with a hydrogen atmosphere (1 atm). After 1 h at rt the reaction mixture was filtered, concentrated in vacuo and purified by flash chromatography (CHCl3:EtOH, 5:1 to 1:1 + NH3; Rf = 0.3 in CHCl3:EtOH 2:1 + 1 drop NH3 conc. sol.) to give the desired product 7 (136 mg, 90%).
1H NMR (500 MHz, D2O): δ 7.56–7.45 (m, 5H), 4.71 (s, 1H, H2), 4.19 (s, 1H, H4), 4.15 (m, 1H, H5), 4.11 (bs, 1H, H3), 3.77–3.63 (bd, J = 13.7 Hz, 1H, H6), 3.53 (bd, J = 13.7 Hz, 1H, H6′).
13C NMR (126 MHz, D2O): δ 133.7 (Ph), 129.1 (2C, Ph), 126.9 (Ph), 70.8 (C3), 67.3 (C4), 65.7 (C5), 58.4 (C2), 46.6 (C6).
HRMS calculated C11H15NO3 + H+ = 210.1125 found: 210.1126.
5-Azido-3-O-benzyl-5-deoxy-1,2-O-isopropylidene-5-C-phenyl-α-D-gluco-pentofuranose (14)
Alcohol 13 (1.66 g, 4.66 mmol) was dissolved in pyridine (25 mL) and TsCl (2.0 g, 10.2 mmol, 2.2 eq.) was added. The reaction mixture was heated to 90 °C and left for 18 h. TLC (ethyl acetate–petroleum ether 1:3; Rf = 0.2) showed full conversion and the reaction was cooled to room temperature followed by dilution with ethyl acetate washing with water and brine. After concentration the crude product was dissolved in DMF (25 mL) and NaN3 (5 eq.) was added. The reaction was stirred at 105 °C for 18 h whereafter TLC (ethyl acetate–petroleum ether 1:3; Rf = 0.45) showed full conversion. The reaction was cooled to room temperature, diluted with water and extracted with ethyl acetate. The combined organic phases were washed with water (5 times) and finally brine. After concentration the crude product was purified by flash chromatography (ethyl acetate–petroleum ether 5:1 to 3:1) to give the product (1.23 g; 69% over 2 steps).
1H NMR (500 MHz, CDCl3) δ 7.48–7.30 (m, 10H, Ar), 5.87 (d, J = 3.7 Hz, 1H, H1), 4.84 (d, J = 10.0 Hz, 1H, H5), 4.76 (d, J = 11.4 Hz, 1H, PhCH2), 4.68 (d, J = 11.4 Hz, 1H, PhCH2), 4.64 (d, J = 3.7 Hz, 1H, H2), 4.33 (dd, J = 10.0, 3.1 Hz, 1H, H4), 4.14 (d, J = 3.1 Hz, 1H, H3), 1.47 (s, 3H, Me), 1.30 (s, 3H, Me).
13C NMR (126 MHz, CDCl3) δ 137.3, 136.9, 128.8, 128.6, 128.6, 128.1, 128.1, 127.8 (12C, 2 Ar), 111.9 (CMe2), 105.3 (C1), 81.9 (C2), 81.6 (C3), 81.5 (C4), 72.4 (PhCH2), 62.9 (C5), 26.9 (Me), 26.3 (Me).
HRMS calculated C21H23N3O4Na = 404.1586 found: 404.1580.
1,5-Dideoxy-1,5-imino-5-phenyl-D-gluco-pentitol (6)
Azide 14 (0.127 g, 0.33 mmol) was treated with TFA and water in THF (5 mL) until the TLC showed full conversion of the starting material (ethyl acetate–petroleum ether 1:3; Rf = 0.45). The solvents were removed in vacuo and the crude was dissolved in ethyl acetate–MeOH (1:1, 5 mL) and Pd/C (ca. 25 mg) was added. The reaction was stirred under a hydrogen atmosphere (1 atm) O.N. TLC (EtOH:CHCl3) showed the formation of the product (Rf = 0.2). Yield = 79 mg (77%).
1H NMR (500 MHz, D2O) δ 7.55–7.48 (m, 5 H, Ph), 4.23 (d, J = 10.8 Hz, 1 H, H1), 4.02 (dd, J = 9.3, 10.8 Hz, 1 H, H2); 3.99 (ddd, J = 5.1, 9.3, 11.6, 1 H, H4), 3.68 (t, J = 9.3 Hz. 1 H, H3), 3.58 (dd, J = 5.1, 12.4 Hz, 1 H, H5), 3.15 (dd, J = 11.6, 12.4 Hz, 1 H, H5′).
13C NMR (126 MHz, D2O) δ 131.9 (Ph), 130.3 (Ph), 129.6 (Ph), 128.3 (Ph), 76.3 (C3), 70.9 (C2), 67.0 (C4), 62.9 (C1), 46.5 (C6).
HRMS calculated C11H15NO3 + H+ = 210.1125 found: 210.1136.
