
Site-specific covalent capture of human O6-alkylguanine-DNA-alkyltransferase using single-stranded intrastrand cross-linked DNA†
D. K.
O'Flaherty
and
C. J.
Wilds
*
Department of Chemistry and Biochemistry, Concordia University, Montréal, Québec H4B1R6, Canada. E-mail: chris.wilds@concordia.ca
First published on 17th November 2016
Abstract
A methodology is reported to conjugate human O6-alkylguanine-DNA-alkyltransferase (hAGT) to the 3′-end of DNA in excellent yields with short reaction times by using intrastrand cross-linked (IaCL) DNA probes. This strategy exploited the substrate specificity of hAGT to generate the desired DNA–protein covalent complex. IaCL DNA linking two thymidine residues, or linking a thymidine residue to a 2′-deoxyguanosine residue (either in a 5′→3′ or 3′→5′ fashion), lacking a phosphodiester linkage at the cross-linked site, were prepared using a phosphoramidite strategy followed by solid-phase synthesis. All duplexes containing the model IaCL displayed a reduction in thermal stability relative to unmodified control duplexes. The O4-thymidine-alkylene-O4-thymidine and the (5′→3′) O6-2′-deoxyguanosine-alkylene-O4-thymidine IaCL DNA adducts were not repaired by any of the AGTs evaluated (human AGT and Escherichia coli homologues, OGT and Ada-C). The (5′→3′) O4-thymidine-alkylene-O6-2′-deoxyguanosine IaCL DNA containing a butylene or heptylene tethers were efficiently repaired by the human variant, whereas Ada-C was capable of modestly repairing the heptylene IaCL adduct. The IaCL strategy has expanded the toolbox for hAGT conjugation to DNA strands, without requiring the presence of a complementary DNA sequence. Finally, hAGT was functionalized with a fluorescently-labelled DNA sequence to demonstrate the applicability of this conjugation method.
Introduction
Bifunctional alkylating agents have the capability of causing DNA damage by generating inter- (ICL) and intrastrand (IaCL) cross-links, mono-adducts, and DNA–protein cross-links (DPC).1,2 Extensive research has been carried out on ICL given their cytotoxicity.3 The introduction of ICL in DNA prevents the unwinding of the DNA strands, a necessary step for vital processes such as replication and transcription. IaCL can also impede cellular proliferation despite residing on only one strand.4,5 For instance, cyclobutane pyrimidine dimers and their Dewar isomers produced by UV-irradiation are common examples of non-drug-induced IaCL DNA, which can lead to carcinogenesis if left unattended.6–8 Exposure of DNA to gamma irradiation produces IaCL DNA between neighboring 2′-deoxyguanosine (dG) and thymidine (dT) residues, which accounts at least in part, to the cytotoxicity of this type of irradiation.9 Moreover, various cancer chemotherapeutic regimens exploit the formation of DNA alkyl lesions to treat certain diseases. Platinum-based chemotherapeutics such as cisplatin are widely utilized for cancer treatment and exert their efficacy mainly via the formation of IaCL DNA lesions.5 Understanding the impact of IaCL on DNA structure, as well as how cells process such DNA damage is an ongoing investigation. As such, there is a considerable value in the development of methodologies for the preparation of species resembling reaction intermediates, which could provide insights on their exerted cytotoxic effects and the mechanism of repair.Minor alkylation at the O6-dG and O4-dT sites are observed when exposing DNA to various alkylating agents, with O6-alkyl dG occurring in higher frequency relative to O4-alkyl dT.10–12 Regardless of their low occurrence, these chemical lesions are chemically stable and are particularly cytotoxic, if left unrepaired, as they severely hinder DNA polymerase activity.13 Both lesions are presumed to cause misinsertions by disrupting the Watson–Crick H-bonding with their natural complements leading to misreading by DNA polymerases during DNA replication. Both O6-MedG·dT and O4MedT·dG adopt nonwobble base pair motifs, which may account for the error-prone behaviors of DNA polymerases.14
O 6-Alkylguanine DNA alkyltransferases (AGTs), proteins of the direct repair pathway, are found in all kingdoms of life and have shown a large variance in substrate specificity in terms of lesion type and site of alkylation.2 They play a pivotal role in maintaining genomic integrity by removing mutagenic lesions found at the O6-position of dG residues, and to a lesser extent the O4-position of dT. It is estimated that about 6650 O6-methyl dG (O6-MedG) lesions are sufficient in causing cell death in human AGT (hAGT) deficient tumour cells.15 The O4-methyl dT (O4MedT) lesion has a greater mutagenic influence in mammalian cell lines, relative to O6-MedG, since this lesion is inefficiently repaired by AGTs or the mismatch repair pathway.16 Moreover, the increased binding affinity of hAGT to DNA containing O4MedT can inhibit NER pathways from successfully removing these lesions.17
The human homologue (hAGT) displays significant repair activity towards O6-alkyl-dG lesions, including mono-adducts such as methyl, benzyl, and 4-oxo- 4-(3-pyridyl)butyl, to name a few.2 AGT-mediated repair has also been associated with cancer resistance mechanisms to a number of chemotherapeutic agents, such as 1,3-bis(2-chloroethyl)-1-nitrosourea18,19 and temozolomide,20,21 known to alkylate the O6-position of dG.22 Determining the scope of repair for AGTs could aid in the development of more potent treatment strategies. Our group has reported the repair by hAGT of ICL linking the O6-atoms of directly opposing 2′-deoxyguanosine residues, as well as in a GNC motif.23,24 Orientation of the alkylene bridge caused little variance in the processing by hAGT. Enhanced repair was observed for the ICL DNA with a heptylene linker, relative to the butylene analogue. The E. coli variant OGT, however, was incapable of removing the ICL between 2′-deoxyguanosine residues. In addition, no repair was detected with cross-links between O4-atoms of thymidine residues, despite the preference of OGT for alkylation at the O4-position of dT compared to the O6-position of dG.25 Recently, we have reported the capability of hAGT to efficiently repair IaCL DNA, between the O6-atoms of two neighbouring dG residues, lacking a phosphate group at the cross-link site. As with the ICL DNA, hAGT was more efficient in repairing the heptylene linkage compared to the butylene IaCL. Interestingly, the IaCL of greater length was virtually entirely consumed by hAGT, whereas repair of the heptylene ICL by a larger excess of protein occurred only to 57%.24 Similar O6-dG-alkylene-O6-dG IaCL containing the intradimer phosphodiester group were not efficiently processed by hAGT.26
Despite its role in maintaining genomic material integrity, hAGT has shown to enhance cytotoxicity of certain bifunctional alkylating agents by efficiently producing DPC adducts. 1,2-Dibromoethane and 1,2,3,4-diepoxybutane, for instance, are capable of inducing AGT-DNA cross-links.27–29 The paradoxical cytotoxicity was attributed to the formation of either a DNA or protein intermediate, capable of readily reacting with its counterpart.30 Given the importance of such conjugates, efficient methodologies for the production of AGT-mediated DPC warrant further investigation.
