
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The first total synthesis of the marine acetylenic alcohol, lembehyne B – a selective inducer of early apoptosis in leukemia cancer cells†
Lilya U.
Dzhemileva
*ab,
Vladimir A.
D'yakonov
*a,
Aleksey A.
Makarov
a,
Evgeny N.
Andreev
a,
Milyausha M.
Yunusbaeva
a and
Usein M.
Dzhemilev
a
aInstitute of Petrochemistry and Catalysis, Russian Academy of Sciences, 141 Prospekt Oktyabrya, 450075, Ufa, Russian Federation. E-mail: DyakonovVA@gmail.com; Fax: +73472842750; Tel: +73472842750
bDepartment of Immunology and Human Reproductive Health Bashkir State Medical University, Lenin Street, 3, 450003, Ufa, Russian Federation. E-mail: Dzhemilev@mail.ru
First published on 8th December 2016
Abstract
The communication reports a new stereoselective method for the synthesis of a natural acetylenic alcohol, lembehyne B. The key stage of the process uses new cross-cyclomagnesiation reaction of aliphatic and oxygenated 1,2-dienes with Grignard reagents in the presence of a catalytic amount of Cp2TiCl2. A study of the cytotoxic properties of lembehyne B on tumor cell lines using flow cytometry demonstrated that this is a selective inducer of early apoptosis of the Jurkat, HL-60 and K562 cell cultures and hypodiploid (sub-G1) sub-population inducer in cell cycle studies for all cell lines used.
Introduction
The Global Ocean occupies more than 70% of the Earth's surface; therefore, flora and fauna inhabiting the Ocean are considered as important sources of biologically active compounds that could serve as the base for the development of modern pharmaceutical agents for treating cancer, neurodegenerative diseases, viral and bacterial infections, and other dangerous human diseases.1 During the last decade alone, about ten thousand new compounds were isolated from marine organisms; some of these compounds are already under Phase I or II clinical trials.2Recently,3 Kobayashi and co-workers isolated, from the Indonesian marine sponge Haliclona sp., micro quantities (10–15 mg kg−1) of lembehynes A–C – acetylenic alcohols (Fig. 1), efficient small-molecule analogues of proteins belonging to the family of neurotrophins, which perform an important function of maintaining the vitality of neurons.4 Hence, lembehynes are considered as potential pharmaceutical drugs for treating neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, and Huntington's chorea.
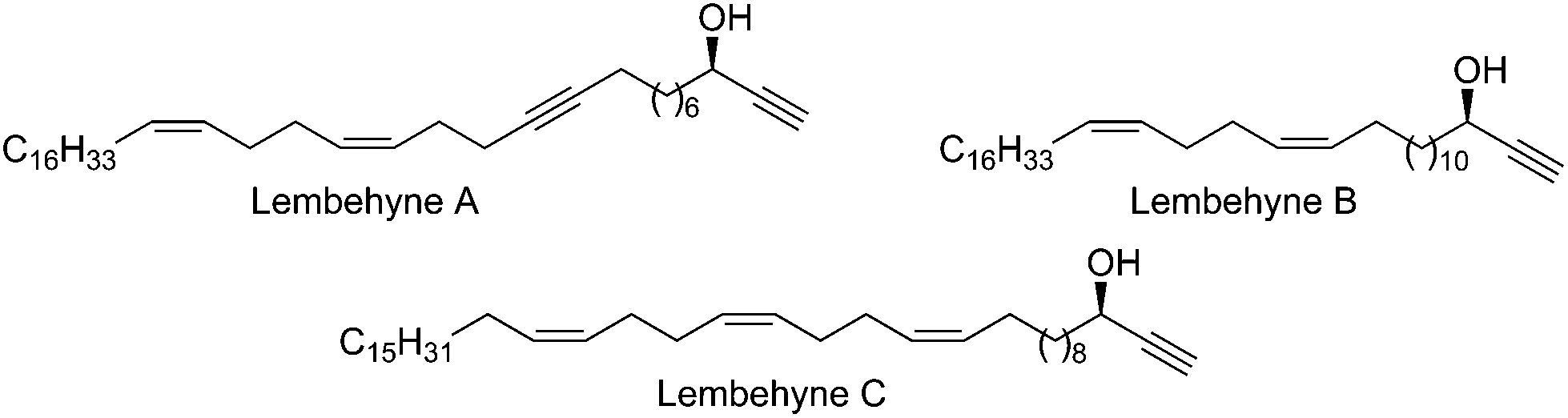 | ||
| Fig. 1 Structures of lembehynes A–C. | ||
The poor accessibility and the lack of efficient methods for the synthesis of natural lembehynes hamper detailed investigation of the properties and preclude taking full advantage of the biomedical potential of these compounds.5