5-Azido-5-deoxy-1,2-O-isopropylidene-3-O-methyl-5-C-phenyl-α-D-gluco-pentofuranose (17)
1,2-O-Isopropylidene-3-O-methyl-α-D-glucofuranose (4.75 g, 20.3 mmol) was dissolved in EtOH:water (9:1) and NaIO4 (6.5 g, 30.4 mmol; 1.5 eq.) was added in portions. After 2 h the reaction mixture was diluted with CH2Cl2, filtered and concentrated in vacuo. The crude was dried by azeotropic co-evaporation using toluene, whereafter it was dissolved in dry THF (30 mL) followed by the addition of PhMgBr (ca. 40 mmol in THF) at 0 °C. The reaction was quenched with NH4Cl (aq.) and extracted with ethyl acetate. The combined organic phases were washed with brine and concentrated in vacuo followed by purification using flash chromatography (ethyl acetate–petroleum ether 1:3; Rf = 0.3). A mixture of the two diastereomers of 16 was isolated (5.00 g; 88%).
The mixture of alcohols (16, 2.00 g; 7.13 mmol) was dissolved in pyridine (30 mL) and TsCl (4.08 g; 21.4 mmol; 3 eq.) was added together with DMAP (87 mg; 0.1 eq.). The reaction was stirred at 90 °C for 2.5 h, additional 2 g of TsCl was added and stirring was continued for 2 days at room temperature. After concentration the crude residue was diluted with ethyl acetate. The ethyl acetate phase was washed with water, HCl (1 M in water), NaHCO3 and finally brine. After concentration in vacuo the residue was purified by flash chromatography (petroleum ether–ethyl acetate 3:1) to give tosylate (1.36 g, 44%). [1H NMR (500 MHz, CDCl3) δ 7.87–7.01 (m, 5H, Ph, Ph′), 6.01 (d, J = 3.9 Hz, 0.25H, H1′), 5.99 (d, J = 3.8 Hz, 0.75H, H1), 5.09 (bd, J = 5.8 Hz, 0.25H, H5′), 5.03 (d, J = 7.8 Hz, 0.75H, H5), 4.56 (2 × d, J = 3.8 Hz, J′ = 3.8 Hz, 1H, H2, H2′), 4.29 (dd, J = 5.8, 3.3 Hz, 0.25H, H4′), 4.24 (dd, J = 7.8, 3.2 Hz, 0.75H, H4), 3.71 (d, J = 3.3 Hz, 0.25H, H3′), 3.42 (s, 0.75H, Me′), 3.31 (d, J = 3.2 Hz, 0.75H, H3), 3.29 (s, 2.25H, Me), 1.48 (s, 2.25H, Me), 1.46 (s, 0.75H, Me′), 1.35–1.30 (m, 6H, Me, Me′). 13C NMR (126 MHz, CDCl3) δ 141.4, 139.7, 128.5, 128.4, 128.1, 127.7, 127.0, 126.0 (Ph, Ph′), 111.9 (CMe2), 111.6 (C′Me2), 105.2 (C1), 105.2 (C1′), 85.2 (C3′), 84.7 (C4), 84.0 (C3), 82.3 (C4′), 81.6 (C2), 81.1 (C2′), 72.4 (C5, C5′), 57.6 (3-OMe′), 57.4 (3-OMe), 26.8 (Me), 26.7 (Me′), 26.3 (Me), 26.2 (Me′).]
1,2-O-Isopropylidene-3-O-methyl-5-C-phenyl-5-O-tosyl-α-L-ido-pentofuranose (0.76 g; 1.74 mmol) was dissolved in DMF (10 mL) and NaN3 (0.454 g, 7 mmol, 4 eq.) was added. The mixture was stirred at 70 °C for 24 h. The reaction mixture was diluted with ethyl acetate and washed with water (4 times) and brine. After concentration in vacuo the residue was purified using flash chromatography (ethyl acetate–petroleum ether 1:4) to give 17 (0.43 g, 80%).
1H NMR (500 MHz, CDCl3) δ 7.42–7.30 (m, 5H, Ar), 5.85 (d, J = 3.7 Hz, 1H, H1), 4.77 (d, J = 10.0 Hz, 1H, H5), 4.61 (d, J = 3.7 Hz, 1H, H2), 4.30 (dd, J = 10.0, 3.1 Hz, 1H, H4), 3.90 (d, J = 3.1 Hz, 1H, H3), 3.53 (s, 3H, OMe), 1.46 (s, 3H, Me), 1.30 (s, 3H, Me).
13C NMR (126 MHz, CDCl3) δ 137.1 (Ar), 128.9 (Ar), 128.9 (Ar), 128.7 (Ar), 127.9 (Ar), 111.9 (CMe2), 105.4 (C1), 83.7 (C3), 81.8 (C4), 81.3 (C2), 63.1 (C5), 57.9 (OMe), 27.0 (Me), 26.4 (Me).
IR: –N3 2104 cm−1.
HRMS calculated C15H19N3O4 + Na+ = 328.1268 found: 328.1288.
