
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxoanion binding to a cyclic pseudopeptide containing 1,4-disubstituted 1,2,3-triazole moieties†
Disha
Mungalpara
a,
Harald
Kelm
b,
Arto
Valkonen
c,
Kari
Rissanen
c,
Sandro
Keller
 d and
Stefan
Kubik
*a
d and
Stefan
Kubik
*a
aTechnische Universität Kaiserslautern, Fachbereich Chemie – Organische Chemie, Erwin-Schrödinger-Straße, 67663 Kaiserslautern, Germany. E-mail: kubik@chemie.uni-kl.de
bTechnische Universität Kaiserslautern, Fachbereich Chemie – Anorganische Chemie, Erwin-Schrödinger-Straße, 67663 Kaiserslautern, Germany
cUniversity of Jyvaskyla, Department of Chemistry, Nanoscience Center, P.O. Box 35, Jyvaskyla FI-40014, Finland
dUniversity of Kaiserslautern, Molecular Biophysics, Erwin-Schrödinger-Str. 13, 67663 Kaiserslautern, Germany
First published on 27th October 2016
Abstract
A macrocyclic pseudopeptide 3 is described featuring three amide groups and three 1,4-disubstituted 1,2,3-triazole units along the ring. This pseudopeptide was designed such that the amide NH groups and the triazole CH groups converge toward the cavity, thus creating an environment well suited for anion recognition. Conformational studies in solution combined with X-ray crystallography confirmed this preorganisation. Solubility of 3 restricted binding studies to organic media such as 5 vol% DMSO/acetone or DMSO/water mixtures with a water content up to 5 vol%. These binding studies demonstrated that 3 binds to a variety of inorganic anions in DMSO/acetone including chloride, nitrate, sulfate, and dihydrogenphosphate anions. In the more competitive DMSO/water mixtures, only affinity to the more strongly coordinating oxoanions is retained. Quantitative binding studies showed that dihydrogen phosphate complexation in DMSO/water involves the dimer of the H2PO4− anion. By contrast, sulfate and hydrogenpyrophosphate complexation involves a stepwise process comprising formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex followed by a 2R:1A complex in which two molecules of 3 (R) bind to a single anion (A). While the second binding equilibrium is associated with a much smaller stability constant in comparison with the first one in the case of sulfate complexation, the two binding constants are of similar magnitude in the case of the hydrogenpyrophosphate anion. Formation of the 2R:1A complex was attributed to the fact that the cavity size and rigidity of 3 prevents saturation of all hydrogen acceptor sites on the anionic guests.
:1 complex followed by a 2R:1A complex in which two molecules of 3 (R) bind to a single anion (A). While the second binding equilibrium is associated with a much smaller stability constant in comparison with the first one in the case of sulfate complexation, the two binding constants are of similar magnitude in the case of the hydrogenpyrophosphate anion. Formation of the 2R:1A complex was attributed to the fact that the cavity size and rigidity of 3 prevents saturation of all hydrogen acceptor sites on the anionic guests.
Introduction
There is hardly a field in organic chemistry today in which 1,2,3-triazole-based building blocks have not made an impact. Formation of 1,2,3-triazoles from azides and alkynes via 1,4-dipolar Huisgen cycloaddition1 has, for example, become a potent ligation strategy with applications in organic synthesis,2 chemical biology,3 supramolecular chemistry,4 and more. The major factor that triggered this development was the discovery of the copper(I)-catalysed variant of the azide–alkyne cycloaddition by Meldal and Sharpless with which drawbacks of the thermal reaction such as low rate and low regioselectivity could be overcome.5 Today, this cycloaddition is considered the prototype of a click-reaction,6 whose importance is progressively increasing also because synthetic approaches to access triazoles are continuously being developed. Examples are the ruthenium(II)-catalysed version that affords 1,5-disubsituted rather than 1,4-disubstituted 1,2,3-triazoles,7 metal-free strain-promoted cycloadditions,8 and more recently organocatalysed triazole syntheses.9In the area of chemical biology, the azide–alkyne cycloaddition benefits from the bioorthogonality of azide and alkyne groups, which allows using this reaction even in vivo.3,8 In addition, 1,2,3-triazoles are mimics of peptide bonds, not only in terms of geometry but also in their electronic properties, so that 1,4-disubstituted and 1,5-disubstituted 1,2,3-triazoles can serve as surrogates for, respectively, trans-peptide and cis-peptide bonds in peptide mimics.10
In supramolecular chemistry, the use of 1,2,3-triazoles often extends beyond simply linking two building blocks because triazoles also feature characteristic recognition elements, namely, the nitrogen atoms for coordination to transition metal ions and a hydrogen bond donor in the form of the CH group.4 The hydrogen bonding ability of 1,2-3-triazoles combined with the ease of their synthesis renders them particularly potent and versatile building blocks for the construction of anion receptors.11 Significant contributions in this area came from the groups of Flood,12 Craig,13 Hecht,14 Beer,15 and Schubert.16 Notably, anion-binding properties of receptors with triazole subunits can be further modulated by converting triazole into triazolium derivatives or by replacing the proton on the CH group with a halogen and making use of halogen-bonding for anion coordination.17 In addition, triazole residues and other anion binding elements, such as carboxamide groups, have also been combined to develop anion receptors.18
In earlier work, we used the 1,2,3-triazole units in cyclic pseudopeptide 1 mainly for structural purposes, namely, to induce conformations similar to those found for cyclopeptide 2 (Fig. 1a).19 This cyclopeptide was shown to possess high affinity for sulfate and iodide anions even in competitive aqueous solvent mixtures.20 Anion affinity turned out to be partly due to the rigid and well preorganised structure of 2 featuring cis-amides at the tertiary amide bonds and a converging arrangement of the NH and proline CαH protons, which serve as hydrogen bond donors. The orientation of the NH groups can be explained by the presence of the pyridine nitrogen atoms that destabilise arrangements with ring nitrogen and carbonyl oxygen atoms located in close proximity (Fig. 1b).
 | ||
| Fig. 1 Structures of macrocyclic receptors 1–4 (top) and effect of the ring nitrogen atom on the orientation of the adjacent amide group (bottom). | ||
Based on these results, pseudopeptide 1 was devised by retaining the pyridine units of 2 and replacing the cis-amides with 1,5-disubstituted 1,2,3-triazole units.19 Because of the structural relationship of cis-amides and 1,5-disubstituted 1,2,3-triazole rings, 1 and 2 were expected to adopt similar overall conformations, which turned out to be the case. As a consequence, 1 also interacts with sulfate and halide anions in competitive aqueous media, although characteristic differences in the binding properties of the two receptors were noted.19
To access new macrocyclic motifs for anion recognition, the approach of incorporating 1,2,3-triazole units along the backbone of such pseudopeptides has now been extended to compound 3 containing 1,4-disubstituted triazole rings. This pseudopeptide should again feature conformations with converging NH groups due to the orienting effects of the pyridine nitrogen atoms. These conformations should, however, substantially differ from the ones of 1 or 2 because of the structural relationship of 1,4-disubstituted triazoles to trans-amides. Thus, 3 conformationally likely resembles cyclopeptides such as 4, which also interact with anions, albeit in organic media.21 The advantages of 3 should be a better preorganisation for anion binding in comparison with 4 and a higher number of hydrogen bond donors with respect to receptors 1, 2, and 4 because the triazole CH groups in 3 could participate in the interactions with the substrate.
Here, we show how the 1,4-disubstituted triazole units affect the conformation of 3 and how the combination of NH and triazole CH hydrogen donor groups along the ring affects the anion binding properties of this pseudopeptide.
