
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemo-enzymatic modification of eukaryotic mRNA
Fabian
Muttach
a,
Nils
Muthmann
a and
Andrea
Rentmeister
*ab
aUniversity of Münster, Department of Chemistry, Institute of Biochemistry, Wilhelm-Klemm-Str. 2, 48149 Münster, Germany. E-mail: a.rentmeister@uni-muenster.de; Fax: +49 251 83-33007; Tel: +49 251 83-33204
bCell-in-Motion Cluster of Excellence (EXC1003-CiM), University of Münster, Germany
First published on 17th November 2016
Abstract
Messenger RNA may not be very abundant in the cell but its central role in gene expression is indisputable. In addition to being the template for translation it can be subject for a variety of regulatory mechanisms affecting gene expression, ranging from simple structural changes to modifications and active transport. To elucidate and potentially control the underlying changes in vitro and in cells, site-specific modification and labeling strategies are required. In this perspective, we introduce chemo-enzymatic concepts for posttranscriptional modification focusing on eukaryotic mRNAs. We describe how eukaryotic mRNA can be enzymatically modified via its 5′ cap. Directions towards chemo-enzymatic mRNA labeling and visualization inside cells are given, taking into account current developments in fluorophore design. Recent achievements and future perspectives will be highlighted in the framework of an honest discussion of existing challenges.
 Fabian Muttach | Fabian Muttach studied Molecular Biotechnology at Heidelberg University from 2009–2014 where he obtained his Bachelor's and Master's degree. During his Master thesis he worked on the chemo-enzymatic synthesis and application of nucleoside-based fluorophores. He then moved to the University of Münster to pursue his PhD studies in the group of Prof. Dr. Andrea Rentmeister. His current research focuses on methyltransferase-based modification strategies for in vitro and cellular applications. |
 Nils Muthmann | Nils Muthmann obtained a Bachelor's degree in Chemistry from the University of Münster. After a six month stay at McMaster University he is currently working on his Master thesis. His work focuses on establishing novel screening methods for engineering methyltransferases. |
 Andrea Rentmeister | Andrea Rentmeister is Professor of Biochemistry with a focus on Biomolecular Label Chemistry in the Department of Chemistry and the Cluster of Excellence “Cells in Motion” at the University of Münster. She received her PhD in 2007 from the University of Bonn under the supervision of Prof. Michael Famulok and was a postdoctoral scholar in the group of Prof. Frances Arnold at the California Institute of Technology from 2007–2010. Her research currently focuses on labeling strategies for RNA that are compatible with living cells. Further interests include protein engineering and natural RNA modifications. She was awarded an Emmy Noether Fellowship in 2011 and the Hoechst “Dozentenpreis” of the Aventis Foundation in 2015. |
Introduction
Labeling RNA is a prerequisite for many types of functional studies yielding insights into the structure and dynamics of these biomolecules1–4 and also finds applications in biotechnology and biomedicine.5–7 Among the considerable number of different classes of RNA, mRNA constitutes merely 1–5% in higher eukaryotes.8 Total numbers of only 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–200000 mRNAs per eukaryotic cell have been reported for yeast9 and mammals,10 respectively. Still, mRNA stands out as it is translated into proteins, yielding approximately 4000 proteins per transcript.11 Particularly in eukaryotes, where mRNAs have longer half-lives (median half-life of 10 hours12) than in prokaryotes, the transcript itself is subject to substantial regulation of gene expression. Mechanisms include changes of the transcript stability (e.g. by miRNAs or by altering the length of the poly(A) tail followed by sequestration into P-bodies), nonsense-mediated decay, subcellular localization and local translation, but also alternative splicing, leading to protein diversification.
000–200000 mRNAs per eukaryotic cell have been reported for yeast9 and mammals,10 respectively. Still, mRNA stands out as it is translated into proteins, yielding approximately 4000 proteins per transcript.11 Particularly in eukaryotes, where mRNAs have longer half-lives (median half-life of 10 hours12) than in prokaryotes, the transcript itself is subject to substantial regulation of gene expression. Mechanisms include changes of the transcript stability (e.g. by miRNAs or by altering the length of the poly(A) tail followed by sequestration into P-bodies), nonsense-mediated decay, subcellular localization and local translation, but also alternative splicing, leading to protein diversification.
