
Total synthesis and confirmation of the revised structures of jiangrines A, C and D†
Zhijiang
Zhang
a and
Bo
Liu
*ab
aKey Laboratory of Green Chemistry & Technology of the Ministry of Education, College of Chemistry, Sichuan University, Chengdu, Sichuan 610064, China. E-mail: chembliu@scu.edu.cn; Fax: +86 28 85413712; Tel: +86 28 85413712
bState Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing 210009, China
First published on 22nd November 2016
Abstract
Our previous total synthesis of the proposed structures of jiangrines C and D shows that the characteristic data of synthetic samples did not match those of the natural ones, prompting us to revise their structures. Accordingly, we now accomplished the total synthesis of jiangrines A, C and D, which confirms our deduction that their molecular skeletons should compose of 2,3-disubstituted pyrrole instead of 2,5-disubstituted pyrrole. Our current synthesis features a silver-catalyzed [3 + 2] cycloaddition between a terminal alkyne and isocyanide, and was completed concisely in only seven linear steps.
Introduction
Jiangrines were isolated by Huang et al. from an actinobacterium, Jiangella gansuensis.1 Biological assays indicated that these pyrrole alkaloids exhibited an inhibitory effect on NO production. The Huang group proposed that jiangrines A, C and D embrace a 2,5-disubstituted pyrrole and a N-tethered phenol, as well as two chiral centers. The proposed structures of these natural alkaloids are depicted in Fig. 1.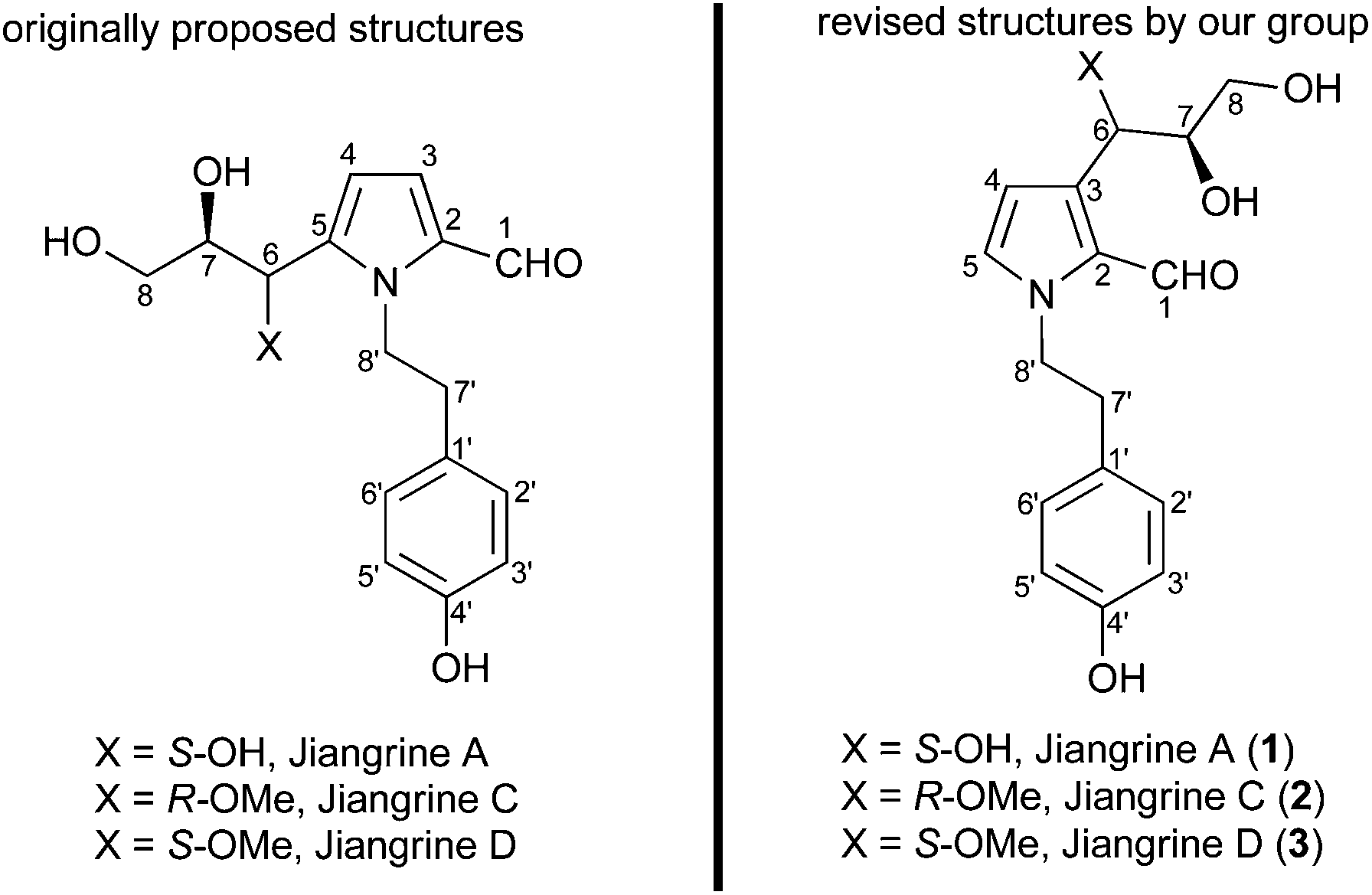 | ||
| Fig. 1 The proposed and revised structures of jiangrines A, C and D. | ||
From our interest in the bioactivity and structures of jiangrines, we initiated and accomplished the total synthesis of the proposed structures of jiangrines C and D.2 However, to our surprise, the characteristic data of our synthetic samples were not in accordance with those of natural jiangrines C and D. This discrepancy and reference to the characteristic data of pyrroles with other substituent patterns made us revise the structures of jiangrines to be 2,3-disubstituted pyrrole (Fig. 1). Herein we would like to present our endeavour to fulfil the total synthesis of the revised structures of jiangrines A, C and D, thus confirming their molecular structures.