Results and discussion
Synthesis
Pseudopeptide 3 was prepared along a similar route as the analogue 1.19 The known monomer 5, featuring a trimethylsilyl-protected alkyne group on one end and a mesylate group on the other, acted as the central building block (Scheme 1). Chain elongation involved initial conversion of 5 into the derivatives 6a and 6b by cleaving the TMS group with tetrabutylammonium fluoride (TBAF) and replacing the mesylate with an azide group under inversion of the configuration at the stereogenic centre, respectively. Compounds 6a and 6b now contained the necessary functional groups to couple them by using a copper(I)-catalysed azide–alkyne cycloaddition. | ||
| Scheme 1 Synthesis of cyclic pseudopeptide 3. | ||
The resulting dimer 7 was chain-elongated along a similar route to the trimer 8, which was subsequently deprotected on the alkyne group and then transformed into the azide. Finally, cyclisation in the presence of copper(I) afforded the desired product 3. Isolation of 3 involved a couple of washing, extraction, and crystallisation steps but did not require chromatography. The isolated yield of product typically amounted to ca. 30%, which is acceptable for such types of macrocyclisation reactions. The alternative approach of using standard peptide chemistry for the synthesis of 3 is not feasible because the required building blocks would contain a stereogenic centre flanked by a carboxylate and a triazole ring, which is typically rather prone to racemisation once the carboxylate group is activated.22
Structural analysis
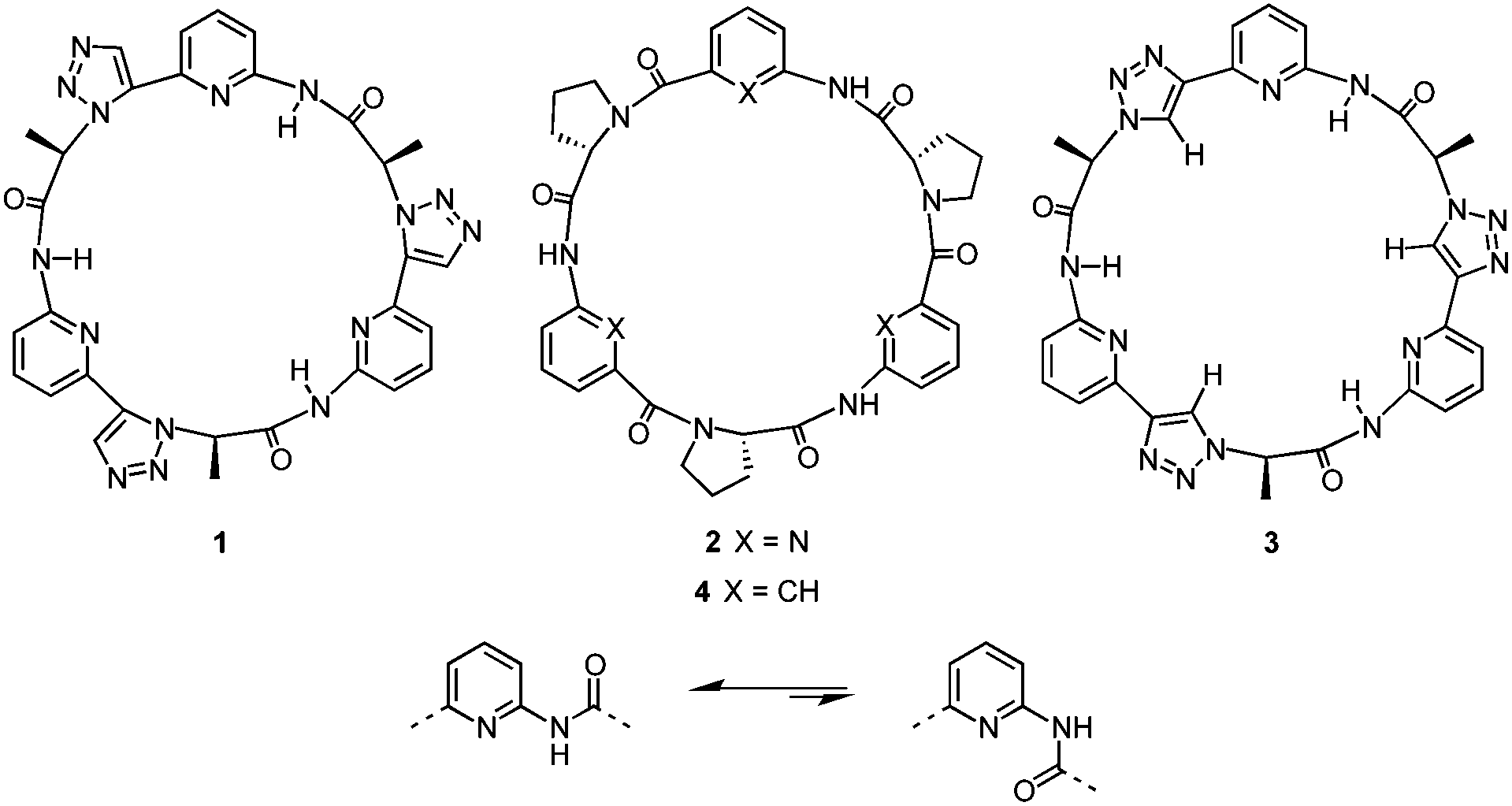
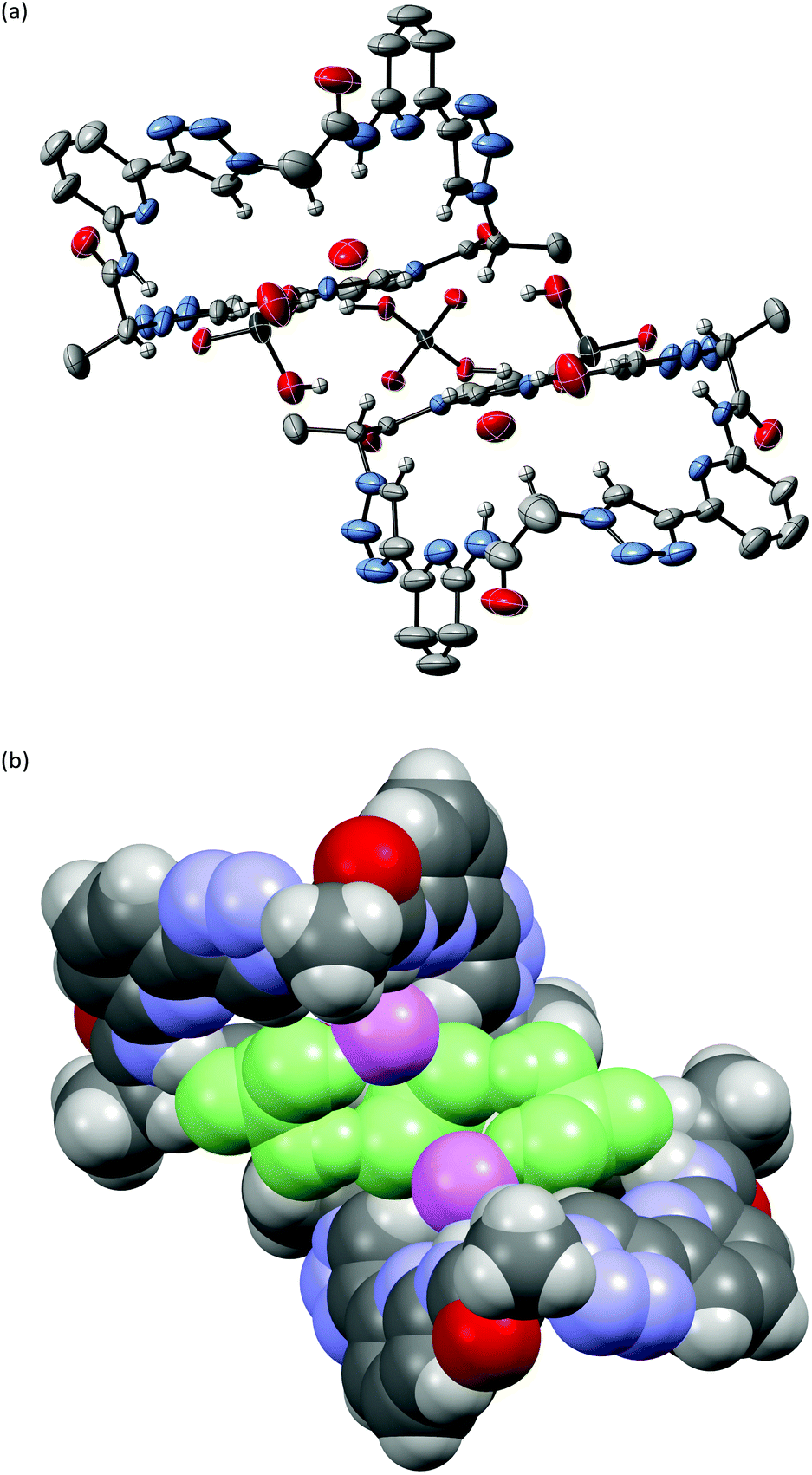
X-ray crystallography of crystals grown from DMSO confirmed the expected constitution of 3. The solid-state structure shows that this pseudopeptide crystallises with 5 molecules of DMSO, one of which resides inside the bowl-shaped cavity defined by the aromatic subunits (Fig. 2).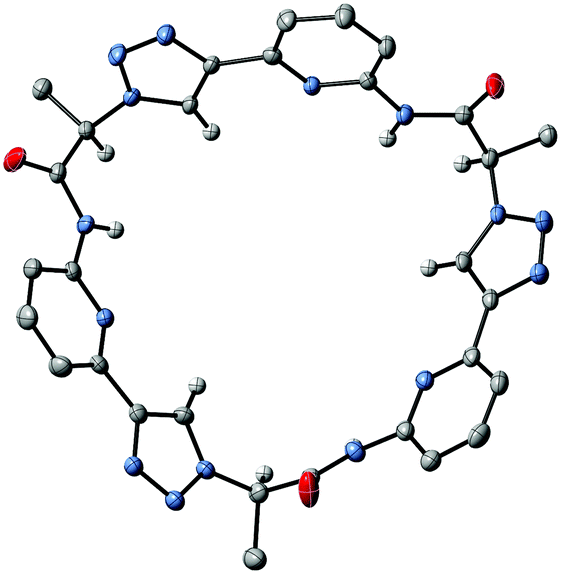 | ||
| Fig. 2 Molecular structure of 3·5DMSO·0.35H2O in the solid state with the thermal ellipsoids shown at the 50% probability level. Solvent molecules and hydrogen atoms except those on the NH and triazole CH groups as well as those on the stereogenic centres C*H are omitted for clarity. | ||
Three DMSO molecules are located on the opposite side of the cavity, interacting with the pseudopeptide via hydrogen bonds between the DMSO oxygen atoms and the pseudopeptide NH groups. The fifth DMSO molecule is disordered with a 0.65:0.35 occupancy ratio. The orientation with the lower abundance shares the space with an additional water molecule. As expected, all three NH protons of 3 point into the direction of the narrow cavity opening as a consequence of the conformational control exerted by the pyridine nitrogen atoms. Importantly, the three 1,2,3-triazole protons point into the same direction as the NH groups, indicating that all six hydrogen donors of 3 could participate in anion binding.
To illustrate the effect of the triazole units in 3 on pseudopeptide conformation, the solid-state structures of compounds 1 and 3 are compared in Fig. 3. Both structures are approximately C3-symmetric and share the converging arrangement of the NH group. They differ in the orientations of the triazole CH groups and in the diameters of the rings, with the one of 3 being larger due to the 1,4-connections on the triazole units. Taking the distance of the ring nitrogen atoms in 1 and 3 as a measure for ring size shows that this distance increases from an averaged 6.16 Å to an averaged 7.31 Å when going from 1 to 3. As the aromatic rings in 3 are also slightly more tilted than in 1, pseudopeptide 3 has a shallower cavity somewhat reminiscent of that found in the N-methylquinuclidinium iodide complex of 4.21 Overall, the solid-state structure provides evidence that 3 should likely be able to interact with anions, but differences with respect to the anion properties of 1 are to be expected.
 | ||
| Fig. 3 Comparison of the solid-state molecular structures of 3·5DMSO·0.35H2O (left) and 1·C3H6O·H2O (right). Solvent molecules and hydrogen atoms except those on the NH and triazole CH groups as well as those on the stereogenic centres C*H are omitted for clarity. | ||
Characterisation of the solution structure was strongly affected by the poor solubility of 3, which was significantly lower than that of 1. While 1 is soluble in a wide range of solvent mixtures ranging from water/methanol mixtures over acetone and DMSO to chloroform, 3 is not soluble in aqueous methanol mixtures. Concentrations sufficiently high for NMR analyses could be obtained only in DMSO or in organic solvents containing at least 5 vol% of DMSO.