The current understanding of the AGT-mediated repair mechanism involves flipping of the damaged nucleotide into the active site, whereby an activated cysteine residue attacks the α-carbon attached to the O6- or O4-atom of dG or dT, respectively.31 DPC covalent complexes between DNA and hAGT have been insightful in enhancing our understanding of the AGT-mediated repair mechanism. The N1,O6-ethanoxanthine modification generates a DPC after hAGT repair and crystallographic studies of this covalent complex have identified critical interactions involved in the repair event.31,32 However, this approach generated a DPC tethered by an ethylene linker and it would be of interest to investigate longer linkers given that AGTs are capable of repairing a wide scope of substrates (butylene and heptylene adducts, for instance).24,25 Towards this end, McManus and co-workers developed a strategy of generating DPCs via the repair of a alkylene ICL between the O6-dG and O4-dT by hAGT, whereby repair solely occurred at the dG nucleotide.33 Unfortunately, the repair reaction conversions for the heptylene ICL substrate was fairly low (25%) after 16 h, using a 30-fold excess of hAGT protein, necessitating an improvement in the methodology to produce DPC species. No appreciable amount of DPC was observed for repair of the butylene ICL analogue. No other AGT variant, apart from hAGT, tested positive for repair, further limiting the versatility of the ICL probe for conjugation.
This study examines the reaction of AGTs with various IaCL DNAs, lacking the phosphodiester linkage, which potentially allow for an efficient production of DPCs tethered at either the 3′- or 5′-end of the DNA strand. The overall approach benefits from improved atom economy, greater reaction yields and is formed rapidly, compared to ICL DNA. The ability of a second AGT molecule reacting with the DPC was observed for the hAGT-mediated repair of the O6-dG-alkylene-O6-dG ICL DNA probes.23,24 To circumvent this issue, we engineered IaCL lacking the intradimer phosphodiester, which is generated a stable DPC species linked to the O4-atom of dT or the O6-atom of dG. Remarkably, these cross-linked adducts were not suitable substrates for another round of repair by a second AGT protein. This avenue provides a versatile and an efficient method to produce DPCs tethered to DNA strands with variable linkage lengths.
Results and discussion
Synthesis of IaCL DNA probes
Bis-alkylating agents, busulfan and hepsulfam, are known to generate butylene IaCL and heptylene ICL adducts, respectively, between purine residues. This served as the basis for the linker lengths chosen in the study herein. Although the specific O6-alkylene-dG or O4-alkylene-dT IaCL lesions have not been identified, formation of other pyrimidine–pyrimidine and pyrimidine–purine adducts in DNA have been previously reported. For instance, pyrimidine(6-4)pyrimidone photoproducts are examples of lesions linking the C4 position of one pyrimidine residue with a neighbouring pyrimidine nucleotide at the 6-position, and are reported to play a significant role in mutagenesis and in inducing human skin cancer.34,35 DNA treated with mechlorethamine has revealed the formation of ICL linking the N3-atoms of mispaired dC residues.36 Gamma irradiation is capable of producing IaCL DNA between adjacent purine and pyrimidine residues, regardless of their orientation (5′-GT or 5′-TG for instance).37–41 These examples prompted us to engineer biologically relevant IaCL mimics to help understand repair by AGTs. Moreover, generating such DNA modifications allowed us to study duplexes containing flexible IaCL lesions that interfere with the Watson–Crick face of the nucleobases.The current study aimed (i) at investigating the ability of certain AGTs to repair a variety of different IaCL DNA, with structures and corresponding labels shown in Fig. 1(A–C). Since the initial report of IaCL DNA between two dG residues demonstrated efficient repair by hAGT (Fig. 1D),42 we envisioned an analogous set of probes suitable for repair by other AGTs such as OGT and Ada-C. The second aim of this study (ii) was to generate appreciable amounts of DPC species with enhanced versatility.
 | ||
| Fig. 1 Structure of the (A) O4-thymidine-alkylene-O4-thymidine, (B) O6-2′-deoxyguanosine-alkylene-O4-thymidine, (C) O4-thymidine-alkylene-O6-2′-deoxyguanosine and (D) O6-2′-deoxyguanosine-alkylene-O6-2′-deoxyguanosine IaCL mimics lacking a phosphodiester linkage between the two residues. The probes shown in (D) have been investigated previously by our group.42 (E) The position of the IaCL mimics in the DNA sequence is shown by the XY (where XY4 has a butylene linker and XY7 a heptylene linker). | ||
The small molecule synthetic details can be found in the ESI,† where the phosphoramidites (7a, 7b, 11a, 11b, 16a, and 16b) were prepared in moderate to good yield using previously reported procedures.42 Purification was achieved using chromatography, which resulted in pure products as demonstrated by the 31P NMR spectra. In all cases, two sharp signals were observed in the region of 147–149 ppm of the 31P NMR spectra, which are diagnostic of phosphoramidites.24,25,42
The IaCL DNA assembly employed either dimer phosphoramidite 7a, 7b, 11a, 11b, 16a, or 16b, which differed in both their nucleobase composition and linker length (butylene compared to heptylene). Linear IaCL DNA assembly was performed according to previously published procedures to prepare duplexes containing similar modifications.25,33,42 The use of “fast-deprotecting” commercially available 3′-O-phosphoramidites was necessary due to the labile nature of O4-alkyl-dT adducts.25 Coupling times were extended to 600 s for cross-linked amidites as opposed to 120 s for standard 3′-O-phosphoramidites, in order to ensure proper coupling of the dimers. To be noted was the addition of 20% (v/v) of THF in ACN to solubilize amidites 11a, 11b, 16a, or 16b. IaCL DNA oligomers were deprotected and cleaved off the CPG solid support via treatment with an anhydrous solution of 0.05 M K2CO3 in methanol for 4 h under gentle rocking. Excess base was neutralized with an equimolar amount of acetic acid and lyophilized, without desalting, in a speed-vacuum concentrator. The resulting pellets were treated with triethylamine trihydrofluoride to remove the remnant silyl protecting groups at 40 °C for 16 h. Initial sonication (2 × 15 s) was used to maximize the reaction. Precipitation of IaCL DNA was achieved with cold n-butanol at −20 °C for 10 min. IaCL oligomers proved slightly soluble in methanol, which was generally more pronounced for heptylene compared to the butylene linked IaCL DNA presumably due of the increased hydrophobicity of longer alkylene linkers. Also, solubility of the oligomers increased with shorter sequences (observed for O4-dT-alkylene-O4-dT IaCL DNA with sequences of 12 nucleotides in length), especially in cases where the modification was engineered in the central region of the sequence (data not shown). The precipitated material was washed with another cold aliquot of n-butanol before purification by SAX-HPLC. Characterization of IaCL DNA by ESI-MS revealed deconvoluted masses in agreement with all expected masses (ESI Fig. S51–S56†).