1,5-Dideoxy-1,5-imino-3-O-methyl-5-C-phenyl-D-gluco-pentitol (18)
Azide 17 (562 mg; 1.84 mmol) was dissolved in AcOH (75% in water) and refluxed for 2 h. TLC (ethyl acetate–petroleum ether 1:4) showed full conversion of the starting material and the solvent was removed under vacuum. The residue was purified by chromatography (ethyl acetate:petroleum ether 1:1) to give 5-azido-5-deoxy-3-O-methyl-5-C-phenyl-D-gluco-pentofuranose (404 mg, 1.52 mmol, 83%). This compound was dissolved in MeOH (20 mL) and Pd/C (162 mg) was added. To the reaction mixture was then added Et3SiH (2.65 g; 22.8 mmol, 15 eq.) in portions via syringe. Pd/C was removed by filtration through Celite, which was then washed carefully with MeOH. The filtrate was concentrated and purified by chromatography (CH2Cl2–MeOH 9:1) to give 18 (144 mg, 42%).
1H NMR (500 MHz, DMSO-d6) δ 7.49–7.35 (m, 5H, Ar), 3.72 (ddd, J = 11.1, 9.1, 5.3 Hz, 1H, H5), 3.64 (dd, J = 10.0, 9.1 Hz, 1H, H3), 3.62 (s, 3H, Me), 3.50 (d, J = 10.0 Hz, 1H, H2), 3.21 (t, J = 9.1 Hz, 1H, H4), 3.14 (dd, J = 12.1, 5.3 Hz, 1H, H6), 2.61 (dd, J = 12.1, 11.1 Hz, 1H, H6′).
13C NMR (126 MHz, DMSO-d6) δ 138.3 (Ar), 127.9 (Ar), 127.3 (Ar), 127.0 (Ar), 87.3 (C4), 73.3 (C3), 69.4 (C5), 64.2 (C2), 58.9 (Me), 48.7 (C6).
[α]D 40.5° (c 1.0, MeOH).
HRMS calculated C12H18NO3 = 224.1281 found: 224.1264.
1,5-Dideoxy-1,5-imino-3-O-methyl-5-C-phenyl-L-ido-pentitol (20)
12 (1.323 g; 4.61 mmol) was dissolved in DMF (25 mL) cooled to 0 °C, where NaH (221 mg; 60% dispersion; 2 eq.) was added followed by MeI (0.6 mL; 1.31 g; 9.22 mmol; 2. eq.). After 1 h the reaction was quenched with water and diluted with EA. The phases were separated and the organic phase was washed with water (5 times) followed by brine and drying over MgSO4. The crude 19 was concentrated on Celite and purified by flash chromatography (petroleum ether:ethyl acetate 1:1). Yield 1.28 g (91%). [1H NMR (500 MHz, CDCl3) δ 7.46–7.29 (m, 5H), 5.99 (d, J = 3.8 Hz, 1H, H1), 4.82 (d, J = 9.5 Hz, 1H, H5), 4.50 (d, J = 3.8 Hz, 1H, H2), 4.33 (dd, J = 9.5, 3.1 Hz, 1H, H4), 3.16 (s, 3H, OMe), 3.11 (d, J = 3.1 Hz, 1H, H3), 1.51 (s, 3H, Me), 1.31 (s, 3H, Me). 13C NMR (126 MHz, CDCl3) δ 136.4 (Ar), 128.8 (Ar), 128.8 (Ar), 127.8 (Ar), 112.0 (CMe2), 105.6 (C1), 83.8 (C3), 83.5 (C4), 81.2 (C2), 64.9 (C5), 57.4 (OMe), 26.9 (Me), 26.3 (Me).]
Azide 19 (1.279 g; 4.19 mmol) was dissolved in acetic acid (75% in water; 75 mL) and refluxed for 1.5 h, where TLC (ethyl acetate–petroleum ether 4:1) showed full conversion of the starting material. The reaction mixture was concentrated in vacuo and the crude product was purified by chromatography (ethyl acetate:petroleum ether 1:1) to give 5-azido-5-deoxy-3-O-methyl-5-C-phenyl-L-ido-pentofuranose (766 mg; 2.89 mmol; 69%). It was redissolved in MeOH (40 mL) and Pd/C (307 mg) was added under a nitrogen atmosphere. Et3SiH (6.91 mL; 5.0 g; 15 eq.) was added in portions via syringe, and the reaction was allowed to stir for 2 h where TLC (ethyl acetate:petroleum ether 1:1 and CH2Cl2:MeOH 9:1) showed full conversion. The reaction mixture was filtered through Celite, which was then washed carefully with MeOH. The filtrate was concentrated in vacuo and purified by chromatography (CH2Cl2:MeOH 9:1). Yield 243 mg (38%).
1H NMR (500 MHz, D2O) δ 7.70–7.39 (m, 5H, Ar), 4.62 (bs, 1H, H2), 4.30 (bs, 1H, H5), 4.25 (d, J < 3 Hz, 1H, H3), 3.79 (dd, J < 3 Hz, 1H, H4), 3.60 (bd, J = 13.7 Hz, 1H, H6), 3.53 (s, 3H, Me), 3.53 (bd, J = 13.7 Hz, 1H, H6′).