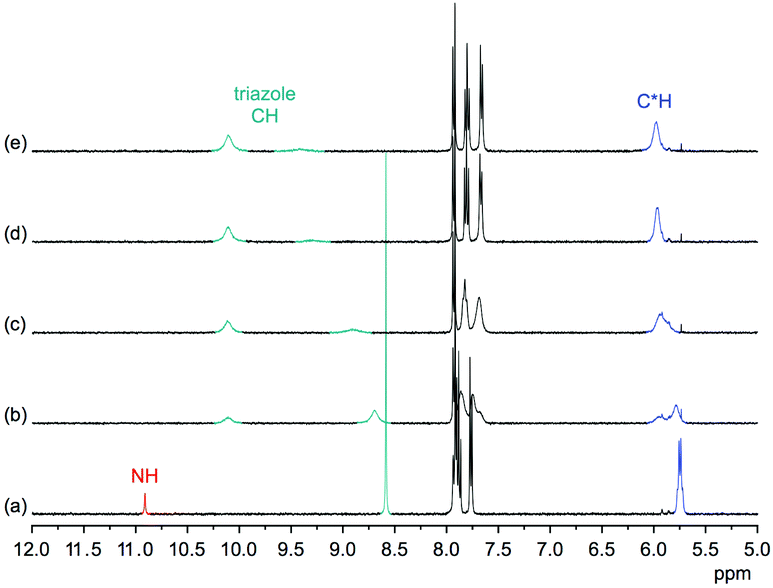
Comparison of the 1H NMR spectra of 3 in 5 vol% DMSO-d6/CDCl3, 5 vol% DMSO-d6/acetone-d6, DMSO-d6, and 2.5 vol% DMSO-d6/D2O showed that the pseudopeptide adopts an averaged C3-symmetric conformation in all of these solvents (Fig. 4a). The signals are sharp, with the exception of those in the spectrum recorded in the chloroform mixture, in which some signal broadening was observed. Increasing the DMSO content of the mixtures caused a pronounced downfield shift of the NH signal, demonstrating the tendency of DMSO molecules to interact with the NH groups of 3, as also observed in the crystal structure. Also the signal of the triazole CH shifts, but the ones of other protons are not affected to a large extent.
 | ||
| Fig. 4 Comparison of the 1H NMR spectra of 3 in 5 vol% DMSO-d6/CDCl3 (a), 5 vol% DMSO-d6/acetone-d6 (b), DMSO-d6 (c), and 2.5 vol% D2O/DMSO-d6 (d) (top) and schematic representation of the crosspeaks found in the NOESY NMR spectrum of 3 in DMSO-d6 (bottom). | ||
The NOESY NMR of 3 in DMSO-d6 exhibits crosspeaks between the signal of the protons on the stereogenic centres of 3 and those belonging to the NH and the triazole CH groups (Fig. 4b). These crosspeaks account for a spatial proximity of the corresponding protons. As no crosspeak between the signal of the NH protons and that of the H3 protons on the aromatic residues was observed, the pseudopeptide seems to adopt an average conformation in solution similar to that found in the solid state.
Qualitative binding studies
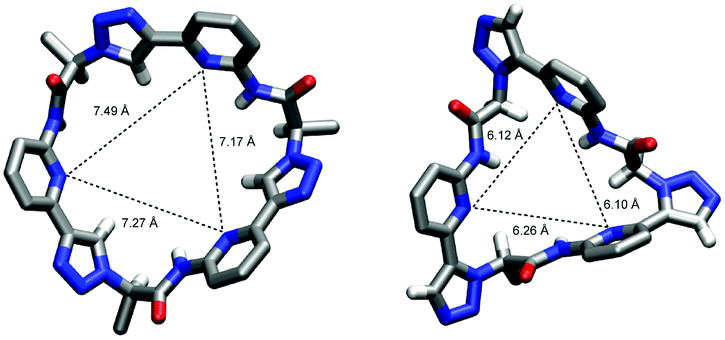
Information about the ability of 3 to interact with anions was obtained by evaluating the effects of various anions on the 1H NMR spectrum of 3. Initially, we used 5 vol% DMSO-d6/acetone-d6 as solvent and recorded 1H NMR spectra of 1 mM solutions of 3 after addition of 5 equiv. of different tetrabutylammonium (TBA) salts (Fig. 5). These investigations showed that the anions mainly cause downfield shifts of three signals, namely, those of the NH, triazole CH, and the C*H on the stereogenic centre. The signal belonging to the peripheral methyl groups of 3 is also affected, but to a significantly smaller extent (not shown). | ||
| Fig. 5 1H NMR spectrum of 3 (1 mM) in 5 vol% DMSO-d6/acetone-d6 in the absence (a) and the presence of 5 equiv. of TBA iodide (b), bromide (c), chloride (d), nitrate (e), dihydrogenphosphate (f), and sulfate (g). The signals of the NH, triazole and C*H protons are marked in red, green, and blue, respectively. | ||
The extents of the signal shifts differ significantly among the different anions. In the halide series, chloride causes the strongest effect and iodide the weakest. For the oxoanions, the anion effect is weakest for nitrate and strongest for sulfate and dihydrogenphosphate (DHP). In the case of the latter two anions, the NH signal is not visible in the spectrum presumably because these anions promote exchange. In addition, the signals of 3 broaden in the presence of sulfate anions.
In the more competitive solvent mixture 2.5 vol% D2O/DMSO-d6, no interactions of halides or nitrate with 3 could be detected, and the only investigated anions that cause changes in the 1H NMR spectrum of 3 are DHP and sulfate anions (see ESI†). Again, mainly the triazole CH and C*H signals shift (the NH signal is not visible in this solvent mixture because of H/D exchange), indicating that the mode of anion binding is not affected by the change of the solvent.
These results indicate that anion binding most likely occurs at the smaller cavity opening of 3, where the NH, triazole CH, and C*H protons converge. Thus, the expected direct participation of the triazole moieties of 3 in anion binding is indeed evident. Assuming that the extents of the signal shifts correlate with binding strength, anion affinity of 3 in 5 vol% DMSO-d6/acetone-d6 reflects the normal coordinating ability of anions, being stronger for more strongly coordinating anions such as the oxoanions sulfate and DHP and the small chloride anion. In the more competitive D2O/DMSO-d6 mixture, anion binding is weakened to such an extent that only complex formation with the oxoanions is retained. Thus, the ability of 1 to bind anions even in highly competitive protic solvents19 is lost upon moving one substituent on each triazole unit from the 5 into the 4 position despite the good preorganisation of 3 for anion binding and the presence of the three additional hydrogen-bond donors along the ring that contribute to anion recognition. This shows how important the unique conformation of 1 (and of 2) is for anion binding in protic media.
Nevertheless, pseudopeptide 3 possesses characteristic properties not observed for 1 or 2 such as an affinity for DHP anions and we therefore concentrated on quantitatively evaluating the interactions of 3 with oxoanions in DMSO.
Quantitative binding studies
:1 equilibrium in 2.5 vol% D2O/DMSO-d6 (see ESI†). The shapes of the binding isotherms subsequently obtained from isothermal titration calorimetry (ITC) and NMR titrations showed that the equilibrium is more complex, however. Although binding is indeed mainly governed by a 1:1 complex, a 2R:1A complex in which two molecules of 3 (R) bind to a single anion (A) has to be considered when the receptor is present in excess.
The results of the ITC titrations are summarized in Table 1. These titrations were performed in different solvent mixtures to elucidate effects of solvent composition on complex stability. Each titration was typically performed in triplicate with freshly prepared solutions of 3 and TBA sulfate, and the thermodynamic parameters were derived by fitting the obtained isotherms to a model that considers binding of up to two receptors (R) to one anion (A), thereby allowing for the formation of both 1:1 and 2R:1A complexes. Although weak binding of a second receptor was evident from the isotherms, only the stability constants of the 1:1 complex could be accurately determined. For the second binding event only an upper limit of K21 could be reasonably estimated.23
| Vol% H2O in H2O/DMSO | logK11a |
logK21b |
c |
c |
|---|---|---|---|---|
|
a Equilibrium constant describing the formation of the 1:1 complex.
b Equilibrium constant describing the formation of the 2R:1A complex from the 1:1 complex.
c Enthalpies and entropies associated with the formation of the 1:1 complex in kJ mol−1.
d Maximum water content of the DMSO (99.6%) used.
e Determined by NMR titration with an estimated error of 10%.