Results and discussion
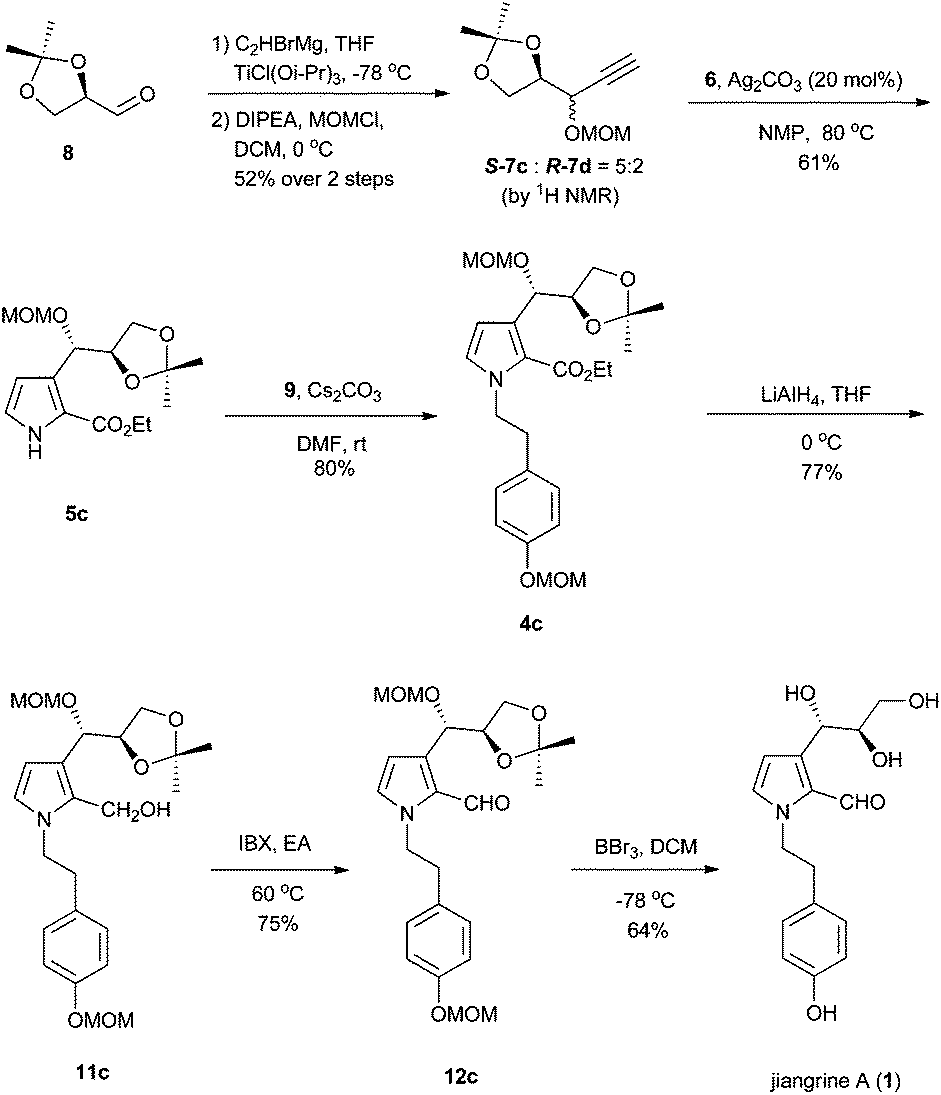
Retrosynthetically, jiangrines could be derived from compound 4 by partial reduction of the ester and global deprotection (Scheme 1). Compound 4 is accessible from the C–N bond formation between compound 5 and the known compound 9, whose preparation has been reported by us from commercially available compound 10.2 Compound 5 could be constructed through a silver-catalyzed [3 + 2] cycloaddition between compounds 6 and 7, a methodology developed by the Lei group.3 Then compound 7 can be readily prepared from (R)-(+)-2,2-dimethyl-1,3-dioxolane-4-carboxaldehyde (8), a widely used chiral building block in organic synthesis.4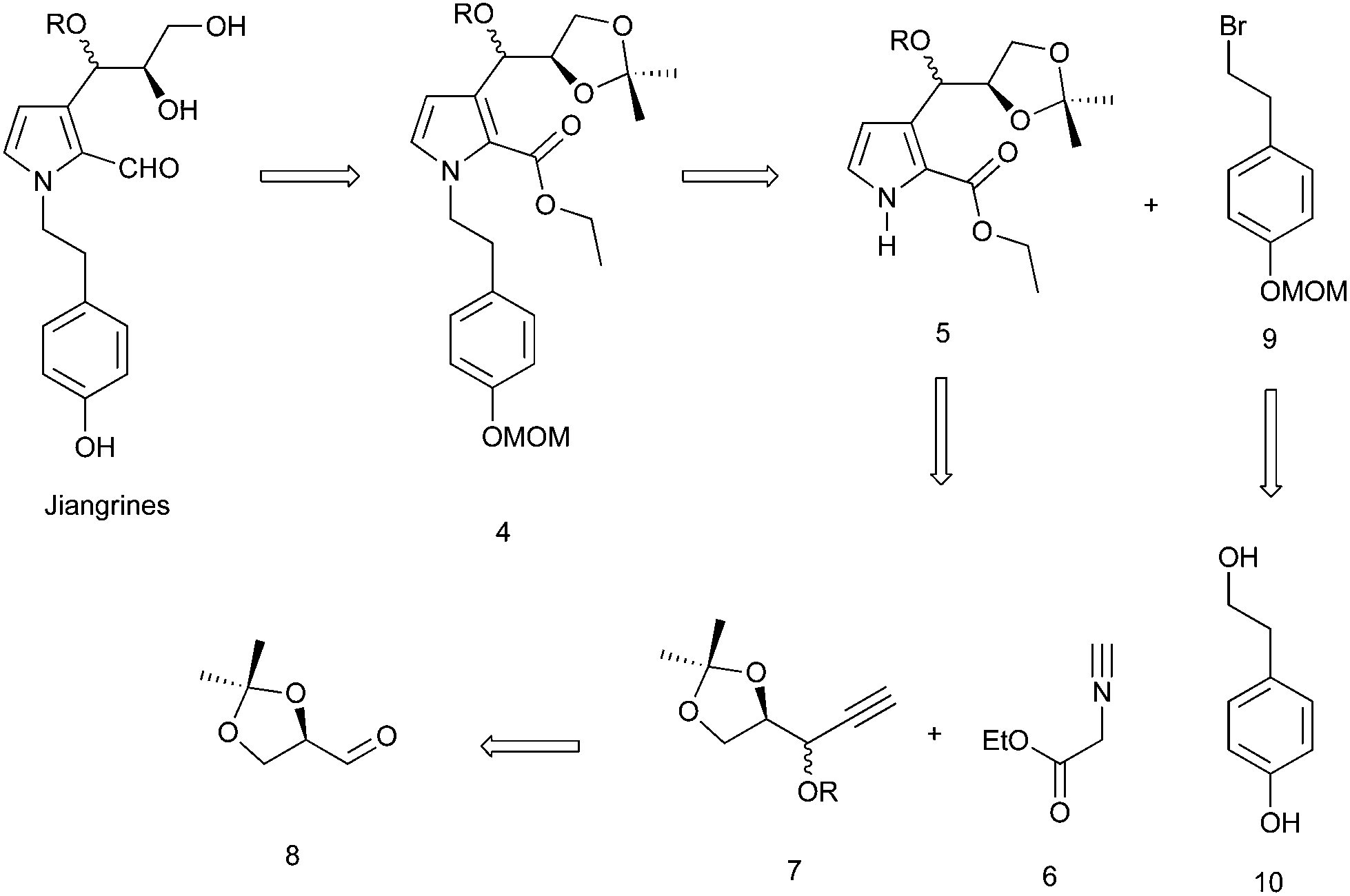 | ||
| Scheme 1 Retrosynthetic analysis. | ||
Accordingly, we started our synthesis by treating 8 with ethynyl magnesium bromide.5 The resultant alcohol was immediately transformed into its methyl ether in the presence of sodium hydride and methyl iodide to give compounds 7a and 7b as a mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) of inseparable diastereomers as determined by 1H NMR analysis (Scheme 2). Methods for the construction of pyrroles have been developed widely by synthetic chemists due to their prevalence in organic molecules.6 The Lei group developed a silver-catalyzed cycloaddition between the terminal alkyne with isocyanide to forge pyrrole.3 Inspired by this methodology, we coupled the diastereomeric mixture of 7a and 7b with ethyl 2-isocyanoacetate 