UV thermal denaturation, circular dichroism, and molecular modeling studies of IaCL DNA
The impact of the different IaCL on DNA duplex stability was assessed by UV thermal denaturation experiments. Results of the experiments are summarized in Fig. 2 (see ESI Fig. S57–S59† for all Tm curves). As previously observed for the O6-dG-alkylene-O6-dG IaCL DNA, a general decrease in Tm value of approximately 16–19 °C relative to the unmodified control was observed in all cases.42 The linker length had little effect on the thermal denaturation profiles across the various duplexes containing the IaCL DNA. All curves displayed a characteristic sigmoidal transition. Interestingly, we observed that the absence of the phosphodiester linkage at the cross-linked site has minimal influence on the duplex stability similar to the O6-dG-alkylene-O6-dG IaCL.26,42 The overall decreased stability likely resulted from H-bonding disruption between the alkylated dT (or dG) and its base-paired 2′-deoxyadenosine (or 2′-deoxycytidine) counterpart. In addition, the decrease in Tm may be a consequence of the bulky alkylene linkers causing local or global perturbations. Circular dichroism (CD) spectroscopy was carried out to evaluate the global structural influence of the IaCL mimics on the DNA duplexes. CD profiles of duplexes containing TG4, TG7 and the unmodified (TG) control are shown in Fig. 3 (see ESI Fig. S60 and S61† for other CD spectra). In all cases, CD profiles displayed characteristic signatures pertaining to the B-DNA family with maxima near 280 nm, cross-overs near 255 nm and minima near 240 nm. More importantly, all the CD profiles of duplexes containing the IaCL were similar to the corresponding unmodified DNA duplexes, which suggested that the presence of the IaCL modification minimally perturbed the DNA duplex, as previously reported.42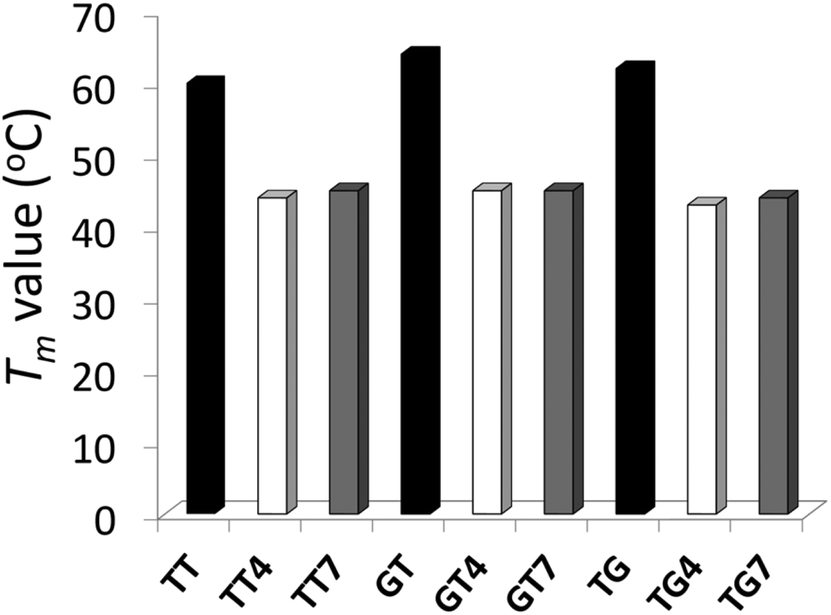 | ||
| Fig. 2 T m values (°C) of duplexes containing TT, TT4, TT7, GT, GT4, GT7, TG, TG4, TG7. Bars in black, colorless and grey represent the unmodified controls, butylene adducts, and heptylene adducts, respectively. Hyperchromicity change (A260) versus temperature (°C) profiles are shown in ESI Fig. S57–S59.† | ||
 | ||
| Fig. 3 Circular dichroism spectra of IaCL duplexes TG4 (⋯), TG7 (———) and unmodified control TG DNA (—). | ||
Molecular modeling was performed on duplexes containing the different IaCL adducts or the unmodified control nucleotides using AMBER force field (see ESI Fig. S62–S64†). The alkylene linkers of these IaCL reside in the major groove of the duplexes. The presence of the alkylene tether seemed to generally disrupt the base pairing between the alkylated nucleobases their complements, relative to the unmodified controls which may account for the decrease in Tm observed. Overall the molecular models suggested minimal global distortions induced by the presence of the IaCL tether, which was in agreement with observations from the CD spectra.