|
||||
| 0.03d | 4.08 (4.00 to 4.15) | <3.2 | 39.7 (37.5 to 42.2) | 62.9 |
| 2.5 | 4.39 (4.36 to 4.42) | <2.4 | −14.1 (−14.4 to −13.8) | 11.0 |
| 4.22e | <2.1e | |||
| 5 | 4.05 (4.00 to 4.09) | <2.3 | −21.6 (−22.7 to −20.6) | 1.5 |
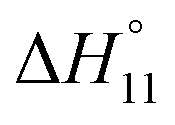
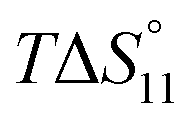
An NMR titration performed in 2.5 vol% D2O/DMSO-d6, whose results are also included in Table 1, yielded similar results. The binding constant K11 resulting from this titration is in excellent agreement with the one obtained by ITC in the same solvent mixture. Moreover, the sigmoidal shape of the binding isotherms resulting from this titration (see ESI†) provided clear evidence for the formation of higher complexes, consistent with the ITC titrations. Unfortunately, also in this case the second, weaker binding constant turned out to be difficult to quantify. Based on the regression analysis one can only safely state that K21 is at least two orders of magnitude smaller than K11. This ratio of the stepwise binding constants shows that substantial amounts of the 2R:1A complex are formed only when 3 is present in excess, which explains why this complex is not visible in the Job plot.24
According to the results of the binding studies, the stability of the 1:1 complex between 3 and a sulfate anion is relatively unaffected by solvent composition. Interestingly, complex stability is slightly higher in 2.5 vol% H2O/DMSO than in the solvent mixtures containing less and more water. Although the difference is small, it mirrors the effect of water on the anion-induced shift of the triazole signal in the NMR spectra. Fig. 6 shows that addition of D2O to a DMSO-d6 solution of the sulfate complex of 3 causes the triazole CH and C*H signals to shift downfield in the 1H NMR spectrum until a D2O content of 2 vol% is reached, with a concomitant sharpening of the signals. Only when further increasing the D2O content of the solution is the expected upfield shift of these signals that indicates weakening of anion-binding being observed. In the absence of TBA sulfate, the 1H NMR spectrum of 3 is practically unaffected when varying the D2O content of the D2O/DMSO-d6 mixture between 0.03% and 5% (see ESI†).
 | ||
| Fig. 6 1H NMR spectrum of 3 (1 mM) in the presence of 5 equiv. of TBA sulfate in D2O/DMSO-d6 mixtures with the D2O content amounting to 0.03 vol% (a), 1 vol% (b), 2 vol% (c), 3 vol% (d) 4 vol% (e), and 5 vol% (f). The signals of the triazole and C*H protons are marked in green and blue, respectively. | ||
The effect of water on the 1H NMR spectrum of the sulfate complex of 3 and the quantitative results of the binding studies indicate that small amounts of water seem to reinforce sulfate complexation. A possible explanation could be that sulfate anions associated with one or more water molecules better fit the available space in the cavity of 3 similarly as in the DHP complex of 3 (vide infra) in which one water molecule was found to bridge hydrogen bond donors along the receptor cavity and oxygen atoms of the anion. Note that under the conditions used for recording the NMR spectra in Fig. 6 (i.e., excess of sulfate), the presence of a higher complex containing more than one pseudopeptide ring can be neglected.
Formation of the 1:1 complex is endothermic in DMSO and becomes increasingly exothermic when the solvent mixture contains more water. Conversely, the large favourable contribution of entropy to complex formation in DMSO decreases. Note that the enthalpies and entropies associated with the formation of the 2R:1A complexes are not considered in Table 1 because the much lower stability of this complex renders their estimation unreliable.
In spite of the clearly visible trends of 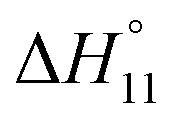 and
and 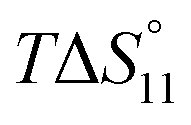 with solvent composition, correlating them with direct receptor–anion interactions or solvation effects is not straightforward because the effects of water molecules involved in complex formation are difficult to estimate.
with solvent composition, correlating them with direct receptor–anion interactions or solvation effects is not straightforward because the effects of water molecules involved in complex formation are difficult to estimate.
These binding studies thus demonstrated that sulfate complexation of receptors 1, 2, and 3 follows related models: all three compounds have a tendency to form complexes with sulfate anions in which two receptor units bind to a single anion. The reasons of forming these higher complexes differ, however. In the case of 2 and to a lesser extent 1, 2R:1A complexes are found in polar protic solvents, and a major driving force of their formation comes from hydrophobic effects between the two receptor rings. Increasing the water content of the solvent thus leads to a strengthening of the second binding step with respect to the first one.19,26 By contrast, sulfate binding of 3 takes place in organic media and is likely caused by the tendency of the anion to saturate its hydrogen-bond acceptor sites. If this cannot be achieved in the 1:1 complex, a second receptor molecule is recruited. Formation of this higher complex is, however, weak in the case of sulfate complexation and becomes even weaker when the water content of the solution increases. Hydrophobic effects between the receptor units in the corresponding 2R:1A complex of 3 are absent in DMSO and probably not even possible because of the significantly different conformations of 1 and 3.
:2A stoichiometry was then confirmed by ITC and NMR titrations as well as X-ray crystallography. Binding constants and thermodynamic parameters derived from titrations in different solvent mixtures are compiled in Table 2.
| Vol% H2O in H2O/DMSO | logK11a |
logK12b |
c |
c |
c |
c |
|---|---|---|---|---|---|---|
|
a Equilibrium constant describing the formation of the 1:1 complex.
b Equilibrium constant describing the formation of the 1R:2A complex from the 1:1 complex.
c Enthalpies and entropies associated with the formation of the 1:1 and 1:2 complexes in kJ mol−1.
d Maximum water content of the DMSO (99.6%) used.
e Determined by NMR titration with an estimated error of 10%.
|
||||||
| 0.03d | 3.85 (3.50 to 4.20) | 3.53 (3.03 to 4.03) | −9.8 (−11.5 to −8.7) | 12.2 | −16.7 (−20.1 to −11.5) | 3.4 |
| 2.5 | 3.46 (3.17 to 3.76) | 3.22 (2.82 to 3.62) | −7.2 (−10.2 to −5.6) | 12.5 | −24.1 (−27.7 to −17.4) | −5.7 |
| 3.21e | 3.10e | |||||
| 5 | 3.57 (3.37 to 3.79) | 2.82 (2.57 to 3.06) | −2.8 (−3.4 to −2.3) | 17.6 | −26.1 (−27.3 to −24.6) | −10.0 |
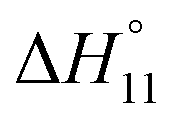
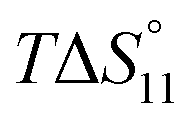
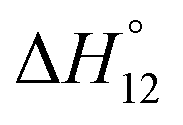
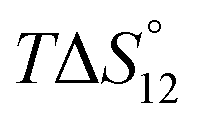
This table shows that the results of ITC and NMR titrations are in very good agreement, this time providing reliable information about the stability constants associated with both binding steps.
In contrast to sulfate binding, the stability constants associated with the 1:1 complex continuously decrease with increasing water content. Moreover, complex formation is exothermic in DMSO and becomes enthalpically less favourable the more water is present. Conversely, the favourable contribution of entropy increases in the same direction.
Once the 1:1 complex is formed, binding of a second DHP anion is favourable as indicated by the fact that the second binding step is strongly exothermic and associated with a stability constant almost equal in size as the one associated with the corresponding 1:1 complex.
The tendency of forming the 1R:2A complexes decreases when increasing the water content of the solvent, a trend that is mainly caused by entropy as the binding enthalpy of forming the 1R:2A complex becomes even more favourable in solvent mixtures containing more water. Because of the continuous decrease of K12, the overall stability of the DHP complex of 3 decreases with increasing water content of the solvent, consistent with the effect of water on the 1H NMR spectrum of this complex (see ESI†). Moreover, binding of the second DHP anion is weakly cooperative in DMSO and 2.5 vol% H2O/DMSO, but not in 5 vol% H2O/DMSO.‡
Structural information about the DHP complex of 3 was obtained from a crystal structure. Crystals were grown by slow evaporation of a solution of 3 (1.2 mM) in DMSO/acetone, 1:1 (v/v) containing 2 equiv. of TBADHP. The arrangement of anions and 3 in these crystals is depicted in Fig. 7. Accordingly, two molecules of 3 bind to three DHP anions in the solid state; the 3/TBADHP ratio therefore deviates from the one found in solution and amounts to 2:3. The conformation of 3 in the complex 3·1.5TBADHP·2.5DMSO·1.7H2O is similar as that observed in 3·5DMSO·0.35H2O shown in Fig. 2 (for an overlay, see ESI†). Thus, complex formation does not require a substantial conformational reorganisation of the pseudopeptide. In 3·1.5TBADHP·2.5DMSO·1.7H2O one of the DMSO molecules resides in the concave cavity of 3, while the others are located outside where also the TBA counterions, one of which is disordered on a symmetry element, can be found. The DHP anions form a tight hydrogen-bonded trimer with short, 2.46 and 2.51 Å, P–O⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P distances between O2⋯O5 and O3⋯O6, respectively (Fig. S2†). Each pseudopeptide ring hydrogen bonds to the terminal anions of the DHP trimer while the central anion is hydrogen bonded to both of the macrocycles. Ten of the twelve oxygen atoms of the DHP trimer interact with hydrogen bond donors along the pseudopeptide rings, two NH groups and three triazole CH groups per ring. With O⋯N distances of 2.76 and 2.92 Å the lengths of the hydrogen bonds to the NH groups are shorter than the ones to the triazole C–H groups, whose O⋯C distances amount to 3.19, 3.24, and 3.53 Å. Two water molecules are also present in the complex, one per pseudopeptide, which bridge the bound anions and the third NH group of each ring. There is no evidence that the pyridine or triazole nitrogen atoms of 3 participate in complex formation as they do not act as hydrogen bond acceptors with the OH groups of the DHP anions.