6 in the presence of 10 mol% of silver carbonate to afford 5a and 5b in 40% and 40% yield respectively, which were separable at this stage. The relative stereochemistry of compound 5a was confirmed by X-ray crystallography. Subsequently, compound 5 was reacted with the known bromide 9 to deliver compound 4. Reduction of ester with diisobutyl aluminium hydride failed to afford the aldehyde 12 in a reasonable yield.7 Thus a two-step procedure involving lithium aluminum hydride reduction and IBX oxidation was applied to achieve 12.
:1) of inseparable diastereomers as determined by 1H NMR analysis (Scheme 2). Methods for the construction of pyrroles have been developed widely by synthetic chemists due to their prevalence in organic molecules.6 The Lei group developed a silver-catalyzed cycloaddition between the terminal alkyne with isocyanide to forge pyrrole.3 Inspired by this methodology, we coupled the diastereomeric mixture of 7a and 7b with ethyl 2-isocyanoacetate 6 in the presence of 10 mol% of silver carbonate to afford 5a and 5b in 40% and 40% yield respectively, which were separable at this stage. The relative stereochemistry of compound 5a was confirmed by X-ray crystallography. Subsequently, compound 5 was reacted with the known bromide 9 to deliver compound 4. Reduction of ester with diisobutyl aluminium hydride failed to afford the aldehyde 12 in a reasonable yield.7 Thus a two-step procedure involving lithium aluminum hydride reduction and IBX oxidation was applied to achieve 12.
