
Dearomatisation approaches to spirocyclic dienones via the electrophilic activation of alkynes†
Aimee K.
Clarke
,
John T. R.
Liddon
,
James D.
Cuthbertson
,
Richard J. K.
Taylor
* and
William P.
Unsworth
 *
*
University of York, Heslington, York YO10 5DD, UK. E-mail: richard.taylor@york.ac.uk; william.unsworth@york.ac.uk
First published on 23rd November 2016
Abstract
Two complementary dearomatising spirocyclisation protocols to generate spirocyclic dienones from anisole and phenol-tethered ynones are described, each proceeding via electrophilic alkyne activation. The first approach focuses on the spirocyclisation of para-substituted anisoles using either SnCl2·2H2O or Cu(OTf)2. The second approach, which enables the spirocyclisation of both ortho- and para-substituted phenols, uses silica-supported AgNO3 to generate similar scaffolds with much greater efficiency. Initial asymmetric studies are also outlined.
Introduction
Spirocyclic dienones are key structural features in numerous bioactive natural products isolated from a variety of trees, plants and bacteria, with representative examples 1–6 shown in Fig. 1.1a–e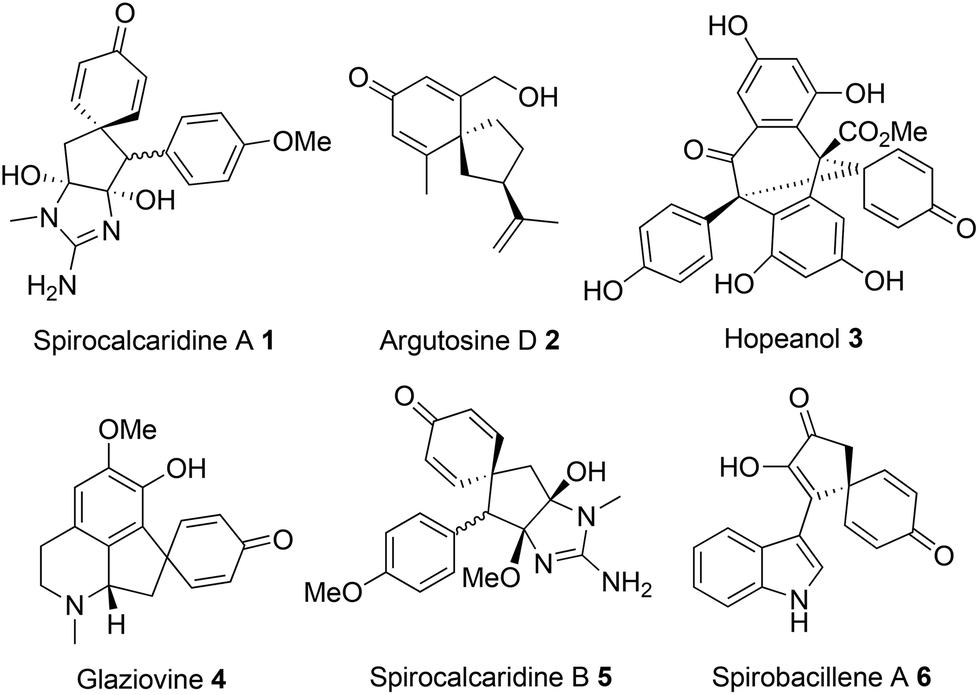 | ||
| Fig. 1 Natural products containing spirocyclic dienone motifs. | ||
A popular approach to synthesise spirocyclic dienones is via the dearomatisation and ipso-cyclisation of a phenol or anisole derivative, with this typically achieved using one of two methods (Scheme 1). The most common method is based on the oxidation of a substituted phenol (Scheme 1a); following oxidation of the phenol, an intramolecular nucleophilic ipso-cyclisation reaction can take place with a range of C-nucleophiles including alkenes/alkynes,2a enamides,2b allyl silanes,2c,d enols/enolates,2e aromatics,2f nitro compounds2g and diazo compounds.2h,i Alternatively, the flow of electron density can be reversed and a substituted phenol/anisole can react directly with a tethered electrophilic species (Scheme 1b); examples of electrophilic ipso-cyclisation with sulfonates,3a–c nitriles,3d epoxides,3e activated allenes,3f activated aryl halides,3g propargyl bromides/carbonates,3h activated alkynes/alkenes3i–k and allylic carbonates3l,m have all been reported.
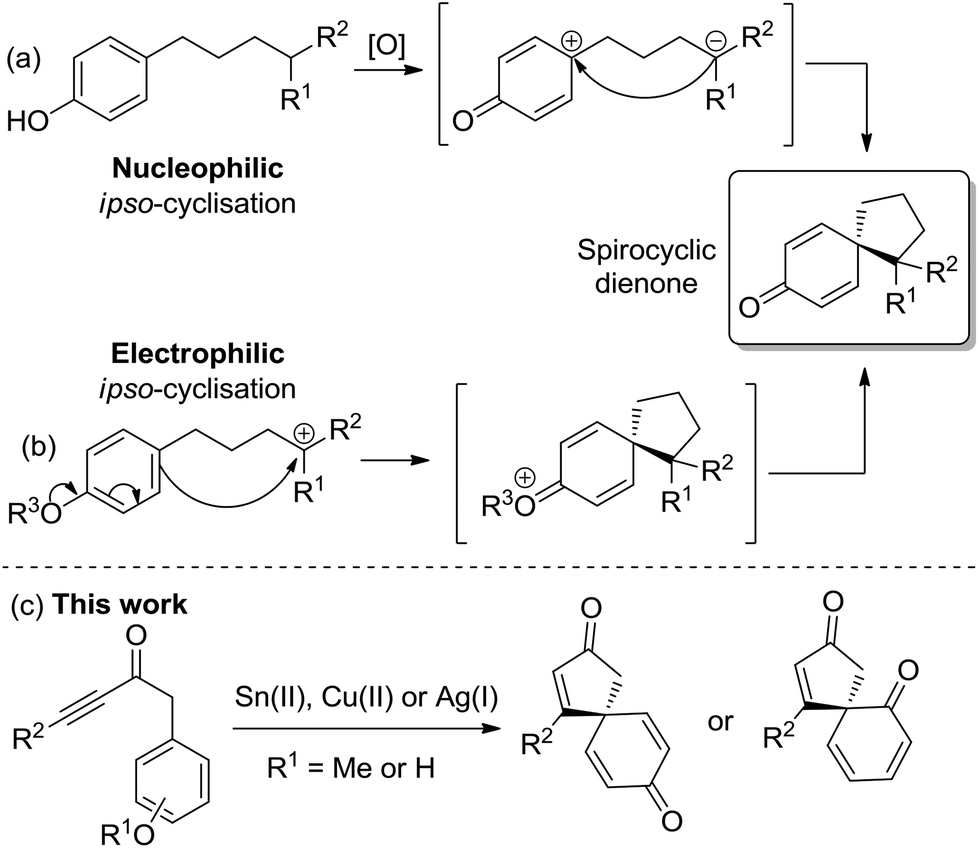 | ||
| Scheme 1 General methods for spirocyclic dienone synthesis. | ||
In this manuscript, two complementary protocols which generate spirocyclic dienones are outlined (Scheme 1c), with both methods promoted by the activation of a tethered alkyne moiety.4 The first approach focuses on the spirocyclisation of para-substituted anisoles using either SnCl2·2H2O or Cu(OTf)2 to activate the alkyne towards nucleophilic attack, while the second uses silica-supported AgNO3 to generate similar scaffolds from analogous phenol precursors with greater efficiency and scope. Substrate scoping studies are described for each reaction series, while comparisons between the two reaction types, synthetic extensions and preliminary asymmetric results are also outlined.
Results and discussion
This research program began during a project to synthesise the spirocyclic natural product spirobacillene A (6, Fig. 1).5,6 In this published work, it was found that treating anisole-tethered ynone 7 with five equivalents of SnCl2·2H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 at RT in CH2Cl2 resulted in its efficient conversion into spirocyclic dienone 8, which was isolated in 89% yield (Scheme 2). The ease and scalability of this key step was instrumental in allowing us to complete the synthesis of spirobacillene A 6, which was published in 2013.5,8
7 at RT in CH2Cl2 resulted in its efficient conversion into spirocyclic dienone 8, which was isolated in 89% yield (Scheme 2). The ease and scalability of this key step was instrumental in allowing us to complete the synthesis of spirobacillene A 6, which was published in 2013.5,8
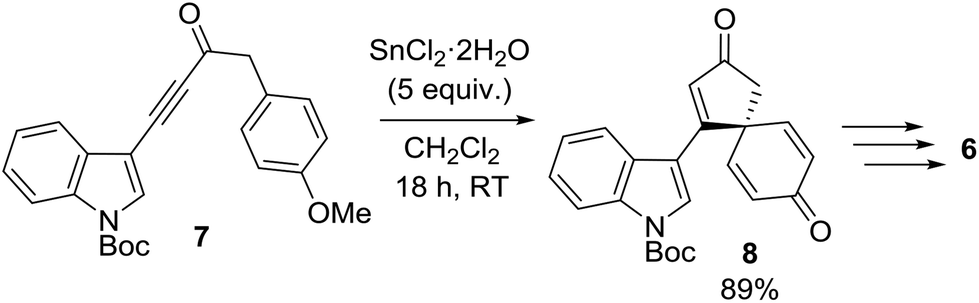 | ||
| Scheme 2 Sn(II)-mediated synthesis of spirobacillene A. | ||
This initial discovery inspired the development of a number of other classes of dearomatising spirocyclisation reactions9 in our group in the following years.10,11 However, prior to this publication, the conversion of ynone 7 into spirocycle 8 remained the only reported reaction of its type in the literature,12 hence it was decided to further optimise this process and to evaluate its scope.
Initial results were disappointing, with unsubstituted and alkyl substituted ynones 9a and 9b both failing to react with SnCl2·2H2O under the conditions used during the total synthesis of spirobacillene A (Table 1, entries 1 and 2). Phenyl substituted ynone 9c also failed to react at room temperature (entry 3), although a small amount of cyclisation was observed upon heating at reflux (entry 4). A plausible explanation for this poor reactivity is that the more electron-rich the alkyne, the more readily it can interact with acidic additives, promoting spirocyclisation. Support for this theory was found when examining the cyclisation of anisole-substituted ynone 9d; under the standard SnCl2·2H2O mediated conditions, a more respectable 75% conversion into spirocyclic dienone 10d was observed (entry 5, 57% isolated yield). Pleasingly, the spirocyclisation was improved significantly by changing the catalyst; full details of reaction optimisation are included in the ESI,† with the highlight being the discovery that Cu(OTf)2 promoted the complete cyclisation of ynone 9d within 1 h, with spirocycle 10d isolated in 80% isolated yield (entry 6). However, Cu(OTf)2 did not lead to any improvement in the reactivity of substrates 9a–9c, forcing us to concede that an electron donating group on the alkyne terminus is a requirement for this transformation.