13C NMR (126 MHz, D2O) δ 133.6 (Ar), 129.1 (Ar), 129.1 (Ar), 126.9 (Ar), 76.4 (C4), 68.4 (C3), 63.2 (C5), 58.7 (C2), 57.8 (Me), 46.9 (C6).
[α]D 56.8° (c 1.0, MeOH).
HRMS calculated C12H18NO3 = 224.1281 found: 224.1301.
3-O-Benzyl-1,5-dideoxy-1,5-imino-5-C-phenyl-D-gluco-pentitol (21)
Azide 14 (0.88 g; 2.31 mmol) was dissolved in 50 mL acetic acid (75% in water) and refluxed for 1.5 h, where TLC (PE–EA 4:1) showed full conversion of the starting material. The reaction was concentrated in vacuo and purified by flash chromatography (ethyl acetate:petroleum ether 1:1) to give 5-azido-3-O-benzyl-5-deoxy-5-C-phenyl-D-gluco-pentofuranose (0.61 mg; 77%). A portion of this material (102 mg; 0.30 mmol) was dissolved in 5 mL MeOH and Pd/C (32 mg) was added under a nitrogen atmosphere followed by Et3SiH (0.9 mL; 4.5 mmol; 15 eq.). The reaction was stirred for 3.5 h where TLC (ethyl acetate:petroleum ether 1:1 and CH2Cl2–MeOH 9:1) showed full conversion. The reaction mixture was filtered through Celite, which was then washed carefully with MeOH. The filtrates were concentrated in vacuo and purified by flash chromatography (CH2Cl2–MeOH 9:1) to give the product (69 mg; 77%).
1H NMR (500 MHz, CDCl3) δ 7.75–7.01 (m, 10H, Ar), 4.94 (d, J = 11.5 Hz, 1H, Bn), 4.81 (d, J = 11.5 Hz, 1H, Bn), 3.74 (ddd, J = 9.7, 9.1, 5.3 Hz, 1H, H5), 3.57 (t, J = 9.1 Hz, 1H, H3), 3.44 (d, J = 9.1 Hz, 1H, H2), 3.32 (t, J = 9.1 Hz, 1H, H4), 3.20 (dd, J = 11.3, 5.3 Hz, 1H), 2.62 (dd, J = 11.3, 9.7 Hz, 1H, H6′).
13C NMR (126 MHz, CDCl3) δ 140.0 (Ar), 138.9 (Ar), 128.8 (Ar), 128.7 (Ar), 128.7 (Ar), 128.3 (Ar), 128.1 (Ar), 128.0 (Ar), 128.0 (Ar), 87.6 (C4), 76.3 (Bn), 75.1 (C3), 71.2 (C5), 66.6 (C2), 50.7 (C6).
HRMS calculated for C18H22NO3 = 300.1594 found: 300.1592.
3-O-Benzyl-N-tert-butoxycarbonyl-1,5-dideoxy-1,5-imino-2,4-di-O-methyl-5-C-phenyl-D-gluco-pentitol (22)
Diol 21 (300 mg; 1.0 mmol) was dissolved in THF:water (1:1; 10 mL), cooled to 0 °C in an ice bath. NaHCO3 (152 mg; 1.1 eq.; 1.1 mmol) was added followed by Boc-anhydride (327 mg; 1.50 mmol, 1.5 eq.) and additional 1.1 eq. of NaHCO3 in 6 mL THF:water (1:1). The reaction was allowed to reach room temperature, where it was kept stirred for 24 h. TLC (CH2Cl2–MeOH 9:1) indicated full conversion, and the reaction mixture was concentrated to remove THF and then diluted with ethyl acetate. The organic phase was washed with water three times, brine, dried over MgSO4 and concentrated in vacuo. Flash chromatography (CH2Cl2–MeOH 19:1) yielded the Boc protected piperidine (289 mg, 72%). Most of this compound (280 mg; 0.70 mmol) was dissolved in dry DMSO (30 mL) and added to degreased NaH (7.4 mmol; 10 eq.) followed by MeI (450 μl; 7.4 mmol; 10 eq.). The reaction was stirred for 2 h, where TLC (ethyl acetate–petroleum ether 1:1) indicated full conversion of the starting material. The reaction was quenched with water and the aqueous phase was extracted with CH2Cl2 (3 × 60 mL). The combined organic phases were washed with water (2 times), brine and dried (MgSO4) followed by concentration in vacuo to give the title product (250 mg; 83%).
1H NMR (500 MHz, CDCl3) δ 7.43–6.94 (m, 10H, Ar), 4.62 (m, 3H, Bn, H2), 4.17 (dd, J = 14.4, 3.1 Hz, 1H, H6), 3.53 (dd, J = 7.8, 3.7 Hz, 1H, H4), 3.46 (ddd, J = 3.7, 3.2, 3.1 Hz, 1H, H5), 3.41 (t, J = 7.8 Hz, 1H), 3.32 (s, 3H, OMe), 3.31 (d, J = 14.4, 3.2 Hz, 0H), 3.16 (s, 3H, OMe), 1.18 (s, 9H, CMe3).