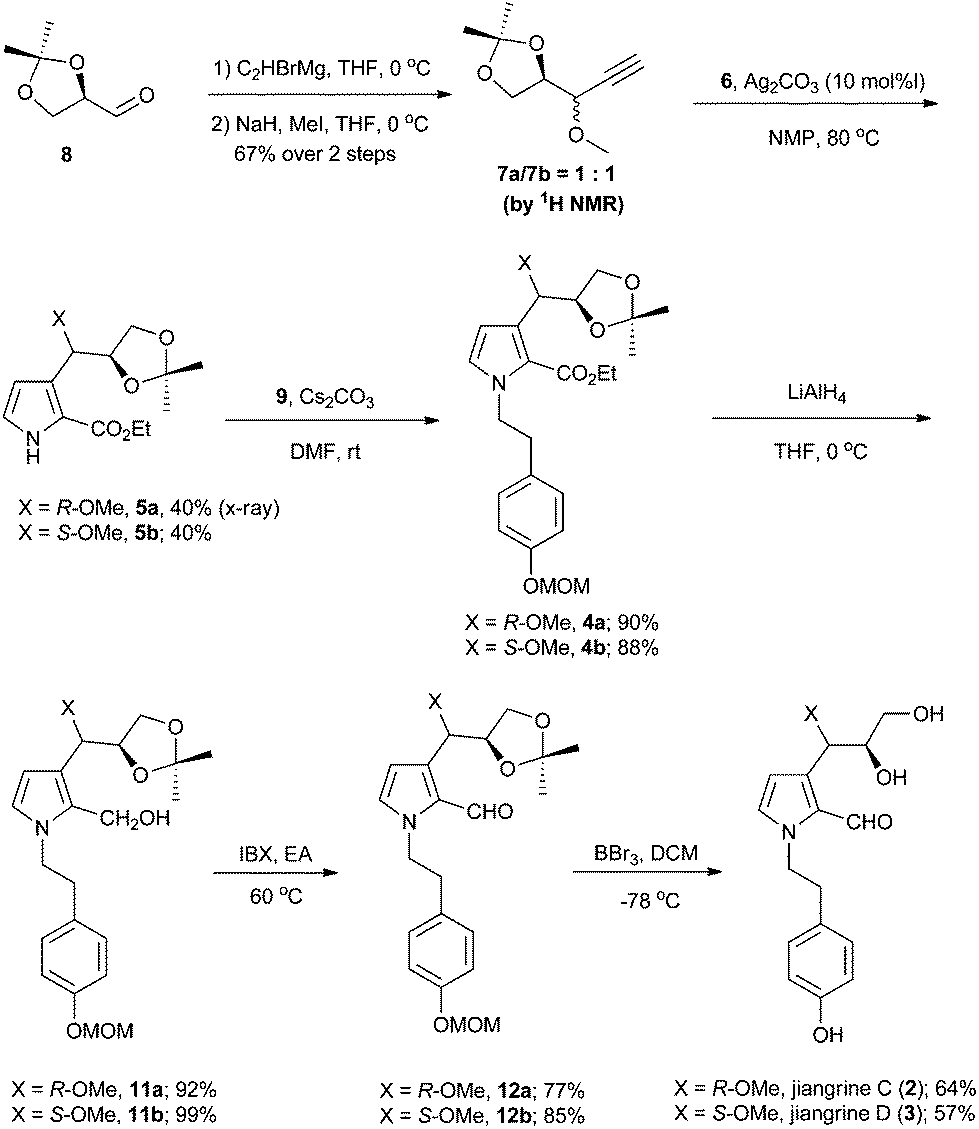 | ||
| Scheme 2 Synthesis of jiangrine C (2) and jiangrine D (3). | ||
Interestingly, when manganese dioxide was used as the oxidant of 11b, we obtained compound 12b′ and the desired 12b (Scheme 3). Application of PCC as the oxidant afforded 12b′ as the sole product, while using Dess–Martin periodinane produced an unidentified major compound other than 12b and 12b′. Finally, compound 12b was smoothly obtained in 77% yield with IBX.8 Although all Brønsted acids attempted could not result in 2 or 3, we eventually achieved target molecules in a moderate yield with boron tribromide. All the characteristic data of our synthetic samples are in good accordance with those of natural jiangrine C (2) and jiangrine D (3), which were obtained as an inseparable mixture from Jiangella gansuensis.1 Thus we successfully revised the structure of the natural alkaloids, jiangrine C (2) and jiangrine D (3), through total chemical synthesis.