|
|
||||
|---|---|---|---|---|
| Entry | Ynone | Reagent | Equiv./time [h]/temp. [°C] | Conversiona,b (isolated yield in brackets) |
| a All reactions were performed using 0.09–0.20 mmol of the ynone 9a–d in CH2Cl2 (0.1 M). b Conversion measured by analysis of the 1H NMR spectra of the unpurified reaction mixture. | ||||
| 1 | 9a | SnCl2·2H2O | 5/20/RT or 45 | No reaction |
| 2 | 9b | SnCl2·2H2O | 5/20/RT or 45 | No reaction |
| 3 | 9c | SnCl2·2H2O | 5/20/RT | No reaction |
| 4 | 9c | SnCl2·2H2O | 5/20/45 | 10% |
| 5 | 9d | SnCl2·2H2O | 5/20/RT | 75% (57%) |
| 6 | 9d | Cu(OTf)2 | 1/1/RT | >95% (80%) |
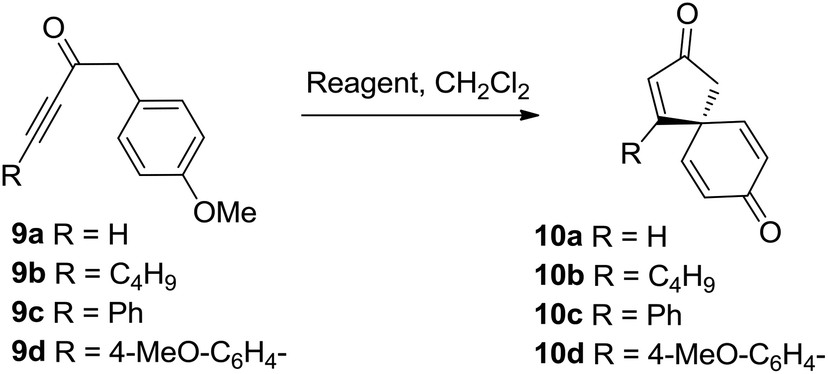
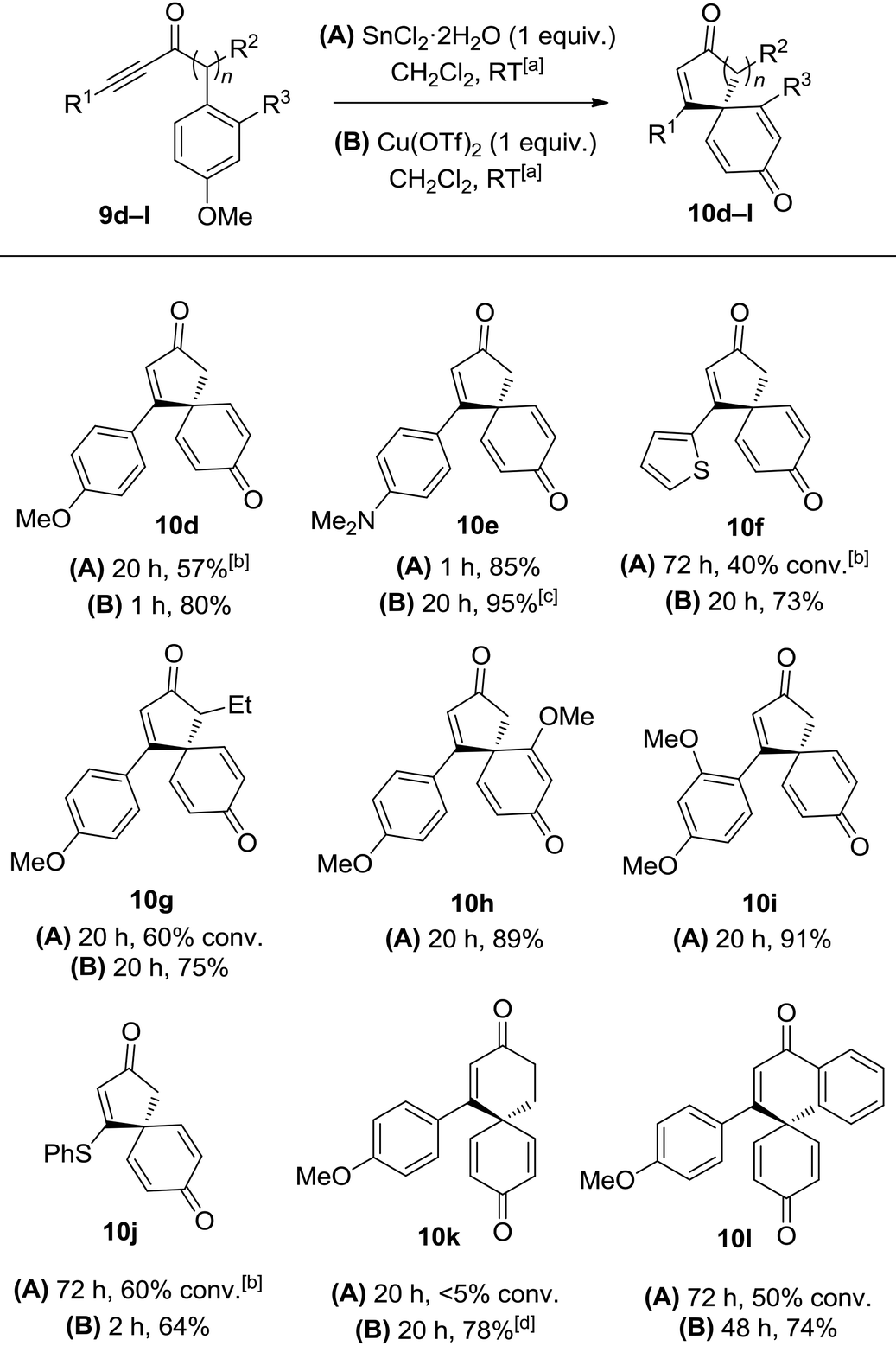
With this in mind, a series of anisoles tethered to electron-rich ynones (9d–l) were made and tested using both SnCl2·2H2O and Cu(OTf)2 to activate the alkyne (Table 2). Spirocycle 10e was formed in just 1 h from aniline-substituted ynone 9e using one equivalent of SnCl2·2H2O. Alternatively, the same product could be made using catalytic Cu(OTf)2 (0.1 equivalents), albeit with a longer reaction time. Thiophene-substituted ynone 9f reacted more slowly; the cyclisation was incomplete following treatment with SnCl2·2H2O at room temperature for 3 days, but proceeded more efficiently with Cu(OTf)2 to afford 10f in a 73% yield. Substitution around either ring system is well tolerated, evidenced by the efficient syntheses of compounds 10g–i. Vinyl sulfide product 10j could also be isolated in a reasonable yield using Cu(OTf)2, demonstrating compatibility with non-aromatic-tethered ynones. Additionally, the reaction is not limited to the synthesis of spirocyclic cyclopentenones; spirocyclic cyclohexenones 10k and 10l were both formed in good yields, although the reactions were slower and thus required additional heating or a longer reaction time.
| a All reactions were performed in CH2Cl2 (0.1 M) at RT with 1 equiv. of reagent unless specified. Where stated, reaction conversions were measured by analysis of the 1H NMR spectra of the unpurified reaction mixture. b 5 equiv. of SnCl2·2H2O used. c 0.1 equiv. of Cu(OTf)2 used. d Reaction performed at 50 °C. |
|---|
|
To summarise this reaction series, para-substituted anisoles tethered to electron-rich ynones can be converted into spirocyclic dienones in high yield using either a Sn(II) or Cu(II) reagent. The simplicity of the synthetic procedure and mild reaction conditions are the most pleasing aspects of this method, although the use of relatively large quantities of Sn(II) and Cu(II) reagents and the requirement to use electron-rich ynones were both identified as areas with potential for improvement.
It was reasoned that both of the above limitations might be addressed by using a more active catalyst. Silver(I) catalysts were identified as particularly promising candidates, given that they have generally been found to be the best catalyst class in related alkyne activation processes,10 but disappointingly, the Ag(I) catalysts tested were ineffective for the spirocyclisation of anisole system 9d. However, by switching the nucleophilic component in the starting material from an anisole to the analogous phenol, the desired spirocyclic product 10d could indeed be formed, and with Ag(I) catalysts now viable for this transformation, significant improvements in the scope and efficiency soon emerged. The use of silica-supported AgNO3 (10 mol%) in CH2Cl2 at RT was found to be a particularly active and convenient catalyst system and was chosen for the substrate scoping studies, which were performed on ynone tethered phenols (11c,d,m–v). For clarity, five of these examples (denoted in the table with a *) were included in an earlier publication,10c while the other seven substrates are novel examples (Table 3). It should also be noted that AgNO3·SiO2 is a much more reactive catalyst than unsupported AgNO3; ynones 11c,d,m,o,q–v did not react in the presence of unsupported AgNO3 and only a 7% conversion to spirocyclic dienone 10n was observed for ynone 11n when using unsupported AgNO3.13
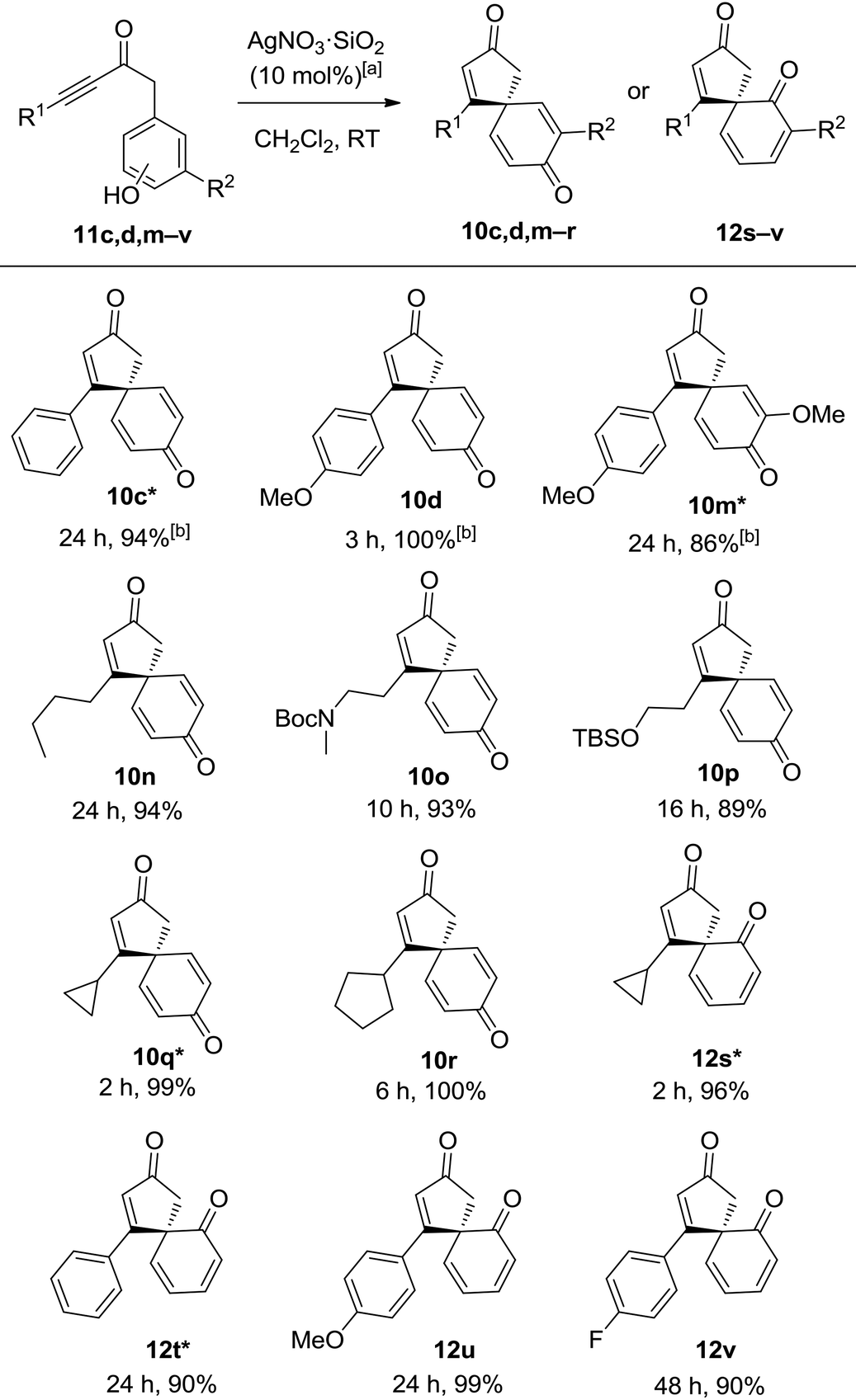
It quickly became apparent that by changing the catalyst and starting material, the requirement for an electron-donating substituent on the ynone had been removed; simple alkyl chains and aromatic substituents in the R1 position were all well tolerated, generating spirocyclic dienone products 10c, 10d, 10n in high yields. Alkyl substituted ynones bearing protected amine and alcohol groups (11o and 11p) also reacted smoothly to furnish their corresponding spirocyclic dienones (10o and 10p). The incorporation of terminal cyclopropane and cyclopentane rings appeared to increase the reactivity of the ynone; dienones 10q and 10r were produced in near-quantitative yields in 2–6 h which is notably faster than most of the other reactions explored in this study. Pleasingly, we were also able to perform the spirocyclisation on ortho-substituted phenols 11s–v; there are relatively few literature examples of dearomatisation and ipso-cyclisation reactions of ortho-substituted phenols, and so the efficient syntheses of chiral spirocyclic products 12s–v in 90–99% yields are especially pleasing.2d,14
The superior reactivity of the Ag(I) mediated reaction system compared to the earlier Sn(II)/Cu(II) reactions is best demonstrated by a direct comparison. Thus, our published synthesis of spirocyclic dienone 8, which is a key intermediate en route to spirobacillene A, required five equivalents of SnCl2·2H2O and 18 h to generate the product in 89% yield.5 In contrast, the same product was generated from phenol 13 in near-quantitative yield in just 7 h using 10 mol% AgNO3·SiO2 (Scheme 3).