Analysis of the lembehyne structure demonstrated that stereoselective formation of the 1Z,5Z-diene moiety present in the lembehyne molecule is the most complicated and multistep part.5 Indeed, in the 11-step total synthesis of lembehyne A, six steps were spent for the preparation of the key synthon, 4Z,8Z-pentacosa-4,8-dien-1-ol.5
Proceeding from the published data, the most popular approaches to the formation of the 1Z,5Z-diene moiety are based on the Wittig reaction, alkene metathesis, alkylation of alkynes, and stereoselective reduction of 1,5-diynes.6
Previously, we developed Ti-catalyzed homo- and cross-cyclomagnesiation reactions of 1,2-dienes with Grignard reagents (Scheme 1), which were successfully used in the strictly stereoselective syntheses of 1Z,5Z-dienes of a specified structure, giant macrocarbocycles, and natural 5Z,9Z-dienoic acids exhibiting antitumor activities.7
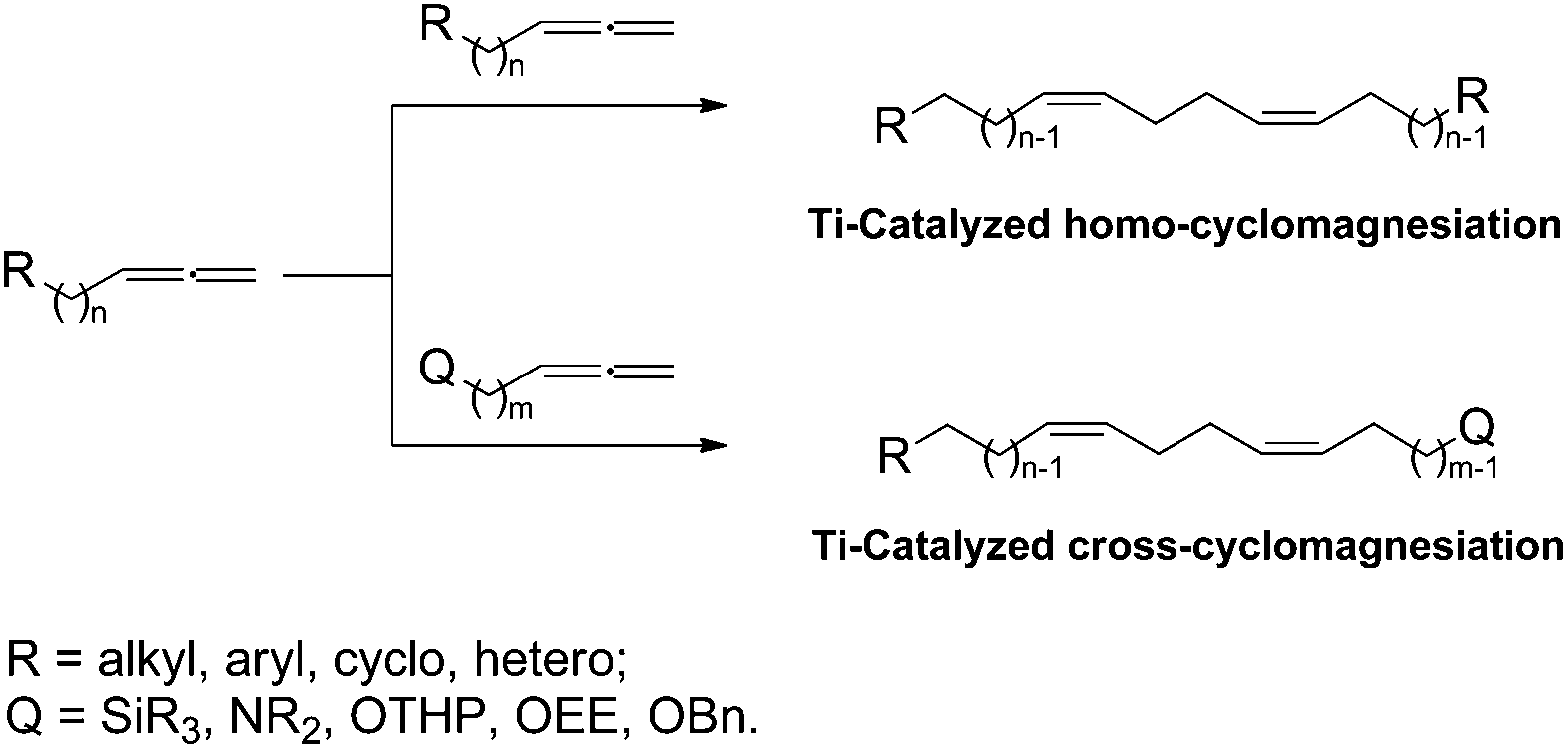 | ||
| Scheme 1 Ti-Catalyzed homo- and cross-cyclomagnesiation. | ||
In view of the foregoing, in this paper we report the development of a preparative stereoselective synthesis of lembehyne B using cross-cyclomagnesiation of aliphatic and oxygenated 1,2-dienes in the key stage of the synthesis, product accumulation and in vitro testing for the antitumor activity on Jurkat, HL-60, and K562 cell lines using flow cytometry.
Results and discussion
By retrosynthetic analysis of the lembehyne B structure, we identified the key monomer: (13Z,17Z)-tetraconta-13,17-dienal, which is accessible through cross-magnesiation of 1,2-nonadecadiene (1) with the tetrahydropyran ether of 13,14-pentadecadienol (2a) or 2-tetradeca-12,13-dien-1-yl-1,3-dioxolane (2b) (Scheme 2). The final steps are intended for the formation of the terminal propargyl moiety to obtain racemic lembehyne B and hydroxy group oxidation followed by stereoselective reduction (Scheme 2).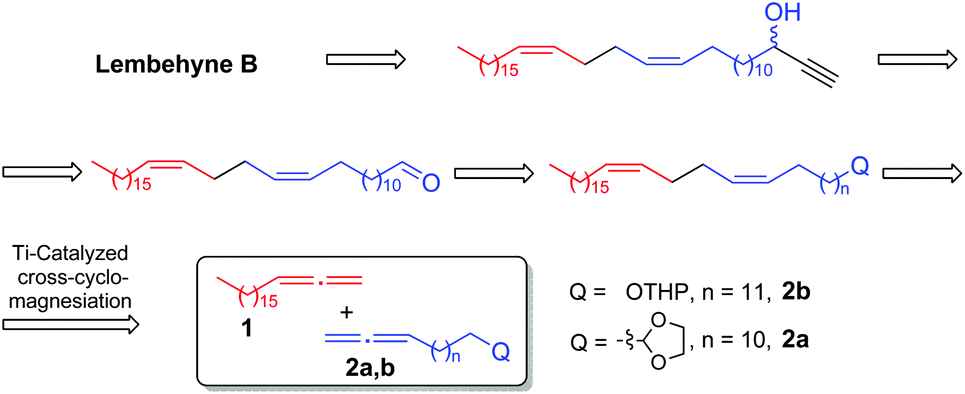 | ||
| Scheme 2 Retrosynthetic analysis of lembehyne B. | ||
In accordance with the developed strategy, 1,2-nonadecadiene (1) and 13,14-pentadecadienol tetrahydropyran ether (2a) were first subjected to cross-cyclomagnesiation on treatment with EtMgBr in the presence of Mg metal and Cp2TiCl2 catalyst (10 mol%). The reaction proceeded via magnesacyclopentane 3a, which was hydrolyzed to give (13Z,17Z)-tetraconta-13,17-dienol tetrahydropyran ether (4) in 88% yield (Scheme 3). Successive tetrahydropyran deprotection8 and Dess–Martin periodinane oxidation of unsaturated alcohol 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 yielded the target (13Z,17Z)-tetraconta-13,17-dienal (6) in ∼64% yield over two steps.
9 yielded the target (13Z,17Z)-tetraconta-13,17-dienal (6) in ∼64% yield over two steps.