AGT repair of IaCL DNA substrates
Four different AGT variants, hAGT, OGT, Ada-C, and an S134P OGT variant were tested for their ability to repair the DNA duplexes containing the different IaCL (TT4, TT7, GT4, GT7, TG4, and TG7). The repair assay was conducted as described in a previous report,42 whereby the successful repair of the IaCL mimic generates a shorter DNA repair product (RP) species and a DPC median product (MP) (Fig. 4), which are observed by denaturing PAGE experiments after reaction times of 16 h at 37 °C.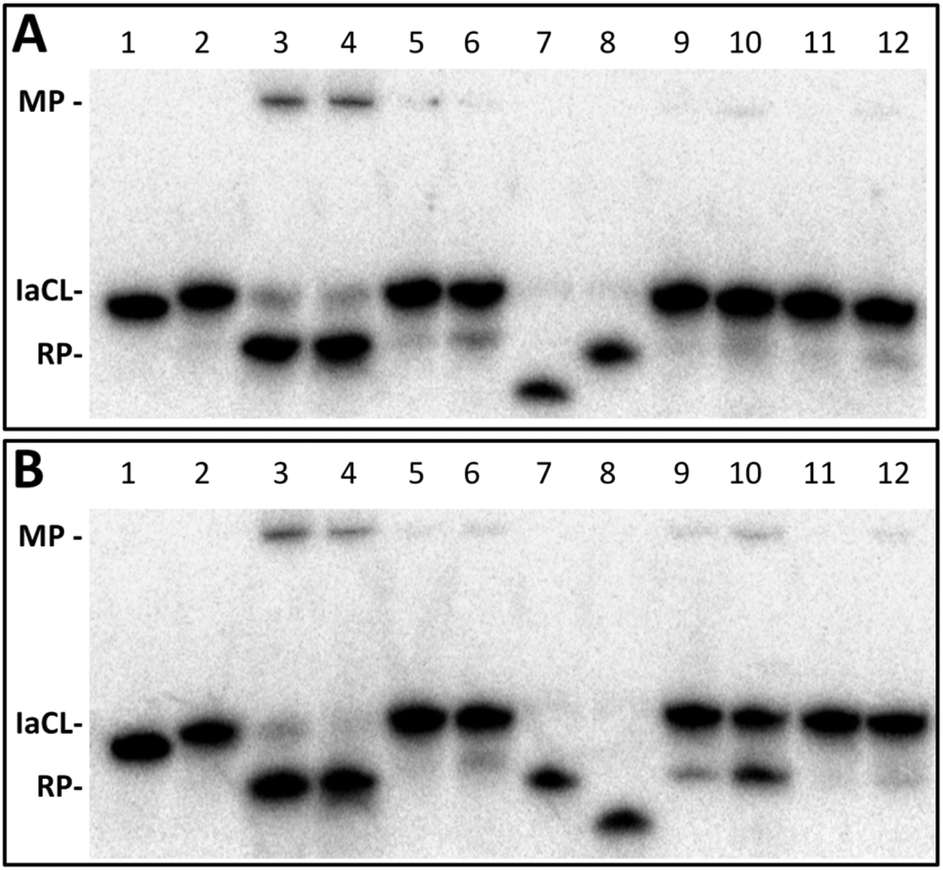 | ||
| Fig. 4 Repair of duplexes containing (A) TG4 and (B) TG7 by hAGT, OGT, Ada-C, and OGT S134P. Denaturing PAGE of repair reactions as described in the experimental section with varying protein concentration (either 5- or 30 fold protein). Panel A; lane 1, 2 pmol control unmodified TG DNA; lane 2, 2 pmol TG4; lane 3, TG4 + 10 pmol hAGT; lane 4, 2 pmol TG4 + 60 pmol hAGT; lane 5, 2 pmol TG4 + 10 pmol OGT; lane 6, 2 pmol TG4 + 60 pmol OGT; lane 7, 2 pmol unmodified 5-mer control DNA 5′-pGGCTT; lane 8, 2 pmol unmodified 10-mer control 5′-pGGC ATC ACC AG; lane 9, 2 pmol TG4 + 10 pmol Ada-C; lane 10, 2 pmol TG4 + 60 pmol Ada-C; lane 11, 2 pmol TG4 + 10 pmol OGT S134P; lane 12, 2 pmol TG4 + 60 pmol OGT S134P. Panel B; identical to panel A except with TG7 instead of TG4, and lane 7, 2 pmol unmodified 10-mer control 5′-pGGC ATC ACC AG; lane 8, 2 pmol unmodified 5-mer control DNA 5′-pGGCTT. | ||
The radioactivity-based repair assay revealed no appreciable repair (<20% repair) of TT4, TT7, GT4, and GT7 at both 5-molar equivalence and 30-molar equivalence of AGT (ESI Fig. S65 and S66†). Lack of repair by Ada-C was not surprising as this protein is generally inactive to adducts larger than a methyl group at the O4- and O6-position of dT and dG, respectively.43,44 It was, however, surprising to observe the lack of OGT-mediated repair of these IaCL DNA, particularly TT4 and TT7, given that OGT is proficient in repairing larger mono-adducts, such as hydroxyl-n-butyl and hydroxy-n-heptyl, at the O4-position of dT.25 The OGT S134P variant,45 which is believed to have a larger active site, was also unable to repair any of these IaCL. Moreover, hAGT proved inefficient at repairing duplexes containing GT4 and GT7 despite being capable of efficiently repairing GG4 and GG7 (see Fig. 1D for structures). Our group has also previously demonstrated repair by hAGT toward an O6-dG-heptylene-O4-dT ICL occurred solely at the O6-atom of dG, with no repair event at the O4-atom of dT.33 The lack of hAGT repair towards GT4 and GT7 suggest that the 3′-phosphodiester linkage plays a critical role in the repair at the dG moiety of the IaCL. This hypothesis is further supported by the efficient repair of TG4 and TG7 by hAGT as illustrated in Fig. 4, which both possess a 3′-phosphodiester linkage at the dG residue (Fig. 1). Crystallographic structures of covalent complexes between modified DNA and hAGT, as well as computational studies, have revealed a critical interaction of Tyr114 with the 3′-phosphodiester in order for efficient repair of O6-alkyl-dG.31,46 We suspect that deleting this residue would severely influence hAGT repair of these IaCL lesions.
Repair profiles of TG4 and TG7 are illustrated in the denaturing gel shown in Fig. 4. Only hAGT was capable of TG4 repair (lanes 3 and 4 in Fig. 4A) with almost complete substrate consumption at 5-fold protein–DNA ratio (10 pmol protein and 2 pmol DNA). As suspected, hAGT was also capable of efficiently repairing the heptylene analogue TG7 (lanes 3 and 4 in Fig. 4B). We were surprised to observe partial repair of TG7 by Ada-C at 30-fold protein equivalence (lane 10 in Fig. 4B). To the extent of our knowledge, this marks the first substrate adduct larger than O6-ethyl-dG capable of undergoing repair by Ada-C,44 albeit at a moderate efficiency (30% substrate consumption reactions of 16 h at 37 °C). The results revealed repair occurring only at the O6-alkylene-dG side of the IaCL, due to the production of the larger DNA RP (Fig. 4B). No repair at thymidinyl moiety of TG7 was observed, which was in agreement with a lack of repair of larger O4-hydroxyalkylene-dT mono-adducts by Ada-C.25 The IaCL strategy may find applications in efficiently generating DNA-Ada cross-linked species that have otherwise been impossible using the ICL approach.33 Our strategy would, however, require optimization given the large protein excess required to initiate repair of TG7.