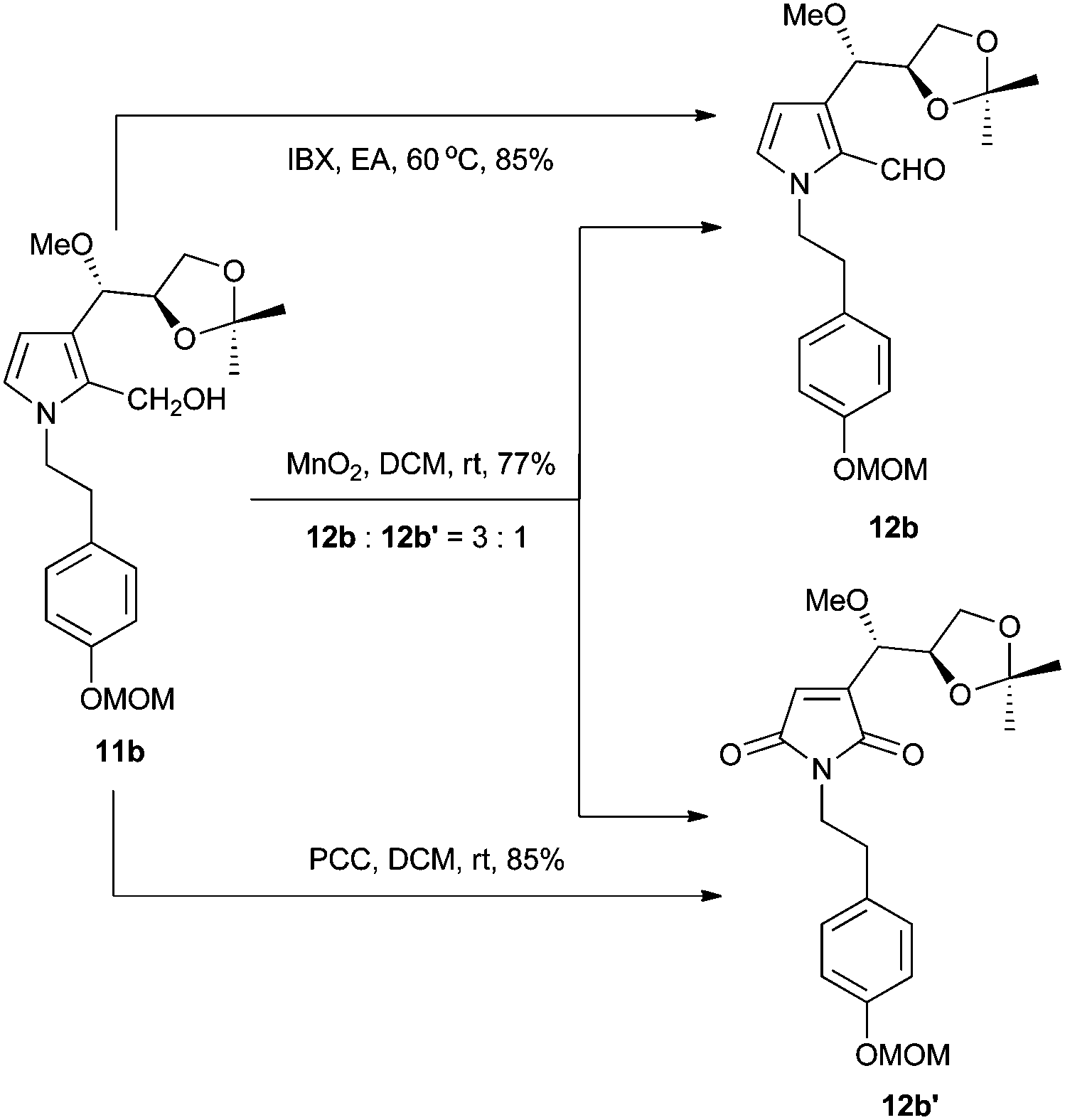 | ||
| Scheme 3 Oxidation of compound 11b. | ||
It is obvious that the direct demethylation of compound 3 would be the most concise way to achieve compound 1. However, we were frustrated to find that all the attempts failed. Thus, to synthesize 1, we had to start with 8 once again. After screening of various metal reagents, we prepared ethynyl titanium by exchanging the corresponding Grignard reagent with chlorotitanium triisopropoxide,9 and then reacted it with 8. The freshly generated hydroxyl was protected as the methoxy methyl ether with DIPEA and MOMCl to afford a 5:2 inseparable diastereomeric mixture with the desired isomer as the major product (Scheme 4). The subsequent transformations follow those in Scheme 2 until the final global deprotection of MOM ether with boron bromide delivered compound 1. All the characteristic data of our synthetic 1 are in good accordance with those reported for jiangrine A, which secured its correct molecular structure. A small discrepancy of optical rotation between the synthetic 1 and the natural sample, [α]22D = +10.8 (c 0.278, MeOH) vs. [α]20D = +18.8 (c 1.14, MeOH), might be ascribed to the presence of an impurity in the natural sample, which was detectable in the original 1H NMR spectrum.1
 | ||
| Scheme 4 Synthesis of jiangrine A (1). | ||
Conclusions
In conclusion, the total synthesis and structural revision of three natural pyrrole alkaloids, jiangrines A, C and D, have been achieved from the readily available starting material, (R)-(+)-2,2-dimethyl-1,3-dioxolane-4-carboxaldehyde in seven steps. Our synthesis features a silver-catalyzed [3 + 2] cycloaddition to build the 2,3-disubstituted pyrrole, which is the first application of this methodology in natural product synthesis.Experimental
Materials and methods
Unless otherwise noted, all reactions were carried out under an argon atmosphere with dry solvents under anhydrous conditions. Tetrahydrofuran (THF) was distilled from sodium–benzophenone; dichloromethane (DCM) was distilled from calcium hydride. N,N′-Dimethyl formamide (DMF) was distilled from calcium hydride. All chemicals were purchased commercially and used without further purification, unless otherwise stated. Flash chromatography was performed using silica gel (200–300 mesh). The reactions were monitored using thin layer chromatography (TLC). Visualization was achieved under a UV lamp (254 nm and 365 nm) and I2 and by developing the plates with p-anisaldehyde, phosphomolybdic acid (PMA) or cerium ammonium molybdate (CAM). 1H and 13C NMR were recorded on a Bruker DRX-400 MHz NMR spectrometer with TMS as the internal standard and were calibrated using a residual undeuterated solvent as an internal reference (CDCl3: 1H NMR = 7.26, 13C NMR = 77.16; CD3OD: 1H NMR = 3.31, 13C NMR = 49.00). The following abbreviations are used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. Coupling constants (J) are reported in hertz (Hz). Optical rotations were measured at the sodium D line with a 100 mm cell path length, and are reported as follows: [α]DT, concentration (g 100 mL−1), and solvent. High resolution mass spectra (HRMS) were recorded by using a Bruker FTMS-7 spectrometer. Infrared (IR) spectra were recorded on a NEXUS 670 FT-IR Fourier transform infrared spectrophotometer and were reported in wavenumbers (cm−1).Synthesis