P distances between O2⋯O5 and O3⋯O6, respectively (Fig. S2†). Each pseudopeptide ring hydrogen bonds to the terminal anions of the DHP trimer while the central anion is hydrogen bonded to both of the macrocycles. Ten of the twelve oxygen atoms of the DHP trimer interact with hydrogen bond donors along the pseudopeptide rings, two NH groups and three triazole CH groups per ring. With O⋯N distances of 2.76 and 2.92 Å the lengths of the hydrogen bonds to the NH groups are shorter than the ones to the triazole C–H groups, whose O⋯C distances amount to 3.19, 3.24, and 3.53 Å. Two water molecules are also present in the complex, one per pseudopeptide, which bridge the bound anions and the third NH group of each ring. There is no evidence that the pyridine or triazole nitrogen atoms of 3 participate in complex formation as they do not act as hydrogen bond acceptors with the OH groups of the DHP anions.
 | ||
| Fig. 7 Molecular structure of 3·1.5TBADHP·2.5DMSO·1.7H2O (a) showing the 2:3 association of 3, the DHP anions, and the water molecules in the solid state with the thermal ellipsoids shown at the 50% probability level. The DMSO molecules, the TBA cations, the minor component of the water disorder, and the hydrogen atoms except those on the NH and triazole CH groups as well as those on the stereogenic centres C*H are omitted for clarity. (b) Shows the same structure as a space-filling model with the DHP trimer in green and the water molecules in pink to better illustrate the arrangement of the binding partners. | ||
While close spatial arrangement of the three anions in this structure may look unusual, the tendency of DHP anions to form such aggregates and even higher ones is well known.27–29 Charge repulsion upon DHP association can obviously be overcompensated by sufficiently strong hydrogen bonding interactions. While isolated DHP aggregates are only weakly stable in a polar medium such as DMSO,29f suitable receptors can cause their stabilisation.29Fig. 7 provides evidence that 3 belongs to this category due to its ring size and proper arrangement of hydrogen bond acceptors. The cavity of 3 seems to be slightly too large to accommodate two DHP anions, necessitating incorporation of an additional water molecule to fill the available space. Formation of 2R:3A complexes is likely entropically unfavourable, which explains why the binding mode is simpler in solution. The solid-state structure does, however, provide a plausible structural rationale for the observed complex stoichiometry in solution because it demonstrates that each pseudopeptide ring is able to interact with altogether two DHP anions.
The detected cooperativity in DHP complex formation indicates that the 1:1 complex between 3 and a DHP anion features a superior binding environment for the incoming anion than the empty pseudopeptide ring. Similar behaviour was reported for other systems.29a,d,e In the case of 3, the water molecule found in the complex could play an additional role. Our results furthermore demonstrate that the presence of water in the medium mainly weakens the second binding event, leading to an overall reduction of complex stability as the water content of the solvent mixture rises. Moreover, the structure of the complex shown in Fig. 7 provides information why complexes containing more than one pseudopeptide ring are not observed in the presence of DHP: in contrast to the sulfate complex of 3, no hydrogen-bond acceptors of the complexed DHP anions remain vacant that would allow interactions with a second pseudopeptide ring.
:1 binding equilibrium.
 | ||
| Fig. 8 1H NMR spectrum of 3 (1 mM) in 2.5 vol% D2O/DMSO-d6 in the absence (a) and the presence of 0.25 (b), 0.50 (c), 0.75 (d), and 1.0 equiv. (e) of TBA HPP. The signals of the NH, triazole, and C*H protons are marked in red, green, and blue, respectively. | ||
The shapes of the binding isotherms of the ITC titration support this assumption. These isotherms exhibit clear steps showing that HPP complexation is associated with several equilibria (see ESI†). The first two steps signify the initial (exothermic) formation of a 2R:1A complex containing two units of 3 and the subsequent transformation of this complex into a 1:1 complex. In the presence of a large excess of anions, a further process takes place possibly involving formation of higher complexes or other aggregation phenomena.
In the absence of detailed knowledge about these higher-order complexes, data fitting was restricted to the concentration regime comprising up to slightly more than one equivalent of HPP, in which the 2R:1A and 1:1 complexes dominate. From this part of the binding isotherm, high binding constants of logK11 = 6.62 and logK21 = 5.64 were calculated for the HPP complex of 3 in 2.5 vol% H2O/DMSO. It should be noted that these constants represent lower estimates because contributions to the binding isotherms from additional equilibria (as suggested by the transitions observed at much higher anion concentrations) were neglected.
Both stability constants are significantly higher than those of the complexes of the other two anions in the same solvent mixture, possibly explaining why the complexation equilibrium of HPP is slow on the NMR timescale. The high stability of the HPP complex can partly be attributed to the threefold charge of the HPP anion and potentially also its better fit into the cavity of 3. As in the case of the sulfate complex, the HPP anion in the 1:1 complex seems to be able to engage in further interactions with a second pseudopeptide molecule, but this interaction is significantly stronger than that of sulfate, leading to a much higher overall stability of the HPP complex (logKT = 12.26).
Replacing DHP with HPP therefore has larger consequences than just replacing the two anions of the 1R:2A DHP complex of 3 by a larger and presumably better-fitting one. Since HPP has a lower degree of protonation than DHP, it has a high propensity to involve more than one pseudopeptide ring in complex formation. Moreover, the threefold negative charge on the HPP anion causes the second binding step to be much stronger than in the case of sulfate.
It should be noted that we also considered TBA trimetaphosphate as potential substrate for 3 because the size and the matching symmetric should render this anion an even better guest than HPP. Although the 1H NMR spectrum of 3 exhibited the typical changes associated with anion binding in the presence of this anion, the binding isotherms obtained in the respective ITC titrations were even more complex than the ones of HPP complexation (see ESI†). While it is likely that stable 1:1 and 2R:1A complexes are again being formed, as also indicated by the corresponding Job plot (see ESI†), additional equilibria contribute so strongly to the binding isotherms that quantification of stability became unreliable.
Conclusions
Complementing the family of anion receptors shown in Fig. 1 by compound 3 improved our understanding of the effects that govern preferred conformations and binding properties of these compounds. While 1 and 2 are structurally closely related because the 1,5-disubstituted triazole units in 1 well mimic the cis-amides found in 2, altering the substitution pattern on the triazole units greatly affects conformational behaviour, solubility, and anion binding properties.In terms of structure, 3 features the predicted converging arrangement of the NH and the triazole CH groups rendering this pseudopeptide well preorganised for anion binding. Moreover, the 1,4-disubstitution pattern of the triazole units causes the cavity diameter of 3 to be slightly larger than that of 1 and the binding site to be relatively exposed to the solvent. The combination and arrangement of two different types of hydrogen-bond donors thus creates an environment particularly suited to host larger oxoanions.
Solubility of 3 restricted binding studies to DMSO/acetone mixtures or to DMSO containing up to 5 vol% of water. In these solvents, anion affinity of 3 is substantial and binding indeed involves both the NH and the triazole CH groups. Moreover, the relatively open cavity of 3 allows the incorporation of protonated anions whose hydrogen-bond donor sites can be arranged away from the receptor as in the case of the DHP complex. By contrast, anion binding of 1 and 2 takes place in a cavity made up by two receptor molecules coming together, which features only hydrogen-bond donors. Receptors 1 and 2 therefore do not interact with phosphate-derived anions. Changing the substitution pattern of the triazole units from the 1,5-disubstitution in 1 to the 1,4-disubstitution in 3 obviously completely alters this selectivity.
In spite of the presence of six anion binding sites along the ring, 3 seems to be unable to fully saturate the acceptor sites on sulfate or HPP anions, presumably because of cavity size and conformational rigidity. As a consequence, 3 tends to form complexes with certain anions in which more than one receptor molecule is involved in complex formation. The ring size of 3 furthermore causes water molecules to be required to mediate anion binding, as evident in the crystal structure of the DHP complex and possibly also the dependence of the stability of the sulfate complex on the water content on the solvent.
Future studies will therefore address improving solubility and simplifying the binding model underlying complex formation of 3. The latter could be achieved, for example, by arranging additional binding sites around the cavity or by designing larger analogues of 3 that completely fold around the anionic guest. These structural modifications could also have a beneficial effect on receptor solubility. Pseudopeptide 3 therefore represents a highly promising scaffold for designing new anion receptors. Work in this context is currently underway.
Experimental
General details
TBA sulfate, TBA DHP, and TBA HPP are commercially available and were used after confirming purity by an elemental analysis. TBA trimetaphosphate was prepared by following a reported procedure.30Analyses were carried out as follows: melting points, Müller SPM-X 300; NMR, Bruker AVANCE III 400 (peak assignments were confirmed by using H,H-COSY and HMQC spectra), spectra were referenced to the residual solvent signals (DMSO-d6: δH = 2.50 ppm, δC = 39.5 ppm); MALDI-TOF-MS, Bruker Ultraflex TOF/TOF; elemental analysis, Elementar vario Micro cube; optical rotation, Perkin Elmer 241 MC digital polarimeter (d = 10 cm); ITC, Microcal VP-ITC.
The following abbreviations are used: TBA, tetrabutylammonium; Epa, 6-ethynylpyridin-2-amine; Lac, CH3CHCO; Tri, 1,2,3-triazole; PyCloP, chlorotripyrrolidinophosphonium hexafluorophosphate; TBTA, tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine); HPP, hydrogenpyrophosphat; DHP, dihydrogenphosphate; TBAF, tetrabutylammonium fluoride.
Syntheses
:1 (v/v) as eluent. The product was recrystallised from hexane/ethyl acetate, 2:1 (v/v) affording white needles. Yield: 13.0 g (38.1 mmol, 56%); m.p. 115–117 °C; [α]25D = −45.4 (c = 1, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ: 11.01 (s, 1H, NH), 8.07 (d, 1H, 3J(H, H) = 8.3 Hz, EpaH(5)), 7.84 (t, 1H, 3J(H, H) = 8.0 Hz, EpaH(4)), 7.31 (dd, 1H, 3J(H, H) = 7.5 Hz, 4J(H, H) = 0.7 Hz, EpaH(3)), 5.23 (q, 1H, 3J(H, H) = 6.7 Hz, LacCH), 3.25 (s, 3H, MsCH3), 1.52 (d, 3H, 3J(H, H) = 6.7 Hz, LacCH3), 0.24 (s, 9H, TMSCH3); 13C NMR (101 MHz, DMSO-d6) δ: 168.2 (CO), 151.4 (EpaC(2)), 140.0 (EpaC(6)), 139.2 (EpaC(4)), 123.1 (EpaC(5)), 114.1 (EpaC(3)), 103.5 (Si–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ), 94.1 (Si–C), 75.2 (LacCH), 38.0 (MsCH3), 18.6 (LacCH3), −0.40 (TMSCH3); MS (MALDI-TOF) m/z (%): [M − CH3SO3H + H]+ 244.9 (97), [M − CH3SO3H + Na]+ 267.0 (35), [M + H]+ 341.1 (100), [M + Na]+ 363.1 (68), [M + K]+ 379.1 (33), [M + C3H6O + H]+ 399.2 (51); elemental analysis calcd (%) for C14H20N2O4SSi: C 49.39, H 5.92, N 8.23, S 9.27; found C 49.17, H 5.91, N 8.16, S 9.42.