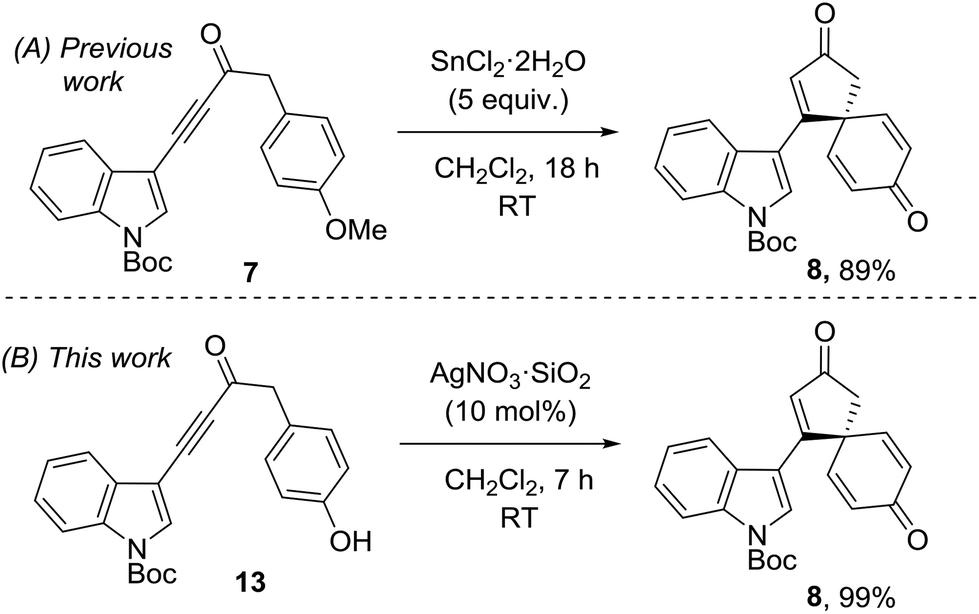 | ||
| Scheme 3 Previous and improved routes to spirobacillene A precursor 8. | ||
The potential of the spirocyclic dienone products to undergo additional complexity generating reactions has also been briefly demostrated; spirocyclic products 10o and 10p were each found to undergo protecting group cleavage and cyclisation in one-pot to furnish novel tricyclic products 14 and 15 as single diastereomers and in reasonable, unoptimised yields (Scheme 4).
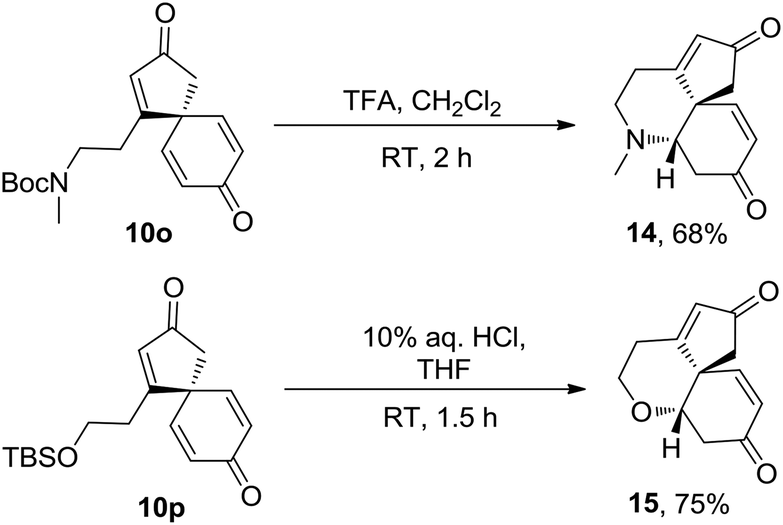 | ||
| Scheme 4 One-pot deprotection and cyclisation of spirocyclic dienones 10o and 10p. | ||
Finally, having shown that ortho-substituted phenols can be converted into chiral spirocyclic dienones, the possibility of performing this reaction asymmetrically has also been briefly examined. Preliminary studies show that spirocyclisation can be achieved with a modest amount of asymmetric induction (23% ee) using the BINOL-based chiral phosphoric acid silver(I) salt 16 (Scheme 5). It is envisaged that optimisation of the reaction conditions and the nature of the silver(I) catalyst15 should lead to improve enatioselectivity in this process.
 | ||
| Scheme 5 Asymmetric spirocyclisation reaction using silver salt of CPA 16. | ||
Conclusions
In summary, two mild and efficient methods for the synthesis of spirocyclic dienones are described. Both were briefly introduced by our group in previous publications, but the significantly expanded substrate scoping studies outlined in this manuscript mean that each can now be considered as general and versatile synthetic approach to this important compound class. Both methods work well on a variety of easy-to-synthesise ynone precursors. The use of catalytic AgNO3·SiO2 to promote the spirocyclisation of ynone tethered phenols is likely to be of particular interest, in view of the excellent isolated yields in this series, the simple product purification and the capacity to recover the active catalyst by filtration.Experimental
Except where stated, all reagents were purchased from commercial sources and used without further purification. 1H NMR and 13C NMR spectra were recorded on a JEOL ECX400 or JEOL ECS400 spectrometer, operating at 400 MHz and 100 MHz respectively. All spectral data were acquired at 295 K. Chemical shifts (δ) are quoted in parts per million (ppm). The residual solvent peak, δH 7.26 and δC 77.0 for CDCl3 and δH 2.50 and δC 39.5 for DMSO-d6. Coupling constants (J) are reported in Hertz (Hz) to the nearest 0.5 Hz. The multiplicity abbreviations used are: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broad). Signal assignment was achieved by analysis of DEPT, COSY, NOESY, HMBC and HSQC experiments where required. Infrared (IR) spectra were recorded on a PerkinElmer UATR 2 Spectrometer as a thin film dispersed from either CH2Cl2 or CDCl3 and are reported in wavenumbers (cm−1). Mass-spectra were obtained by the University of York Mass Spectrometry Service, using electrospray ionisation (ESI) on a Bruker Daltonics, Micro-tof spectrometer. Melting points were determined using Gallenkamp apparatus and are uncorrected. Reactions were monitored using thin layer chromatography (TLC), which was carried out on Merck silica gel 60F254 pre-coated aluminium foil sheets and were visualised using UV light (254 nm) and stained with basic aqueous potassium permanganate. Flash column chromatography was carried out using slurry packed Fluka silica gel (SiO2), 35–70 μm, 60 Å, under a light positive pressure, eluting with the specified solvent system. Compounds 9d16 and 9e10a were prepared according to literature procedures.General experimental
General procedure A: Weinreb amide synthesis I
To a suspension of acid (1.00 mmol) in DCM (2 mL) at RT was added CDI (220 mg, 1.20 mmol). A homogeneous solution quickly formed, and was stirred at RT for 1 h, after which time MeNH(OMe)·HCl (107 mg, 1.10 mmol) and stirring continued for a further 2 h. The crude reaction mixture was then poured into water (10 mL) and basified to ca. pH 10 with 2 M aq. NaOH extracted with EtOAc (3 × 30 mL) and washed with 10% aq. HCl (15 mL). The organic extracts were dried over MgSO4 and concentrated in vacuo, affording the Weinreb amide product which was used without further purification.General procdure A2: Weinreb amide synthesis II
To a stirred solution of acid (1.00 mmol), MeNH(OMe)·HCl (107 mg, 1.10 mmol) and DIPEA (0.52 mL, 3.00 mmol) in CH2Cl2 (2.5 mL) was added T3P 50% in EtOAc (955 mg, 1.50 mmol). The solution was stirred at RT until completion was observed by TLC. The reaction mixture was poured into water (10 mL) and acidified using 10% aq. HCl (5 mL). The organics were collected and the aqueous extracted with EtOAc (3 × 30 mL). The organics were combined, washed with aq. 2 M NaOH (10 mL), brine (10 mL), dried over MgSO4 and concentrated in vacuo to afford the Weinreb amide product which was used without further purification.General procedure B: ynone formation I
To a solution of terminal alkyne (1.50 mmol) in THF (10 mL) at −78 °C was added n-BuLi (0.875 mL, 1.4 mmol, 1.6 M in hexanes). The resulting yellow solution was stirred at −78 °C for 30 min, then transferred via cannula to a cooled (−78 °C) solution of Weinreb amide (1.00 mmol) in THF (5 mL). The mixture was stirred at −78 °C for 5 min then warmed to −10 °C and stirred for a further 1 h. The reaction was then re-cooled to −78 °C and quenched with sat. aq. NH4Cl (30 mL), allowed to warm to RT, diluted with water (70 mL), extracted with EtOAc (3 × 100 mL), dried over MgSO4 and concentrated in vacuo. Purification by flash column chromatography (10:1 petrol:EtOAc to elute the excess alkyne, then 5:1 petrol:EtOAc to elute the product) afforded the ynone product.