13C NMR (126 MHz, CDCl3) δ 155.5 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 142.0 (Ar), 138.5 (Ar), 128.4 (Ar), 128.3 (Ar), 128.3 (Ar), 127.6 (Ar), 127.0 (Ar), 126.5 (Ar), 83.6 (C4), 83.4 (C3), 81.4 (C5), 80.1 (CMe3), 72.9 (Bn), 60.1 (OMe), 59.6 (C2), 56.6 (OMe), 40.8 (C6), 28.3 (CMe3).
O), 142.0 (Ar), 138.5 (Ar), 128.4 (Ar), 128.3 (Ar), 128.3 (Ar), 127.6 (Ar), 127.0 (Ar), 126.5 (Ar), 83.6 (C4), 83.4 (C3), 81.4 (C5), 80.1 (CMe3), 72.9 (Bn), 60.1 (OMe), 59.6 (C2), 56.6 (OMe), 40.8 (C6), 28.3 (CMe3).
HRMS calculated for C25H33NO5Na = 450.2256 found: 450.2247.
1,5-Dideoxy-1,5-imino-2,4-di-O-methyl-5-C-phenyl-D-gluco-pentitol (23)
Compound 22 (235 mg; 0.55 mmol) was dissolved in 10 mL MeOH:acetic acid (1:1) and the solution was saturated with hydrogen gas using a balloon equipped with a long needle. The atmosphere in the flask was when exchanged with nitrogen and Pd(OH)2 (50 mg) was added followed by flushing with hydrogen gas. After 24 h under a hydrogen atmosphere TLC showed that the reaction was incomplete and an additional 50 mg Pd(OH)2 was added. This was repeated after additional 24 h, whereafter the reaction was left for further 72 h with 50 mg fresh catalyst. The crude reaction mixture was filtered through Celite, which was then washed carefully with MeOH. The filtrate was concentrated in vacuo and purified by flash chromatography (petroleum ether:ethyl acetate 1:1 + 1% Et3N) to give the debenzylated product (130 mg; 70%). Some of this product (57 mg; 0.15 mmol) was treated with TFA (400 μl) in CH2Cl2 (4 mL). After 18 h TLC (ethyl acetate–petroleum ether 4:1 + 1% Et3N) showed full conversion, and the reaction was filtered and concentrated in vacuo to give the title product. Yield: 60 mg (quantitative).
1H NMR (500 MHz, CDCl3) δ 7.59–7.40 (m, 5H, Ar), 4.27 (d, J = 10.3 Hz, 1H, H5); 3.83 (dd, J = 9.2, 10.3 Hz, 1H, H4), 3.82 (dd, J = 12.5, 5.2 Hz, 1H, H1a); 3.79 (dd, J = 9.2, 9.2 Hz, 1H, H3), 3.73 (ddd, J = 11.2, 9.2, 5.2 Hz, 1H, H2), 3.56 (s, 3 H, Me), 3.12 (s, 3H, Me), 3.08 (dd, J = 12.5, 11.2 Hz).
13C NMR (126 MHz, D2O, DMSO log tube) δ 131.0 (Ar), 129.4 (Ar), 128.7 (Ar), 127.3 (Ar), 80.0 (C3), 75.5 (C2), 74.0 (C4), 60.9 (C5), 59.2 (Me), 57.6 (Me), 43.0 (C1).
[α]D 33.4° (c 1.0, MeOH).
HRMS calculated for C13H20NO3 = 238.1438 found: 238.1437.
5-Azido-3-O-benzyl-5-deoxy-1,2-O-isopropylidene-5-C-phenyl-α-L-ido-pentofuranose (24)
Compound 12 (105 mg; 0.36 mmol) was dissolved in dry DMF (2.5 mL) and cooled to 0 °C in an ice bath, and NaH (60% dispersion) (19 mg; 0.79 mmol; 2.2 eq.) was added. BnCl (0.5 mL) was added under an atmosphere of nitrogen, and the reaction was stirred for 1 h at 0 °C followed by 1.5 h at room temperature, where TLC (petroleum ether:ethyl acetate 3:1) showed full conversion. The reaction was quenched with water and diluted with ethyl acetate. The phases were separated, and the organic phase was washed with water (3 times), brine and dried over MgSO4, filtered and concentrated in vacuo. The crude residue was purified by flash chromatography (petroleum ether–ethyl acetate 6:1) to give the product (128 mg; 93%).
1H NMR (500 MHz, chloroform-d) δ 7.41–7.22 (m, 10H, Ar), 6.04 (d, J = 3.8 Hz, 1H, H1), 4.88 (d, J = 9.6 Hz, 1H, H5), 4.57 (d, J = 3.8 Hz, 1H, H2), 4.49 (dd, J = 9.6, 3.2 Hz, 1H, H4), 4.43 (d, J = 11.3 Hz, 1H, Bn), 4.09 (d, J = 11.3 Hz, 1H, Bn), 3.47 (d, J = 3.2 Hz, 1H, H3), 1.55 (s, 3H, Me), 1.32 (s, 3H, Me).