:1 (v/v) as eluent. The product was obtained as a white powder. Yield: 1.4 g (5.4 mmol, 92%), MS (MALDI-TOF) m/z (%): [M + H]+ 268.9 (100), [M + Na]+ 291.0 (10), [M + K]+ 279.1 (17).
:1 (v/v) as eluent. Yield: 1.50 g (5.3 mmol, 95%), MS (MALDI-TOF) m/z (%): [M − N2+H]+ 260.0 (31), [M − N2+Na]+ 282.0 (55), [M + H]+ 288.0 (33), [M + Na]+ 310.0 (100), [M + K]+ 326.0 (74).
:1 (v/v) (150 mL), followed by the addition of a solution of TBTA (143 mg, 270 μmol, 5 mol%), Cu(MeCN)4PF6 (101 mg, 270 μmol, 5 mol%), and sodium ascorbate (107 mg, 540 μmol, 10 mol%) in t-BuOH/H2O, 1:1 (v/v) (20 mL). The reaction mixture was stirred at 25 °C for 20 h and extracted with ethyl acetate three times (60 mL). The combined organic layers were washed with water twice and dried over MgSO4. The solvent was evaporated under vacuum, and the crude product was purified by column chromatography using ethyl acetate/hexane, 1:2 (v/v) as eluent. The product was obtained as a white solid. Yield: 2.2 g (4.0 mmol, 76%); m.p. 109–111 °C; [α]25D = −162.1 (c = 0.1, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ: 11.41 (s, 1H, NH), 10.77 (s, 1H, NH), 8.63 (s, 1H, TriH), 7.99–8.05 (m, 2H, EpaH(3)), 7.93 (t, 1H, 3J(H, H) = 7.9 Hz, EpaH(4)), 7.83 (t, 1H, 3J(H, H) = 8.0 Hz, EpaH(4)), 7.78 (dd, 1H, 3J(H, H) = 7.4 Hz, 4J(H, H) = 0.8 Hz, EpaH(5)), 7.31 (d, 1H, 3J(H, H) = 7.5 Hz, EpaH(5)), 5.75 (q, 1H, 3J(H, H) = 7.1 Hz, LacCH), 5.34 (q, 1H, 3J(H, H) = 6.5 Hz, LacCH), 3.26 (s, 3H, MsCH3), 1.85 (d, 3H, 3J(H, H) = 7.1 Hz, LacCH3), 1.55 (d, 3H, 3J(H, H) = 6.7 Hz, LacCH3), 0.25 (s, 9H, TMSCH3); 13C NMR (101 MHz, DMSO-d6) δ: 168.2 (CO), 168.1 (CO), 151.5 (EpaC(2)), 151.1 (EpaC(2)), 148.5 (EpaC(6)), 146.4 (TriC(4)), 140.0 (EpaC(6)), 139.5 (EpaC(4)), 139.3 (EpaC(4)), 123.2 (EpaC(5)), 122.8 (TriC(5)), 115.9 (EpaC(5)), 113.9 (EpaC(3)), 113.0 (EpaC(3)), 103.4 (Si–C), 94.2 (Si–C), 75.3 (LacC), 58.8 (LacC), 38.1 (MsCH3), 18.7 (LacCH3), 17.9 (LacCH3), −0.4 (TMSCH3); MS (MALDI-TOF) m/z (%): [M − N2 − CH3SO3H + H]+ 432.2 (16), [M − CH3SO3H + H]+ 460.3 (100), [M − N2 + H]+ 528.3 (17), [M + H]+ 556.3 (13), [M + Na]+ 578.3 (30), [M + K]+ 594.3 (14); elemental analysis calcd (%) for C24H29N7O5SSi: C 51.87, N 17.64, H 5.26, S 5.77 found C 51.60, N 17.34, H 5.40, S 5.60.
:1 (v/v) as eluent. The product was obtained as a white powder. Yield: 2.6 g (5.4 mmol, 92%), MS (MALDI-TOF) m/z (%): [M − CH3SO3H + H]+ 388.2 (100), [M + H]+ 484.3 (35), [M + Na]+ 506.3 (48), [M + K]+ 522.3 (8).
:1 (v/v) (200 mL), followed by the addition of a solution of TBTA (143 mg, 270 μmol, 5 mol%), Cu(MeCN)4PF6 (101 mg, 270 μmol, 5 mol%), and sodium ascorbate (107 mg, 540 μmol, 10 mol%) in t-BuOH/H2O, 1:1 (v/v) (20 mL). The reaction mixture was stirred at 25 °C for 20 h and extracted with ethyl acetate three times (60 mL). The combined organic layers were washed with water twice and dried over MgSO4. Ethyl acetate was evaporated under vacuum, and the crude product was purified by column chromatography using ethyl acetate/hexane, 2:1 (v/v) as eluent. Pure product was obtained as a white solid. Yield: 3.2 g (4.1 mmol, 75%); m.p. 195–200 °C (dec.); [α]25D = −215.1 (c = 0.1, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ: 11.39 (s, 1H, NH), 11.24 (s, 1H, NH), 10.78 (s, 1H, NH), 8.65 (s, 1H, TriH), 8.64 (s, 1H, TriH), 7.91–8.05 (m, 5H, EpaH(4) + EpaH(3)), 7.83 (t, 1H, 3J(H, H) = 8.0 Hz, EpaH(4)), 7.76–7.80 (m, 2H, EpaH(5)), 7.30 (d, 1H, 3J(H, H) = 7.5 Hz, EpaH(5)), 5.85 (q, 1H, 3J(H, H) = 6.4 Hz, LacCH), 5.75 (q, 1H, 3J(H, H) = 7.1 Hz, LacCH), 5.31 (q, 1H, 3J(H, H) = 6.7 Hz, LacCH), 3.25 (s, 3H, MsCH3), 1.87 (d, 3H, 3J(H, H) = 7.2 Hz, LacCH3), 1.85 (d, 3H, 3J(H, H) = 7.2 Hz, LacCH3), 1.54 (d, 3H, 3J(H, H) = 6.7 Hz, LacCH3), 0.24 (s, 9H, 3J(H, H) = 7.1 Hz, TMSCH3); 13C NMR (101 MHz, DMSO-d6) δ: 168.2 (CO), 168.1 (CO), 151.5 (EpaC(2)), 151.2 (EpaC(2)), 151.1 (EpaC(2)), 148.6 (EpaC(6)), 148.5 (EpaC(6)), 146.5 (TriC(4)), 146.4 (TriC(4)), 140.1 (EpaC(6)), 139.6 (EpaC(4)), 139.5 (EpaC(4)), 139.4 (EpaC(4)), 123.2 (EpaC(5)), 122.8 (TriC(5)), 122.8 (TriC(5)), 116.0 (EpaC(5)), 115.9 (EpaC(5)), 114.0 (EpaC(3)), 113.0 (EpaC(3)), 112.9 (EpaC(3)), 103.5 (Si–C), 94.2 (Si–C), 75.3 (LacC), 58.9 (LacC), 58.8 (LacC), 38.1 (MsCH3), 18.7 (LacCH3), 18.0 (LacCH3), 17.9 (LacCH3), −0.4 (TMSCH3); MS (MALDI-TOF) m/z (%):[M − CH3SO3H + H]+ 675.6 (100), [M − CH3SO3H + H2 + H]+ 677.7 (94), [M − CH3SO3H + H2 + Na]+ 699.7 (46), [M + H]+ 771.7 (27), [M + Na]+ 593.7 (17), [M + C3H6O + H]+ 829.8 (37); elemental analysis calcd (%) for C34H38N12O6SSi: C 52.97, N 21.80, H 5.97, S 4.16 found C 52.59, N 21.51, H 5.00, S 3.90.
:1 (v/v) (50 mL). The resulting solution was added dropwise to a suspension of TBTA (19 mg, 36 μmol, 10 mol%), sodium ascorbate (3.5 mg, 18 μmol, 5 mol%), and Cu(MeCN)4PF6 (13 mg, 35 μmol, 10 mol%) in t-BuOH/H2O, 1:1 (v/v) (250 mL) over a period of 30 min at 25 °C. The reaction mixture was stirred at 35 °C, and progress was followed by HPLC. Additional solid TBTA (9 mg, 18 μmol, 5 mol%) and Cu(MeCN)4PF6 (7 mg, 18 μmol, 5 mol%) were added every 24 h until HPLC indicated full conversion, typically after 3 d. Ethyl acetate was added and the aqueous layer removed. The organic solvent was evaporated, and the residue was washed several times with acetone to remove the catalyst and TBTA and dried. The product was obtained analytically pure after recrystallisation from DMSO. Yield: 65 mg (101 μmol, 29%); m.p. > 200 °C (dec); [α]25D = +6.94 (c = 0.1, DMSO); 1H NMR (400 MHz, DMSO-d6) δ: 10.91 (s, 3H, NH), 8.59 (s, 3H, TriH), 7.94 (d, 2H, 3J(H, H) = 8.0 Hz, EpaH(4)), 7.89 (t, 3H, 3J(H, H) = 7.8 Hz, EpaH(5)), 7.77 (d, 3H, 3J(H, H) = 7.6 Hz, EpaH(3)), 5.76 (q, 3H, 3J(H, H) = 6.7 Hz, LacCH), 1.85 (d, 9H, 3J(H, H) = 6.9 Hz, LacCH3); 13C NMR (101 MHz, DMSO-d6) δ: 166.6 (CO), 151.2 (EpaC(2)), 148.4 (EpaC(6)), 146.3 (TriC(4)), 139.6 (EpaC(4)), 123.4 (TriC(5)), 116.0 (EpaC(5)), 113.1 (EpaC(3)), 59.1 (LacC), 16.1 (LacCH3); MS (MALDI-TOF) m/z (%): [M + H]+ 646.5 (34), [M + Na]+ 668.5 (100), [M + K]+ 684.6 (19); elemental analysis calcd (%) for C30H27N15O3·DMSO·2H2O: C 50.58, N 27.65, H 4.91, S 4.22 found C 50.27, N 27.43, H 4.62, S 4.02.