The plethora of posttranscriptional regulatory options for eukaryotic mRNAs emphasizes the importance of labeling mRNAs to make them accessible to functional studies, particularly in living cells, where dynamic processes take place. The low level abundance of mRNAs at the same time denotes challenges resulting from low signal to background ratios.
In addition to making mRNAs “visible” for various biophysical measurements, site-specific modifications of mRNA hallmarks may, in principle, be used to selectively inhibit one out of several interactions and their respective downstream pathways in mRNA metabolism. Such an approach aims to selectively control the function of mRNAs at various processing stages independent of their sequence and would provide an alternative to sequence-dependent approaches for posttranscriptional regulation of gene expression.
Various RNA labeling concepts have been developed during the past few years, and listing all of them clearly goes beyond the scope of this perspective. We will introduce the concept of chemo-enzymatic strategies for RNA labeling and focus on posttranscriptional modification approaches involving enzymatic modification of endogenous (i.e. untagged) mRNA. The main benefits of these chemo-enzymatic approaches are that (i) mRNA with an unaltered sequence can be addressed and (ii) labeling occurs by small organic dyes which should only minimally perturb the mRNA properties. For alternative concepts, e.g. hybridization-based probes or RNA-binding proteins we would like to refer the reader to recent review articles.13–16
Posttranscriptional chemo-enzymatic labeling
In a chemo-enzymatic RNA labeling approach, a small functional moiety (e.g. bearing an alkyne or azido group) is enzymatically installed – either co- or posttranscriptionally. This reactive handle is subsequently converted in a bioorthogonal reaction—i.e. a reaction that does not interfere with biological processes.17,18 In addition to being inert to functionalities found in cells, these reactions must be highly selective, fast, and not produce toxic byproducts.Chemo-enzymatic approaches combine the high specificity of enzymes with high-yielding chemical reactions and are therefore ideally suited for the precise modification of biomolecules in vitro and in cells. Intracellular labeling of RNA by chemo-enzymatic approaches is feasible, because enzymes are predestined to function under physiological conditions and several bioorthogonal reactions have been developed to a stage where they function also inside living cells.19 The two-step strategy provides flexibility regarding the type of label to be attached. In addition to reporters for biophysical measurements, affinity labels or crosslinkers are conceivable tags. Last but not least, the effect of the enzymatically introduced small modification may also serve as a selective modulator of intracellular interactions.
Fig. 1 illustrates the concept of posttranscriptional chemo-enzymatic RNA labeling. The alternative co-transcriptional chemo-enzymatic labeling relies on the same two-step principle but enzymatic incorporation of modified monomeric building blocks during transcription or polyadenylation.20–23
 | ||
| Fig. 1 Concept of posttranscriptional chemo-enzymatic labeling. A small reactive moiety is enzymatically transferred from an appropriate cosubstrate to the target site. This modification is chosen to make the RNA amenable to a selective labeling reaction, typically via click-chemistry. The enzymatic step ensures high specificity, the bioorthogonal reaction provides flexibility regarding the reporter group. | ||
To achieve selective posttranscriptional enzymatic modification, an RNA modifying enzyme acting on a defined recognition motif or target site is needed (Fig. 1). Structural elements derived from tRNAs, which undergo substantial posttranscriptional modifications, have proven useful baits for RNA-modifying enzymes, but require addition of a substrate tag thereby preventing modification of endogenous RNAs.24,25
We will focus on methyltransferase-based modification and labeling strategies harnessing analogs of the natural cosubstrate S-adenosyl-L-methionine (AdoMet). AdoMet is the second most abundant cosubstrate in nature after ATP but can also be chemically synthesized from S-adenosylhomocysteine by simple nucleophilic substitution. This synthesis route can be used to generate a variety of AdoMet analogs, if the appropriate substrate with a leaving group (typically bromide or triflate) is available.26,27 These AdoMet analogs are often still accepted by methyltransferases, which display different degrees of (co-)substrate promiscuity. Depending on the architecture and cosubstrate promiscuity of the methyltransferase, modifications of different size, steric demands and functional groups can be placed site-specifically on the target biomolecule. Typical examples include alkene, alkyne, and azido functionalities but also involve benzylic residues that have been transferred by small molecule,28,29 protein,30 DNA26,27- and RNA methyltransferases.31,32
Methyltransferase-based labeling features several advantages compared to other labeling methods. They rely only on a small recognition motif in the endogenous sequence, thereby circumventing the need to change the target molecule by addition of a tag.33,34 This allows for the installation of a modification with minimal structural perturbation. Additionally, methyltransferases are highly regiospecific enabling – in case of mRNA – modification of the 5′ cap even at different positions.31,35
In the field of methyltransferase-based RNA labeling, modification of a tRNA(Phe) was the first report of using a synthetic AdoMet analog.36 Sequence-specific labeling of RNA was achieved by using a box C/D small ribonucleoprotein RNA 2′-O-methyltransferase.37 In both reports, an alkyne functionality was introduced and then reacted in vitro in a copper-catalyzed azide–alkyne cycloaddition (CuAAC). Building on this concept, we established two approaches for site-specific modification of eukaryotic mRNAs.