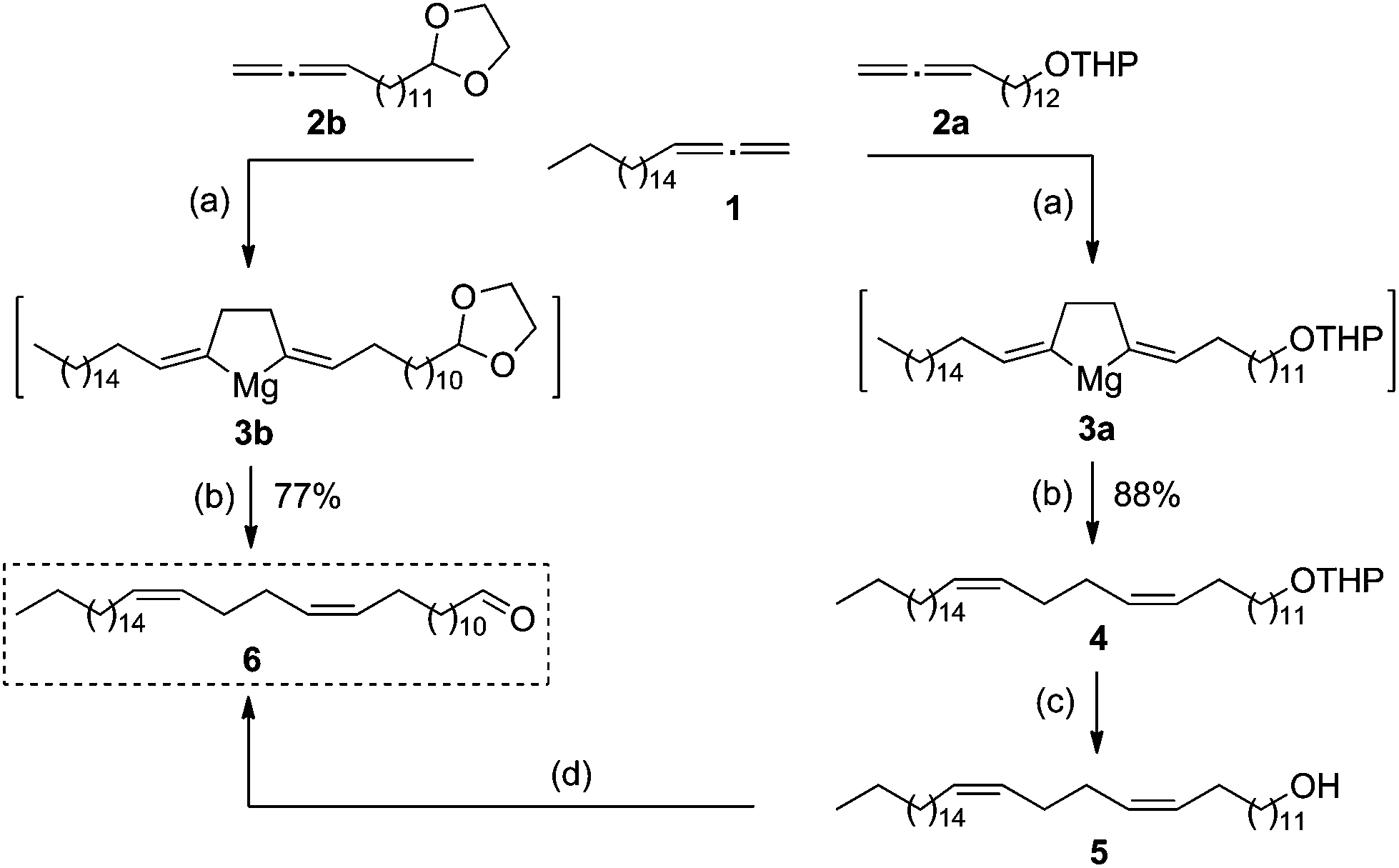 | ||
| Scheme 3 Ti-Catalyzed cross-cyclomagnesiation of aliphatic and oxygenated 1,2-dienes in the synthesis of (13Z,17Z)-tetraconta-13,17-dienal (6). Reagents and condition: (a) EtMgBr, Mg, Cp2TiCl2, Et2O, rt; (b) H+; (c) p-TsOH, MeOH/CHCl3, 77%; (d) Dess–Martin periodinane, THP, rt, 83%. | ||
In turn, cross-magnesiation of 1,2-nonadecadiene (1) and 2-tetradeca-12,13-dien-1-yl-1,3-dioxolane (2b) with EtMgBr in the presence of Mg metal (halide ion acceptor) and a catalytic amount of Cp2TiCl2 (10 mol%) (1:2b:EtMgBr:Mg:[Ti] = 12:10:30:20:0.1, Et2O, 20–22 °C, 7 h) furnishes magnesacyclopentane 3b, which is acid-hydrolyzed to afford (13Z,17Z)-tetraconta-13,17-dienal (6) in ∼77% yield in one preparative step (Scheme 3).
Thus, the yield of (13Z,17Z)-tetraconta-13,17-dienal was 64% according to the former route and 77% according to the latter one.
At the final step, aldehyde 6 was made to react with lithium trimethylsilylacetylenide,10 prepared beforehand by the reaction of equimolar amounts of trimethylsilylacetylene and n-BuLi in THF, the reaction affording silane 7 in 90% yield over a period of 3 days at room temperature. The trimethylsilyl deprotection on treatment with tetrabutylammonium fluoride (TBAF) in THF over a period of 4 h furnished racemic lembehyne B (8) in nearly quantitative yield (Scheme 4).