13C NMR (126 MHz, CDCl3) δ 136.9 (Ar), 136.0 (Ar), 128.7 (2 × C, Ar), 128.4 (Ar), 127.9 (Ar), 127.7 (Ar), 127.6 (Ar), 111.8 (CMe2), 105.3 (C1), 83.4 (C3), 81.6 (C4), 81.5 (C2), 71.8 (Bn), 64.7 (C5), 26.8 (Me), 26.2 (Me).
HRMS calculated C21H23N3O4 + Na+ = 404.1586 found: 404.1556.
3-O-Benzyl-1,5-dideoxy-1,5-imino-5-C-phenyl-D-gluco-pentitol (25)
Compound 24 (461 mg; 1.21 mmol) was dissolved in 60 mL 75% aqueous acetic acid and refluxed for 2 h, where TLC (petroleum ether:ethyl acetate 4:1) showed full conversion. After concentration in vacuo the residue was purified by flash chromatography (petroleum ether:ethyl acetate 2:1) to give 5-azido-3-O-benzyl-5-deoxy-5-C-phenyl-L-ido-pentofuranose (292 mg; 71%). This was then dissolved in MeOH (15 mL) and Pd/C (10%; 90 mg) was added followed by Et3SiH (2mL; 12.8 mmol; 15 eq.). After 2 h TLC (petroleum ether:ethyl acetate 1:1 and CH2Cl2: MeOH 9:1) showed full conversion and the reaction was filtered through Celite, which was then carefully washed with MeOH. After concentration in vacuo the residue was purified by flash chromatography (CH2Cl2–MeOH 14:1) to give 25 (101 mg, 39%).
1H NMR (500 MHz, CDCl3) δ 7.43–7.19 (m, 10H, Ar), 4.61 (d, J = 11.9 Hz, 1H, Bn), 4.57 (d, J = 11.9 Hz, 1H, Bn), 4.02 (bs, 1H, H2), 3.78 (bs, 2H, H4, H5), 3.74 (bs, 1H, H3), 3.09 (m, 2H, H6, H6′).
13C NMR (126 MHz, CDCl3) δ 139.9 (Ar), 138.2 (Ar), 128.7 (Ar), 128.6 (Ar), 128.0 (Ar), 127.7 (Ar), 127.7 (Ar), 127.2 (Ar), 76.0 (C4), 72.3 (Bn), 70.9 (C3), 66.2 (C5), 59.0 (C2), 48.3 (C6).
HRMS calculated C18H22NO3 = 300.1594 found: 300.1597.
3-O-Benzyl-N-tert-butoxycarbonyl-1,5-dideoxy-1,5-imino-2,4-di-O-methyl-5-C-phenyl-L-ido-pentitol (26)
Compound 25 (101 mg; 0.34 mmol) was dissolved in 3 mL THF–water (1:1) and cooled to 0 °C in an ice bath. Na2CO3 (40 mg, 0.37 mmol, 1.1 eq.) was added to the solution followed by Boc anhydride (110 mg, 0.50 mmol, 1.5 eq.) together with additional 40 mg Na2CO3 dissolved in 2 mL THF–water (1:1). The reaction mixture was left at 0 °C for 30 min and thereafter at room temperature for 23 h where TLC (CH2Cl2–MeOH 9:1) showed full conversion. THF and water were partly removed by evaporation and the rest were diluted with ethyl acetate followed by washing the organic phase with water (3 times) and brine before drying it (MgSO4). Concentration in vacuo and subsequent purification by flash chromatography (CH2Cl2–MeOH 29:1) gave the Boc derivative (104 mg; 0.26 mmol; 77%). This intermediate was redissolved in dry DMSO (10 mL) and added to a flask containing degreased NaH (110 mg as 60% dispersion; 10 eq.) together with MeI (170 μl; 2.72 mmol; 10 eq.). The reaction was stirred for 1.5 h where TLC (ethyl acetate–petroleum ether 1:1) showed full conversion and was then quenched with water. The reaction mixture was extracted with CH2Cl2, and the combined organic phases were washed with water (2 times) and brine followed by drying over MgSO4 and concentration in vacuo to give the title product 26 (99 mg; 89%).
1H NMR (500 MHz, CDCl3) δ 7.54–7.39 (m, 2H), 7.37–7.11 (m, 8H), 5.70 (s, 0.5H), 5.38 (s, 0.5H), 4.86–4.70 (m, 2H), 4.22 (s, 0.5H), 3.94 (s, 0.5H), 3.76 (s, 1H), 3.54–3.44 (m, 1H), 3.45–3.13 (m, 7H), 2.47 (m, 1H), 1.38 (m, 9H).