To a suspension of NaH (60% in mineral oil, 0.870 g, 21.7 mmol) in THF (70 mL) was added a solution of the above mixture in THF (10 mL) dropwise at 0 °C. Then methyl iodide (2.2 mL, 36.2 mmol) was added dropwise. After stirring at room temperature for 2 h, the reaction was quenched with a saturated NH4Cl solution (20 mL). The aqueous layer was extracted with ethyl acetate (3 × 50 mL). The combined organic layer was dried over anhydrous sodium sulphate and evaporated. The residue was purified with a silica column using petroleum ether–ethyl acetate (20:1) as the eluent to give an inseparable diastereomeric mixture of compounds 7a and 7b (1.15 g, 67% over 2 steps) as a pale yellow oil. 1H NMR analysis showed that the ratio of 7a to 7b was 1:1.
To a suspension of the mixture 7 (7a and 7b, 0.620 g, 3.64 mmol) and Ag2CO3 (0.100 g, 0.364 mmol) in NMP (20 mL) was added compound 6 (0.618 g, 5.47 mmol) dropwise at room temperature. The reaction mixture was stirred at 80 °C, and monitored by TLC, after 8 h. The reaction was quenched with a saturated NH4Cl solution (20 mL). The aqueous layer was extracted with ethyl acetate (4 × 40 mL). The combined organic layer was washed with saturated aqueous NaCl (2 × 50 mL) and dried over anhydrous sodium sulphate and evaporated. The residue was purified with a silica column using petroleum ether–ethyl acetate (7:1) as the eluent to give 5a (0.413 g, 40% yield) and 5b (0.408 g, 40% yield).
IR (thin film): 3323, 2984, 2934, 2822, 1698, 1562, 1421, 1370, 1320, 1258, 1156, 1128, 849, 786 cm−1. 1H NMR (400 MHz, CDCl3) δ 9.22 (s, 1H), 6.90 (t, J = 2.8 Hz, 1H), 6.35 (t, J = 2.8 Hz, 1H), 5.01 (d, J = 5.2 Hz, 1H), 4.39–4.30 (m, 3H), 4.02–3.93 (m, 2H), 3.27 (s, 3H), 1.38–1.34 (m, 6H), 1.32 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 161.2, 128.8, 122.5, 120.7, 110.1, 109.5, 78.5, 76.1 (s), 66.2, 60.4, 57.0, 26.4, 25.5, 14.6. HRMS (ESI): m/z [M + Na]+ calcd for C14H21NNaO5: 306.1312, found: 306.1315.
:1) as the eluent to give 4a (0.3621 g, 90% yield) as a colorless oil. [α]26D = −48.6 (c 0.296, CH2Cl2). IR (thin film): 2983, 2932, 1697, 1512, 1419, 1233, 1209, 1153, 1103, 1005 cm−1. 1H NMR (400 MHz, CDCl3) δ 6.95–6.89 (m, 4H), 6.57 (d, J = 2.8 Hz, 1H), 6.10 (d, J = 2.8 Hz, 1H), 5.13 (s, 2H), 5.00 (d, J = 8.0 Hz, 1H), 4.54–4.37 (m, 2H), 4.36–4.24 (m, 3H), 3.65–3.56 (m, 2H), 3.46 (s, 3H), 3.23 (s, 3H), 2.94 (t, J = 7.2 Hz, 2H), 1.46 (s, 3H), 1.43–1.35 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 161.4, 156.1, 131.7, 130.2, 129.9, 128.9, 119.9, 116.4, 110.2, 107.6, 94.6, 79.7, 78.1, 66.2, 60.3, 56.3, 56.1, 51.6, 37.5, 26.9, 25.9, 14.5. HRMS (ESI): m/z [M + Na]+ calcd for C24H33NNaO7: 470.2149, found: 470.2153.