Chemo-enzymatic strategy for site-specific modification of eukaryotic mRNA
Eukaryotic mRNAs have two major hallmarks: the 5′ cap and the poly(A)-tail at the 3′ end. The 5′ cap consists of an inverted 7-methylguanosine which is connected through a triphosphate bridge to the mRNA. These structural features protect the eukaryotic mRNAs from degradation by exoribonucleases, but also interact with numerous binding partners as quality control and to coordinate fundamental processes, such as splicing, mRNA export from the nucleus, and translation. Several viruses have evolved different mechanisms to acquire a 5′ cap, including a cap snatching mechanism found in influenza.38 Site-specific cap manipulation may thus not only feature mRNA labeling and isolation approaches but also dissecting the roles of cap interacting proteins.We have reported on the identification and engineering of different 5′ cap methyltransferases. The trimethylguanosine synthase variants V34A and T34A from Giardia lamblia and Giardia intestinalis (GlaTgs-V34A and GinTgs-T34A) accept AdoMet analogs bearing alkene-, alkyne-, or azido-moieties and transfer the respective side-chains to the exocyclic amino group of the mRNA cap (Fig. 2).31,39 In combination with subsequent click reactions this proved to be an efficient strategy for mRNA cap labeling in vitro. Not surprisingly, we noticed that the enzymatic transfer efficiency of Tgs enzymes decreased with the size of the cosubstrate side-chain (Fig. 2), indicative of steric constraints in the substrate binding pocket. This limitation, which we are currently addressing by protein engineering, does not occur for the N7 cap methyltransferase from Encephalitozoon cuniculi (Ecm1), as we recently discovered.35 Ecm1 converted all AdoMet analogs tested so far roughly as fast as the natural cosubstrate AdoMet (Fig. 2) and thus provides access to novel cap modifications which will likely give access to new applications.
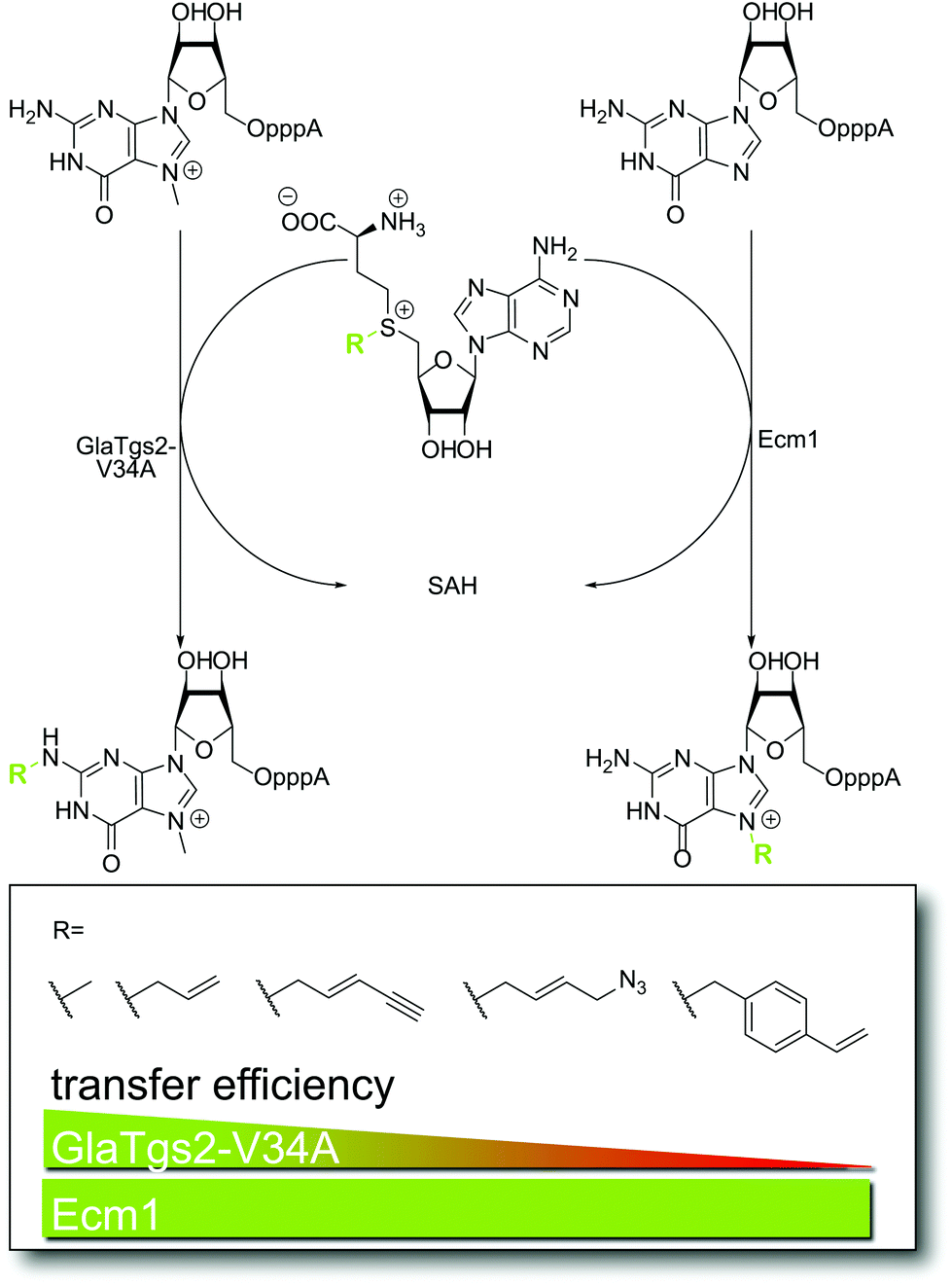 | ||
| Fig. 2 Enzymatic step of methyltransferase-based labeling approach for the eukaryotic mRNA cap in vitro. For GlaTgs-V34A, transfer efficiencies decrease with increasing steric demand of the introduced moiety (e.g. alkene, alkyne, azido and vinylbenzyl functions), whereas Ecm1 is equally efficient regardless of the size of the group transferred. | ||