Time course assays were used to gain insights on the kinetics of the reaction with AGTs and the results are summarized in Fig. 5 (see ESI Fig. 68† for gels). Substrate TG4 (Fig. 5A) was processed by hAGT significantly slower compared to TG7 (Fig. 5B), which was in agreement with repair trends observed with other IaCL42 as well as ICL24 DNA repair by hAGT. Approximately 25% of TG4 IaCL substrate remained after a reaction period of 60 min with 5-fold excess hAGT, whereas similar levels were reached after only 10 min for TG7. Interestingly, the TG4 IaCL substrate was repaired more efficiently in comparison to the GG4 IaCL DNA. For instance, TG4 reached reaction plateau (20% substrate remaining) after approximately 90 min, whereas it took hAGT approximately 480 min to reach similar levels for GG4.42 This trend was not observed for the heptylene analogues (TG7versusGG7), with substrate levels reaching approximately 20% after 15 min. This may be due to the energetics involved in rotating the α-carbon of the O6-alkylated-dG in a favorable orientation to facilitate nucleophilic attack by the Cys145 thiolate anion. There are reports that suggest orientation of the alkyl group at the O6-atom of dG influences the repair by hAGT.47 Perhaps the alkylated dG nucleobase is more easily rotated in the case of TG4 compared to GG4, because pyrimidines are capable of adopting a variety of different conformations, thus conferring added flexbility.48 The added flexibility may aid in the repair of the more-constrained butylene IaCL DNA, consistent with greater amount of repair observed for TG4versusGG4. A similar effect may not necessarily translate to the identical benefit for the longer heptylene tether, as longer linkers are inherently more flexible (due to the additional rotatable bonds). This may explain why similar hAGT repair was observed for TG7 and GG7. We have recently solved the NMR solution structure of an O4-2′-deoxyuridine-heptylene-O4-2′-deoxyuridine ICL DNA duplex (PDB code 2LZV), which revealed that the α-carbon of the O4-alkylated pyrimidine adopted an unexpected E conformation between the α-carbon and the C4-O4 bond. Perhaps such a conformation is contributing to the effects observed with the TG IaCl versusGG IaCL. The formation of median product (MP) in the case of TG4 and TG7, similar to observations previously made for GG4 and GG7, suggested that reaction conditions could be optimized for the efficient production of DPCs.
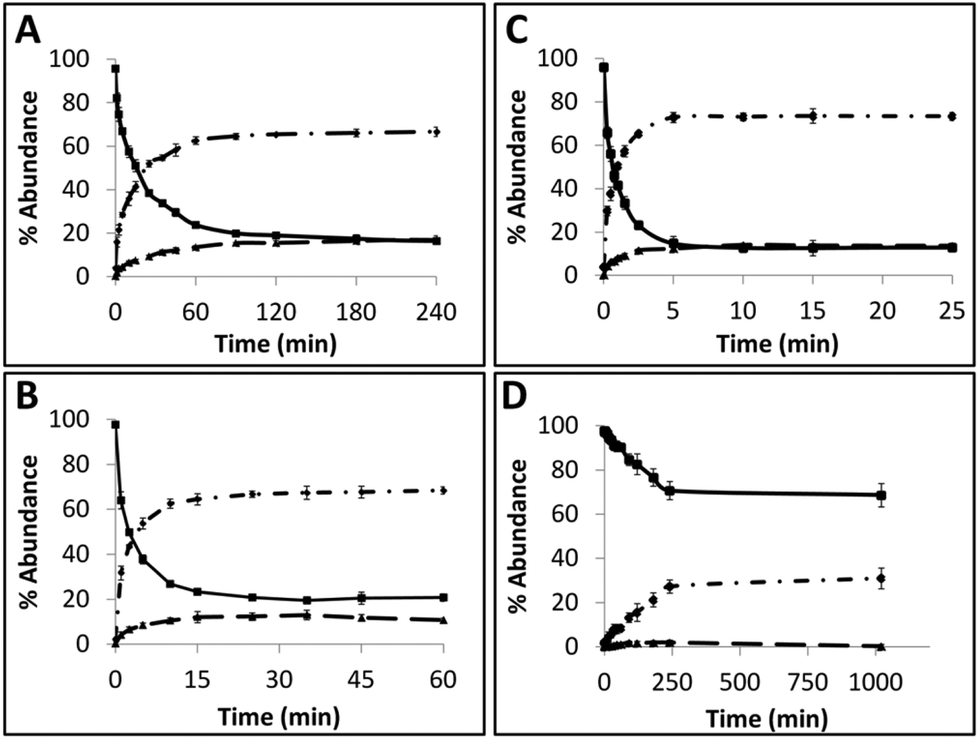 | ||
| Fig. 5 Time course repair assay of TG4 (A) and TG7 (B) by hAGT, at 5-molar equivalence protein, displaying greater repair of TG7 by hAGT. Time course repair assay of TG7 by hAGT (C) and Ada-C (D) at 30-molar equivalence protein, displaying greater repair of TG7 by hAGT compared to Ada-C. Graphical illustrations display abundances [%] of MP (———), RP (—•—) and substrate (—) over time (min). | ||
Time course assays revealed that Ada-C repaired TG7 slower relative to hAGT (Fig. 5C and D, respectively, see ESI Fig. S68† for gels). At 30-fold protein equivalences, TG7 levels reached about 70% after reaction times of 4 h by Ada-C, whereas similar amounts were detected after only 15 s with hAGT. These results were consistent with the narrow substrate scope for Ada-C (O6-methyl-dG, O6-ethyl-dG, and O4-methyl-dT)44 relative to the wide substrate range previously reported for hAGT.2,42 Unfortunately, no considerable amounts of MP were detected for Ada-C mediated repair (Fig. 4B and 5D).
Studies from our group and others have suggested that repair by AGTs may be dependent on conformations of the alkyl group in order for efficient transfer of the alkyl group to occur.25,33,47,49 This effect may not be as pronounced in freely rotating adducts or smaller appendages that have a narrower range of orientations. The DNA–protein complex must be such that the α-carbon of the DNA lesion resides in a favorable orientation to allow attack by the cysteine thiolate anion.31 It should be noted that there could be factors other than alkyl appendage conformation that affect substrate discrimination. For instance, inefficient repair was observed for the O6-methyl-riboguanosine within a DNA oligomer by hAGT.50 The poor repair was accredited to a potential clash between the 2′-hydroxyl group and an amino acid near the active site of hAGT.31 Added flexibility from the lack of the phosphodiester group in this IaCL mimic may explain the proficient repair observed by hAGT and mediocre repair by Ada-C. Also, the lack of a 3′-phosphodiester linkage prevents efficient repair by hAGT of O6-alkylene-dG (such as those found in GT4 and GT7), which may simply be due to a Tyr114 residue that is otherwise interacting with the 3′-phosphodeister group. It is unclear if the alkylene linker orientation, presumably pointing towards the 3′-end of the sequence, in GT4 and GT7 adopts an adverse conformation.