General procedure B2: ynone formation II
To a stirred solution of alkyne (3.00 mmol) in THF (3 mL) at −78 °C under argon was added n-BuLi (1.00 mL, 2.5 mmol, 2.5 M in hexanes) dropwise. The mixture was stirred for 30 min at −78 °C and then transferred via cannula to a −78 °C solution of Weinreb amide (1.00 mmol) in THF (5 mL). Upon complete transfer the mixture was warmed to RT and stirred for the specified amount of time. The reaction was quenched with sat. aq. NH4Cl (30 mL), diluted with water (70 mL) and extracted with EtOAc (3 × 100 mL). The organics were combined, washed with brine (100 mL), dried over MgSO4, concentrated in vacuo and purified by flash column chromatography to afford the ynone product.General procedure C: spirocyclisation using Sn(II)/Cu(II)
To a solution of ynone (1.00 mmol) in CH2Cl2 (10 mL) was added an acid catalyst (0.1–5.0 equiv.). The resulting suspension was stirred at the specified temperature until completion was observed by TLC, before adding an excess of solid K2CO3 and stirring for an additional 10 min. The mixture was then filtered, rinsed with CH2Cl2 and concentrated in vacuo. Purification by flash column chromatography afforded the spirocyclic product.General procedure C2: spirocyclisation using AgNO3·SiO2
To a solution of ynone (1 mmol) in CH2Cl2 (10 mL) was added AgNO3·SiO2 (0.01–0.1 equiv., 1 wt% AgNO3 on SiO2). The mixture was stirred at the specified temperature until completion was observed by TLC. The reaction mixture was filtered, washing the catalyst with EtOAc (10 mL), then concentrated in vacuo to afford the spirocyclic product.Compound synthesis
:1 petrol:EtOAc) afforded the title compound10d as a brown solid (19 mg, 80%); mp. 135–137 °C; Rf 0.20 (1:1 petrol:EtOAc); νmax (thin film)/cm−1 1669, 1639, 1579, 1541, 1488, 1234, 1163, 1013, 848, 822; δH (400 MHz, CDCl3) 2.75 (2 H, s), 3.81 (3 H, s), 6.46 (2 H, d, J = 10.0), 6.62 (1 H, s), 6.85 (2 H, d, J = 9.0), 6.95 (2 H, d, J = 10.0), 7.48 (2 H, d, J = 9.0); δC (100 MHz, CDCl3) 46.8, 51.0, 55.4, 114.3, 125.3, 127.5, 129.4, 130.0, 152.0, 162.3, 173.1, 184.7, 203.0; HRMS (ESI+): Found: 289.0825; C17H14NaO3 (MH+) Requires 289.0835 (3.5 ppm error). Spectroscopic data matched those previously reported in the literature.16
:1 petrol:EtOAc) afforded the title compound10e as a yellow solid (29 mg, 95%); mp. 199–201 °C; Rf 0.20 (1:1 petrol:EtOAc); νmax (thin film)/cm−1 1656, 1636, 1581, 1548, 1500, 1353, 1182, 1156, 719; δH (400 MHz, CDCl3) 2.72 (2 H, s), 3.02 (6 H, s), 6.46 (2 H, d, J = 10.0), 6.55 (1 H, s), 6.58 (2 H, d, J = 9.0), 6.98 (2 H, d, J = 10.0), 7.43 (2 H, d, J = 9.0); δC (100 MHz, CDCl3) 39.9, 46.7, 50.9, 111.3, 119.9, 124.0, 129.2, 129.5, 152.3, 152.9, 173.3, 185.0, 203.0; HRMS (ESI+): Found: 280.1334; C18H18NO2 (MH+) Requires 280.1332 (0.8 ppm error). Spectroscopic data matched those previously reported in the literature.10a
:1 petrol:EtOAc); νmax (thin film)/cm−1 2148, 1636, 1586, 1489, 1230, 1019, 704; δH (400 MHz, CDCl3) 3.80 (3 H, s), 3.85 (2 H, s), 6.90 (2 H, d, J = 8.5), 7.03 (1 H, dd, J = 5.0, 3.5) 7.22 (2 H, d, J = 8.5), 7.40 (1 H, d, J = 3.5), 7.47 (1 H, d, J = 5.0); δC (100 MHz, CDCl3) 50.9, 55.2, 86.7, 92.4, 114.1, 119.6, 125.1, 127.6, 130.8, 131.8, 136.7, 158.9, 189.1; HRMS (ESI+): Found: 257.0625; C15H13O2S (MH+) Requires 257.0631 (1.7 ppm error).
:1 petrol:EtOAc) afforded the title compound10f as a pale yellow oil (28 mg, 73%); Rf 0.50 (1:1 petrol:EtOAc); νmax (thin film)/cm−1 1691, 1661, 1576, 1246, 1027, 907, 859, 724; δH (400 MHz, CDCl3) 2.77 (2 H, s), 6.49 (2 H, d, J = 10.0), 6.58 (1 H, s), 6.92 (2 H, d, J = 10.0), 7.03 (1 H, dd, J = 4.5, 3.5) 7.32 (1 H, d, J = 3.5), 7.52 (1 H, d, J = 4.5); δC (100 MHz, CDCl3) 46.3, 50.9, 127.3, 128.7, 130.2, 130.5, 131.4, 135.8, 150.9, 166.3, 184.7, 202.6; HRMS (ESI+): Found: 243.0480; C14H11O2S (MH+) Requires 243.0474 (−2.3 ppm error).
:1 petrol:EtOAc); νmax (thin film)/cm−1 2188, 1658, 1601, 1508, 1248, 1058, 1027, 831, 540; δH (400 MHz, CDCl3) 0.91 (3 H, t, J = 7.5), 1.77–1.89 (1 H, m), 2.16–2.27 (1 H, m), 3.63–3.67 (1 H, m), 3.79 (3 H, s), 3.82 (3 H, s), 6.85 (2 H, d, J = 9.0), 6.89 (2 H, d, J = 9.0), 7.24 (2 H, d, J = 9.0), 7.41 (2 H, d, J = 9.0); δC (100 MHz, CDCl3) 12.0, 24.8, 55.2, 55.4, 61.7, 87.5, 93.5, 111.9, 114.1, 114.2, 129.7, 130.0, 135.0, 158.9, 161.5, 188.4; HRMS (ESI+): Found: 309.1490; C20H21O3 (MH+) Requires 309.1485 (−1.6 ppm error).
:1 petrol:EtOAc) afforded the title compound10g as a pale brown oil (43 mg, 75%); Rf 0.50 (1:1 petrol:EtOAc); νmax (thin film)/cm−1 1696, 1661, 1590, 1509, 1257, 1177, 1030, 908, 832, 725; δH (400 MHz, CDCl3) 0.97 (3 H, t, J = 7.5), 1.25–1.35 (1 H, m), 1.75–1.84 (1 H, m), 2.66 (1 H, t, J = 7.0), 3.81 (3 H, s), 6.47 (1 H, dd, J = 10.0, 1.5), 6.56 (1 H, s), 6.56 (1 H, dd, J = 10.0, 1.5), 6.80 (1 H, dd, J = 10.0, 3.0), 6.84 (2 H, d, J = 9.0), 6.96 (1 H, dd, J = 10.0, 3.0), 7.44 (2 H, d, J = 9.0); δC (100 MHz, CDCl3) 12.6, 20.0, 55.4, 55.8, 58.9, 114.3, 125.6, 126.9, 129.3, 130.0, 131.6, 151.0, 152.2, 162.1, 171.5, 185.3, 205.1; HRMS (ESI+): Found: 295.1324; C19H19O3 (MH+) Requires 295.1329 (1.6 ppm error).
:1 petrol:EtOAc); νmax (thin film)/cm−1 2184, 1664, 1601, 1589, 1508, 1256, 1211, 1124, 1071, 1027, 835, 786, 575; δH (400 MHz, CDCl3) 3.77–3.84 (11 H, m), 6.46–6.50 (2 H, m), 6.84 (2 H, d, J = 8.5), 7.11 (1 H, d, J = 8.5), 7.37 (2 H, d, J = 8.5); δC (100 MHz, CDCl3) 46.0, 55.3, 55.3, 55.4, 87.7, 92.7, 98.5, 104.2, 111.9, 114.2, 115.1, 131.7, 135.1, 158.7, 160.4, 161.5, 186.2; HRMS (ESI+): Found: 311.1284; C19H19O4 (MH+) Requires 311.1278 (2.1 ppm error).
:1 petrol:EtOAc, then 1:1 petrol:EtOAc) afforded the title compound10h as a white solid (51 mg, 89%); mp. 169–171 °C; Rf 0.19 (1:1 petrol:EtOAc); νmax (thin film)/cm−1 1689, 1652, 1588, 1360, 1244, 1177, 1027, 989, 858, 840; δH (400 MHz, CDCl3) 2.61 (1 H, d, J = 18.0), 2.86 (1 H, d, J = 18.0), 3.65 (3 H, s), 3.79 (3 H, s), 5.76 (1 H, d, J = 1.5), 6.34 (1 H, dd, J = 10.0, 1.5), 6.61 (1 H, s), 6.64 (1 H, d, J = 10.0), 6.83 (2 H, d, J = 9.0), 7.43 (2 H, d, J = 9.0); δC (100 MHz, CDCl3) 47.7, 52.5, 55.4, 56.2, 103.4, 114.3, 125.1, 128.3, 128.4, 128.9, 147.4, 162.1, 171.6, 175.5, 187.2, 203.6; HRMS (ESI+): Found: 297.1112; C18H17O4 (MH+) Requires 297.1121 (1.9 ppm error).
:1 petrol:EtOAc); νmax (thin film)/cm−1 2185, 1652, 1600, 1505, 1296, 1244, 1210, 1026, 819; δH (400 MHz, CDCl3) 3.79 (3 H, s), 3.82 (3 H, s), 3.84–3.87 (5 H, m), 6.39 (1 H, d, J = 2.0), 6.45 (1 H, dd, J = 8.5, 2.0), 6.86 (2 H, d, J = 8.5), 7.25 (2 H, d, J = 8.5), 7.34 (1 H, d, J = 8.5); δC (100 MHz, CDCl3) 51.3, 55.2, 55.5, 55.7, 91.3, 91.9, 98.2, 101.5, 105.4, 114.0, 125.6, 130.9, 136.5, 158.7, 163.2, 163.6, 185.5; HRMS (ESI+): Found: 311.1272; C19H19O4 (MH+) Requires 311.1278 (1.9 ppm error).
:1 petrol:EtOAc, then 1:1 petrol:EtOAc) afforded the title compound10i as a pale brown solid (48 mg, 91%); mp. 172–174 °C; Rf 0.19 (1:1 petrol:EtOAc); νmax (thin film)/cm−1 1688, 1660, 1605, 1551, 1233, 1213, 1026, 859; δH (400 MHz, CDCl3) 2.67 (2 H, s), 3.80 (3 H, s), 3.84 (3 H, s), 6.37 (1 H, dd, J = 8.5, 2.5), 6.40 (2 H, d, J = 10.0), 6.46 (1 H, d, J = 2.5), 6.95 (2 H, d, J = 10.0), 6.97 (1 H, s), 7.26 (1 H, d, J = 8.5); δC (100 MHz, CDCl3) 46.1, 52.4, 55.4, 55.5, 98.9, 104.4, 115.0, 129.2, 130.2, 131.6, 152.9, 160.1, 163.2, 168.8, 184.9, 204.7; HRMS (ESI+): Found: 297.1113; C18H17O4 (MH+) Requires 297.1121 (2.6 ppm error).
:1 petrol:EtOAc); νmax (thin film)/cm−1 2115, 1651, 1510, 1246, 1176, 1120, 738, 686; δH (400 MHz, CDCl3) 3.79 (2 H, s), 3.80 (3 H, s), 6.89 (2 H, d, J = 8.5), 7.17–7.35 (7 H, m); δC (100 MHz, CDCl3) 50.2, 55.2, 87.6, 100.7, 114.2, 125.0, 127.0, 127.8, 129.5, 129.6, 130.9, 158.9, 183.3; HRMS (ESI+): Found: 283.0786; C17H15O2S (MH+) Requires 283.0787 (0.1 ppm error).