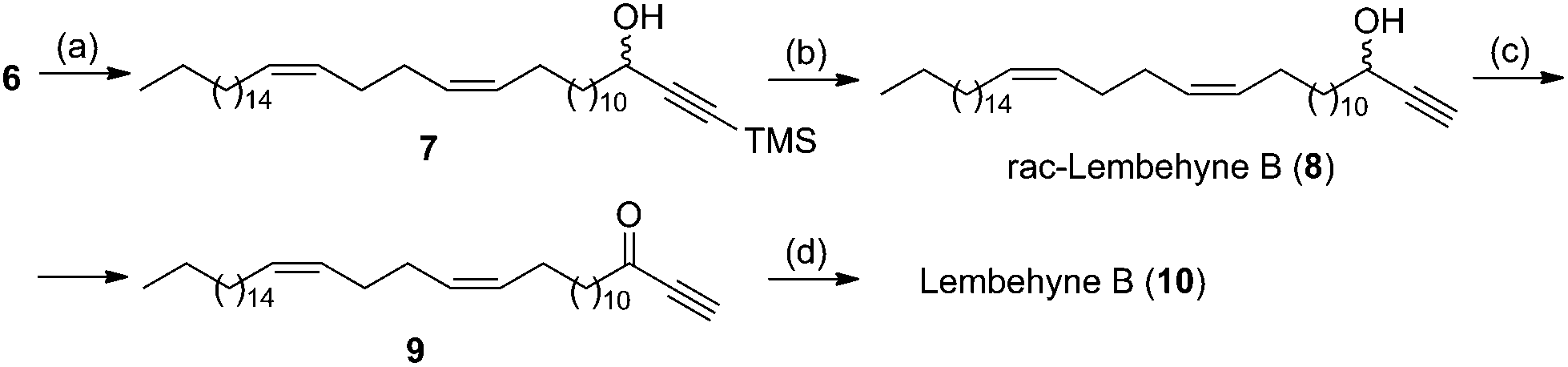 | ||
| Scheme 4 Synthesis of lembehyne B. Reagents and condition: (a) lithium trimethylsilylacetylenide, THF, rt, 90%; (b) TBAF, THF, rt, 99%; (c) Dess–Martin periodinane, THF, rt, 86%; (d) Alpine-borane, THF, rt, 84%. | ||
Using racemic lembehyne B, we synthesized its natural stereomer with the 3R-configuration of the hydroxy group at the C-3 carbon atom. First, Dess–Martin periodinane oxidation of alkynol 8 in CH2Cl2 at room temperature gave (15Z,19Z)-hexaconta-15,19-dien-1-yl-3-one (9) in 86% yield over a period of 1 h (Scheme 4). The stereoselective reduction of ketone 9 was performed using the B-3-pinanyl-9-borabicyclo[3.3.1]nonane (Alpine-borane) reagent,11 prepared beforehand from (+)-α-pinene (98% ee) and 9-borabicyclo[3.3.1]nonane. This gave lembehyne B (10) with enantiomeric excess of 95% in 84% yield.
During the last 10–15 years, several hundred natural alkynes, exhibiting high antitumor activities, were isolated from macro- and microalgae, freshwater- and marine cyanobacteria, and other marine organisms.2 Meanwhile, no data on the antitumor activity of natural lembehynes are available from the literature. Therefore, we performed the first in vitro tests of lembehyne B for the antitumor activity on leukemia cells (Jurkat, HL-60 and K562). The tests included determination of IC50, study of cell viability and effect on the cell cycle by means of flow cytofluorometry.
The quantitative and qualitative analyses of cell viability, cell cycle, and apoptosis-inducing activity of lembehyne B were performed using the Guava Nexin Reagent and Guava Cell Cycle (Millipore).
The in vitro cytotoxic activity of lembehyne B against Jurkat, HL-60, and K562 human leukemia cells was studied using Guava ViaCount (Millipore). Lembehyne B in concentrations of 0.5 to 4 μM exhibited a clear-cut cytotoxic effect against all types of cancer cells we studied. However, the highest IC50 value was found for K562 cells (3 μM), whereas for Jurkat and HL-60, the IC50 values were 2 μM and 2.2 μM, respectively.
The lembehyne B-induced apoptosis in Jurkat, HL-60, and K562 cell cultures was estimated by detection of phosphatidylserine externalization on the plasmatic membrane after treatment of cell cultures with the test compound. It is noteworthy that the effect of lembehyne on the induction of apoptosis is more pronounced in Jurkat cells than in other cells, which is consistent with high cytotoxicity of the compound against this cell line. As can be seen from Fig. 2, exposure of Jurkat tumor cell culture to lembehyne B results in a considerable dose-dependent increase in the amount of apoptotic cells occurring at early stage of apoptosis. The highest percentage of early apoptosis (72.6%) is observed at 2 μM concentration of the test compounds. In particular, as shown in Fig. 2, for Jurkat cells, both early and late stages of apoptosis are enhanced as compared with HL60 (P ≤ 0.0001) or K562 (P ≤ 0.0001).
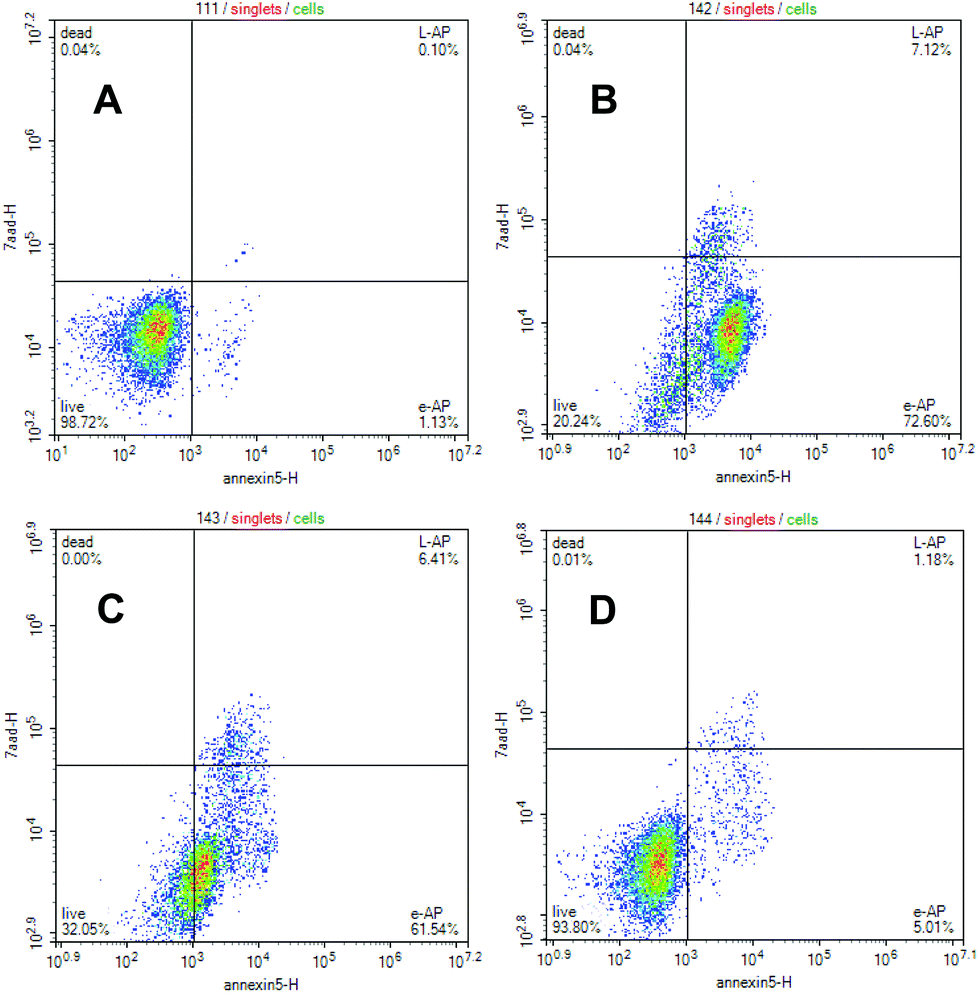 | ||
| Fig. 2 Jurkat cells treated with different concentrations of lembehyne B (10) were double-stained with annexine V/PI and analyzed by flow cytometry. (A) Control; (B) 10, 2 μM; (C) 10, 1 μM; (D) 10, 0.5 μM. | ||
According to cell cycle data determined by Guava Cell Cycle Reagent (Fig. 3), lembehyne B is a potent inducer of hypodiploid cell population (sub-G1 phase) in all three cell lines after the appropriate treatment with the test compounds. No significant differences were identified between cell-cycle distribution of cell subpopulations for lembehyne B-treated Jurkat, HL-60, and K562 cells.