), 94.1 (Si–C), 75.2 (LacCH), 38.0 (MsCH3), 18.6 (LacCH3), −0.40 (TMSCH3); MS (MALDI-TOF) m/z (%): [M − CH3SO3H + H]+ 244.9 (97), [M − CH3SO3H + Na]+ 267.0 (35), [M + H]+ 341.1 (100), [M + Na]+ 363.1 (68), [M + K]+ 379.1 (33), [M + C3H6O + H]+ 399.2 (51); elemental analysis calcd (%) for C14H20N2O4SSi: C 49.39, H 5.92, N 8.23, S 9.27; found C 49.17, H 5.91, N 8.16, S 9.42.
:1 (v/v) as eluent. The product was obtained as a white powder. Yield: 1.4 g (5.4 mmol, 92%), MS (MALDI-TOF) m/z (%): [M + H]+ 268.9 (100), [M + Na]+ 291.0 (10), [M + K]+ 279.1 (17).
:1 (v/v) as eluent. Yield: 1.50 g (5.3 mmol, 95%), MS (MALDI-TOF) m/z (%): [M − N2+H]+ 260.0 (31), [M − N2+Na]+ 282.0 (55), [M + H]+ 288.0 (33), [M + Na]+ 310.0 (100), [M + K]+ 326.0 (74).
:1 (v/v) (150 mL), followed by the addition of a solution of TBTA (143 mg, 270 μmol, 5 mol%), Cu(MeCN)4PF6 (101 mg, 270 μmol, 5 mol%), and sodium ascorbate (107 mg, 540 μmol, 10 mol%) in t-BuOH/H2O, 1:1 (v/v) (20 mL). The reaction mixture was stirred at 25 °C for 20 h and extracted with ethyl acetate three times (60 mL). The combined organic layers were washed with water twice and dried over MgSO4. The solvent was evaporated under vacuum, and the crude product was purified by column chromatography using ethyl acetate/hexane, 1:2 (v/v) as eluent. The product was obtained as a white solid. Yield: 2.2 g (4.0 mmol, 76%); m.p. 109–111 °C; [α]25D = −162.1 (c = 0.1, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ: 11.41 (s, 1H, NH), 10.77 (s, 1H, NH), 8.63 (s, 1H, TriH), 7.99–8.05 (m, 2H, EpaH(3)), 7.93 (t, 1H, 3J(H, H) = 7.9 Hz, EpaH(4)), 7.83 (t, 1H, 3J(H, H) = 8.0 Hz, EpaH(4)), 7.78 (dd, 1H, 3J(H, H) = 7.4 Hz, 4J(H, H) = 0.8 Hz, EpaH(5)), 7.31 (d, 1H, 3J(H, H) = 7.5 Hz, EpaH(5)), 5.75 (q, 1H, 3J(H, H) = 7.1 Hz, LacCH), 5.34 (q, 1H, 3J(H, H) = 6.5 Hz, LacCH), 3.26 (s, 3H, MsCH3), 1.85 (d, 3H, 3J(H, H) = 7.1 Hz, LacCH3), 1.55 (d, 3H, 3J(H, H) = 6.7 Hz, LacCH3), 0.25 (s, 9H, TMSCH3); 13C NMR (101 MHz, DMSO-d6) δ: 168.2 (CO), 168.1 (CO), 151.5 (EpaC(2)), 151.1 (EpaC(2)), 148.5 (EpaC(6)), 146.4 (TriC(4)), 140.0 (EpaC(6)), 139.5 (EpaC(4)), 139.3 (EpaC(4)), 123.2 (EpaC(5)), 122.8 (TriC(5)), 115.9 (EpaC(5)), 113.9 (EpaC(3)), 113.0 (EpaC(3)), 103.4 (Si–C), 94.2 (Si–C), 75.3 (LacC), 58.8 (LacC), 38.1 (MsCH3), 18.7 (LacCH3), 17.9 (LacCH3), −0.4 (TMSCH3); MS (MALDI-TOF) m/z (%): [M − N2 − CH3SO3H + H]+ 432.2 (16), [M − CH3SO3H + H]+ 460.3 (100), [M − N2 + H]+ 528.3 (17), [M + H]+ 556.3 (13), [M + Na]+ 578.3 (30), [M + K]+ 594.3 (14); elemental analysis calcd (%) for C24H29N7O5SSi: C 51.87, N 17.64, H 5.26, S 5.77 found C 51.60, N 17.34, H 5.40, S 5.60.
:1 (v/v) as eluent. The product was obtained as a white powder. Yield: 2.6 g (5.4 mmol, 92%), MS (MALDI-TOF) m/z (%): [M − CH3SO3H + H]+ 388.2 (100), [M + H]+ 484.3 (35), [M + Na]+ 506.3 (48), [M + K]+ 522.3 (8).
:1 (v/v) (200 mL), followed by the addition of a solution of TBTA (143 mg, 270 μmol, 5 mol%), Cu(MeCN)4PF6 (101 mg, 270 μmol, 5 mol%), and sodium ascorbate (107 mg, 540 μmol, 10 mol%) in t-BuOH/H2O, 1:1 (v/v) (20 mL). The reaction mixture was stirred at 25 °C for 20 h and extracted with ethyl acetate three times (60 mL). The combined organic layers were washed with water twice and dried over MgSO4. Ethyl acetate was evaporated under vacuum, and the crude product was purified by column chromatography using ethyl acetate/hexane, 2:1 (v/v) as eluent. Pure product was obtained as a white solid. Yield: 3.2 g (4.1 mmol, 75%); m.p. 195–200 °C (dec.); [α]25D = −215.1 (c = 0.1, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ: 11.39 (s, 1H, NH), 11.24 (s, 1H, NH), 10.78 (s, 1H, NH), 8.65 (s, 1H, TriH), 8.64 (s, 1H, TriH), 7.91–8.05 (m, 5H, EpaH(4) + EpaH(3)), 7.83 (t, 1H, 3J(H, H) = 8.0 Hz, EpaH(4)), 7.76–7.80 (m, 2H, EpaH(5)), 7.30 (d, 1H, 3J(H, H) = 7.5 Hz, EpaH(5)), 5.85 (q, 1H, 3J(H, H) = 6.4 Hz, LacCH), 5.75 (q, 1H, 3J(H, H) = 7.1 Hz, LacCH), 5.31 (q, 1H, 3J(H, H) = 6.7 Hz, LacCH), 3.25 (s, 3H, MsCH3), 1.87 (d, 3H, 3J(H, H) = 7.2 Hz, LacCH3), 1.85 (d, 3H, 3J(H, H) = 7.2 Hz, LacCH3), 1.54 (d, 3H, 3J(H, H) = 6.7 Hz, LacCH3), 0.24 (s, 9H, 3J(H, H) = 7.1 Hz, TMSCH3); 13C NMR (101 MHz, DMSO-d6) δ: 168.2 (CO), 168.1 (CO), 151.5 (EpaC(2)), 151.2 (EpaC(2)), 151.1 (EpaC(2)), 148.6 (EpaC(6)), 148.5 (EpaC(6)), 146.5 (TriC(4)), 146.4 (TriC(4)), 140.1 (EpaC(6)), 139.6 (EpaC(4)), 139.5 (EpaC(4)), 139.4 (EpaC(4)), 123.2 (EpaC(5)), 122.8 (TriC(5)), 122.8 (TriC(5)), 116.0 (EpaC(5)), 115.9 (EpaC(5)), 114.0 (EpaC(3)), 113.0 (EpaC(3)), 112.9 (EpaC(3)), 103.5 (Si–C), 94.2 (Si–C), 75.3 (LacC), 58.9 (LacC), 58.8 (LacC), 38.1 (MsCH3), 18.7 (LacCH3), 18.0 (LacCH3), 17.9 (LacCH3), −0.4 (TMSCH3); MS (MALDI-TOF) m/z (%):[M − CH3SO3H + H]+ 675.6 (100), [M − CH3SO3H + H2 + H]+ 677.7 (94), [M − CH3SO3H + H2 + Na]+ 699.7 (46), [M + H]+ 771.7 (27), [M + Na]+ 593.7 (17), [M + C3H6O + H]+ 829.8 (37); elemental analysis calcd (%) for C34H38N12O6SSi: C 52.97, N 21.80, H 5.97, S 4.16 found C 52.59, N 21.51, H 5.00, S 3.90.