:1 petrol:EtOAc, then 1:1 petrol:EtOAc) afforded the title compound10j as a pale orange solid (43 mg, 64%); mp. 113–115 °C; Rf 0.25 (1:1 petrol:ethyl acetate); νmax (thin film)/cm−1 1687, 1665, 1548, 860, 752; δH (400 MHz, CDCl3) 2.74 (2 H, s), 5.63 (1 H, s), 6.47 (2 H, d, J = 10.0), 6.79 (2 H, d, J = 10.0), 7.42–7.49 (5 H, m); δC (100 MHz, CDCl3) 46.2, 51.3, 125.8, 128.4, 130.2, 130.4, 130.6, 134.5, 149.2, 182.5, 184.5, 199.8; HRMS (ESI+): Found: 269.0621; C16H13O2S (MH+) Requires 269.0631 (3.0 ppm error).
:1 petrol:EtOAc); νmax (thin film)/cm−1 2185, 1659, 1601, 1508, 1293, 1245, 117, 1084, 1026, 830; δH (400 MHz, CDCl3) 2.92–3.02 (4 H, m), 3.78 (3 H, s), 3.83 (3 H, s), 6.83 (2 H, d, J = 8.5), 6.89 (2 H, d, J = 8.5), 7.14 (2 H, d, J = 8.5), 7.51 (2 H, d, J = 8.5); δC (100 MHz, CDCl3) 29.2, 47.1, 55.2, 55.4, 87.7, 92.3, 111.6, 113.9, 114.3, 129.3, 132.4, 135.1, 158.0, 161.6, 187.1; HRMS (ESI+): Found: 295.1317; C19H19O3 (MH+) Requires 295.1329 (3.7 ppm error).
:1 petrol:EtOAc) afforded the title compound10k as a pale yellow solid (46 mg, 78%); mp. 165–167 °C; Rf 0.25 (1:1 petrol:EtOAc); νmax (thin film)/cm−1 1657, 1623, 1603, 1510, 1243, 1178, 1030, 859, 731; δH (400 MHz, CDCl3) 2.25 (2 H, t, J = 6.5), 2.68 (2 H, t, J = 6.5), 3.78 (3 H, s), 6.30 (1 H, s), 6.42 (2 H, d, J = 10.0), 6.79 (2 H, d, J = 9.0), 7.06 (2 H, d, J = 10.0), 7.19 (2 H, d, J = 9.0); δC (100 MHz, CDCl3) 34.0, 37.7, 45.5, 55.3, 114.0, 127.7, 128.1, 130.1, 130.4, 152.6, 159.2, 161.0, 184.7, 196.9; HRMS (ESI+): Found: 281.1179; C18H17O3 (MH+) Requires 281.1172 (2.5 ppm error).
:1 petrol:EtOAc, then 5:1 petrol:EtOAc) afforded the title compound9l as a pale yellow oil (141 mg, 43%); Rf 0.70 (1:1 petrol:EtOAc); νmax (thin film)/cm−1 2184, 1620, 1599, 1508, 1289, 1248, 1027, 999, 830, 760; δH (400 MHz, CDCl3) 3.77 (3 H, s), 3.80 (3 H, s), 6.79 (2 H, d, J = 9.0), 6.94 (2 H, d, J = 8.5), 7.22 (2 H, d, J = 9.0), 7.35 (2 H, d, J = 8.5), 7.40–7.45 (2 H, m), 7.53–7.58 (1 H, m), 7.92 (1 H, d, J = 7.5); δC (100 MHz, CDCl3) 55.3, 55.4, 88.9, 94.9, 112.0, 113.8, 114.0, 127.0, 129.9, 130.7, 130.9, 131.9, 132.9, 135.0, 138.1, 142.3, 159.4, 161.4, 180.8; HRMS (ESI+): Found: 343.1324; C23H19O3 (MH+) Requires 343.1329 (1.0 ppm error).
:1 petrol:EtOAc) afforded the title compound10l as a pale yellow oil (47 mg, 74%); Rf 0.25 (1:1 petrol:EtOAc); νmax (thin film)/cm−1 1655, 1602, 1510, 1331, 1251, 1031, 836; δH (400 MHz, CDCl3) 3.797 (3 H, s), 6.44 (2 H, d, J = 9.5), 6.72 (1 H, s), 6.74 (2 H, d, J = 9.5), 6.82 (2 H, d, J = 9.0), 7.25–7.29 (3 H, m), 7.48–7.57 (2 H, m), 8.25 (1 H, d, J = 7.0); δC (100 MHz, CDCl3) 50.7, 55.3, 113.7, 127.3, 128.2, 128.7, 128.9, 129.6, 130.0, 130.0, 130.2, 133.2, 138.3, 150.0, 156.0, 160.6, 183.6, 185.2; HRMS (ESI+): Found: 329.1170; C22H17O3 (MH+) Requires 329.1172 (0.8 ppm error).
:1 EtOAc:hexane); νmax (thin film)/cm−1 3264, 1631, 1614, 1594, 1515, 1446, 1233, 1172, 1002, 798; δH (400 MHz, CDCl3) 3.22 (3 H, s), 3.65 (3 H, s), 3.70 (2 H, s), 6.68 (2 H, d, J = 8.5), 7.07 (2 H, d, J = 8.5); δC (100 MHz, CDCl3) 32.3, 38.2, 61.3, 115.6, 125.9, 130.4, 155.2, 173.3; HRMS (ESI+): Found: 218.0788; C10H13NNaO3 (MNa+) Requires 218.0788 (−0.3 ppm error), Found: 196.0975; C10H14NO3 (MH+) Requires 196.0968 (−3.2 ppm error).
:1 hexane:EtOAc, then 1:1 hexane:EtOAc) afforded the title compound11c as a pale yellow solid (135 mg, 56%); mp 96–98 °C; Rf 0.51 (6:4 hexane:EtOAc); νmax (thin film)/cm−1 3368, 2202, 1655, 1514, 1224, 1079, 758, 688; δH (400 MHz, CDCl3) 3.87 (2 H, s), 5.42 (1 H, br s), 6.83–6.88 (2 H, m), 7.16–7.21 (2 H, m), 7.33–7.39 (2 H, m), 7.42–7.50 (3 H, m); δC (100 MHz, CDCl3) 51.3, 87.7, 93.3, 115.7, 119.7, 125.1, 128.6, 130.9, 131.1, 133.1, 155.1, 186.1; HRMS (ESI+): Found: 259.0731; C16H12NaO2 (MNa+) Requires 259.0730 (−0.6 ppm error), Found: 237.0919; C16H13O2 (MH+) Requires 237.0910 (−3.8 ppm error).
:1 hexane:EtOAc); νmax (thin film)/cm−1 3068, 1693, 1658, 1592, 1251, 859, 764; δH (400 MHz, CDCl3) 2.80 (2 H, s), 6.49 (2 H, d, J = 10.0), 6.71 (1 H, s), 6.96 (2 H, d, J = 10.0), 7.34–7.40 (2 H, m), 7.42–7.48 (1 H, m), 7.49–7.54 (2 H, m); δC (100 MHz, CDCl3) 46.9, 51.2, 127.4, 129.0, 129.9, 130.0, 131.6, 132.9, 151.4, 173.9, 184.7, 203.3; HRMS (ESI+): Found: 259.0732; C16H12NaO2 (MNa+) Requires 259.0730 (−0.9 ppm error). Spectroscopic data matched those previously reported in the literature.25
:1 hexane:EtOAc, then 7:3 hexane:EtOAc) afforded the title compound11d as a yellow solid (502 mg, 76%); mp 86–88 °C; Rf 0.62 (1:1 hexane:EtOAc); νmax (thin film)/cm−1 3353, 2195, 1651, 1600, 1510, 1254, 1170, 1076, 834; δH (400 MHz, CDCl3) 3.83 (3 H, s), 3.85 (2 H, s), 5.52 (1 H, br s), 6.86 (4 H, m), 7.17 (2 H, d, J = 8.0), 7.41 (2 H, d, J = 8.5); δC (100 MHz, CDCl3) 51.1, 55.4, 87.8, 94.7, 111.5, 114.3, 115.6, 125.4, 131.1, 135.2, 155.1, 161.7, 186.3; HRMS (ESI+): Found: 289.0839; C17H14NaO3 (MNa+) Requires 289.0835 (−1.4 ppm error).
:1 hexane:EtOAc); νmax (thin film)/cm−1 3316, 2939, 1639, 1514, 1432, 1271, 1200, 1151, 1033; δH (400 MHz, CDCl3) 3.19 (3 H, s), 3.62 (3 H, s), 3.70 (2 H, s), 3.87 (3 H, s), 5.35 (1 H, br s), 6.75 (1 H, d, J = 8.0), 6.82–6.86 (2 H, m); δC (100 MHz, CDCl3) 32.2, 38.8, 55.8, 61.3, 111.7, 114.2, 122.1, 126.5, 144.5, 146.5, 172.7; HRMS (ESI+): Found: 248.0884; C11H15NNaO4 (MNa+) Requires 248.0893 (3.7 ppm error), Found: 226.1070; C11H16NO4 (MH+) Requires 226.1074 (1.6 ppm error).
:1 hexane:EtOAc, then 7:3 hexane:EtOAc) afforded the title compound11m as a yellow solid (443 mg, 58%); mp 61–63 °C; Rf 0.20 (7:3 hexane:EtOAc); νmax (thin film)/cm−1 3437, 2195, 1655, 1601, 1509, 1254, 1237, 1170; δH (400 MHz, CDCl3) 3.84 (5 H, s), 3.89 (3 H, s), 5.63 (1 H, br s), 6.78–6.95 (5 H, m), 7.43 (2 H, d, J = 8.5); δC (100 MHz, CDCl3) 51.7, 55.4, 55.9, 87.7, 94.1, 111.6, 112.0, 114.3, 114.5, 122.8, 125.2, 135.1, 144.9, 146.6, 161.7, 185.7; HRMS (ESI+): Found: 319.0939; C18H16NaO4 (MNa+) Requires 319.0941 (0.7 ppm error).
:2 hexane:EtOAc) afforded the title compound10m an off-white oil (50.9 mg, 86%); Rf 0.14 (1:1 hexane:EtOAc); νmax (thin film)/cm−1 1691, 1664, 1636, 1603, 1587, 1509, 1258, 1208, 1177, 831; δH (400 MHz, CDCl3) 2.78 (1 H, d, J = 18.5), 2.86 (1 H, d, J = 18.5), 3.66 (3 H, s), 3.83 (3 H, s), 5.86 (1 H, d, J = 2.5), 6.50 (1 H, d, J = 9.5), 6.61 (1 H, s), 6.85 (2 H, d, J = 8.5), 6.97 (1 H, dd, J = 9.5, 2.5), 7.48 (2 H, d, J = 8.5); δC (100 MHz, CDCl3) 48.0, 51.6, 55.2, 55.4, 114.3, 119.4, 125.3, 127.1, 129.0, 129.4, 151.7, 152.4, 162.2, 173.7, 180.1, 203.4; HRMS (ESI+): Found: 319.0947; C18H16NaO4 (MNa+) Requires 319.0941 (−2.0 ppm error).