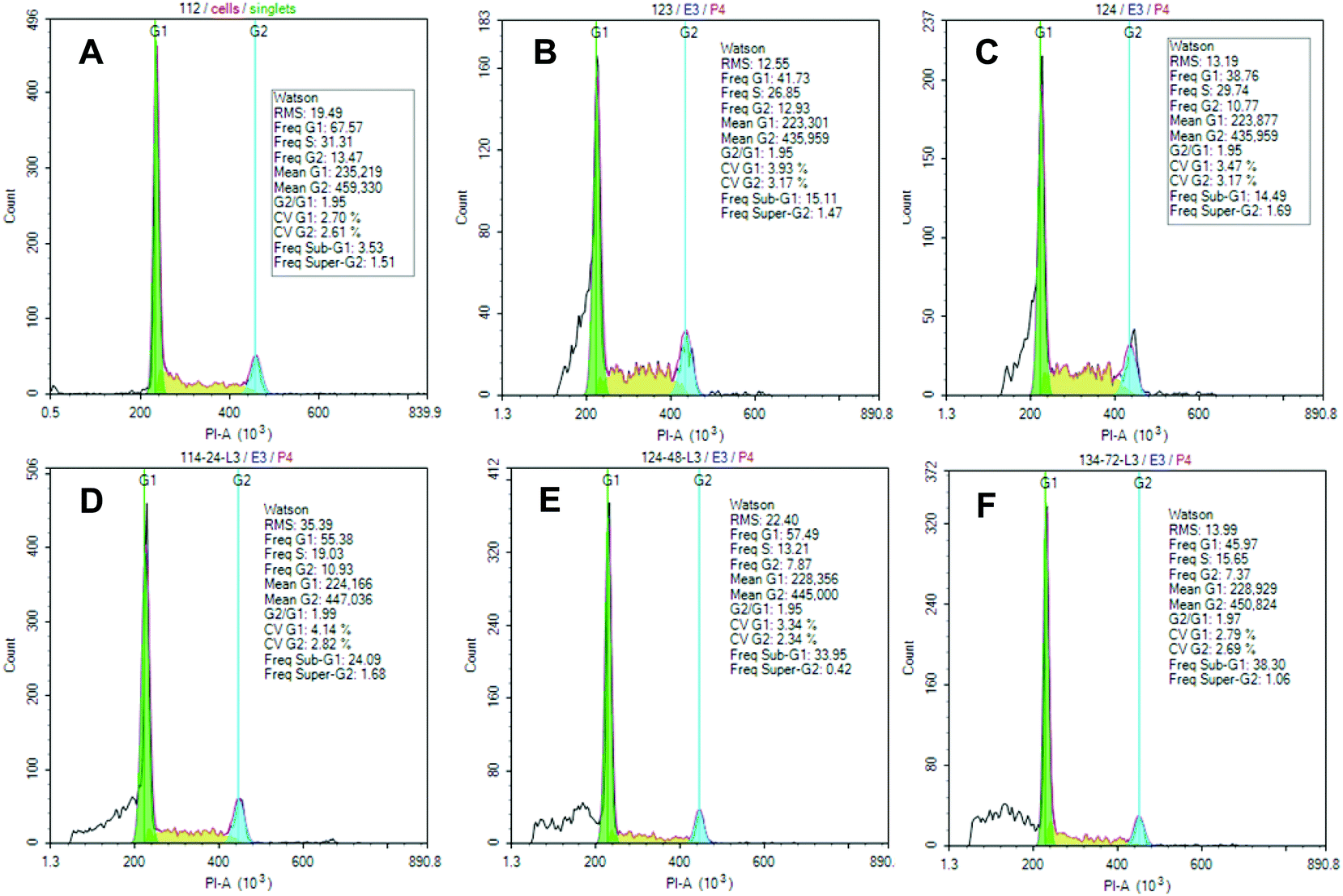 | ||
| Fig. 3 Cell cycle phases for Jurkat cell line. Upper row: (A) control, (B) 10, 2 μL, (C) 10, 1 μL. Lower row: (10, 2 μL): (D) 24 h, (E) 48 h, (F) 72 h. | ||
Conclusions
Thus, an original stereoselective method for the synthesis of lembehyne B was developed for the first time. The key stage of the synthesis was based on the new reaction that we developed, namely, Ti-catalyzed cross-cyclomagnesiation of oxygenated aliphatic 1,2-dienes with Grignard reagents. We believe that this method bears huge synthetic potential for the preparation of stereochemically pure lembehynes and their analogues by varying the structure of the starting 1,2-dienes. By means of flow cytometry, it was shown for the first time that lembehyne B is a selective early apoptosis inducer for the Jurkat, HL-60, and K562 cell cultures. Currently, active research along this line is in progress in order to implement stereoselective methods for the synthesis of the whole range of natural lembehynes and their analogues for performing, in particular, full-scale pharmacological investigations of the biological activity and structure–activity relationships.Experimental section
General methods
1H and 13C NMR spectra were obtained using a Bruker Ascend 500 spectrometer in CDCl3 operating at 500 MHz for 1H and 125 MHz for 13C and Bruker AVANCE 400 spectrometer in CDCl3 operating at 400 MHz for 1H and 100 MHz for 13C. Mass spectra were obtained with a MALDI TOF/TOF spectrometer in a α-cyano-4-hydroxycinnamic acid matrix. Elemental analyses were measured on a 1106 Carlo Erba apparatus. Individuality and purity of the synthesized compounds were controlled using TLC on Sorbfil plates; anisic aldehyde in acetic acid was used as a developer. Column chromatography was carried out on Acrus silica gel (0.060–0.200 mm). Flow cytometry studies were obtained using a NovoCyte™ 2000 FlowCytometry System (ACEA). All solvents were dried (1,4-dioxane, tetrahydrofuran, diethyl ether over Na) and freshly distilled before use. All reactions were carried out under a dry argon atmosphere. Aliphatic and oxygenated 1,2-dienes have been synthesized according procedures.7g,10,12,13Methods of synthesis and characterization data of all compounds
Cross-cyclomagnesiation of nonadeca-1,2-diene (1) and 2-(pentadeca-13,14-dien-1-yloxy)tetrahydro-2H-pyran (2a) with EtMgBr in the presence of Mg metal and Cp2TiCl2 catalyst
Diethyl ether (30 mL), nonadeca-1,2-diene (1) (1.27 g, 4.8 mmol), 2-(pentadeca-13,14-dien-1-yloxy)tetrahydro-2H-pyran (2a) (1.23 g, 4.0 mmol), EtMgBr (16.0 mmol) (as 1.5 M solution in Et2O), Mg powder (0.29 g, 12.0 mmol) and Cp2TiCl2 (0.1 g, 0.4 mmol) were placed in a glass reactor with stirring under argon (∼0 °C). The reaction mixture was warmed-up to room temperature (20–22 °C) and stirred for 6 h. The reaction mixture was treated with a 5% solution of NH4Cl in H2O (20 mL) and extracted with diethyl ether (2 × 100 mL). The combined organic phases were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Silica gel column chromatography (hexane/EtOAc (35/1)) of the residue gave compound 5 (1.98 g, 88%) as a pale yellow oily liquid.THP-deprotection of ether (4) was carried out with p-TsOH in CH2Cl2/MeOH using known method8
The oxidation of the alcohol (5) with Dess–Martin periodinane was carried out according known procedure9
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–C