:H2O = 10:1 solution (10 mL) and 1 M NaOH solution (5 mL). The precipitate was filtered and evaporated. The residue was purified with a silica column using petroleum ether–ethyl acetate (6:1) as the eluent to give 11a (0.202 g, 92% yield) as a pale yellow oil. [α]25D = −26.6 (c 0.436, CH2Cl2). IR (thin film): 2933, 1613, 1512, 1370, 1233, 1153, 1079, 1002, 922, 827, 713 cm−1. 1H NMR (400 MHz, CDCl3) δ 6.91 (s, 4H), 6.57 (d, J = 2.8 Hz, 1H), 6.01 (d, J = 2.8 Hz, 1H), 5.13 (s, 2H), 4.43–4.35 (m, 3H), 4.23 (d, J = 7.2 Hz, 1H), 4.16–4.06 (m, 2H), 3.79 (dd, J = 8.4, 6.4 Hz, 1H), 3.61 (dd, J = 8.4, 6.8 Hz, 1H), 3.45 (s, 3H), 3.24 (s, 3H), 2.97 (td, J = 7.2, 1.6 Hz, 2H), 2.04 (t, J = 5.6 Hz, 1H), 1.36 (d, J = 9.6 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 156.2, 131.8, 130.4, 129.9, 121.4, 118.2, 116.5, 110.0, 107.2, 94.6, 79.0, 78.4, 66.5, 56.4, 56.1, 53.5, 48.6, 37.7, 26.6, 25.7. HRMS (ESI): m/z [M + Na]+ calcd for C22H31NNaO6: 428.2044, found: 428.2050.
:1) as the eluent to give 12a (0.081 g, 77% yield) as a pale yellow oil. [α]25D = −41.6 (c 0.416, CH2Cl2). IR (thin film): 2985, 2932, 2896, 2826, 1657, 1512, 1437, 1373, 1234, 1153, 1075, 1004, 824 cm−1. 1H NMR (400 MHz, CDCl3) δ 9.87 (s, 1H), 6.99–6.94 (m, 2H), 6.93–6.83 (m, 2H), 6.61 (d, J = 2.4 Hz, 1H), 6.08 (d, J = 2.4 Hz, 1H), 5.13 (s, 2H), 4.54 (d, J = 7.6 Hz, 1H), 4.51–4.41 (m, 2H), 4.41–4.32 (m, 1H), 3.71 (dd, J = 8.4, 6.4 Hz, 1H), 3.54 (dd, J = 8.4, 7.6 Hz, 1H), 3.46 (s, 3H), 3.31 (s, 3H), 2.95 (t, J = 7.2 Hz, 2H), 1.41 (s, 3H), 1.38 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 179.0, 156.2, 134.2, 131.4, 131.0, 130.0, 128.3, 116.5, 110.4, 108.9, 94.7, 79.1, 78.6, 66.4, 56.9, 56.1, 51.7, 37.0, 26.7, 25.7. HRMS (ESI): m/z [M + Na]+ calcd for C22H29NNaO6: 426.1887, found: 426.1890.
:1) as the eluent to give 2 (0.014 g, 64% yield) as a colorless viscous oil. [α]26D = −40.6 (c 0.918, CH2Cl2). IR (thin film): 3426, 2978, 1643, 1448, 1212, 1116, 1048, 973 cm−1. 1H NMR (400 MHz, CD3OD) δ 9.76 (d, J = 0.6 Hz, 1H), 6.89 (d, J = 8.4 Hz, 2H), 6.82 (d, J = 2.4 Hz, 1H), 6.64 (d, J = 8.8 Hz, 2H), 6.11 (d, J = 2.4 Hz, 1H), 4.60 (d, J = 6.8 Hz, 1H), 4.47 (t, J = 7.0 Hz, 2H), 3.72 (td, J = 6.0, 4.0 Hz, 1H), 3.52 (dd, J = 11.2, 4.0 Hz, 1H), 3.36–3.32 (m, 1H), 3.27 (s, 3H), 2.90 (t, J = 7.2 Hz, 2H). 13C NMR (100 MHz, CD3OD) δ 180.9, 157.1, 137.6, 132.9, 130.9, 130.2, 129.4, 116.2, 110.0, 79.0, 76.9, 63.9, 57.1, 52.4, 37.9. HRMS (ESI): m/z [M + Na]+ calcd for C17H21NNaO5: 342.1312, found: 342.1317.
To a solution of the above mixture in DCM (50 mL) were added DIPEA (8.26 mL, 50.0 mmol) and MOMCl (3.04 mL, 40.0 mmol) dropwise at 0 °C. The mixture was stirred at room temperature for 23 h; the reaction was quenched with a saturated NaHCO3 solution (20 mL). The aqueous layer was extracted with ethyl acetate (4 × 50 mL). The combined organic layer was dried over anhydrous sodium sulphate and evaporated. The residue was purified with a silica column using petroleum ether–ethyl acetate (10:1) as the eluent to give an inseparable diastereomeric mixture of compounds 7c and 7d (0.520 g, 52% over 2 steps) as a pale yellow oil. An aliquot of this sample was collected and detected by 1H NMR, showing that the ratio of 7c to 7d was 5:2.