Cap modifications may provide access to controlling biological functions of mRNAs
The 5′ cap of eukaryotic mRNA is essential for efficient cap-dependent translation of the mRNA and protects against degradation. In the case of cap-dependent translation, the cap-binding protein eIF4E plays a key role for initiation.40 It assembles the adapter protein eIF4G and the poly(A)-binding protein PAB to the mRNA cap, leading to the recruitment of the 40S ribosomal subunit via other eIFs.41In vitro a remarkable difference (five orders of magnitude) in binding affinity of eIF4E to the naturally occurring 7-methylguanosine cap and a cap missing the methylation is observed.42 This has been attributed to a need for discrimination against GTP which is found in millimolar concentrations inside the cell.43 It is well known that synthetic modifications of the mRNA cap regulate translational efficiencies. Darzynkiewicz et al. have synthesized various β-globin mRNAs with N7-substituted cap structures and could increase or decrease translational efficiencies measured by in vitro translation assays.44 Likewise, synthetic cap analogs competed with native capped mRNAs and – when added to an in vitro translation system – inhibited translation depending on the cap analog modifications.45We built on these observations that artificially introduced modifications at the cap can modulate translational efficiencies. The high co-substrate promiscuity of Ecm1 (Fig. 2) that we recently discovered, allowed for the efficient enzymatic in vitro preparation of differently capped reporter mRNAs coding for GFP or luciferase.35 Using GlaTgs2-V34A, the enzymatic preparation of N2-modified capped mRNAs was also possible. We tested how efficiently these reporter mRNAs were translated both in an in vitro translation system and in cells transfected with the cap-modified mRNA and observed that different modifications at either of the two positions reduced translation to a different degree. Control of cap modification as achieved by enzymatic modifications may thus become a suitable tool for regulation of mRNA translation inside cells and thereby present a new post-transcriptional regulatory strategy.