CH–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2), 2.42–2.45 (2H, m, OCH–C2), 5.39–5.40 (4H, m, C), 9.78–9.79 (1H, m, OC). 13C NMR (100.62 MHz, CDCl3) δ: 202.9 (CH), 130.4 (CH), 129.1 (CH), 43.9 (CH2), 31.9 (CH2), 29.2–29.7 (signals of 22C, CH2), 27.4 (2C, CH2), 27.3 (2C, CH2), 22.7 (CH2), 22.1 (CH2), 14.1 (CH3).
2), 2.42–2.45 (2H, m, OCH–C2), 5.39–5.40 (4H, m, C), 9.78–9.79 (1H, m, OC). 13C NMR (100.62 MHz, CDCl3) δ: 202.9 (CH), 130.4 (CH), 129.1 (CH), 43.9 (CH2), 31.9 (CH2), 29.2–29.7 (signals of 22C, CH2), 27.4 (2C, CH2), 27.3 (2C, CH2), 22.7 (CH2), 22.1 (CH2), 14.1 (CH3).
Cross-cyclomagnesiation of nonadeca-1,2-diene (1) and 2-tetradeca-12,13-dien-1-yl-1,3-dioxolane (2b) with EtMgBr in the presence of Mg metal and Cp2TiCl2 catalyst
Diethyl ether (30 mL), nonadeca-1,2-diene (1) (1.27 g, 4.8 mmol), 2-tetradeca-12,13-dien-1-yl-1,3-dioxolane (2b) (1.06 g, 4.0 mmol), EtMgBr (30.0 mmol) (as 1.5 M solution in Et2O), Mg powder (0.48 g, 20.0 mmol) and Cp2TiCl2 (0.1 g, 0.4 mmol) were placed in a glass reactor with stirring under argon (∼0 °C). The reaction mixture was warmed-up to room temperature (20–22 °C) and stirred for 6 h. The reaction mixture was treated with a 10% solution of HCl in H2O (20 mL) and extracted with diethyl ether (2 × 100 mL). The combined organic phases were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Silica gel column chromatography (hexane/EtOAc (35/1)) of the residue gave dienal 6 (1.50 g, 77%) as a pale yellow oily liquid.Procedure for preparation of alkyne (7)
To a solution of trimethylsilyl acetylene 0.58 g (6 mmol) in THF (10 ml) was added dropwise a solution of 4 ml n-BuLi (1.5 M in hexane) at −40 °C. The solution was stirred for 1 h at −40 to 0 °C. Then the solution was added dropwise to THF solution of 1.5 g (3.08 mmol) dienal (7) at −10 °C. The reaction mixture was warmed-up to room temperature (20–22 °C) and stirred for 3 days. The reaction mixture was treated with a 5% solution of NH4Cl in H2O (20 mL) and extracted with diethyl ether (2 × 100 mL). The combined organic phases were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Silica gel column chromatography of the residue gave compound 7 (1.63 g, 90%) as a pale yellow oily liquid.CH–C2), 4.37 (1H, t, HO–C, J = 5 Hz), 5.38–5.41 (4H, m, C). 13C NMR (100.62 MHz, CDCl3) δ: 130.4 (CH), 129.2 (CH), 106.9 (C), 89.3 (CH), 62.9 (CH), 37.7 (CH2), 31.9 (CH2), 29.2–29.8 (signals of 22C, CH2), 27.4 (2C, CH2), 27.3 (2C, CH2), 25.1 (CH2), 22.7 (CH2), 14.1 (CH3), −0.11 (3C, CH3).
Procedure for preparation of rac-lembehyne B (8)
To a solution of alkyne (7) 1.17 g (2 mmol) in THF (10 ml) was added TBAF (1 M in THF, 1.2 equiv.) at 0 °C, then the solution was stirred for 4 h at room temperature. The reaction mixture was treated with saturated aq. NaCl and extracted with diethyl ether (2 × 50 mL). The combined organic phases were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Silica gel column chromatography of the residue gave compound 8 (1.07 g, 99%) as a colorless waxy solid.The oxidation of the alcohol (8) with Dess–Martin periodinane was carried out according9
2), 2.04–2.10 (8H, m, CH–C2), 2.58–2.62 (2H, m, OCH–C2), 3.22 (1H, s, C), 5.39–5.42 (4H, m, C). 13C NMR (100.62 MHz, CDCl3) δ: 187.5 (C), 130.4 (CH), 130.3 (CH), 129.1 (2C, CH), 81.5 (C), 78.2 (CH), 45.5 (CH2), 31.9 (CH2), 29.3–29.7 (signals of 19C, CH2), 28.9 (CH2), 27.4 (2C, CH2), 27.3 (2C, CH2), 22.7 (CH2), 23.8 (CH2), 14.1 (CH3).