To a suspension of the mixture 7 (7c and 7d, 0.172 g, 0.860 mmol) and Ag2CO3 (0.047 g, 0.172 mmol) in NMP (6 mL) was added compound 6 (0.1459 g, 1.290 mmol) dropwise at room temperature. The reaction mixture was stirred at 80 °C, and monitored by TLC, after 4 h. The reaction was quenched with a saturated NH4Cl solution (20 mL). The aqueous layer was extracted with ethyl acetate (4 × 50 mL). The combined organic layer was washed with saturated aqueous NaCl (2 × 50 mL) and dried over anhydrous sodium sulphate and evaporated. The residue was purified with a silica column using petroleum ether–ethyl acetate (6:1) as the eluent to give 5c (0.163 g, 61%) as a pale yellow oil. Its absolute configuration is confirmed by comparing its similar chemical shifts and coupling constants of 1H NMR with those of compound 5b. [α]27D = +124.7 (c 0.190, CHCl3). IR (thin film): 3316, 2985, 2923, 2879, 1699, 1562, 1419, 1366, 1321, 1261, 1128, 1033, 910, 846, 786 cm−1. 1H NMR (400 MHz, CDCl3) δ 9.09 (s, 1H), 6.89 (t, J = 2.4 Hz, 1H), 6.38 (t, J = 2.4 Hz, 1H), 5.44 (d, J = 4.8 Hz, 1H), 4.61 (dd, J = 10.0, 6.4 Hz, 2H), 4.40 (dd, J = 11.2, 6.4 Hz, 1H), 4.33 (q, J = 7.2 Hz, 2H), 3.97 (d, J = 6.8 Hz, 2H), 3.37 (s, 3H), 1.44–1.29 (m, 9H). 13C NMR (100 MHz, CDCl3) δ 161.1, 128.5, 122.2, 120.4, 110.5, 109.5, 94.8, 78.5, 70.9, 65.9, 60.6, 55.8, 26.4, 25.6, 14.6. HRMS (ESI): m/z [M + Na]+ calcd for C15H23NNaO6: 336.1418, found: 336.1416.
Acknowledgements
We acknowledge financial support from the NSFC (21290180, 21322205, 21321061 and 21672153). The support from the Open Fund of State Key Laboratory of Natural Medicines in China Pharmaceutical University is acknowledged (no. SKLNMKF201601).Notes and references
- L. Han, C. Cao, Y. Jiang, P. Guan, J. Liu, L. Li, L. Xu and X. Huang, J. Nat. Prod., 2014, 77, 2605 CrossRef CAS PubMed.
- Z. Zhang and B. Liu, Sci. China: Chem., 2016, 59, 1205 CrossRef CAS.
- M. Gao, C. He, H. Chen, R. Bai, B. Cheng and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 6958 CrossRef CAS PubMed.
- C. R. Schmid and J. D. Bryant, Org. Synth., 1993, 72, 6 Search PubMed.
- C. Bonini, L. Chiummiento, M. Pullez, G. Solladié and F. Colobert, J. Org. Chem., 2004, 69, 5015 CrossRef CAS PubMed.
- (a) Y. Nishibayashi, M. Yoshikawa, Y. Inada, M. D. Milton, M. Hidai and S. Uemura, Angew. Chem., Int. Ed., 2003, 42, 2681 CrossRef CAS PubMed; (b) O. V. Larionov and A. de Meijere, Angew. Chem., Int. Ed., 2005, 44, 5664 CrossRef CAS PubMed; (c) S. Kamijo, C. Kanazawa and Y. Yamamoto, J. Am. Chem. Soc., 2005, 127, 9260 CrossRef CAS PubMed; (d) R. Martín, M. R. Rivero and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 7079 CrossRef PubMed; (e) E. M. Beck, N. P. Grimster, R. Hatley and M. J. Gaunt, J. Am. Chem. Soc., 2006, 128, 2528 CrossRef CAS PubMed; (f) D. J. St. Cyr and B. A. Arndtsen, J. Am. Chem. Soc., 2007, 129, 12366 CrossRef PubMed; (g) S. J. Hwang, S. H. Cho and S. Chang, J. Am. Chem. Soc., 2008, 130, 16158 CrossRef CAS PubMed; (h) X. Fu, J. Chen, G. Li and Y. Liu, Angew. Chem., Int. Ed., 2009, 48, 5500 CrossRef CAS PubMed; (i) M. Fujiwara, M. Kawatsura, S. Hayase, M. Nanjo and T. Itoha, Adv. Synth. Catal., 2009, 351, 123 CrossRef CAS; (j) Y. D. Lu and B. A. Arndtsen, Angew. Chem., Int. Ed., 2008, 47, 5430 CrossRef CAS PubMed; (k) X. Tang, L. Huang, C. Qi, W. Wu and H. Jiang, Chem. Commun., 2013, 49, 9597 RSC.
- I. A. Sayyed, K. Alex, A. Tillack, N. Schwarz, A. Spannenberg, D. Michalik and M. Beller, Tetrahedron, 2008, 64, 4590 CrossRef CAS.
- H. Deng, W. Cao, Z. Zhang and B. Liu, Org. Biomol. Chem., 2016, 14, 6225 CAS.
- G. Casiraghi, M. Cornia, G. Rassu, C. Del Sante and P. Spanu, Tetrahedron, 1992, 48, 5619 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1510892. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob02379g |
| This journal is © The Royal Society of Chemistry 2017 |