Labeling reactions for modified RNA – from test tube to living cells
For fluorescence or affinity labeling of enzymatically introduced reactive handles, the choice of the chemical labeling reaction is crucial for efficient modification of mRNA. A range of bioorthogonal reactions was developed in recent years. Among those, the most important reactions are the strain-promoted azide–alkyne cycloaddition (SPAAC) of azides and strained alkynes and tetrazine ligations between tetrazines and strained alkenes or bicyclononynes. Since a detailed description of bioorthogonal reactions goes beyond the scope of this perspective we will focus on RNA labeling in cells and refer to excellent reviews regarding the chemistry itself.18,19,46We could react reporter-mRNAs bearing azido-functionalized caps with strained alkyne-fluorophore conjugates both in vitro and in living cells transfected with the RNA construct.35 To achieve this, it was necessary to optimize the uptake and washing conditions of the fluorophore-reagent. To improve the signal-to-noise ratio, fluorogenic reactions such as the tetrazine ligation are especially promising (Fig. 3). Typically, a 5–20-fold fluorescence increase is achieved depending on the fluorescent dye–tetrazine conjugate.21,47 Tetrazine ligations on chemically and enzymatically synthesized RNAs were already successfully performed both in vitro21 and in cells transfected with the RNA construct.48
 | ||
| Fig. 3 Perspectives for the RNA labeling step in living cells. Turn-on fluorophores generate products with increased fluorescence quantum yield after tetrazine ligation with strained alkenes or bicyclononynes. | ||
In vitro, we could also react 5′ caps bearing vinylbenzyl groups in the tetrazine ligation and in the fluorogenic photoclick reaction.49 However, these reactions were not successful up to now in living cells. Limitations are probably the rate constant of the (unstrained) vinylbenzyl group in tetrazine ligations and the low yield of the photoclick reaction. For the future, the use of strained alkenes and bicyclononyne (Fig. 3) together with enhanced turn-on properties of fluorescent (water-soluble) dyes will be important to improve RNA detection.
Challenges for enzymatic modification of endogenous mRNA inside cells: conclusions and outlook
So far, we described strategies for postsynthetic enzymatic modification and subsequent labeling in bioorthogonal reactions. First studies showed already that these reactions can be successfully implemented in living cells to visualize mRNA. However, the modified mRNA was introduced into these cells by lipofection (Fig. 4). Implementing also the enzymatic modification step inside living cells would represent a major advancement in the field. Current approaches for intracellular enzymatic modification have relied on structural features appended to the RNA of interest.24,25 Enzymatic modification of an unaltered endogenous mRNA would be a big improvement. This goal is in reach with methyltransferases, however, several challenges have to be considered. | ||
| Fig. 4 Schematic summary of achievements and challenges regarding chemo-enzymatic mRNA modification inside cells. (A) AdoMet and its analogs are not cell permeable but can be generated in situ and in cells from methionine analogs. These amino acids are taken up by the cell and converted to the respective AdoMet analog by a MAT variant (MAT-Var).51,52 (B) Methyltransferases, including Tgs enzymes, can make use of the AdoMet analog as cosubstrates and transfer non-natural groups to their targets. This has been demonstrated in vitro and for protein methyltransferases also in cells.31,53 For RNA-modification in cells, the target site accessibility and the cosubstrate preference remain to be elucidated. Alternatively, the cap-modified mRNA can be introduced into cells via transfection (e.g. lipofection). (C) The modification at the 5′ cap can be used to tune biological functions of mRNAs, as demonstrated for translation in a proof-of-concept study.35 (D) Modifications at the mRNA cap allow for subsequent fluorescent labeling by intracellular click-reactions.35 | ||