The stereoselective reduction of ketone 9 with B-3-pinanyl-9-borabicyclo[3.3.1]nonane (Alpine-borane reagent) was carried out according procedure11
The spectral data of compound 10 were identical to previously published for lembehyne B.3c2), 2.04–2.10 (8H, m, CH–C2), 4.39 (1H, td, J = 7.0, 2.0 Hz), 5.39–5.41 (4H, m, C). 13C NMR (100.62 MHz, CDCl3) δ: 130.4 (2C, CH), 129.2 (2C, CH), 85.0 (C), 72.8 (CH), 62.4 (CH), 37.7 (CH2), 31.9 (CH2), 29.3–29.7 (signals of 19C, CH2), 29.2 (CH2), 27.4 (2C, CH2), 27.3 (2C, CH2), 25.0 (CH2), 22.7 (CH2), 14.1 (CH3).
Biology
Cell culture
Human leukemia cells Jurkat, HL-60, K562 were obtained from the HPA Culture Collections (UK). The cell lines was cultured in RPMI (Gibco BRL) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin solution at 37 °C in a humidified incubator under a 5% CO2 atmosphere. Testing compounds was dissolved in DMSO and diluted with the tissue culture medium before use.Viability and apoptosis
In vitro cytotoxicity was assessed using a standard MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) colorimetric assay.14 The IC50 value was determined from plots of % viability against the dose of the compound added. The percentage of cell growth inhibition was calculated as 100 minus (mean cell culture optical density in the test/mean cell culture optical density in the control) multiplied by 100. The value obtained for the first three wells without addition of the test compound (control triplet), which was measured in parallel for each test compound, was taken to be 100%. The mean value and the error of mean were calculated separately for each concentration of the test compound. The results were used to plot the cell viability (%) versus the test compound concentration, and the dose inhibiting the cell viability by 50% (IC50) and the standard error (SE) of IC50 were calculated using the GraphPad Prism 7.0 software (GraphPad Inc., USA).Apoptosis was determined by flow cytometric analysis of Annexin V and 7-aminoactinomycin D staining. Briefly, 200 μl of Guava Nexin reagent (Millipore, Bedford, MA, USA) was added to 5 × 105 cells in 200 μl, and the cells were incubated with the reagent for 20 min at room temperature in the dark. At the end of incubation, the cells were analyzed on NovoCyte™ 2000 FlowCytometry System (ACEA).
Cell cycle analysis
Cell cycle was analyzed using the method of propidium iodide staining. Briefly, cells were plated in 24-well round bottom plates at a density 10 × 105 cells per well, centrifuged at 450g for 5 minutes, and fixed with ice-cold 70% ethanol for 24 hour at 0 °C. Cells were then washed with PBS and incubated with 250 μl of Guava Cell Cycle Reagent (Millipore) for 30 minutes at room temperature in the dark. Samples were analyzed on NovoCyte™ 2000 FlowCytometry System (ACEA).Acknowledgements
This work was financially supported by the Russian Science Foundation (Grant No. 16-13-10172). The structural studies of the synthesized compounds were performed with the use of Collective Usage Centre “Agidel” at the Institute of Petrochemistry and Catalysis of RAS. The biological studies of muricadienin were done in the Center for Molecular Design and Drug Bioscreening at the Institute of Petrochemistry and Catalysis of RAS that was created with the financial support of the Russian Science Foundation.Notes and references
- (a) T. L. Simmons, E. Andrianasolo, K. McPhail, P. Flatt and W. H. Gerwick, Mol. Cancer Ther., 2005, 4, 333–342 CAS; (b) G. M. Nicholas and A. J. Phillips, Nat. Prod. Rep., 2006, 23, 79–99 RSC; (c) J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2006, 23, 26–78 RSC; (d) C. Chakraborty, C.-H. Hsu, Z.-H. Wen and C.-S. Lin, Curr. Top. Med. Chem., 2009, 9, 1536–1545 CrossRef CAS PubMed; (e) I. Bhatnagar and S.-K. Kim, Mar. Drugs, 2010, 8, 2702–2720 CrossRef CAS PubMed; (f) R. Montaser and H. Luesch, Future Med. Chem., 2011, 3, 1475–1489 CrossRef CAS PubMed; (g) G. Schwartsmann, A. B. da Rocha, R. G. S. Berlinck and J. Jimeno, Lancet Oncol., 2001, 2, 221–225 CrossRef CAS PubMed.
- (a) A. Siddiq and V. Dembitsky, Anti-Cancer Agents Med. Chem., 2008, 8, 132–170 CrossRef CAS; (b) Z.-F. Zhou, M. Menna, Y.-S. Cai and Y.-W. Guo, Chem. Rev., 2015, 115, 1543–1596 CrossRef CAS PubMed; (c) N. Fusetani, H. Y. Li, K. Tamura and S. Matsunaga, Tetrahedron, 1993, 49, 1203–1210 CrossRef CAS; (d) G. Nuzzo, M. L. Ciavatta, G. Villani, E. Manzo, A. Zanfardino, M. Varcamonti and M. Gavagnin, Tetrahedron, 2012, 68, 754–760 CrossRef CAS; (e) A. D. Patil, W. C. Kokke, S. Cochran, T. A. Francis, T. Tomszek and J. W. Westley, J. Nat. Prod., 1992, 55, 1170–1177 CrossRef CAS PubMed; (f) J. H. Shin, Y. W. Seo, K. W. Cho, J.-R. Rho and V. J. Paul, Tetrahedron, 1998, 54, 8711–8720 CrossRef CAS; (g) K. Watanabe, Y. Tsuda, Y. Yamane, H. Takahashi, K. Iguchi, H. Naoki, T. Fujita and R. W. M. Van Soest, Tetrahedron Lett., 2000, 41, 9271–9276 CrossRef CAS; (h) S. Tsukamoto, H. Kato, H. Hirota and N. Fusetani, J. Nat. Prod., 1997, 60, 126–130 CrossRef CAS; (i) S. Ohta, H. Okada, H. Kobayashi, J. M. Oclarit and S. Ikegami, Tetrahedron Lett., 1993, 34, 5935–5938 CrossRef CAS; (j) D. Listunov, V. Maraval, R. Chauvin and Y. Génisson, Nat. Prod. Rep., 2015, 32, 49–75 RSC.