13C NMR (126 MHz, CDCl3) δ 155.0 (CO), 139.0, 138.1 (broad), 137.4 (broad), 128.6 (broad), 128.4, 128.4, 128.1 (broad), 128.0, 127.6, 127.2, 83.0 (broad), 82.1 (broad), 81.3 (broad), 80.7 (broad), 80.6 (CMe3), 80.3 (broad), 75.3 (broad), 74.7 (broad), 58.8 (broad), 58.5 (broad), 55.3 (broad), 53.7 (broad), 41.8 (broad), 41.0 (broad), 28.4 (CMe3).
[α]D 27.0° (c 1.0, CHCl3).
HRMS calculated for C25H33NO5Na = 450.2251 found: 450.2249.
1,5-Dideoxy-1,5-imino-2,4-di-O-methyl-5-C-phenyl-L-ido-pentitol (27)
26 (84 mg; 0.20 mmol) was dissolved in MeOH:AcOH (1:1; 3 mL) and the solution was saturated with hydrogen gas before 50 mg Pd(OH)2 was added under an atmosphere of nitrogen. The reaction was left for 20 h under a hydrogen atmosphere. TLC (petroleum ether–ethyl acetate 1:1 + 1% Et3N) showed full conversion, and the reaction was filtered through Celite, which was then washed carefully with MeOH. The solution was concentrated in vacuo to give the debenzylated product (50 mg; 76%). [1H NMR (500 MHz, CDCl3) δ 7.46–7.34 (m, 5H, Ar), 4.22 (bs, 1H, H2), 4.22 (bm, 1H, H4), 3.46 (bm, 1H, H3), 3.44 (s, 3H, Me), 3.33 (bm, J = 3.6 Hz, 1H, H5), 3.10 (s, 3H, Me), 3.09–3.04 (bm, J = 14.4, 3.6 Hz, 2 H, H6, H6′). 13C NMR (126 MHz, DMSO-d6) δ 138.6 (Ar), 127.5 (Ar), 126.3 (Ar), 126.1 (Ar), 80.5 (C3), 76.5 (C4), 65.5 (C5), 57.5 (Me), 55.7 (C2), 55.6 (Me), 41.7 (C6), [α]D 39.9° (c 1.0, CHCl3).]
The Boc-protected intermediate (50 mg; 0.15 mmol) was treated with TFA (400 μl) in CH2Cl2 (4 ml containing 1% water) and concentrated to give the title product 27 (60 mg, quantitative).
1H NMR (500 MHz, D2O) δ 7.61–7.52 (m, 5H, Ar), 4.76 (d, J = 1.9 Hz, 0H), 4.43 (dd, J = 3.1, 1.4 Hz, 1H), 3.83–3.74 (m, 3H), 3.62 (d, J = 2.3 Hz, 1H), 3.60–3.57 (m, 0H), 3.54 (d, J = 1.9 Hz, 3H), 3.18 (d, J = 1.9 Hz, 3H).
13C NMR (126 MHz, D2O) δ 163.1, 162.8 (CO, TFA), 138.3 (Ar), 128.6 (Ar), 127.6 (Ar), 127.1 (Ar), 116.4 (q, J = 292 Hz, 1 C, TFA), 80.9, 76.8, 65.9, 58.7 (Me), 56.9 (C1), 56.8 (Me), 42.6 (C6).
[α]D 23.8° (c 1.0, MeOH).
HRMS calculated for C13H20NO3 = 238.1438 found: 238.1439.
Acknowledgements
We acknowledge the Danish National Research Council (FNU) and the University of Copenhagen for support.Notes and references
- (a) L. P. Hammett, Chem. Rev., 1935, 17, 125 CrossRef CAS; (b) R. W. Taft, J. Am. Chem. Soc., 1957, 79, 1045 CrossRef CAS; (c) R. W. Taft and R. D. Topsom, Prog. Phys. Org. Chem., 1987, 16, 1 CrossRef.
- (a) J. Clark and D. D. Perrin, Q. Rev., Chem. Soc., 1964, 18, 295 RSC; (b) S. Inouye, Chem. Pharm. Bull., 1968, 16, 1134 CrossRef CAS.
- (a) H. H. Jensen, L. Lyngbye, A. Jensen and M. Bols, Chem. – Eur. J., 2002, 8, 1218 CrossRef CAS PubMed; (b) H. H. Jensen, L. Lyngbye and M. Bols, Angew. Chem., Int. Ed., 2001, 40, 3447 CrossRef CAS.
- (a) H. H. Jensen and M. Bols, J. Chem. Soc., Perkin Trans 1, 2001, 905 RSC; (b) H. H. Jensen, A. Jensen, R. Hazell and M. Bols, J. Chem. Soc., Perkin Trans. 1, 2002, 1190 RSC.
- (a) A. V. Samoshin, I. S. Veselov, L. Huynh, A. K. Shestakova, V. A. Chertkov, G. V. Grishina and V. V. Samoshin, Tetrahedron Lett., 2011, 52, 5375 CrossRef CAS; (b) A. V. Samoshin, H. Joo, A. Y. Korneichuk, I. S. Veselov, G. V. Grishina and V. V. Samoshin, Tetrahedron Lett., 2013, 54, 1020 CrossRef CAS; (c) A. V. Samoshin, I. S. Veselov, V. A. Chertkov, A. A. Yaroslavov, G. V. Grishina, N. M. Samoshina and V. V. Samoshin, Tetrahedron Lett., 2013, 54, 5600 CrossRef CAS; (d) V. V. Samoshin, B. Brazdova, V. A. Chertkov, D. E. Gremyachinskiy, A. K. Shestakova, E. K. Dobretsova, L. P. Vatlina, J. Yuan and H. J. Schneider, ARKIVOC, 2005, 129 CAS; (e) B. Brazdova, N. R. Zhang, V. V. Samoshin and X. Guo, Chem. Commun., 2008, 4774 RSC; (f) Y. Terui and K. Tori, J. Chem. Soc., Perkin Trans. 2, 1975, 127 RSC; (g) E. D. Goddard-Borger, M. B. Tropak, S. Yonekawa, C. Tysoe, D. J. Mahuran and S. G. Withers, J. Am. Chem. Soc., 2012, 55, 2737 CAS.
- J. O. Olsen, S. P. A. Sauer, C. M. Pedersen and M. Bols, Org. Biomol. Chem., 2015, 13, 3116 Search PubMed.
- L. Svansson, B. D. Johnston, J.-H. Gu, B. Patrick and B. M. Pinto, J. Am. Chem. Soc., 2000, 122, 1270 CrossRef.
- (a) D. C. Lankin, N. S. Chandrakumar, S. N. Rao, D. P. Spangler and J. P. Snyder, J. Am. Chem. Soc., 1993, 115, 3356 CrossRef CAS; (b) J. P. Snyder, N. S. Chandrakumar, H. Sato and D. C. Lankin, J. Am. Chem. Soc., 2000, 122, 544 CrossRef CAS.
- The piperidinium ion is still destabilized by the electronegative substituent, but the axial configuration is relatively more stabilized compared to the equatorial counterpart.
- B. Bernet, U. Piantini and A. Vasella, Carbohydr. Res., 1990, 204, 11 CrossRef CAS.
- (a) H. H. Jensen and M. Bols, Acc. Chem. Res., 2006, 39, 259 CrossRef CAS PubMed; (b) M. Heuckendorff, C. M. Pedersen and M. Bols, Chem. – Eur. J., 2010, 16, 13982 CrossRef CAS PubMed.
- (a) M. Bols, X. Liang and H. H. Jensen, J. Org. Chem., 2002, 67, 8970 CrossRef CAS PubMed; (b) M. N. Namchuk, J. D. McCarter, A. Becalski, T. Andrews and S. G. Withers, J. Am. Chem. Soc., 2000, 122, 1270 CrossRef CAS.
- In the carbohydrate studies it has been found that methyl 3,6-anhydroglucoside hydrolyzes 446 times faster than the corresponding methyl α-D-glucoside, more than the expected 100 times as estimated on the basis of σs values, which is probably due to the fact that besides being all axial the compound has one oxygen less. See: C. McDonnell, O. L. Lopez, P. Murphy, J. F. Bolanos, R. Hazell and M. Bols, J. Am. Chem. Soc., 2004, 126, 12374 CrossRef CAS PubMed.
- (a) C. M. Pedersen, L. U. Nordstrøm and M. Bols, J. Am. Chem. Soc., 2007, 129, 9222 CrossRef CAS PubMed; (b) M. Heuckendorff, C. M. Pedersen and M. Bols, J. Org. Chem., 2012, 77, 5559 CrossRef CAS PubMed.
- H. H. Jensen, L. U. Nordstrøm and M. Bols, J. Am. Chem. Soc., 2004, 126, 9205 CrossRef CAS PubMed.
- R. U. Lemieux and J. Howard, Can. J. Chem., 1963, 41, 308–316 CrossRef CAS.
- K. R. C. Prakash and S. P. Rao, Tetrahedron, 1993, 49, 1505–1510 CrossRef CAS.
- C. A. Grob and M. G. Schlageter, Helv. Chim. Acta, 1976, 59, 264 CrossRef CAS.
- The difference of 1.3 is also perfectly in accordance with the difference in base strength between benzylamine (pKa 9.34) and methylamine (pKa 10.66).
- (a) F. Carey and R. J. Sundberg, Advanced Organic Chemistry, Kluever Academic Publishers, New York, 4th edn, 2002 Search PubMed; (b) E. L. Eliel and M. C. Knoeber, J. Am. Chem. Soc., 1968, 90, 3444 CrossRef CAS.
- (a) H. S. Aaron, G. E. Wicks Jr. and C. P. Rader, J. Org. Chem., 1964, 29, 2248 CrossRef CAS; (b) H. O. House, H. C. Miller, C. G. Pitt and P. P. Wickham, J. Org. Chem., 1963, 28, 2407 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob02427k |
| This journal is © The Royal Society of Chemistry 2017 |