:1 hexane:EtOAc, then 7:3 hexane:EtOAc) afforded the title compound11n as a yellow oil (181 mg, 82%); Rf 0.76 (1:1 hexane:EtOAc); νmax (thin film)/cm−1 3373, 2959, 2933, 2209, 1652, 1514, 1224, 796; δH (400 MHz, CDCl3) 0.89 (3 H, t, J = 7.5), 1.35 (2 H, qt, J = 7.5, 7.5), 1.48 (2 H, tt, J = 7.5, 7.0), 2.32 (2 H, t, J = 7.0), 3.74 (2 H, s), 5.93 (1 H, br s), 6.80 (2 H, d, J = 8.0), 7.09 (2 H, d, J = 8.0); δC (100 MHz, CDCl3) 13.4, 18.6, 21.8, 29.5, 51.3, 80.7, 97.3, 115.6, 124.9, 130.9, 155.1, 186.7; HRMS (ESI+): Found: 239.1050; C14H16NaO2 (MNa+) Requires 239.1043 (−3.2 ppm error).
:1 hexane:EtOAc); νmax (thin film)/cm−1 2959, 2929, 2875, 2857, 1720, 1696, 1657, 1614, 1599, 1255, 1232, 862; δH (400 MHz, CDCl3) 0.90 (3 H, t, J = 7.5), 1.33 (2 H, qt, J = 7.5, 7.5), 1.52 (2 H, tt, J = 7.5, 7.5), 2.10 (2 H, t, J = 7.5), 2.65 (2 H, s), 6.22 (1 H, s), 6.45 (2 H, d, J = 9.0), 6.66 (2 H, d, J = 9.0); δC (100 MHz, CDCl3) 13.7, 22.2, 28.8, 29.5, 45.0, 52.6, 130.58, 130.61, 150.0, 181.6, 184.9, 204.6; HRMS (ESI+): Found: 239.1043; C14H16NaO2 (MNa+) Requires 239.1043 (−0.3 ppm error), Found: 217.1219; C14H17O2 (MH+) Requires 217.1223 (2.0 ppm error). Spectroscopic data matched those previously reported in the literature.26
:1 hexane:EtOAc, then 1:1 hexane:EtOAc) afforded the title compound11o as a yellow oil (241 mg, 74%); Rf 0.62 (1:1 hexane:EtOAc); νmax (thin film)/cm−1 3331, 2977, 2212, 1663, 1515, 1395, 1366, 1225, 1145, 730; δH (400 MHz, CDCl3) 1.47 (9 H, s), 2.54 (2 H, t, J = 7.0), 2.86 (3 H, s), 3.36 (2 H, t, J = 7.0), 3.71 (2 H, s), 6.07 (1 H, br s), 6.81 (2 H, d, J = 8.0), 7.08 (2 H, d, J = 8.0); δC (100 MHz, CDCl3) 18.6, 28.4, 35.0, 47.2, 51.2, 80.3, 81.7, 92.9, 115.8, 124.8, 130.9, 155.6, 155.6, 185.4; HRMS (ESI+): Found: 340.1522; C18H23NNaO4 (MNa+) Requires 340.1519 (−0.9 ppm error). Note: majority of peaks broadened in 1H NMR spectrum due to presence of rotamers.
:1 hexane:EtOAc); νmax (thin film)/cm−1 2975, 1720, 1688, 1662, 1624, 1615, 1392, 1365, 1165, 1144, 860; δH (400 MHz, CDCl3) 1.43 (9 H, s), 2.31 (2 H, t, J = 7.0), 2.62 (2 H, s), 2.81 (3 H, s), 3.39 (2 H, t, J = 7.0), 6.20 (1 H, s), 6.43 (2 H, d, J = 9.5), 6.65–6.73 (2 H, br m); δC (100 MHz, CDCl3) 27.2, 28.4, 34.2, 45.1, 47.1, 52.8, 80.0, 130.9, 132.0, 149.4, 155.5, 176.9, 184.5, 203.8; HRMS (ESI+): Found: 340.1522; C18H23NNaO4 (MNa+) Requires 340.1519 (−0.8 ppm error). Note: majority of peaks broadened in 1H NMR spectrum due to presence of rotamers.
:1 hexane:EtOAc, then 6:4 hexane:EtOAc) to afford the title compound11p as a yellow oil (367 mg, 84%); Rf 0.56 (1:1 hexane:EtOAc); νmax (thin film)/cm−1 3379, 2953, 2929, 2857, 2213, 1670, 1514, 1253, 1106, 836, 795, 778; δH (400 MHz, CDCl3) 0.06 (6 H, s), 0.88 (9 H, s), 2.53 (2 H, t, J = 7.0), 3.71 (2 H, t, J = 7.0), 3.73 (2 H, s), 6.77 (2 H, app. d, J = 8.5), 7.06 (2 H, app. d, J = 8.5); δC (100 MHz, CDCl3) −5.2, 18.4, 23.5, 25.9, 51.3, 60.8, 81.5, 93.7, 115.7, 124.8, 131.0, 155.2, 186.3; HRMS (ESI+): Found: 341.1536; C18H26NaO3Si (MNa+) Requires 341.1543 (2.3 ppm error).
:2 hexane:EtOAc, then 1:1 hexane:EtOAc) afforded the title compound10p as an orange solid (49.0 mg, 89%); Rf 0.42 (1:1 hexane:EtOAc); mp 76–80 °C; νmax (thin film)/cm−1 2928, 2857, 1721, 1698, 1657, 1621, 1254, 1102, 864, 834, 775; δH (400 MHz, CDCl3) 0.01 (6 H, s), 0.85 (9 H, s), 2.28 (2 H, dt, J = 6.0, 1.5), 2.26 (2 H, s), 3.75 (2 H, t, J = 6.0), 6.31 (1 H, t, J = 1.5), 6.42 (2 H, app. d, J = 10.0), 6.64 (2 H, app. d, J = 10.0); δC (100 MHz, CDCl3) −5.35, 18.3, 25.9, 32.3, 44.9, 52.7, 60.6, 130.8, 132.0, 149.8, 178.0, 184.9, 204.8; HRMS (ESI+): Found: 341.1541; C18H26NaO3Si (MNa+) Requires 341.1543 (0.7 ppm error).
:1 hexane:EtOAc) afforded the title compound11q as a white solid (276 mg, 90%); mp 81–83 °C; Rf 0.59 (1:1 hexane:EtOAc); νmax (thin film)/cm−1 3367, 2201, 1647, 1514, 1222; δH (400 MHz, CDCl3) 0.80–0.85 (2 H, m), 0.92–0.98 (2 H, m), 1.31–1.39 (1 H, m), 3.71 (2 H), 5.30 (1 H, br s), 6.80 (2 H, d, J = 8.0), 7.09 (2 H, d, J = 8.0); δC (100 MHz, CDCl3) −0.3, 9.9, 51.1, 76.5, 101.6, 115.5, 125.3, 130.9, 154.9, 186.0; HRMS (ESI+): Found: 223.0734; C13H12NaO2 (MNa+) Requires 223.0730 (−2.1 ppm error), Found: 201.0906; C13H13O2 (MH+) Requires 201.0910 (1.9 ppm error).
:1 hexane:EtOAc); νmax (thin film)/cm−1 1689, 1666, 1624, 1605, 1401, 1252, 860; δH (400 MHz, CDCl3) 0.77–0.82 (2 H, m), 1.11–1.17 (2 H, m), 1.18–1.24 (1 H, m), 2.64 (2 H, s), 5.75 (1 H, s), 6.45 (2 H, d, J = 10.0), 6.72 (2 H, d, J = 10.0); δC (100 MHz, CDCl3) 11.0, 13.8, 45.0, 52.8, 123.5, 130.6, 150.0, 184.9, 185.6, 204.2; HRMS (ESI+): Found: 223.0733; C13H12NaO2 (MNa+) Requires 223.0730 (−1.7 ppm error), Found: 201.0906; C13H13O2 (MH+) Requires 201.0910 (2.2 ppm error).
:1 hexane:EtOAc, then 7:3 hexane:EtOAc) afforded the title compound11r as a pale yellow oil (207 mg, 89%); Rf 0.74 (1:1 hexane:EtOAc); νmax (thin film)/cm−1 3376, 2961, 2871, 2205, 1650, 1514, 1224, 1172; δH (400 MHz, CDCl3) 1.50–1.76 (6 H, m), 1.83–1.97 (2 H, m), 2.73 (1 H, tt, J = 7.5, 7.5), 3.74 (2 H, s), 5.49 (1 H, br s), 6.80 (2 H, d, J = 8.0), 7.10 (2 H, d, J = 8.0); δC (100 MHz, CDCl3) 25.1, 30.0, 33.0, 51.3, 80.2, 101.3, 115.5, 125.2, 130.9, 154.9, 186.6; HRMS (ESI+): Found: 251.1041; C15H16NaO2 (MNa+) Requires 251.1043 (0.7 ppm error).
:1 hexane:EtOAc); νmax (thin film)/cm−1 2958, 1697, 1657, 1621, 1609, 1403, 1249, 864; δH (400 MHz, CDCl3) 1.37–1.50 (2 H, m), 1.52–1.67 (2 H, m), 1.68–1.82 (2 H, m), 1.83–1.95 (2 H, m), 2.30 (1 H, tt, J = 8.0, 8.0), 2.64 (2 H, s), 6.22 (1 H, s), 6.44 (2 H, d, J = 10.0), 6.69 (2 H, d, J = 10.0); δC (100 MHz, CDCl3) 25.5, 34.8, 40.2, 45.0, 53.0, 129.0, 130.5, 150.0, 185.0, 186.8, 204.7; HRMS (ESI+): Found: 251.1033; C15H16NaO2 (MNa+) Requires 251.1043 (3.9 ppm error), Found: 229.1215; C15H17O2 (MH+) Requires 229.1223 (3.7 ppm error).
:1 hexane:EtOAc, then 1:1 hexane:EtOAc) afforded the title compoundS5 as a white solid (788 mg, 31%); mp 63–65 °C; Rf 0.39 (1:1 hexane:EtOAc); νmax (thin film)/cm−1 3260, 1628, 1596, 1456, 1246, 1000, 753; δH (400 MHz, CDCl3) 3.24 (3 H, s), 3.80 (3 H, s), 3.87 (2 H, s), 6.85 (1 H, dd, J = 8.0, 7.5), 6.99 (1 H, d, J = 8.0), 7.09 (1 H, d, J = 7.5), 7.19 (1 H, dd, J = 8.0, 8.0), 9.50 (1 H, s); δC (100 MHz, CDCl3) 32.0, 35.1, 62.0, 118.2, 120.2, 120.9, 129.1, 130.9, 156.8, 173.5; HRMS (ESI+): Found: 218.0794; C10H13NNaO3 (MNa+) Requires 218.0788 (−3.0 ppm error), Found: 196.0967; C10H14NO3 (MH+) Requires 196.0968 (−0.8 ppm error).
:1 hexane:EtOAc) afforded the title compound11s as an off-white solid (452 mg, 88%); mp 98–100 °C; Rf 0.78 (1:1 hexane:EtOAc); νmax (thin film)/cm−1 3369, 2202, 1649, 1458, 1269, 755; δH (400 MHz, CDCl3) 0.87–0.92 (2 H, m), 0.97–1.04 (2 H, m), 1.37–1.45 (1 H, m), 3.85 (2 H, s), 6.70 (1 H, br s), 6.88–6.93 (2 H, m), 7.11 (1 H, app. d, J = 7.5), 7.19 (1 H, app. dd, J = 8.0, 8.0); δC (100 MHz, CDCl3) −0.1, 10.2, 47.5, 76.8, 103.2, 117.1, 120.6, 120.9, 129.2, 131.3, 154.9, 187.2; HRMS (ESI+): Found: 223.0738; C13H12NaO2 (MNa+) Requires 223.0730 (−3.6 ppm error), Found: 201.0918; C13H13O2 (MH+) Requires 201.0910 (−3.8 ppm error).