- (a) S. Aoki, K. Matsui, K. Tanaka, R. Satari and M. Kobayashi, Tetrahedron, 2000, 56, 9945–9948 CrossRef CAS; (b) S. Aoki, K. Matsui, T. Takata, W. Hong and M. Kobayashi, Biochem. Biophys. Res. Commun., 2001, 289, 558–563 CrossRef CAS PubMed; (c) S. Aoki, K. Matsui, H. Wei, N. Murakami and M. Kobayashi, Tetrahedron, 2002, 58, 5417–5422 CrossRef CAS.
- (a) Y.-A. Barde, Neuron, 1989, 2, 1525–1534 CrossRef CAS PubMed; (b) S. Korsching, J. Neurosci., 1993, 13, 2739 CAS.
- N. Murakami, T. Nakajima and M. Kobayashi, Tetrahedron Lett., 2001, 42, 1941–1943 CrossRef CAS.
- (a) P. L. Mena, O. Pilet and C. Djerassi, J. Org. Chem., 1984, 49, 3260–3264 CrossRef CAS; (b) D. Raederstorff, A. Y. L. Shu, J. E. Thompson and C. Djerassi, J. Org. Chem., 1987, 52, 2337–2346 CrossRef CAS; (c) T. Nemoto, G. Yoshino, M. Ojika and Y. Sakagami, Tetrahedron, 1997, 53, 16699–16710 CrossRef CAS; (d) E. D. Reyes and N. M. Carballeira, Synthesis, 1997, 1195–1198 CrossRef CAS; (e) N. M. Carballeira, A. Emiliano, N. Hernández-Alonso and F. A. González, J. Nat. Prod., 1998, 61, 1543–1546 CrossRef CAS PubMed; (f) N. M. Carballeira, A. Emiliano and A. Guzmán, Chem. Phys. Lipids, 1999, 100, 33–40 CrossRef CAS PubMed; (g) N. M. Carballeira, J. E. Betancourt, E. A. Orellano and F. A. González, J. Nat. Prod., 2002, 65, 1715–1718 CrossRef CAS PubMed; (h) B. A. Kulkarni, A. Sharma, S. Gamre and S. Chattopadhyay, Synthesis, 2004, 595–599 CAS; (i) N. M. Carballeira, R. O'Neill and D. Silva, Chem. Phys. Lipids, 2008, 156, 41–44 CrossRef CAS PubMed; (j) I. Liblikas, R. Mozūraitis, E. M. Santangelo, R. Noreika and A.-K. Borg-Karlson, Chem. Biodiversity, 2009, 6, 1388–1403 CrossRef CAS PubMed; (k) J. Adrian and C. B. W. Stark, Org. Lett., 2014, 16, 5886–5889 CrossRef CAS PubMed; (l) J. Adrian and C. B. W. Stark, Eur. J. Org. Chem., 2016, 4607–4610 CrossRef; (m) J. Adrian and C. B. W. Stark, J. Org. Chem., 2016, 81, 8175–8186 CrossRef CAS PubMed; (n) J. Schmidt, J. Adrian and C. B. W. Stark, Org. Biomol. Chem., 2015, 13, 8173–8176 RSC; (o) R. A. Kunkalkar, D. Laha and R. A. Fernandes, Org. Biomol. Chem., 2016, 14, 9072–9079 RSC.
- (a) U. M. Dzhemilev, V. A. D'yakonov, L. O. Khafizova and A. G. Ibragimov, Tetrahedron, 2004, 60, 1287–1291 CrossRef CAS; (b) V. A. D'yakonov, A. A. Makarov, A. G. Ibragimov, L. M. Khalilov and U. M. Dzhemilev, Tetrahedron, 2008, 64, 10188–10194 CrossRef; (c) V. A. D'yakonov, A. A. Makarov, E. Kh. Makarova and U. M. Dzhemilev, Tetrahedron, 2013, 69, 8516–8526 CrossRef; (d) V. A. D'yakonov, A. A. Makarov, L. U. Dzhemileva, E. Kh. Makarova, E. K. Khusnutdinova and U. M. Dzhemilev, Chem. Commun., 2013, 49, 8401–8403 RSC; (e) V. A. D'yakonov, L. U. Dzhemileva, A. A. Makarov, A. R. Mulyukova, D. S. Baev, E. K. Khusnutdinova, T. G. Tolstikova and U. M. Dzhemilev, Med. Chem. Res., 2016, 25, 30–39 CrossRef; (f) V. A. D'yakonov, L. U. Dzhemileva, A. A. Makarov, A. R. Mulyukova, D. S. Baev, E. K. Khusnutdinova, T. G. Tolstikova and U. M. Dzhemilev, Bioorg. Med. Chem. Lett., 2015, 25, 2405–2408 CrossRef PubMed; (g) U. M. Dzhemilev, V. A. D'yakonov, R. A. Tuktarova, L. U. Dzhemileva, S. R. Ishmukhametova, M. M. Yunusbaeva and A. de Meijere, J. Nat. Prod., 2016, 79, 2039–2044 CrossRef CAS PubMed.
- O. Kuisle, E. Quiñoá and R. Riguera, J. Org. Chem., 1999, 64, 8063–8075 CrossRef CAS PubMed.
- S. D. Meyer and S. L. Schreiber, J. Org. Chem., 1994, 59, 7549–7552 CrossRef CAS.
- L. Brandsma, Synthesis of Acetylenes, Allenes and Cumulenes: Methods and Techniques, Elsevier SPC, Amsterdam-Oxford-New York, 2004, p. 470 Search PubMed.
- M. M. Midland, A. Tramontano, A. Kazubski, R. S. Graham, D. J. S. Tsai and D. B. Cardin, Tetrahedron, 1984, 40, 1371–1380 CrossRef CAS.
- C. E. Janßen and N. Krause, Eur. J. Org. Chem., 2005, 2322–2329 CrossRef.
- S. Chanthamath, H. W. Chua, S. Kimura, K. Shibatomi and S. Iwasa, Org. Lett., 2014, 16, 3408–3411 CrossRef CAS PubMed.
- K. Du, J. Liang, Y. Wang, J. Kou, C. Qian, L. Jia and H. Chao, Dalton Trans., 2014, 43, 17303–17316 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob02346k |
| This journal is © The Royal Society of Chemistry 2017 |