Until recently, one of the major obstacles for implementing methyltransferase-based labeling strategies in living cells was the lack of cell permeability of the required AdoMet analogs. In nature, AdoMet is produced from methionine and ATP by a methionine adenosyltransferase (MAT). Catalytically active residues are highly conserved in MATs among different kingdoms of life, highlighting their essential role for the organism.50 We and others have used engineered human MAT that accepted methionine analogs and produced AdoMet analogs in situ. However, this reaction shows strong product inhibition. By combining the MAT reaction with the N2-cap modification reaction of GlaTgs-V34A, we circumvented product inhibition by direct conversion of the intermediate AdoMet analog. We achieved mRNA cap modification starting from methionine analogs in one pot both in buffer and in eukaryotic cell lysate.51,52 Since methionine analogs are cell permeable amino acids, this strategy can be used to generate AdoMet analogs in cells (Fig. 4A). Wang et al. could already demonstrate the intracellular production of AdoMet analogs in HEK cells carrying the plasmid coding for the MAT variant.53
It is reasonable to expect that AdoMet analogs – in particular the ones with small transferable side-chains – will be also accepted by a range of wild-type methyltransferases (MTases) exhibiting relaxed substrate specificity. The problem of off-targets effects can be largely circumvented if the molecule class of interest (e.g. mRNA) is isolated. However, for applications aiming at imaging RNA in living cells, this labeling approach will have to be pushed towards cosubstrates that are not tolerated by most wild-type MTases, at least not those that have relevant activity.
Finally, the accessibility of the mRNA cap inside cells may be limited because several proteins are known to bind to it in different cellular compartments. The best studied and most important cap-binding proteins in eukaryotic cells are the cap-binding complex54 in the nucleus and the aforementioned eIF4E in the cytoplasm.43 It remains to be elucidated how these proteins will impact the access of the methyltransferases to the mRNA cap inside cells. Similarly, natural methylation in cells may block the mRNA cap and render it inaccessible for further modification at the respective position.
In summary, chemo-enzymatic approaches have been established to label eukaryotic mRNAs with small organic fluorophores and to tune their biological function in vitro and in cells (Fig. 4C and D). It is conceivable to establish also the enzymatic modification step in cells based on the enzymatic generation of AdoMet analogs from cell permeable methionine analogs (Fig. 4A). The biggest remaining challenges for this application are cap accessibility in cells and background resulting from off-target labeling (Fig. 4B). The concept of introducing small functional groups suggests that perturbations of the natural functions will be minimal. However, the effect of cap modifications on binding to partners other than eIF4E is currently unknown and has to be tested. E.g. controls with previously established RNA labeling techniques based on hybridization or on extension with long tags (reviewed in Rath et al.13 and Mannack et al.14) will be important to ensure unaltered mRNA localization. Nevertheless, the available mRNA modification toolkit already provides exciting new options for labeling modified mRNAs in living cells, for enriching mRNAs from cells as well as a new avenue for tuning gene expression via translation efficiency.
Acknowledgements
A. R. thanks the DFG for support by an Emmy Noether fellowship (RE2796/2-1) and the Fonds der Chemischen Industrie (Dozentenpreis). F. M. is supported by the DFG Priority Programme SPP1784 (RE2796/3-1).References
- G. Sicoli, F. Wachowius, M. Bennati and C. Höbartner, Angew. Chem., Int. Ed., 2010, 49, 6443–6447 CrossRef CAS PubMed.
- B. Puffer, C. Kreutz, U. Rieder, M. O. Ebert, R. Konrat and R. Micura, Nucleic Acids Res., 2009, 37, 7728–7740 CrossRef CAS PubMed.
- K. Halbmair, J. Seikowski, I. Tkach, C. Höbartner, D. Sezer and M. Bennati, Chem. Sci., 2016, 7, 3172–3180 RSC.
- K. Fauster, M. Hartl, T. Santner, M. Aigner, C. Kreutz, K. Bister, E. Ennifar and R. Micura, ACS Chem. Biol., 2012, 7, 581–589 CrossRef CAS PubMed.