:1 hexane:EtOAc); νmax (thin film)/cm−1 1694, 1660, 1632, 1607, 1557, 1200, 862; δH (400 MHz, CDCl3) 0.68–0.79 (2 H, m), 0.97–1.09 (2 H, m), 1.20–1.28 (1 H, m), 2.39 (1 H, d, J = 18.0), 2.77 (1 H, d, J = 18.0), 5.72 (1 H, s), 6.23 (1 H, d, J = 9.5), 6.28 (1 H, d, J = 9.0), 6.47 (1 H, dd, J = 9.0, 5.5), 7.19 (1 H, ddd, J = 9.5, 5.5, 1.5); δC (100 MHz, CDCl3) 11.0, 11.9, 13.2, 46.8, 62.7, 122.8, 123.9, 126.7, 142.5, 142.8, 184.5, 200.3, 206.2; HRMS (ESI+): Found: 223.0732; C13H12NaO2 (MNa+) Requires 223.0730 (−1.3 ppm error), Found: 201.0907; C13H13O2 (MH+) Requires 201.0910 (1.3 ppm error).
:1 hexane:EtOAc, then 7:3 hexane:EtOAc) afforded the title compound11t as a yellow solid (1.33 g, 93%); mp 106–108 °C; Rf 0.67 (6:4 hexane:EtOAc); νmax (thin film)/cm−1 3333, 2982, 2202, 1661, 1489, 1458, 1156, 753; δH (400 MHz, CDCl3) 4.01 (2 H, s), 6.27 (1 H, br s), 6.94–6.98 (2 H, m), 7.19–7.26 (2 H, m), 7.39 (2 H, m), 7.45–7.50 (1 H, m), 7.51–7.55 (2 H, m); δC (100 MHz, CDCl3) 47.5, 87.8, 94.0, 116.8, 119.6, 120.4, 121.0, 128.6, 129.3, 131.1, 131.5, 133.3, 154.8, 187.0; HRMS (ESI+): Found: 259.0722; C16H12NaO2 (MNa+) Requires 259.0730 (3.1 ppm error), Found: 237.0914; C16H13O2 (MH+) Requires 237.0910 (−1.5 ppm error).
:1 hexane:EtOAc, then 1:1 hexane:EtOAc) afforded the title compound12t a pale yellow oil (103 mg, 90%); Rf 0.22 (7:3 hexane:EtOAc); νmax (thin film)/cm−1 1694, 1659, 1595, 1195, 760; δH (400 MHz, CDCl3) 2.54 (1 H, d, J = 18.0), 2.78 (1 H, d, J = 18.0), 6.34 (1 H, d, J = 10.0), 6.44–6.49 (2 H, m), 6.77 (1 H, s), 7.24–7.30 (1 H, m), 7.30–7.38 (4 H, m), 7.38–7.44 (1 H, m); δC (100 MHz, CDCl3) 48.3, 60.5, 121.8, 126.5, 127.3, 129.0, 129.5, 131.5, 132.3, 142.3, 144.6, 173.7, 200.2, 204.6; HRMS (ESI+): Found: 259.0723; C16H12NaO2 (MNa+) Requires 259.0730 (2.5 ppm error), Found: 237.0906; C16H13O2 (MH+) Requires 237.0910 (1.9 ppm error).
The title compound was also prepared in an enantioenriched form using CPA 1615 (56% conv., 23% ee, see Scheme 4); [α]21D = +5.6 (c = 0.2, CHCl3).
:1 hexane:EtOAc, then 7:3 hexane:EtOAc) afforded the title compound11u as a yellow solid (478 mg, 70%); mp 108–110 °C; Rf 0.29 (7:3 hexane:EtOAc); νmax (thin film)/cm−1 3364, 2193, 1645, 1599, 1508, 1254, 1080, 834; δH (400 MHz, CDCl3) 3.85 (3 H, s), 3.99 (2 H, s), 6.64 (1 H, s), 6.87–6.96 (4 H, m), 7.17–7.24 (2 H, m), 7.49 (2 H, d, J = 8.0); δC (100 MHz, CDCl3) 47.6, 55.4, 87.0, 95.8, 111.2, 114.4, 117.1, 120.7, 121., 129.2, 131.5, 135.5, 154.9, 162.0, 187.2; HRMS (ESI+): Found: 289.0833; C17H14NaO3 (MNa+) Requires 289.0835 (0.6 ppm error).
:1 hexane:EtOAc); νmax (thin film)/cm−1 1689, 1659, 1602, 1587, 1510, 1262, 1179, 1026, 833, 731; δH (400 MHz, CDCl3) 2.51 (1 H, d, J = 18.0), 2.75 (1 H, d, J = 18.0), 3.81 (3 H, s), 6.34 (1 H, d, J = 9.5), 6.45–6.47 (2 H, m), 6.68 (1 H, s), 6.85 (2 H, d, J = 8.5), 7.25–7.31 (3 H, m); δC (100 MHz, CDCl3) 48.3, 55.4, 60.4, 114.4, 121.5, 124.8, 126.5, 127.2, 129.2, 142.3, 145.0, 162.2, 173.4, 200.5, 204.5; HRMS (ESI+): Found: 289.0834; C17H14NaO3 (MNa+) Requires 289.0835 (0.4 ppm error), Found: 267.1004; C17H15O3 (MH+) Requires 267.1016 (4.2 ppm error).
:1 hexane:EtOAc, then 7:3 hexane:EtOAc) afforded the title compound11v as a yellow solid (430 mg, 66%); mp 115–117 °C; Rf 0.46 (7:3 hexane:EtOAc); νmax (thin film)/cm−1 3357, 2203, 1651, 1598, 1505, 1458, 1234, 1082, 838, 754; δH (400 MHz, CDCl3) 3.99 (2 H, s), 6.25 (1 H, s), 6.89–6.97 (2 H, m), 7.08 (2 H, dd, 3JHH = 8.5, 3JHF 8.5), 7.18–7.25 (2 H, m), 7.51 (2 H, dd, 3JHH = 8.5, 4JHF = 5.5); δC (100 MHz, CDCl3) 47.4, 87.7, 92.8, 115.7 (d, 4JCF = 4.0), 116.2 (d, 2JCF = 22.0), 116.8, 120.4, 121.1, 129.3, 131.5, 135.6 (d, 3JCF = 8.5), 154.7, 164.1 (d, 1JCF = 255), 186.8; HRMS (ESI+): Found: 277.0628; C16H11FNaO2 (MNa+) Requires 277.0635 (2.5 ppm error).
:2 EtOAc:hexane) afforded the title compound12v a yellow oil (88.7 mg, 90%); Rf 0.36 (1:1 hexane:EtOAc); νmax (thin film)/cm−1 1694, 1659, 1601, 1582, 1508, 1238, 1193, 1163, 836; δH (400 MHz, CDCl3) 2.53 (1 H, d, J = 18.0), 2.76 (1 H, d, J = 18.0), 6.33 (1 H, d, J = 9.5), 6.42–6.50 (2 H, m), 6.70 (1 H, s), 7.03 (2 H, dd, 3JHH = 8.5, 3JHF 8.5), 7.25–7.29 (1 H, m), 7.29–7.34 (2 H, m); δC (100 MHz, CDCl3) 48.4, 60.5, 116.2 (d, 2JCF = 22.0), 121.9, 126.5, 128.6 (d, 4JCF = 3.0), 129.2, 129.5 (d, 3JCF = 8.5), 142.4, 144.3, 164.3 (d, 1JCF = 254), 172.3, 200.1, 204.3; HRMS (ESI+): Found: 277.0642; C16H11FNaO2 (MNa+) Requires 277.0635 (−2.4 ppm error), Found: 255.0818; C16H12FO2 (MH+) Requires 255.0816 (−0.7 ppm error).
:1 hexane:EtOAc, then 8:2 hexane:EtOAc) afforded the title compound13 as a yellow oil (306 mg, 80%); Rf 0.37 (7:3 hexane:EtOAc); νmax (thin film)/cm−1 3374, 2980, 2188, 1743, 1369, 1232, 1150, 1072; δH (400 MHz, CDCl3) 1.70 (9 H, s), 3.89 (2 H, s), 4.98 (1 H, br s), 6.87 (2 H, d, J = 8.0), 7.23 (2 H, d, J = 8.0), 7.32 (1 H, dd, J = 8.0, 7.5), 7.38 (1 H, dd, J = 8.0, 7.5), 7.51 (1 H, d, J = 8.0), 7.91 (1 H, s), 8.14 (1 H, d, J = 8.0); δC (100 MHz, CDCl3) 28.2, 51.2, 85.3, 86.8, 92.4, 100.5, 115.5, 115.8, 120.1, 123.8, 125.7, 125.8, 129.9, 131.2, 133.1, 134.8, 148.6, 155.2, 185.3; HRMS (ESI+): Found: 398.1357; C23H21NNaO4 (MNa+) Requires 398.1363 (1.5 ppm error). Note: some peaks broadened in 13C NMR spectrum due to presence of rotamers.
:1 hexane:EtOAc); νmax (thin film)/cm−1 1742, 1694, 1662, 1595, 1370, 1351, 1228, 1148, 1109, 861, 732; δH (400 MHz, CDCl3) 1.63 (9 H, s), 2.76 (2 H, s), 6.50 (2 H, d, J = 9.5), 6.91 (1 H, s), 6.97 (2 H, d, J = 9.5), 7.34–7.45 (2 H, m), 7.78 (1 H, d, J = 8.0), 7.93 (1 H, s), 8.25 (1 H, d, J = 8.0); δC (100 MHz, CDCl3) 28.0, 45.6, 51.9, 85.4, 114., 115.7, 120.3, 124.2, 125.7, 127.8, 128.2, 128.4, 129.7, 135.8, 148.4, 151.9, 165.9, 184.3, 203.4. Note: some peaks broadened in 13C NMR spectrum due to presence of rotamers. Spectroscopic data matched those previously reported in the literature.5
:1 EtOAc:MeOH) to afford the title compound14 as a colourless oil (29.2 mg, 66%); Rf 0.47 (9:1 EtOAc:MeOH); νmax (thin film)/cm−1 2790, 1709, 1684, 1632, 1209; δH (400 MHz, CDCl3) 2.23–2.32 (4 H, m), 2.45–2.54 (2 H, m), 2.58–2.75 (4 H, m), 2.87 (1 H, dd, J = 16.0, 2.5), 3.10 (1 H, ddd, J = 11.0, 5.5, 2.5), 6.00 (1 H, s), 6.09 (1 H, d, J = 10.0), 6.41 (1 H, dd, J = 10.0, 2.5); δC (100 MHz, CDCl3) 29.7, 40.0, 42.1, 45.9, 49.4, 56.7, 70.2, 127.7, 129.2, 149.8, 181.3, 196.1, 204.9; HRMS (ESI+): Found: 218.1170; C13H16NO2 (MH+) Requires 218.1176 (2.5 ppm error).
:2 hexane:EtOAc, then 8:2 EtOAc:hexane) to afford the title compound15 as an off-white solid (15.0 mg, 75%); Rf 0.47 (EtOAc); mp 115–120 °C; νmax (thin film)/cm−1 3564, 2961, 2925, 2856, 1706, 1682, 1629, 1404, 1234, 1204, 1060, 1009, 781, 701; δH (400 MHz, CDCl3) 2.40 (1 H, d, J = 18.5), 2.55 (1 H, d, J = 18.5), 2.60–2.75 (4 H, m), 3.50 (1 H, dt, J = 11.5, 3.0), 3.81–3.83 (1 H, m), 4.22 (1 H, ddd, J = 11.5, 5.5, 2.5), 6.08 (1 H, d, J = 1.5), 6.13 (1 H, d, J = 10.0), 6.42 (1 H, dd, J = 10.0, 3.0); δC (100 MHz, CDCl3) 30.1, 41.5, 44.3, 48.9, 68.1, 81.5, 128.8, 129.7, 148.2, 179.5, 195.1, 204.3; HRMS (ESI+): Found: 227.0680; C12H12NaO3 (MNa+) Requires 227.0679 (−0.6 ppm error).