:1 (v/v) (50 mL). The resulting solution was added dropwise to a suspension of TBTA (19 mg, 36 μmol, 10 mol%), sodium ascorbate (3.5 mg, 18 μmol, 5 mol%), and Cu(MeCN)4PF6 (13 mg, 35 μmol, 10 mol%) in t-BuOH/H2O, 1:1 (v/v) (250 mL) over a period of 30 min at 25 °C. The reaction mixture was stirred at 35 °C, and progress was followed by HPLC. Additional solid TBTA (9 mg, 18 μmol, 5 mol%) and Cu(MeCN)4PF6 (7 mg, 18 μmol, 5 mol%) were added every 24 h until HPLC indicated full conversion, typically after 3 d. Ethyl acetate was added and the aqueous layer removed. The organic solvent was evaporated, and the residue was washed several times with acetone to remove the catalyst and TBTA and dried. The product was obtained analytically pure after recrystallisation from DMSO. Yield: 65 mg (101 μmol, 29%); m.p. > 200 °C (dec); [α]25D = +6.94 (c = 0.1, DMSO); 1H NMR (400 MHz, DMSO-d6) δ: 10.91 (s, 3H, NH), 8.59 (s, 3H, TriH), 7.94 (d, 2H, 3J(H, H) = 8.0 Hz, EpaH(4)), 7.89 (t, 3H, 3J(H, H) = 7.8 Hz, EpaH(5)), 7.77 (d, 3H, 3J(H, H) = 7.6 Hz, EpaH(3)), 5.76 (q, 3H, 3J(H, H) = 6.7 Hz, LacCH), 1.85 (d, 9H, 3J(H, H) = 6.9 Hz, LacCH3); 13C NMR (101 MHz, DMSO-d6) δ: 166.6 (CO), 151.2 (EpaC(2)), 148.4 (EpaC(6)), 146.3 (TriC(4)), 139.6 (EpaC(4)), 123.4 (TriC(5)), 116.0 (EpaC(5)), 113.1 (EpaC(3)), 59.1 (LacC), 16.1 (LacCH3); MS (MALDI-TOF) m/z (%): [M + H]+ 646.5 (34), [M + Na]+ 668.5 (100), [M + K]+ 684.6 (19); elemental analysis calcd (%) for C30H27N15O3·DMSO·2H2O: C 50.58, N 27.65, H 4.91, S 4.22 found C 50.27, N 27.43, H 4.62, S 4.02.
The measurements were carried out at 25 °C using a reference power of 25 μJ s−1, a filter period of 2 s, a stirrer speed of 307 rpm. Other experimental parameters of the individual titrations are specified in the ESI.† Automated baseline assignment and peak integration of raw thermograms were accomplished by singular value decomposition and peak-shape analysis using NITPIC.32a Estimation of best-fit parameter values by weighted nonlinear least-squares fitting and calculation of 68.3% confidence intervals were performed with the public-domain software SEDPHAT,32b as explained in detail elsewhere.32c,d
Acknowledgements
We thank Dr M. Meyer, Institut de Chimie Moléculaire de l’ Université de Bourgogne (ICMUB), Dijon for help with the HypNMR software and Dr R. Goddard, Max-Planck-Institut für Kohlenforschung, Mülheim/Ruhr for performing preliminary crystallographic studies. K.R. thanks the Academy of Finland for financial support (Project Nos. 263256 and 292746).Notes and references
- R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1967, 100, 2494–2507 CrossRef CAS.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS PubMed.
- B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- B. C. Boren, S. Narayan, L. K. Rasmussen, L. Zhang, H. Zhao, Z. Lin, G. Jia and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 8923–8930 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- (a) C. G. S. Lima, A. Ali, S. S. van Berkel, B. Westermann and M. W. Paixão, Chem. Commun., 2015, 51, 10784–10796 RSC; (b) J. John, J. Thomas and W. Dehaen, Chem. Commun., 2015, 51, 10797–10806 RSC.
- (a) D. S. Pedersen and A. Abell, Eur. J. Org. Chem., 2011, 2399–2411 CrossRef CAS; (b) I. E. Valverde and T. L. Mindt, Chimia, 2013, 67, 262–266 CrossRef CAS PubMed.
- Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262–1271 RSC.
- Y. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649–2652 CrossRef CAS PubMed.
- H. Juwarker, J. M. Lenhardt, D. M. Pham and S. L. Craig, Angew. Chem., Int. Ed., 2008, 47, 3740–3743 CrossRef CAS PubMed.
- R. M. Meudtner and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 4926–4930 CrossRef CAS PubMed.
- N. L. Kilah, M. D. Wise, C. J. Serpell, A. L. Thompson, N. G. White, K. E. Christensen and P. D. Beer, J. Am. Chem. Soc., 2010, 132, 11893–11895 CrossRef CAS PubMed.
- B. Schulze, C. Friebe, M. D. Hager, W. Günther, U. Köhn, B. O. Jahn, H. Görls and U. S. Schubert, Org. Lett., 2010, 12, 2710–2713 CrossRef CAS PubMed.
- (a) M. Erdélyi, Chem. Soc. Rev., 2012, 41, 3547–3557 RSC; (b) T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC; (c) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed; (d) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed; (e) D. Bulfield and S. M. Huber, Chem. – Eur. J., 2016, 22, 14434–14450 CrossRef CAS PubMed.
- (a) Y.-J. Li, L. Xu, W.-L. Yang, H.-B. Liu, S.-W. Lai, C.-M. Che and Y.-L. Li, Chem. – Eur. J., 2012, 18, 4782–4790 CrossRef CAS PubMed; (b) L. Xu, Y. Li, Y. Yu, T. Liu, S. Cheng, H. Liu and Y. Li, Org. Biomol. Chem., 2012, 10, 4375–4380 RSC; (c) L. Cao, R. Jiang, Y. Zhu, X. Wang, Y. Li and Y. Li, Eur. J. Org. Chem., 2014, 2687–2693 CrossRef CAS.
- M. R. Krause, R. Goddard and S. Kubik, J. Org. Chem., 2011, 76, 7084–7095 CrossRef CAS PubMed.
- (a) S. Kubik, R. Goddard, R. Kirchner, D. Nolting and J. Seidel, Angew. Chem., Int. Ed., 2001, 40, 2648–2651 CrossRef CAS; (b) S. Kubik and R. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5127–5132 CrossRef CAS PubMed.
- S. Kubik and R. Goddard, J. Org. Chem., 1999, 64, 9475–9486 CrossRef CAS.
- J. Zabrocki, J. B. Dunbar Jr., K. W. Marshall, M. V. Toth and G. R. Marshall, J. Org. Chem., 1992, 57, 202–209 CrossRef CAS.
- J. Broecker, C. Vargas and S. Keller, Anal. Biochem., 2011, 418, 307–309 CrossRef CAS PubMed.
- F. Ulatowski, K. Dąbrowa, T. Bałakier and J. Jurczak, J. Org. Chem., 2016, 81, 1746–1756 CrossRef CAS PubMed.
- G. Kemmer and S. Keller, Nat. Protoc., 2010, 5, 267–281 CrossRef CAS PubMed.
- Z. Rodriguez-Docampo, S. I. Pascu, S. Kubik and S. Otto, J. Am. Chem. Soc., 2006, 128, 11206–11210 CrossRef CAS PubMed.
- (a) N. Ohama, M. Machida, T. Nakamura and Y. Kunifuji, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 962–964 CrossRef; (b) J. M. Karle and I. L. Karle, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 1605–1608 CrossRef; (c) M. A. Hossain, M. Işıklan, A. Pramanik, M. A. Saeed and F. R. Fronczek, Cryst. Growth Des., 2012, 12, 567–571 CrossRef CAS PubMed; (d) A. Rajbanshi, S. Wan and R. Custelcean, Cryst. Growth Des., 2013, 13, 2233–2237 CrossRef CAS.
- (a) F. Rull, A. Del Valle, F. Sobron and S. Veintemillas, J. Raman Spectrosc., 1989, 20, 625–631 CrossRef CAS; (b) R. H. Wood and R. F. Platford, J. Solution Chem., 1975, 4, 977–982 CrossRef CAS.
- (a) V. Amendola, M. Boiocchi, D. Esteban-Gómez, L. Fabbrizzi and E. Monzani, Org. Biomol. Chem., 2005, 3, 2632–2639 RSC; (b) P. S. Lakshminarayanan, I. Ravikumar, E. Suresh and P. Ghosh, Chem. Commun., 2007, 5214–5216 RSC; (c) P. Dydio, T. Zieliński and J. Jurczak, Org. Lett., 2010, 12, 1076–1078 CrossRef CAS PubMed; (d) V. Blažek, N. Bregović, K. Mlinarić-Majerski and N. Basarić, Tetrahedron, 2011, 67, 3846–3857 CrossRef; (e) V. Blažek, K. Molčanov, K. Mlinarić-Majerski, B. Kojić-Prodić and N. Basarić, Tetrahedron, 2013, 69, 517–526 CrossRef; (f) N. Bregović, N. Cindro, L. Frkanec, K. Užarević and V. Tomišić, Chem. – Eur. J., 2014, 20, 15863–15871 CrossRef PubMed.
- S. Mohamady and S. D. Taylor, Org. Lett., 2013, 15, 2612–2615 CrossRef CAS PubMed.
- C. Frassineti, S. Ghelli, P. Gans, A. Sabatini, M. S. Moruzzi and A. Vacca, Anal. Biochem., 1995, 231, 374–382 CrossRef CAS PubMed.
- (a) S. Keller, C. Vargas, H. Zhao, G. Piszczek, C. A. Brautigam and P. Schuck, Anal. Chem., 2012, 84, 5066–5073 CrossRef CAS PubMed; (b) J. C. D. Houtman, P. H. Brown, B. Bowden, H. Yamaguchi, E. Appella, L. E. Samelson and P. Schuck, Protein Sci., 2007, 16, 30–42 CrossRef CAS PubMed; (c) G. Krainer, J. Broecker, C. Vargas, J. Fanghänel and S. Keller, Anal. Chem., 2012, 84, 10715–10722 CrossRef CAS PubMed; (d) G. Krainer and S. Keller, Methods, 2015, 76, 116–123 CrossRef CAS PubMed.
- G. Ercolani, J. Am. Chem. Soc., 2003, 125, 16097–16103 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, and mass spectra of compounds 5, 7, 8, 3; NOESY NMR spectrum of 3; 1H NMR spectra of qualitative binding studies; Job plots and results of NMR titrations and of selected ITC titrations; details of crystal structures. CCDC 1504361 and 1505706. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob02172g |
| ‡ For completely independent binding steps one would expect a 4 times smaller association constant K12 for the second binding step with respect to K11 of the 1:1 complex. The reason is that the rate constant of forming the 1:1 complex is statistically two times higher than that of forming the 1:2 complex, while the dissociation rate constant of the 1:2 complex is twice that of the 1:1 complex. Since the ratio of the rate constant of formation and the rate constant of dissociation gives the equilibrium constant, it follows that K12 = K11/4 in the absence of cooperativity.33 |
| This journal is © The Royal Society of Chemistry 2017 |