Identification of GG4, GG7, TG4 and TG7 repair products by SDS-PAGE and ESI-MS
The radioactivity-based assay described above mainly monitors DNA species. SDS-PAGE and mass spectrometric characterization were performed in order to identify proteinaceous species. We conducted repair reactions of AGT protein (600 pmol) and IaCL DNA (600 pmol), either single-stranded or part of a duplex. GG4 and GG7 were utilized as positive controls. As previously demonstrated for GG4 and GG7,42 hAGT-mediated repair of TG4 and TG7 was not significantly affected by the lack, or presence, of the complementary DNA strand (data not shown). The Coomassie blue-stained gel shown in Fig. 6 displays almost quantitative conversion of hAGT to a slower migrating (presumably the DPC) species when reacted with single-stranded GG4, GG7, TG4, and TG7 (lanes 2, 3, 5, and 6, respectively) for 16 h at 37 °C. Furthermore, excellent reaction conversions were observed when shortening the reaction times to only one hour (ESI Fig. S69†). DPC formation occurred in a more-efficient fashion compared to AGT-cross-linking using the N1,O6-ethano-2′-deoxyxanthosine modification, which was performed with longer reaction times and higher protein-to-DNA ratio.32 No appreciable amount of DPC, however, was detected by reacting Ada-C (600 pmol) with TG7 (600 pmol) as shown in lane 8 of Fig. 6.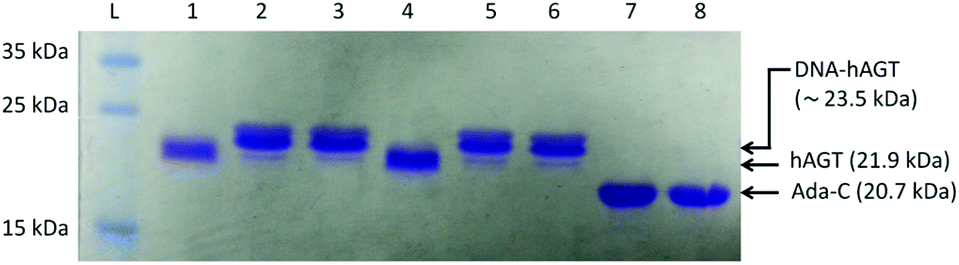 | ||
| Fig. 6 15% SDS-PAGE (Coomassie stained) of hAGT mediated repair of single-stranded GG4, GG7, TG4, and TG7, and Ada-C mediated repair of TG7. Repair of 600 pmol single-stranded IaCL DNA by 600 pmol protein for 16 h at 37 °C: lane L, unstained protein molecular weight marker; lane 1 unmodified control (GG) DNA + hAGT; lane 2 GG4 + hAGT reaction; lane 3 GG7 + hAGT reaction; lane 4 unmodified control (TG) DNA + hAGT; lane 5 TG4 + hAGT reaction; lane 6 TG7 + hAGT reaction; lane 7 unmodified control (TG) + Ada-C; lane 8 TG7 + Ada-C. Note that there is no appreciable amounts of DNA-Ada-C cross-link formation (lane 8). | ||
The DPC species were characterized by ESI-MS and the spectra of complexes produced from reacting hAGT with GG4, GG7, TG4, and TG7 for 30 min at 37 °C are shown in Fig. 7 (Panels A, B, C, and D, respectively). The spectra of butylene-linked products displayed a minor peak attributed to unreacted hAGT (21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 876 Da) and a major peak corresponding to the butylene DPC. The heptylene analogues, on the other hand, reveal the presence of only one major peak attributed to the heptylene DPC species. It should be noted that the absence of another peak with higher mass values (∼25 kDa) precludes the efficient formation of the other DPC reaction products (i.e. hAGT reacting with the 5′-residue of the IaCL). These results support the importance of the 3′-phosphodiester linkage in the repair mechanism by hAGT, as previously hypothesized.31,46 Moreover, these results are consistent with previous studies that demonstrate repair of an O6-dG-alkylene-O6-dG ICL DNA, but no repair towards an O4-dT-alkylene-O4-dT ICL DNA, by hAGT.23–25 The mixed O6-dG-heptylene-O4-dT ICL DNA was only repaired by hAGT at the dG residue33 similar to TG4 and TG7, but not GT4 and GT7 which both remained inert. The repair by hAGT of the TG4 and TG7 IaCL is illustrated in Fig. 8A.
876 Da) and a major peak corresponding to the butylene DPC. The heptylene analogues, on the other hand, reveal the presence of only one major peak attributed to the heptylene DPC species. It should be noted that the absence of another peak with higher mass values (∼25 kDa) precludes the efficient formation of the other DPC reaction products (i.e. hAGT reacting with the 5′-residue of the IaCL). These results support the importance of the 3′-phosphodiester linkage in the repair mechanism by hAGT, as previously hypothesized.31,46 Moreover, these results are consistent with previous studies that demonstrate repair of an O6-dG-alkylene-O6-dG ICL DNA, but no repair towards an O4-dT-alkylene-O4-dT ICL DNA, by hAGT.23–25 The mixed O6-dG-heptylene-O4-dT ICL DNA was only repaired by hAGT at the dG residue33 similar to TG4 and TG7, but not GT4 and GT7 which both remained inert. The repair by hAGT of the TG4 and TG7 IaCL is illustrated in Fig. 8A.
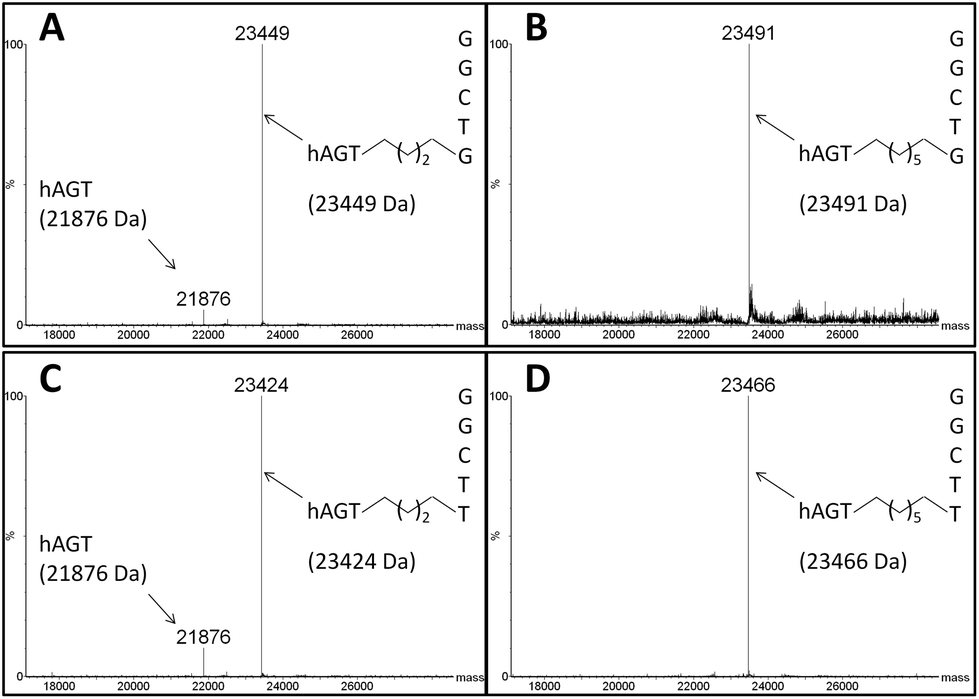 | ||
| Fig. 7 Identification of hAGT-DNA covalent complex by ESI-MS. Product identified from reaction of hAGT with single-stranded (A) GG4, (B) GG7, (C) TG4, (D) TG7. Values in brackets represent the calculated masses. | ||
 | ||
| Fig. 8 (A) hAGT-mediated repair of TG4 and TG7 IaCL. (B) Fate of TG4, TG7, GG4, and GG7 repair by hAGT producing a DPC species and a released strand. The addition of the label is optional. | ||
Cellular DPC production has become an increased concern due to their diversity and elevated cytotoxicity.51 Our IaCL approach has demonstrated a facile methodology to generate hAGT/DNA cross-linked species with excellent reaction conversions, particularly in comparison to the conjugation method using repair of ICL adducts. We have expanded the versatility of the current method by the introduction of different size alkylene tethers to form DPCs. In addition, site-specific hAGT cross-linking was possible to the O6-atom of dG or the O4-atom of dT, a feature possible due to the necessity of the 3′-phosphodiester group for efficient hAGT-mediated repair of O6-alkyl-dG adducts. There are no current methodologies, to the extent of our knowledge, to efficiently generate a DPC between hAGT and an O6-alkylene-dG residue. The N1,O6-ethanoxanthosine avenue generates an ethylene bridge between Cys145 and the N1-atom of the xanthosine nucleotide.31,32 As a proof-of-concept, we synthesized an oligomer with fluorescein tags at the 5′-end of the GG4 or GG7 DNA oligomer (as shown in Fig. 8B). Cross-linking experiments yielded a fluorescently labelled DPC species, produced with similar yields as above (ESI Fig. S70†). Given the programmability of DNA, we can envision this new “tagging” technology as having a DNA spacer between the hAGT molecule and any label compatible with DNA and protein (bio)chemistry. This would allow for added versatility of the resulting DPC species.