Acknowledgements
The authors would like to thank the University of York (A. K. C., J. D. C., W. P. U.), the EPSRC (J. T. R. L. EP/M018601/01) and the Leverhulme Trust (for an Early Career Fellowship, ECF-2015-013, W. P. U.) for financial support. We would also like to thank Jack Partington, Harry Kirby, Jamie Rose and Richard Bickford-Smith (University of York undergraduate students) for assistance during preliminary studies.Notes and references
- (a) R. A. Edrada, C. C. Stessman and P. Crews, J. Nat. Prod., 2003, 66, 939 CrossRef CAS PubMed; (b) H. M. Ge, C. Xu, X. T. Wang, B. Huang and R. X. Tan, Eur. J. Org. Chem., 2006, 5551 CrossRef; (c) P. Cheng, Y. Ma, S. Yao, Q. Zhang, E. Wang, M. Yan, X. Zhang, F. Zhang and J. Chen, Bioorg. Med. Chem. Lett., 2007, 17, 5316 CrossRef CAS PubMed; (d) J.-J. Fu, J.-J. Qin, Q. Zeng, Y. Huang, H. Z. Jin and W.-D. Zhang, Chem. Pharm. Bull., 2010, 58, 1263 CrossRef CAS PubMed; (e) H. B. Park, Y.-J. Kim, J. K. Lee, K. R. Lee and H. C. Kwon, Org. Lett., 2012, 14, 5002 CrossRef CAS PubMed.
- (a) M.-A. Beaulieu, K. C. Guérard, G. Maertens, C. Sabot and S. Canesi, J. Org. Chem., 2011, 76, 9460 CrossRef CAS PubMed; (b) T. Honda and H. Shigehisa, Org. Lett., 2006, 8, 657 CrossRef CAS PubMed; (c) K. C. Nicolaou, D. J. Edmonds, A. Li and G. S. Tria, Angew. Chem., Int. Ed., 2007, 46, 3942 CrossRef CAS PubMed; (d) C. Zheng, L. Wang, J. Li, L. Wang and D. Z. Wang, Org. Lett., 2013, 15, 4046 CrossRef CAS PubMed; (e) A. S. Kende, K. Koch and C. A. Smith, J. Am. Chem. Soc., 1988, 110, 2210 CrossRef CAS; (f) T. Dohi, Y. Minamitsuji, A. Maruyama, S. Hirose and Y. Kita, Org. Lett., 2008, 10, 3559 CrossRef CAS PubMed; (g) S.-K. Hong, H. Kim, Y. Seo, S. H. Lee, J. K. Cha and Y. G. Kim, Org. Lett., 2010, 12, 3954 CrossRef CAS PubMed; (h) C. Iwata, T. Fusaka, T. Fujiwara, K. Tomita and M. Yamada, J. Chem. Soc., Chem. Commun., 1981, 463 RSC; (i) S. Ishiwata, K. Itakura and K. Misawa, Chem. Pharm. Bull., 1970, 18, 1219 CrossRef CAS.
- (a) P. Magnus, K. D. Marks and A. Meis, Tetrahedron, 2015, 71, 3872 CrossRef CAS; (b) N. A. McGrath, E. S. Bartlett, S. Sittihan and J. T. Njardarson, Angew. Chem., Int. Ed., 2009, 48, 8543 CrossRef CAS PubMed; (c) J. D. McChesney and R. A. Swanson, J. Org. Chem., 1982, 47, 5201 CrossRef CAS; (d) R. P. Gajewski, Tetrahedron Lett., 1976, 17, 4125 CrossRef; (e) O. Hares, D. Hobbs-Mallyon and D. A. Whiting, J. Chem. Soc., Perkin Trans. 1, 1993, 1481 RSC; (f) A. S. K. Hashmi, L. Schwarz and M. Bolte, Tetrahedron Lett., 1998, 39, 8969 CrossRef CAS; (g) S. Rousseaux, J. García-Fortanet, M. A. Del Aguila Sanchez and S. L. Buchwald, J. Am. Chem. Soc., 2011, 133, 9282 CrossRef CAS PubMed; (h) T. Nemoto, R. Wu, Z. Zhao, T. Yokosaka and Y. Hamada, Tetrahedron, 2013, 69, 3403 CrossRef CAS; (i) T. Dohi, T. Nakae, Y. Ishikado, D. Kato and Y. Kita, Org. Biomol. Chem., 2011, 9, 6899 RSC; (j) L. Luo, H. Zheng, J. Liu, H. Wang, Y. Wang and X. Luan, Org. Lett., 2016, 18, 2082 CrossRef CAS PubMed; (k) W.-T. Wu, R.-Q. Xu, L. Zhang and S.-L. You, Chem. Sci., 2016, 7, 3427 RSC; (l) T. Nemoto, Y. Ishige, M. Yoshida, Y. Kohno, M. Kanematsu and Y. Hamada, Org. Lett., 2010, 12, 5020 CrossRef CAS PubMed; (m) Q.-F. Wu, W.-B. Liu, C.-X. Zhuo, Z.-Q. Rong, K.-Y. Ye and S.-L. You, Angew. Chem., Int. Ed., 2011, 50, 4455 CrossRef CAS PubMed.
- (a) Y. Yamamoto, I. D. Gridnev, N. T. Patil and T. Jin, Chem. Commun., 2009, 5075 RSC; (b) C. J. V. Halliday and J. M. Lynam, Dalton Trans., 2016, 45, 12611 RSC.
- W. P. Unsworth, J. D. Cuthbertson and R. J. K. Taylor, Org. Lett., 2013, 15, 3306 CrossRef CAS PubMed.
- For the synthesis of other spirocyclic natural products developed by our group, see: (a) W. P. Unsworth, K. A. Gallagher, M. I. Jean, J. P. Schmidt, L. J. Diorazio and R. J. K. Taylor, Org. Lett., 2013, 15, 262 CrossRef CAS PubMed; (b) C. L. Moody, V. Franckevičius, P. Drouhin, J. E. M. N. Klein and R. J. K. Taylor, Tetrahedron Lett., 2012, 53, 1897 CrossRef CAS.
- For synthetic applications of SnCl2·2H2O, see: A. N. Cayley, K. A. Gallagher, C. Ménard-Moyon, J. P. Schmidt, L. J. Diorazio and R. J. K. Taylor, Synthesis, 2008, 3846 CAS.
- For another synthesis of spirobacillene A, see: H. Yang, J. Feng and Y. Tang, Chem. Commun., 2013, 49, 6442 RSC.
- For recent reviews on dearomatising spirocyclisation methods, see: (a) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed; (b) S. P. Roché and J.-J. Y. Tréguier, Tetrahedron, 2015, 71, 3549 CrossRef; (c) M. J. James, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Chem. – Eur. J., 2016, 22, 2856 CrossRef CAS PubMed.
- For examples based on alkyne activation, see: (a) M. J. James, J. D. Cuthbertson, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Angew. Chem., Int. Ed., 2015, 54, 7640 CrossRef CAS PubMed; (b) M. J. James, R. E. Clubley, K. Y. Palate, T. J. Procter, A. C. Wyton, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Org. Lett., 2015, 17, 4372 CrossRef CAS PubMed; (c) A. K. Clarke, M. J. James, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Angew. Chem., Int. Ed., 2016, 55, 1379 Search PubMed; (d) J. T. R. Liddon, M. J. James, A. K. Clarke, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Chem. – Eur. J., 2016, 22, 8777 CrossRef CAS PubMed.
- For examples based on other activation modes, see: M. J. James, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Angew. Chem., Int. Ed., 2016, 55, 9671 CrossRef CAS PubMed.
- This refers to the direct conversion of an anisole-tethered ynone into a spirocyclic dienone.
- AgNO3·SiO2 is also a far more effecient catalyst than Cu(OTf)2 on this reaction system, with no reaction observed when ynone 11c was treated with 10 mol% Cu(OTf)2 at RT for 3d. The reactions were not performed in the dark and to the best of our knowledge, are insensitive to light.
- For dearomatisation using ortho-substituted phenols/napthols, see: (a) J. C. Green and T. R. R. Pettus, J. Am. Chem. Soc., 2011, 133, 1603 CrossRef CAS PubMed; (b) A. Rudolph, P. H. Bos, A. Meetsma, A. J. Minnaaed and B. L. Feringa, Angew. Chem., Int. Ed., 2011, 50, 5834 CrossRef CAS PubMed; (c) A. Seoane, N. Casanova, N. Quiñones, J. L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2014, 136, 7607 CrossRef CAS PubMed; (d) W.-T. Wu, L. Zhang and S.-L. You, Chem. Soc. Rev., 2016, 45, 1570 RSC; (e) J. Shao, L. Li, J. Zhang, J. Hu, J. Xue and Y. Li, RSC Adv., 2016, 6, 31363 RSC.
- (a) G. L. Hamilton, E. J. Kang, M. Mba and F. D. Toste, Science, 2007, 317, 496 CrossRef CAS PubMed; (b) M. Terada, F. Li and Y. Toda, Angew. Chem., Int. Ed., 2014, 53, 235 CrossRef CAS PubMed.
- P. B. Koswatta, J. Das, M. Yousufuddin and C. J. Lovely, Eur. J. Org. Chem., 2015, 2603 CrossRef CAS PubMed.
- R. Wu, J. S. Schumm, D. L. Pearson and J. M. Tour, J. Org. Chem., 1996, 61, 6906 CrossRef CAS PubMed.
- H. Nemoto, T. Kawano, N. Ueji, M. Bando, M. Kido, I. Suzuki and M. Shibuya, Org. Lett., 2000, 2, 1015 CrossRef CAS PubMed.
- A. H. Mermerian and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 5604 CrossRef CAS PubMed.
- S. F. Nielsen, A. Kharazmi and S. B. Christensen, Bioorg. Med. Chem., 1998, 6, 937 CrossRef CAS PubMed.
- H. Nemoto, T. Kawano, N. Ueji, M. Bando, M. Kido, I. Suzuki and M. Shibuya, Org. Lett., 2000, 2, 1015 CrossRef CAS PubMed.
- K. Miura, H. Saito, N. Fujisawa, D. Wang, H. Nishikori and A. Hosomi, Org. Lett., 2001, 3, 4055 CrossRef CAS PubMed.
- D. M. Rudzinski, C. B. Kelly and N. E. Leadbeater, Chem. Commun., 2012, 48, 9610 RSC.
- Y. Wang, K. Zhou, Q. Lan and X.-S. Wang, Org. Biomol. Chem., 2015, 13, 353 CAS.
- R. A. Haack and K. R. Beck, Tetrahedron Lett., 1989, 30, 1605 CrossRef CAS.
- F. T. Boyle, O. Hares, Z. S. Matusiak, W. Li and D. A. Whiting, J. Chem. Soc., Perkin Trans. 1, 1997, 2707 RSC.
- H. F. Sneddon, M. J. Gaunt and S. V. Ley, Org. Lett., 2003, 5, 1147 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimisation table and NMR spectra. See DOI: 10.1039/c6ob02426b |
| This journal is © The Royal Society of Chemistry 2017 |