- E. Paredes, M. Evans and S. R. Das, Methods, 2011, 54, 251–259 CrossRef CAS PubMed.
- X. Ni, M. Castanares, A. Mukherjee and S. E. Lupold, Curr. Med. Chem., 2011, 18, 4206–4214 CrossRef CAS PubMed.
- C. Meyer, U. Hahn and A. Rentmeister, J. Nucleic Acids, 2011, 2011, 904750 Search PubMed.
- J. S. Mattick, J. Exp. Biol., 2007, 210, 1526–1547 CrossRef PubMed.
- F. Miura, N. Kawaguchi, M. Yoshida, C. Uematsu, K. Kito, Y. Sakaki and T. Ito, BMC Genomics, 2008, 9, 574–574 CrossRef PubMed.
- E. Shapiro, T. Biezuner and S. Linnarsson, Nat. Rev. Genet., 2013, 14, 618–630 CrossRef CAS PubMed.
- B. Futcher, G. I. Latter, P. Monardo, C. S. McLaughlin and J. I. Garrels, Mol. Cell Biol., 1999, 19, 7357–7368 CrossRef CAS PubMed.
- E. Yang, E. van Nimwegen, M. Zavolan, N. Rajewsky, M. Schroeder, M. Magnasco and J. E. Darnell Jr., Genome Res., 2003, 13, 1863–1872 CrossRef CAS PubMed.
- A. K. Rath and A. Rentmeister, Curr. Opin. Biotechnol., 2015, 31, 42–49 CrossRef CAS PubMed.
- L. V. Mannack, S. Eising and A. Rentmeister, F1000Research 2016, 5(F1000 Faculty Rev), 775, DOI:10.12688/f1000research.8151.1.
- J. M. Holstein and A. Rentmeister, Methods, 2016, 98, 18–25 CrossRef CAS PubMed.
- D. Schulz and A. Rentmeister, ChemBioChem, 2014, 15, 2342–2347 CrossRef CAS PubMed.
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16–20 CrossRef CAS PubMed.
- A. A. Sawant, A. A. Tanpure, P. P. Mukherjee, S. Athavale, A. Kelkar, S. Galande and S. G. Srivatsan, Nucleic Acids Res., 2016, 44, e16 CrossRef PubMed.
- C. Domnick, F. Eggert and S. Kath-Schorr, Chem. Commun., 2015, 51, 8253–8256 RSC.
- C. Y. Jao and A. Salic, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15779–15784 CrossRef CAS PubMed.
- M. L. Winz, A. Samanta, D. Benzinger and A. Jäschke, Nucleic Acids Res., 2012, 40, e78 CrossRef CAS PubMed.
- F. Li, J. Dong, X. Hu, W. Gong, J. Li, J. Shen, H. Tian and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 4597–4602 CrossRef CAS PubMed.
- S. C. Alexander, K. N. Busby, C. M. Cole, C. Y. Zhou and N. K. Devaraj, J. Am. Chem. Soc., 2015, 137, 12756–12759 CrossRef CAS PubMed.
- C. Dalhoff, G. Lukinavicius, S. Klimasauskas and E. Weinhold, Nat. Chem. Biol., 2006, 2, 31–32 CrossRef CAS PubMed.
- S. Willnow, M. Martin, B. Lüscher and E. Weinhold, ChemBioChem, 2012, 13, 1167–1173 CrossRef CAS PubMed.
- B. J. C. Law, A.-W. Struck, M. R. Bennett, B. Wilkinson and J. Micklefield, Chem. Sci., 2015, 6, 2885–2892 RSC.
- S. Singh, J. Zhang, T. D. Huber, M. Sunkara, K. Hurley, R. D. Goff, G. Wang, W. Zhang, C. Liu, J. Rohr, S. G. Van Lanen, A. J. Morris and J. S. Thorson, Angew. Chem., Int. Ed., 2014, 53, 3965–3969 CrossRef CAS PubMed.
- M. Luo, ACS Chem. Biol., 2012, 7, 443–463 CrossRef CAS PubMed.
- D. Schulz, J. M. Holstein and A. Rentmeister, Angew. Chem., Int. Ed., 2013, 52, 7874–7878 CrossRef CAS PubMed.