The IaCL may find applications in molecular nanotechnology devices as novel functional irreversible switches. There has been a growing interest in exploiting the activity AGTs for uses as nanosensors in DNA origami,52 protein tagging,53 and molecular imaging54 to name a few. The proposed IaCL switch, once inserted into a system, would benefit from orthogonality with virtually no response to other switching mechanisms like pH, UV-irradiation and toehold-mediated mechanisms. Moreover, we have shown that the efficient repair of the TG and GG IaCL are fast and reliable in producing only one DNA–protein complex. Our IaCL may be an efficient avenue to conjugate a hAGT molecule to a device or cleaving a particular DNA segment, which is part of the system, all depending on the directionality of the TG IaCL.
Experimental
Please see ESI† for details concerning the synthesis and characterization of nucleosides and oligonucleotides, UV thermal denaturation experiments, circular dichroism (CD) spectroscopy, protein over-expression and purification, repair assay details, ESI-MS identification of DPC species and additional figures.Conclusions
The introduction of a variety of different IaCL DNA mimics were prepared using a straightforward synthetic methodology in purity and scales sufficient for biophysical and biochemical studies. Our model IaCL adducts all decreased the thermal stability of duplexes by about 15–19 °C, relative to unmodified control, by UV thermal denaturation experiments. Circular dichroism spectroscopy revealed minimal global perturbation of the DNA structure relative to control duplexes. A series of well-defined hAGT-DNA covalent complexes were generated via either a butylene or heptylene linker, to either an O4-alkylated thymidinyl or an O6-alkylated 2′-deoxyguanidyl residue. We identified a novel substrate for Ada-C (TG7), with no evident amount of DPC observed using the assays described herein. Probes GT4 and GT7 remained inert to the AGTs tested, with no observable formation of 5′-tethered DPC complexes. The reason for the substrate discrimination by hAGT was hypothesized to arise from (1) the lack of the 3′-phosphodiester linkage of the O6-alkylated dG residue and/or (2) the α-carbon adopting a non-favourable conformation which prevented successful repair by hAGT. The use of IaCL DNA substrates affords an attractive versatile method for the production of site-specific hAGT-DNA covalent complex.Acknowledgements
We are grateful to Dr Anthony E. Pegg (Pennsylvania State University) for the plasmids encoding the wild-type hAGT, OGT and Ada-C genes. We are also grateful to Dr Francis McManus, Lauralicia Sacre and Dr Meena Kathiresan for helpful discussions concerning the repair assays and protein over-expression. We thank Dr Anne Noronha for helpful discussions and Alain Tessier for assistance concerning LC-MS experiments. This research was supported by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC, Grant No. 299384-2011) and the Canada Research Chair (CRC, Grant No. 950-213807) program. D.K.O. was the recipient of a Canada Graduate Scholarship (CGS) from NSERC and a Graduate Award from the Groupe de Recherche Axé sur la Structure des Protéines (GRASP).References
- N. Kondo, A. Takahashi, K. Ono and T. Ohnishi, J. Nucleic Acids, 2010, 543531 Search PubMed.
- A. E. Pegg, Chem. Res. Toxicol., 2011, 24, 618–639 CrossRef CAS PubMed.
- D. M. Noll, T. M. Mason and P. S. Miller, Chem. Rev., 2006, 106, 277–301 CrossRef CAS PubMed.
- T. Iwamoto, Y. Hiraku, S. Olkawa, H. Mizutani, M. Kojima and S. Kawanishi, Cancer Sci., 2004, 95, 454–458 CrossRef CAS PubMed.
- A. Eastman, Biochemistry, 1986, 25, 3912–3915 CrossRef CAS PubMed.
- Y. H. You, P. E. Szabó and G. P. Pfeifer, Carcinogenesis, 2000, 21, 2113–2117 CrossRef CAS PubMed.
- G. P. Pfeifer and A. Besaratinia, Photochem. Photobiol. Sci., 2012, 11, 90–97 CAS.
- D. E. Brash, J. A. Rudolph, J. A. Simon, A. Lin, G. J. McKenna, H. P. Baden, A. J. Halperin and J. Pontén, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 10124–10128 CrossRef CAS.
- S. Bellon, D. Gasparutto, C. Saint-Pierre and J. Cadet, Org. Biomol. Chem., 2006, 4, 3831–3837 CAS.
- D. T. Beranek, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1990, 231, 11–30 CrossRef CAS PubMed.
- B. Singer and J. M. Essigmann, Carcinogenesis, 1991, 12, 949–955 CrossRef CAS PubMed.
- B. Singer, Cancer Res., 1986, 46, 4879–4885 CAS.
- G. T. Pauly, S. H. Hughes and R. C. Moschel, Carcinogenesis, 1998, 19, 457–461 CrossRef CAS PubMed.
- P. F. Swann, Mutat. Res., 1990, 233, 81–94 CrossRef CAS PubMed.
- A. Rasouli-Nia, Sibghat-Ullah, R. Mirzayans, M. C. Paterson and R. S. Day III, Mutat. Res., 1994, 314, 99–113 CAS.
- S. M. Bronstein, T. R. Skopek and J. A. Swenberg, Cancer Res., 2011, 52, 2008–2011 Search PubMed.
- L. Samson, S. Han and J. C. Marquis, Carcinogenesis, 1997, 18, 919–924 CrossRef CAS PubMed.
- T. P. Brent and J. S. Remack, Nucleic Acids Res., 1988, 16, 6779–6788 CrossRef CAS PubMed.
- E. Gonzaga, P. Margison, P. M. Potter, T. P. Brent, D. Yu, D. B. Ludlum and A. Rafferty, Cancer Res., 1992, 52, 6052–6058 Search PubMed.
- M. J. M. Darkes, G. L. Plosker and B. Jarvis, Am. J. Cancer, 2002, 1, 55–80 CrossRef CAS.
- S. R. Wedge, J. K. Porteous and E. S. Newlands, Cancer Chemother. Pharmacol., 1997, 40, 266–272 CrossRef CAS PubMed.
- M. E. Dolan and A. E. Pegg, Clin. Cancer Res., 1997, 3, 837–847 CAS.
- Q. Fang, A. M. Noronha, S. P. Murphy, C. J. Wilds, J. L. Tubbs, J. A. Tainer, G. Chowdhury, F. P. Guengerich and A. E. Pegg, Biochemistry, 2008, 47, 10892–10903 CrossRef CAS PubMed.
- F. P. McManus, Q. Fang, J. D. M. Booth, A. M. Noronha, A. E. Pegg and C. J. Wilds, Org. Biomol. Chem., 2010, 8, 4414–4426 CAS.
- F. P. McManus, D. K. O'Flaherty, A. M. Noronha and C. J. Wilds, Org. Biomol. Chem., 2012, 10, 7078–7090 CAS.
- D. K. O'Flaherty and C. J. Wilds, Chem. – Asian J., 2016, 11, 576–583 CrossRef PubMed.
- R. Loeber, M. Rajesh, Q. Fang, A. E. Pegg and N. Tretyakova, Chem. Res. Toxicol., 2006, 19, 645–654 CrossRef CAS PubMed.
- H. Liu, M. Xu-Welliver and A. E. Pegg, Mutat. Res., 2000, 452, 1–10 CrossRef CAS PubMed.
- N. Abril, F. L. LuqueRomero, M. J. PrietoAlamo, J. A. Rafferty, G. P. Margison and C. Pueyo, Carcinogenesis, 1997, 18, 1883–1888 CrossRef CAS PubMed.
- L. Liu, A. E. Pegg, K. M. Williams and F. P. Guengerich, J. Biol. Chem., 2002, 277, 37920–37928 CrossRef CAS PubMed.
- D. S. Daniels, T. T. Woo, K. X. Luu, D. M. Noll, N. D. Clarke, A. E. Pegg and J. A. Tainer, Nat. Struct. Mol. Biol., 2004, 11, 714–720 CAS.
- D. M. Noll and N. D. Clarke, Nucleic Acids Res., 2001, 29, 4025–4034 CAS.
- F. P. McManus, A. Khaira, A. M. Noronha and C. J. Wilds, Bioconjugate Chem., 2013, 24, 224–233 CrossRef CAS PubMed.
- H. Yokoyama and R. Mizutani, Int. J. Mol. Sci., 2014, 15, 20321–20338 CrossRef CAS PubMed.
- A. Gentil, F. Le Page, A. Margot, C. W. Lawrence, A. Borden and A. Sarasin, Nucleic Acids Res., 1996, 24, 1837–1840 CrossRef CAS PubMed.
- P. Rojsitthisak, N. Jongaroonngamsang, R. M. Romero and I. S. Haworth, PLoS One, 2011, 6, e20745 CAS.
- H. C. Box, H. B. Patrzyc, J. B. Dawidzik, J. C. Wallace, H. G. Freund, H. Iijima and E. E. Budzinski, Radiat. Res., 2000, 153, 442–446 CrossRef CAS PubMed.
- H. C. Box, E. E. Budzinski, J. B. Dawidzik, J. S. Gobey and H. G. Freund, Free Radical Biol. Med., 1997, 23, 1021–1030 CrossRef CAS PubMed.
- C. Gu and Y. Wang, Biochemistry, 2004, 43, 6745–6750 CrossRef CAS PubMed.
- Y. Jiang, H. Hong, H. Cao and Y. Wang, Biochemistry, 2007, 46, 12757–12763 CrossRef CAS PubMed.
- S. Bellon, J. L. Ravanat, D. Gasparutto and J. Cadet, Chem. Res. Toxicol., 2002, 15, 598–606 CrossRef CAS PubMed.
- D. K. O'Flaherty and C. J. Wilds, Chem. – Eur. J., 2015, 21, 10522–10529 CrossRef PubMed.
- L. Samson, J. Thomale and M. F. Rajewsky, EMBO J., 1988, 7, 2261–2267 CAS.
- R. J. Graves, B. F. Li and P. F. Swann, Carcinogenesis, 1989, 10, 661–666 CrossRef CAS PubMed.
- F. P. McManus and C. J. Wilds, ChemBioChem, 2014, 15, 1966–1977 CrossRef CAS PubMed.
- J. Hu, A. Ma and A. R. Dinner, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4615–4620 CrossRef CAS PubMed.
- K. Abdu, M. K. Aiertza, O. J. Wilkinson, J. A. Grasby, P. Senthong, A. C. Povey, G. P. Margison and D. M. Williams, Chem. Commun., 2012, 48, 11214–11216 RSC.
- K. Wiechelman and E. R. Taylor, J. Biomol. Struct. Dyn., 1998, 15, 1181–1194 CAS.
- F. P. McManus and C. J. Wilds, Toxicol. Res., 2013, 2, 158–162 RSC.
- A. E. Pegg, K. Morimoto and M. E. Dolan, Chem.–Biol. Interact., 1988, 65, 275–281 CrossRef CAS PubMed.
- J. Stingele and S. Jentsch, Nat. Rev. Mol. Cell Biol., 2015, 16, 455–460 CrossRef CAS PubMed.
- M. Tintoré, I. Gállego, B. Manning, R. Eritja and C. Fàbrega, Angew. Chem., Int. Ed., 2013, 52, 7747–7750 CrossRef PubMed.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed.
- N. B. Cole and J. G. Donaldson, ACS Chem. Biol., 2012, 7, 464–469 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, experimental procedures, compound characterization, modified oligomer characterization, additional UV thermal denaturation experiments, additional circular dichroism experiments, and additional repair assays. See DOI: 10.1039/c6ob02246d |
| This journal is © The Royal Society of Chemistry 2017 |