- J. M. Holstein, D. Schulz and A. Rentmeister, Chem. Commun., 2014, 50, 4478–4481 RSC.
- G. Pljevaljcic, F. Schmidt and E. Weinhold, ChemBioChem, 2004, 5, 265–269 CrossRef CAS PubMed.
- P. A. Boriack-Sjodin and K. K. Swinger, Biochemistry, 2016, 55, 1557–1569 CrossRef CAS PubMed.
- J. M. Holstein, L. Anhäuser and A. Rentmeister, Angew. Chem., Int. Ed., 2016, 55, 10899–10903 CrossRef CAS PubMed.
- Y. Motorin, J. Burhenne, R. Teimer, K. Koynov, S. Willnow, E. Weinhold and M. Helm, Nucleic Acids Res., 2011, 39, 1943–1952 CrossRef CAS PubMed.
- M. Tomkuviene, B. Clouet-d'Orval, I. Cerniauskas, E. Weinhold and S. Klimasauskas, Nucleic Acids Res., 2012, 40, 6765–6773 CrossRef CAS PubMed.
- S. Reich, D. Guilligay, A. Pflug, H. Malet, I. Berger, T. Crepin, D. Hart, T. Lunardi, M. Nanao, R. W. H. Ruigrok and S. Cusack, Nature, 2014, 516, 361–366 CrossRef CAS PubMed.
- J. M. Holstein, D. Stummer and A. Rentmeister, Protein Eng., Des. Sel., 2015, 28, 179–186 CrossRef CAS PubMed.
- H. Matsuo, H. Li, A. M. McGuire, C. M. Fletcher, A. C. Gingras, N. Sonenberg and G. Wagner, Nat. Struct. Biol., 1997, 4, 717–724 CrossRef CAS PubMed.
- S. Karaki, C. Andrieu, H. Ziouziou and P. Rocchi, Adv. Protein Chem. Struct. Biol., 2015, 101, 1–26 CAS.
- A. Niedzwiecka, J. Marcotrigiano, J. Stepinski, M. Jankowska-Anyszka, A. Wyslouch-Cieszynska, M. Dadlez, A. C. Gingras, P. Mak, E. Darzynkiewicz, N. Sonenberg, S. K. Burley and R. Stolarski, J. Mol. Biol., 2002, 319, 615–635 CrossRef CAS PubMed.
- T. von der Haar, J. D. Gross, G. Wagner and J. E. McCarthy, Nat. Struct. Mol. Biol., 2004, 11, 503–511 CAS.
- E. Darzynkiewicz, J. Stepinski, I. Ekiel, C. Goyer, N. Sonenberg, A. Temeriusz, Y. Jin, T. Sijuwade, D. Haber and S. M. Tahara, Biochemistry, 1989, 28, 4771–4778 CrossRef CAS PubMed.
- E. Darzynkiewicz, I. Ekiel, P. Lassota and S. M. Tahara, Biochemistry, 1987, 26, 4372–4380 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- K. Lang, L. Davis, J. Torres-Kolbus, C. Chou, A. Deiters and J. W. Chin, Nat. Chem., 2012, 4, 298–304 CrossRef CAS PubMed.
- A. M. Pyka, C. Domnick, F. Braun and S. Kath-Schorr, Bioconjugate Chem., 2014, 25, 1438–1443 CrossRef CAS PubMed.
- J. M. Holstein, D. Stummer and A. Rentmeister, Chem. Sci., 2015, 6, 1362–1369 RSC.
- G. D. Markham and M. A. Pajares, Cell. Mol. Life Sci., 2009, 66, 636–648 CrossRef CAS PubMed.
- F. Muttach and A. Rentmeister, Angew. Chem., Int. Ed., 2016, 55, 1917–1920 CrossRef CAS PubMed.
- F. Muttach and A. Rentmeister, Methods, 2016, 107, 3–9 CrossRef CAS PubMed.
- R. Wang, K. Islam, Y. Liu, W. Zheng, H. Tang, N. Lailler, G. Blum, H. Deng and M. Luo, J. Am. Chem. Soc., 2013, 135, 1048–1056 CrossRef CAS PubMed.
- T. Gonatopoulos-Pournatzis and V. H. Cowling, Biochem. J., 2014, 457, 231–242 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |