
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantum chemical study and isothermal titration calorimetry of β-cyclodextrin complexes with mianserin in aqueous solution†
Anna
Ignaczak
 *a,
Bartłomiej
Pałecz
b and
Sylwia
Belica-Pacha
b
*a,
Bartłomiej
Pałecz
b and
Sylwia
Belica-Pacha
b
aDepartment of Theoretical and Structural Chemistry, Faculty of Chemistry, University of Lodz, Pomorska 163/165, 90-236 Lodz, Poland. E-mail: anignacz@uni.lodz.pl
bUnit of Biophysical Chemistry, Department of Physical Chemistry, Faculty of Chemistry, University of Lodz, Pomorska 165, 90-236 Lodz, Poland
First published on 6th January 2017
Abstract
β-Cyclodextrin (β-CD) is studied as a carrier of the drug mianserin (MIA). β-CD with MIA adducts with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1 stoichiometry are investigated in vacuo and in water using quantum chemical methods: PM6 and B3LYP/6-31G(d,p). An effect of the dispersion correction GD2 and the basis set superposition error on the complexation energies is also evaluated. Additionally, the interaction between MIA hydrochloride and β-CD in aqueous solution at 298.15 K is examined experimentally by isothermal titration calorimetry. Interaction parameters, such as the binding constant, enthalpy, entropy and Gibbs free energy, are presented. Analysis of the obtained data led to the following conclusions: the interaction of MIA with β-CD is rather strong; there is no significant energetic difference between the 1:1 complexes of β-CD with S-MIA and R-MIA enantiomers; the 2:1 (β-CD:MIA) adduct is energetically more favorable than 1:1; the complex formation of MIA + β-CD is enthalpy and entropy driven.
:1 and 2:1 stoichiometry are investigated in vacuo and in water using quantum chemical methods: PM6 and B3LYP/6-31G(d,p). An effect of the dispersion correction GD2 and the basis set superposition error on the complexation energies is also evaluated. Additionally, the interaction between MIA hydrochloride and β-CD in aqueous solution at 298.15 K is examined experimentally by isothermal titration calorimetry. Interaction parameters, such as the binding constant, enthalpy, entropy and Gibbs free energy, are presented. Analysis of the obtained data led to the following conclusions: the interaction of MIA with β-CD is rather strong; there is no significant energetic difference between the 1:1 complexes of β-CD with S-MIA and R-MIA enantiomers; the 2:1 (β-CD:MIA) adduct is energetically more favorable than 1:1; the complex formation of MIA + β-CD is enthalpy and entropy driven.
Introduction
One of the commonly used psychotropic drugs is mianserin, classified as a noradrenergic and specific serotonergic antidepressant,1,2 mostly used as its hydrochloride salt3 (MIA; Fig. 1a and d). Mianserin hydrochloride as a psychotropic drug disturbs the interaction of noradrenaline with 5-HT receptors by preventing its reabsorption. It blocks presynaptic receptors (α2-adrenoreceptors) responsible for the binding of serotonin and the mechanism of mianserin action allows for prolonged action of both hormones (noradrenaline and serotonin) improving the mood of a person suffering from depression.4–7 In pharmacological tests of antidepressant activity4 a higher potency is present for the (S)-(+)-mianserin optical enantiomer. A pharmacokinetic study with mianserin hydrochloride indicated that the maximum anticipated oral availability was calculated to be in the range of 34–49%.8 The relatively low extent of absorption can partially be due to incomplete absorption from the gastro-intestinal tract and as a consequence of a hepatic first-pass effect.9 Bioavailability studies show that one class of substances capable of reducing the toxicity of drugs and concomitantly increase their availability is cyclodextrins.10,11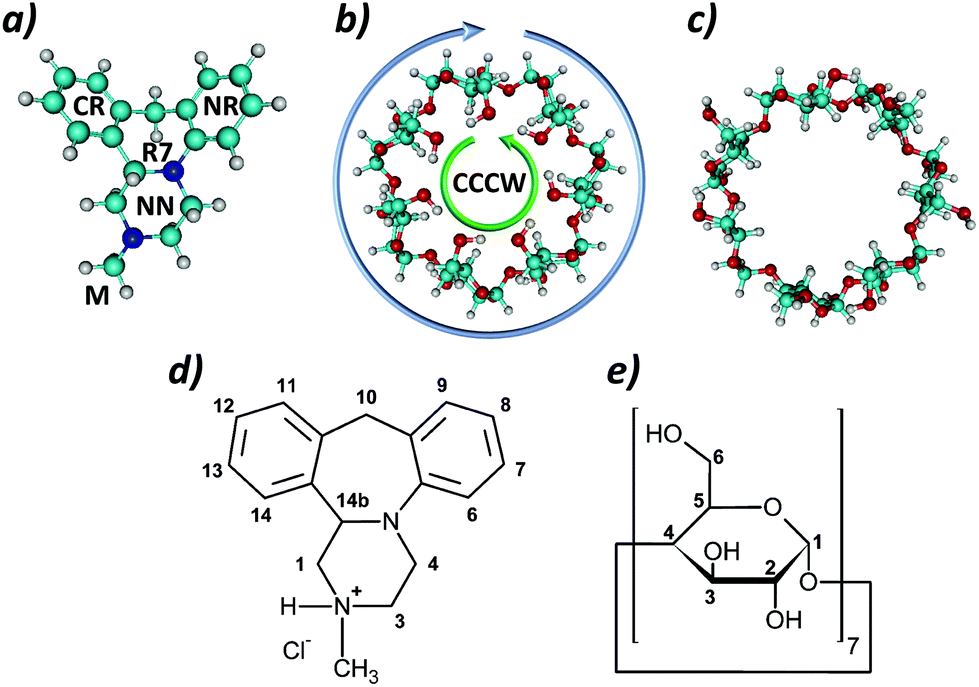 | ||
| Fig. 1 Structures of: (a) mianserin, (b) β-CD-CCCW, (c) β-CD-EXP, and numbering of carbon atoms in: (d) mianserin hydrochloride; (e) β-cyclodextrin. | ||
Some experimental studies on the effects of cyclodextrins on mianserin hydrochloride have been carried out in the past.12,13 From 13C NMR spectra (Job's plots) it was concluded13 that the 1:1 complex of MIA with β-CD predominates in D2O solution, but the parameters of interaction, such as the complex formation constant and other thermodynamic parameters, have not been reported. To our knowledge no computational study was performed on this system. Herein we report the results of our theoretical studies on the structures and complexation energies of 1:1 and 1:2 (MIA:β-CD) adducts performed with the semiempirical method PM6 and the density functional theory (DFT) B3LYP with the 6-31G(d,p) basis set. Moreover, using isothermal titration calorimetry (ITC) we have evaluated the thermodynamic parameters of the complex formation between β-CD and mianserin hydrochloride molecules in the real system. Because commercially available mianserin hydrochloride is a racemic mixture, we have chosen the racemate to use in our experimental examinations presented below.
Method of calculations
The structures of MIA and β-CD used in quantum chemical calculations are shown in Fig. 1. Two different forms of β-CD were tested (Fig. 1b and c). The first one, denoted below as β-CD-CCCW, is the highly ordered geometry reported in ref. 14 as the ground state structure in vacuo. The second one, denoted as β-CD-EXP, is the structure obtained from experiment.15 Since the latter structure contained only the heavy atoms of β-CD, hydrogens were added using the Hyperchem program.16In our study we focus on the comparison of different possible structures of the MIA–β-CD complex, which are shown in Fig. 2. As can be seen, in the case of the 1:1 complex eleven most distinctive orientations of the MIA molecule with respect to β-CD are considered. For the 1:2 complex only two orientations (CAPS A and B in Fig. 2) are taken into account, in both MIA is encapsulated inside the dimer formed by two β-CD molecules.
 | ||
| Fig. 2 Orientations of MIA and CD cone in the 1:1 and 1:2 complexes tested in quantum calculations. The labels CR, NR and M refer to different fragments of the MIA molecule (see Fig. 1a), FΛ and FV indicate its two “flat” orientations, while 1, 2 and S – different sides of the CD cone. | ||
To explore the strength of interaction in each of these orientations with β-CD-CCCW the following procedure was used: the first stage involved calculations made with the semiempirical method PM6 using the Mopac program.17 This method has been applied in several earlier studies on inclusion complexes with cyclodextrins.18–27 It allows to handle hydrogen bonds28 and, as shown in ref. 18–21, predicts structural properties of such systems which are coherent with experimental results. In our calculations mianserin was placed as shown in Fig. 2 and from each initial orientation 108 different structures were produced using the Hyperchem program by a systematic rotation of MIA around each axis, changing the angle stepwise by 10°, giving in total 1188 structures of the 1:1 complex to evaluate. For all structures full PM6 optimization (no constrains) was performed. A similar procedure was employed for the 1:2 complex – in this case two different orientations of MIA were used as starting points (see Fig. 2); thus, in total 216 structures were probed.
In the next step, from all PM6 optimized 1:1 complexes eleven structures were selected, each corresponding to one of the 11 orientations shown in Fig. 2. Of course, within the whole set generated, for each orientation there is a number of similar structures differing in energies due to small geometry modifications; in each such subset the lowest energy structure was selected as a representative of the given orientation. A similar selection was made for the two structures CAPS A and CAPS B of the 1:2 complex.
The thirteen chosen structures served as starting geometries for the calculations performed using the hybrid DFT functional B3LYP,29,30 the 6-31G(d,p) basis set and the ultrafine integration grid. All DFT calculations were performed using the Gaussian09 package.31 For each system full DFT optimization was performed in vacuo for the two enantiomers: (S)-(+)-mianserin and (R)-(–)-mianserin. Additionally, for the DFT optimized structures an effect of the solvent on the interactions was explored by performing optimizations in the presence of water treated with the polarizable continuum model (PCM) and default parameters defined in the Gaussian program. In this part of calculations the structures optimized in vacuo constituted starting configurations for optimizations in water.
For the lowest energy structures vibrational analysis was performed to verify that these are true minima and from the results the thermodynamic properties of these complexes were obtained. As discussed extensively in the literature, the accuracy of thermodynamic parameters obtained from quantum calculations can be improved by applying various corrections.32–36 At the present stage of our study only one correction was introduced. Namely, in the Gaussian program the frequency calculations are made assuming that the molecule is in the gas-phase at a pressure of 1 atm, which at 298 K corresponds to 24.79 dm3, while in solution it should be 1 dm3.36 As described in detail in ref. 36, the solvation free energy change associated with moving a solute from a standard-state gas-phase to a standard-state solution-phase is equal to 7.913 kJ mol−1. Therefore in the present work the total free energies obtained in water were corrected by this value. Indeed, it is recognised that a static correction such as this is a general one and would not be able to fully reproduce the correction requisite for each structure; future work undertaking this is encouraged.
The B3LYP results for the complexes containing the β-CD-EXP molecule were obtained from full optimization of the structures made by replacing β-CD-CCCW with β-CD-EXP in the B3LYP-optimized complexes [S-MIA]–[β-CD-CCCW] and [β-CD-CCCW]–[S-MIA]–[β-CD-CCCW]. Finally, for selected B3LYP optimized structures were performed single point calculations using the B3LYP-GD2 functional, which includes the empirical dispersion model of Grimme.37 The same basis set was used as with the B3LYP method. At this stage additionally the basis set superposition errors (BSSE) using the counterpoise correction (CP)38 were calculated.
Experimental
Mianserin hydrochloride (1,2,3,4,10,14b-hexahydro-2-methyl-dibenzo[c,f]pyrazino[1,2-a]azepine hydrochloride; CAS number 21535-47-7; m.w. = 300.83 g mol−1) and β-cyclodextrin (cyclomaltoheptaose; CAS number 7585-39-9; m.w. = 1134.98 g mol−1) were purchased from Sigma-Aldrich with a purity >99%.Isothermal calorimetric titrations were performed on a MicroCal VP-ITC (USA) instrument employing aqueous solutions at a temperature of 298.15 K. The ITC instrument was composed of two cells: the sample cell (1.4275 ml) and the reference cell filled with water. Water used in calorimetric measurements was distilled three times and degassed prior to experiments. The titration of a ligand – cyclodextrin aqueous solution – to the sample cell with the drug solution was made stepwise with pre-set 380 s intervals. A typical titration involved 55 injections (5 μl each) of a 14 mM β-cyclodextrin solution into the cell containing a 0.5 mM mianserin hydrochloride solution from a syringe acting also as a stirrer, which rotated at a speed of 307 rpm. The first injection was not considered for the analysis. The titration of β-cyclodextrin solution into water and water into mianserin hydrochloride solution was repeated for the same concentrations of solutions used in the main experiment in order to correct the raw data by subtraction of the heat of dilution. During the isothermal titration of the drug solution with cyclodextrin, the released or adsorbed heat associated with each injection was compensated by the power supplied to the sample cell and the feedback power was directly proportional to the heat-flow, dQ/dt, which was recorded as an output signal. After integration of the heat-flow, the calculated heat was analyzed as a function of the cyclodextrin/drug ratio and the data were fitted by a non-linear least squares method using the ORIGIN software yielding the stoichiometric coefficient of binding (n), the equilibrium binding constant (Ka), and the standard changes in the enthalpy of complex formation (ΔH°), as well as the standard changes in entropy (ΔS°) and in the free energy (ΔG°). The parameters were obtained as average values from the four independent experiments.
Results and discussion
Quantum chemical calculations
The structures of β-cyclodextrin and mianserin have been studied in the past using both experimental and quantum chemical methods.3,14,15,39–43 It was shown that the favored structure for mianserin is that with the piperazine ring NN (Fig. 1a) adopting a chair conformation with the methyl group M in the equatorial orientation. Within the central R7 ring there is an anti-periplanar relationship between the lone pair of the nitrogen atom and the H(14b) hydrogen atom attached to the carbon at the chiral center.3 The number of possible conformers for β-CD is much larger due to many possible orientations of the hydroxyl groups. Snor et al.14 performed DFT calculations for different symmetric conformations in the gas phase, in which rims of hydrogen bonds were formed by hydroxyl groups on both primary and secondary carbon atoms. Under anhydrous conditions two low energy structures were identified. In both, one β-CD entry point is closed by the rim of hydrogen bonds at primary hydroxyl groups in the counterclockwise (CC) orientation while, on the other side of the CD ring, the orientation of the secondary hydroxyl groups can be either counterclockwise (CC) or clockwise (CW) (Fig. 1b). The lowest energy conformer CCCW (denoted as β-CD-CCCW) was used in the first part of our studies on the interaction of MIA with β-CD.It is known, however, that the structure of β-CD in biological systems is very different from its ground state in vacuo, as it is strongly perturbed by interactions with other molecules.15 Therefore, in the second part of our theoretical studies as a model was used the cyclodextrin structure β-CD-EXP extracted from its complex with a cyclodextrin glycosyltransferase.15 The geometry of this molecule is entirely different from that in the ground state: the rim of hydrogen bonds formed by primary hydroxyl groups is disrupted and the entry point on their side is open (Fig. 1c).
First we analyzed whether the interaction of the molecule β-CD-CCCW with the two enantiomers, S and R, of MIA differs significantly. In Table 1 the results obtained with semiempirical PM6 and density functional B3LYP methods are presented. For all systems the interaction energy between MIA and β-CD was calculated as the difference between the optimized complex and its isolated components: ΔE = Ecomplex − EMIA − Eβ-CD for 1:1 and ΔE = Ecomplex − EMIA − 2Eβ-CD for 1:2 stoichiometry. Here EMIA and Eβ-CD denote the ground state energies of isolated MIA and β-CD-CCCW. In the case of β-CD-EXP the Eβ-CD reference energy was obtained by optimizing the β-CD-EXP structure extracted from the lowest energy (FΛ1) optimized complex [S-MIA]–[β-CD-EXP].
:1 and 1:2 adducts of mianserin with β-CD-CCCW and β-CD-EXP obtained in vacuo and in water (PCM) for each MIA orientation shown in Fig. 2. For the β-CD-CCCW case the results are presented for enantiomers R and S of MIA in its chair conformer. Additionally the results are shown for the [β-CD]–[β-CD] dimer (the same β-CD orientation as in CAPS A and B). All values are in kJ mol−1
| Orientations | PM6 R | PM6 S | B3LYP R | B3LYP S | B3LYP S | B3LYP S | B3LYP S |
|---|---|---|---|---|---|---|---|
| ΔE | ΔE | ΔE | ΔE | ΔE | ΔE | ΔE | |
| β-CD-CCCW | β-CD-CCCW | β-CD-CCCW | β-CD-CCCW | β-CD-CCCW | β-CD-EXP | β-CD-EXP | |
| Vacuum | Vacuum | Vacuum | Vacuum | Water | Vacuum | Water | |
| CR1 | −66.15 | −67.62 | −41.15 | −53.20 | −48.52 | −49.27 | −36.59 |
| NR1 | −51.00 | −61.67 | −53.41 | −45.66 | −38.27 | −55.69 | −46.21 |
| M1 | −60.00 | −62.26 | −16.04 | −24.34 | −19.85 | −19.75 | −13.94 |
| FΛ1 | −47.86 | −44.42 | −44.98 | −53.70 | −46.36 | −62.58 | −51.28 |
| FV1 | −65.40 | −64.14 | −22.67 | −23.94 | −19.65 | −26.96 | −24.51 |
| S | −32.57 | −36.63 | −23.11 | −22.23 | −14.55 | −28.53 | −20.25 |
| CR2 | −25.33 | −24.28 | −17.93 | −17.55 | −16.15 | −56.35 | −42.26 |
| NR2 | −18.92 | −24.79 | −18.21 | −23.42 | −21.47 | −22.24 | −12.85 |
| M2 | −31.90 | −33.45 | −24.90 | −29.39 | −30.03 | −25.13 | −14.98 |
| FΛ2 | −32.36 | −25.58 | −28.50 | −23.42 | −21.47 | −57.22 | −47.16 |
| FV2 | −24.95 | −24.58 | −19.37 | −19.18 | −17.89 | −32.02 | −20.18 |
| CAPS A | −250.04 | −250.96 | −249.63 | −278.15 | −228.55 | −274.48 | −198.73 |
| CAPS B | −252.42 | −257.66 | −227.77 | −218.55 | |||
| [β-CD]–[β-CD] dimer | −203.60 | −314.61 | −267.56 | −321.31 | −253.72 |
For the 1:1 guest–host complex both methods, PM6 and B3LYP, predict stronger interaction of mianserin with the wider rim of the β-CD-CCCW cone, with the complexation energies being of similar order of magnitude. There are however some differences as to the preferred orientation. The striking discrepancy appears for M1 and FV1 configurations, for which the PM6 interaction seems to be strongly overestimated. Also, the PM6 method for both enantiomers predicts the same orientation CR1 to be favored, while the higher level method indicates two different configurations.
According to the B3LYP results, for the R-MIA enantiomer the most stable is the structure obtained from the initial NR1 configuration. The next preferred orientation for this enantiomer is FΛ1, which is the most stable for the S-MIA enantiomer. For this isomer two orientations, FΛ1 and CR1, yield very similar interaction energies, because in these cases the final optimized structures are essentially the same. All B3LYP optimized structures and the corresponding total electronic energies are provided in the ESI.† As can be seen in Table 1, there is some symmetry in the complexation energies FΛ1, CR1 and NR1 when the two enantiomers are compared. The interaction energies for other conformations are much weaker. The stronger interactions in the three complexes are due to the formation of hydrogen bonds between one hydroxyl group of β-CD and one of the nitrogen atoms in MIA. In vacuo the following distances are found in the FΛ1 [S-MIA]–[β-CD-CCCW] complex: dOH = 0.996 Å, dN⋯H = 1.83 Å, dN⋯O = 2.81 Å, while the O–H⋯N angle is equal to 165°. According to the Jeffrey's categorization,44,45 this corresponds to moderately strong hydrogen bonds.
The most stable structures optimized in vacuo for both enantiomers are presented in Fig. 3a and b. Much stronger complexation energies are obtained for the 1:2 complex (Fig. 3c). This results from the formation of hydrogen bonds between the hydroxyl groups on the secondary carbons of two cyclodextrin molecules, which is seen also in ΔE for the dimer β-CD–β-CD (Table 1). The inclusion of mianserin causes some deformation of the β-CD molecules; therefore, the complexation energy for the 1:2 complex is less than that for the [β-CD]–[β-CD] dimer, but the overall balance still gives a quite large energy gain when the system [β-CD]–[MIA]–[β-CD] is formed. The B3LYP results in Table 1 show that for both enantiomers of MIA the structure CAPS A in vacuo is much more stable than CAPS B, therefore the latter was omitted in our further studies.
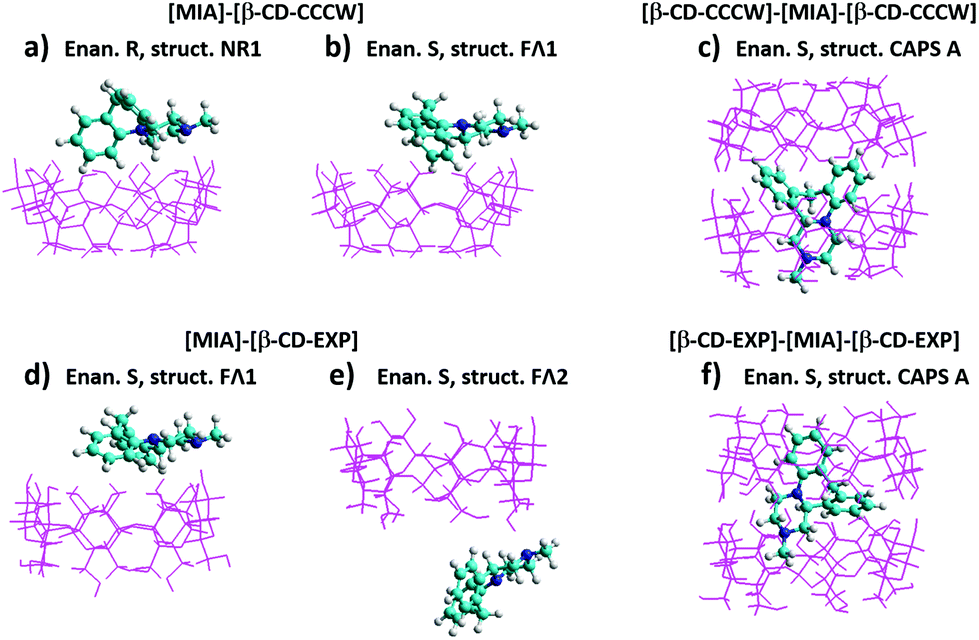 | ||
| Fig. 3 Structures found for the lowest energy complexes (1:1 and 1:2) optimized in vacuo at the B3LYP/6-31G(d,p) theory level. | ||
Subsequently, we analyzed two additional aspects of MIA–β-CD complexation, namely the interaction of MIA with the structure β-CD-EXP and an effect of solvent. The results of these calculations are also presented in Table 1. Since the complexation energies for the two enantiomers were highly similar in vacuo, this part of our studies was limited to the S-enantiomer. The results show that water (PCM) in general decreases complexation energies by several kJ mol−1, but does not change the overall trends. The optimized structures are similar to those found in vacuo.
The complexation Gibbs free energies ΔG and enthalpies ΔH obtained for the most stable structures with the B3LYP method are given in Table 2. They have been calculated using formulas similar to these defining ΔE. All ΔG values for the 1:1 complex are positive, suggesting that the reaction would not be spontaneous. In the case of [S-MIA]–[β-CD-CCCW] the smallest free energies of 14 kJ mol−1 are obtained in vacuo for each enantiomer, R and S, of mianserin, although they correspond to different structures. For β-CD-EXP the free energy of formation of the complex with S-MIA in the FΛ2 configuration is significantly smaller than for the corresponding FΛ1 structure, and this is further lowered by the presence of solvent. The results obtained for the 1:2 stoichiometry suggest that formation of such an inclusion complex is thermodynamically preferable, also for the conformer β-CD-CCCW in vacuo.
| Structure | Conditions | ΔH (B3LYP) | ΔG (B3LYP) | ΔH (B3LYP-GD2/BSSE) | ΔG (B3LYP-GD2/BSSE) | |
|---|---|---|---|---|---|---|
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 complex :1 complex
|
||||||
| [S-MIA]–[β-CD-CCCW] | FΛ1 | Vacuum | −46.13 | 14.07 | −88.25 | −28.05 |
| [S-MIA]–[β-CD-CCCW] | NR1 | Vacuum | −38.26 | 19.32 | −77.10 | −19.53 |
| [R-MIA]–[β-CD-CCCW] | FΛ1 | Vacuum | −37.30 | 22.82 | −79.06 | −18.94 |
| [R-MIA]–[β-CD-CCCW] | NR1 | Vacuum | −46.13 | 14.06 | −88.59 | −28.40 |
| [S-MIA]–[β-CD-EXP] | FΛ1 | Vacuum | −54.26 | 10.74 | −87.92 | −22.93 |
| [S-MIA]–[β-CD-EXP] | FΛ2 | Vacuum | −49.09 | 2.29 | −65.21 | −13.83 |
| [S-MIA]–[β-CD-CCCW] | FΛ1 | Water | −37.86 | 15.05 | −89.92 | −37.02 |
| [S-MIA]–[β-CD-CCCW] | NR1 | Water | −30.27 | 18.22 | −78.97 | −30.48 |
| [R-MIA]–[β-CD-CCCW] | FΛ1 | Water | −31.18 | 21.93 | −81.40 | −28.29 |
| [R-MIA]–[β-CD-CCCW] | NR1 | Water | −37.02 | 15.19 | −89.47 | −37.25 |
| [S-MIA]–[β-CD-EXP] | FΛ1 | Water | −42.62 | 7.31 | −86.40 | −36.47 |
| [S-MIA]–[β-CD-EXP] | FΛ2 | Water | −39.38 | 1.36 | −65.38 | −24.64 |
|
1:2 complex
|
||||||
| [β-CD-CCCW]–[S-MIA]–[β-CD-CCCW] | CAPS A | Vacuum | −262.29 | −78.24 | −444.38 | −260.33 |
| [β-CD-EXP]–[S-MIA]–[β-CD-EXP] | CAPS A | Vacuum | −252.99 | −53.52 | −456.31 | −256.84 |
| [β-CD-CCCW]–[S-MIA]–[β-CD-CCCW] | CAPS A | Water | −206.70 | −34.54 | −450.08 | −277.92 |
| [β-CD-EXP]–[S-MIA]–[β-CD-EXP] | CAPS A | Water | −176.10 | 2.72 | −460.34 | −281.52 |
The magnitude of corrections for the dispersion forces, which can play an important role in these systems, was investigated by performing single point calculations using the B3LYP-GD2 functional.29,30,37 Into these calculations were included also the BSSE corrections, which were not taken into account in the B3LYP results. The complexation energies obtained from the B3LYP-GD2 calculations and the CP values calculated with the same method were used to correct the enthalpy and free energy by the dispersion and BSSE effects, and the results are presented in the last two columns of Table 2.
The calculations reveal that for the systems studied the dispersion contribution to the interaction energies is very large: for example in vacuo it is −87 kJ mol−1 for the complex [S-MIA]–[β-CD-CCCW] (FΛ1 orientation) and −411 kJ mol−1 for the corresponding 1:2 complex (CAPS A). Although the BSSE has an opposite effect, the final B3LYP-GD2 complexation energies are much more negative than the uncorrected B3LYP values. As a result, the B3LYP-GD2/BSSE free energies are negative for all structures, indicating spontaneous formation of such complexes. One may note in Table 2 that for the [S-MIA]–[β-CD-EXP] complex the B3LYP and B3LYP-GD2 methods indicate different orientations as being thermodynamically most favored. However, our evaluation of the dispersion effect on the complexation energies is approximate, based on the structures obtained from the B3LYP optimization.
Isothermal titration calorimetry (ITC)
Thermodynamic parameters in the real system can be obtained experimentally using isothermal titration calorimetry. Simple isothermal titration of the drug by the cyclodextrin solution gives an output signal, which is directly proportional to the heat-flow, dQ/dt (ref. 46) (Fig. 4a), which depends on the cyclodextrin/drug ratio (Fig. 4b). The values of the association constant Ka and other thermodynamic parameters, listed in Table 3, were obtained using the model referred to as one-set of independent sites47 or a single-class binding model.48,49 In this model all binding sites are considered identical as no distinction between the interacting sites is made47 and the corresponding equilibrium constants are the same for all binding sites.48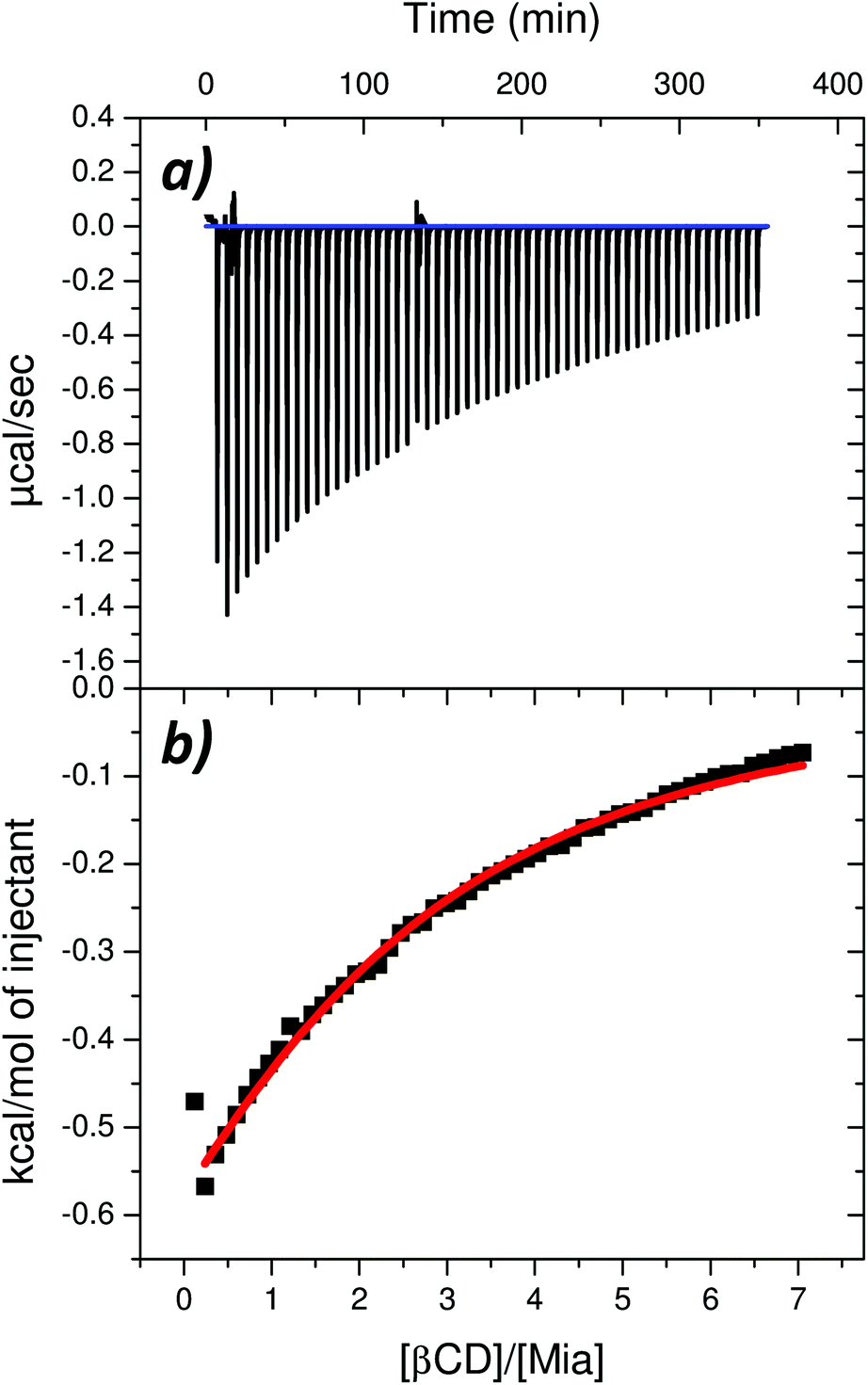 | ||
| Fig. 4 (a) Thermal effects of the titration of a 0.5 mM solution of mianserin hydrochloride (in cell) with a 14 mM solution of β-cyclodextrin (in a syringe). (b) Integrated thermal effect of β-cyclodextrin–drug interaction corrected by the dilution effect of the drug and the β-cyclodextrin. The solid red line is the best non-linear least-squares fit of the data. | ||
As indicated by the value of the association constant (Table 3), which is >1000 M−1, the interaction between the drug and the cyclodextrin molecules is rather strong (the stability of the complex is high) and the complexation process is spontaneous – the value of the Gibbs free energy of binding is less than zero. Moreover, the stoichiometry of the complex formed between mianserin hydrochloride and β-cyclodextrin is close to the ratio of 1:2 (Table 3) with a negligible amount of a co-existing mixture of complexes with 1:1 stoichiometry and even 1:3.50 These results agree qualitatively with the ΔG computed values shown in Table 2, which are also negative and much larger for the 1:2 complex than for 1:1. The significant difference in the magnitude of the computed and measured ΔG may result from the fact that the theoretical model considers just MIA, not its hydrochloride, and also that it does not explicitly include water molecules. Formation of hydrogen bonds between water, MIA and β-CD before their association will be competitive to the complexation considered in the quantum calculations, and obviously must affect the reaction thermodynamics. The complex can be formed only if some solvent molecules directly interacting with the ligand and the macromolecules are released, and this corresponds to certain changes in both enthalpy and entropy of solvation. The final outcome in the real system results from the balance between contributions coming from the β-CD–MIA complex formation, the solvation of substrates and products, as well as competitive reactions, such as the formation of the β-CD dimers or larger aggregates. The complexation process in the real system is exothermic, as confirmed by the negative change in the standard enthalpy value (ΔH° < 0) of the host–guest interactions (Table 3), but the entropic effects of binding dominate over the enthalpic effects (|ΔH°| < |TΔS°|). The positive and higher value of entropic effects consisting mostly of the solvation and conformational entropy indicates that the binding process is entropy-driven. Upon complex formation between the β-CDs and MIA the number of conformations of the ligand and the macromolecules is reduced in comparison with the number of conformations of the free components. This corresponds to a decrease in the conformational entropy and contributes unfavorably to the value of Gibbs free energy of binding, as observed in the computed values (Table 2).
Since the PCM model does not include explicit solute–solvent interactions, the positive value of TΔS° obtained from ITC is expected to result from the solvation entropy contribution. Solvent molecules that interact electrostatically or by hydrogen-bonding with the ligand or with the macromolecules are restricted and have low entropy.46 Their release corresponds to a favorable increase in solvation entropy. Moreover, in aqueous solution, in the direct vicinity of the non-polar groups of the drug and β-cyclodextrin molecules, the hydrogen bonds between water molecules are reinforced as a result of hydrophobic hydration.51–56 When the MIA molecule associates with the β-CD molecules, restricted and highly ordered water molecules hydrogen-bonded or being in the direct vicinity of non-polar groups of the drug or inside the β-CD are released into the bulk water, with a large increase in entropy as a result.57
It should be mentioned that the predominant 1:2 stoichiometry found in the present work contradicts the earlier 13C NMR result of 1:1.13 The difference can be due to the way of the stoichiometry determination in the cited literature. It is based on the chemical shift of the only one carbon atom in position 6 in the NR aromatic ring of the MIA molecule,13 whereas our result is based on a holistic approach taking into account all positions of the carbon atoms of MIA and β-CD molecules. Moreover, the authors13 acknowledged that a sandwich-like MIA–CD complex with 1:2 stoichiometry is also expected when the MIA molecule is complexed via rings CR and NR simultaneously with two different CD molecules.
Conclusions
The main conclusions drawn from our results are as follows:(a) The quantum calculations show that energetically there is no difference between the 1:1 complexes formed by R and S enantiomers of mianserin.
(b) Formation of hydrogen bonds between one of the hydroxyl groups of cyclodextrin and a nitrogen atom of mianserin has a major impact on the computed complexation energies. While in the case of β-CD-CCCW such bonds can be created only by hydroxyl groups on secondary carbon atoms, the β-CD-EXP can interact with MIA from both sides of the cone, i.e. via hydroxyl groups on primary and secondary carbon atoms. This suggests that in real systems 2:1 complexes (MIA:β-CD) could also be formed.
(c) The lowest energy structures of 1:1 complexes indicated by B3LYP calculations are not typical inclusion complexes – no part of mianserin is located inside the β-CD cavity, thus mianserin remains rather suspended over the gate to the cone. However, our preliminary calculations at the B3LYP-GD2 level suggest that this conclusion should be further re-examined by performing full geometry optimization using the dispersion corrected functional.
(d) Both methods B3LYP and B3LYP-GD2 show that formation of hydrogen bonds between two cyclodextrins is energetically favorable and it has a stabilizing effect on the 1:2 mianserin–cyclodextrin complex, despite some energy loss required to deform the β-CD structure. The same 1:2 stoichiometry was found to be predominant in the real system in our ITC measurement.
(e) The quantum chemical calculation results suggest that the [β-CD]–[β-CD] dimer formation is energetically beneficial. Moreover, formation of bigger structures built from cyclodextrin rings similar to polyrotaxanes is possible58,59 and competitive to the complexation of mianserin with cyclodextrin.
(f) Both computed (B3LYP-GD2) and ITC measured results show that the formation of complexes is an exothermic and exergonic process. The opposite sign of the computed and measured values of ΔS indicates a significant role of the solvation entropy in the process.
Of course, the computed results and trends discussed above have been obtained at a certain theory level, defined by the chosen methods. It would be desirable to verify/refine them by applying other approaches, such as the stochastic search of the configurational space and more advanced quantum methods, and we hope that such complementary research will be undertaken in future.
Acknowledgements
This research was supported in part by the PL-Grid Infrastructure.References
- W. J. Van Der Burg, I. L. Bonta, J. Delobelle, C. Ramon and B. Vargaftig, J. Med. Chem., 1970, 13, 35–39 CrossRef CAS PubMed.
- B. B. Vargaftig, J. L. Coignet, C. J. de Vos, H. Grijsen and I. L. Bonta, Eur. J. Pharmacol., 1971, 16, 336–346 CrossRef CAS PubMed.
- S. G. Sagdinc and A. E. Sahinturk, J. Mol. Struct., 2012, 1021, 53–62 CrossRef CAS.
- R. M. Brogden, R. C. Heel, T. M. Speight and G. S. Avery, Drugs, 1978, 16, 273–301 CrossRef CAS PubMed.
- P. A. Palha, L. Ferreira, A. B. Abreu-Lima, C. Ramalhao and R. Fernandez, Clin. Ther., 1985, 142, 87–88 Search PubMed.
- J. Pawlowska, Z. Czarnocki, K. Wojtasiewicz and J. K. Maurin, Tetrahedron: Asymmetry, 2003, 14, 3335–3342 CrossRef CAS.
- R. S. Farag, M. S. Afifi and M. M. Abd-Rabow, Int. J. Pharm. Sci. Res., 2011, 2, 1197–1203 CAS.
- C. J. Timmer, S. Pourbaix, J. P. Desager, M. Sclavons and C. Harvengt, Eur. J. Drug Metab. Pharmacokinet., 1985, 10, 315–323 CrossRef CAS PubMed.
- O. Huber and J. Szejtli, Inclusion Science; Volume 5, 1988, Proceedings of the Fourth International Symposium on Cyclodextrins; Munich, West Germany.
- J.-Q. Zhang, K. Li, K.-M. Jiang, Y.-W. Cong, S.-P. Pu, X.-G. Xie, Y. Jin and J. Lin, RSC Adv., 2016, 6, 17074–17082 RSC.
- S. I. Farag Badawy, M. M. Ghorab and C. M. Adeyeye, Int. J. Pharm., 1996, 145, 137–143 CrossRef.
- C. B. Eap, K. Powell and P. Baumann, J. Chromatogr. Sci., 1997, 35, 315–320 CAS.
- B. Chankvetadze, G. Endresz, D. Bergenthal and G. Blaschke, J. Chromatogr., A, 1995, 717, 245–253 CrossRef CAS.
- W. Snor, E. Liedl, P. Weiss-Greiler, A. Karpfen, H. Viernstein and P. Wolschann, Chem. Phys. Lett., 2007, 441, 159–162 CrossRef CAS.
- The 3CGT.pdb file provided by Protein Data Bank, http://www.rcsb.org/pdb/explore/explore.do?structureId=3CGT CrossRef PubMed, from A. K. Schmidt, S. Cottaz, H. Driguez and G. E. Schulz, Biochemistry, 1998, 27, 5909–5915 CrossRef PubMed.
- HyperChem™ Professional 8.0.10, Hypercube, Inc., 1115 NW 4th Street, Gainesville, Florida 32601, USA Search PubMed.
- J. J. P. Stewart, MOPAC2012, Stewart Computational Chemistry, Colorado Springs, CO, USA, http://OpenMOPAC.net, 2012 Search PubMed.
- M. I. Sancho, E. Gasull, S. E. Blanco and E. A. Castro, Carbohydr. Res., 2011, 346, 1978–1984 CrossRef CAS PubMed.
- K. Elasaad, B. Norberg and J. Wouters, Supramol. Chem., 2012, 24, 312–324 CrossRef CAS.
- L. A. Fredin and T. C. Allison, J. Phys. Chem. A, 2016, 120, 2135–2143 CrossRef CAS PubMed.
- T. Ishizu, H. Tsutsumi, H. Yamamoto and K. Harano, Magn. Reson. Chem., 2009, 47, 283–287 CrossRef CAS PubMed.
- M. V. Araujo, O. F. L. Macedoa, C. Nascimento, L. Conegero, L. Barreto, L. E. Almeida, N. Costa and I. F. Gimenez, Spectrochim. Acta, Part A, 2009, 72, 165–170 CrossRef PubMed.
- I. Djilani, L. Nouar, F. H. S. Madi, A. Bouhadiba and D. Khatmi, C. R. Chim., 2013, 16, 696–703 CrossRef.
- N. Lachi, D. Khatmi and R. Djemil, C. R. Chim., 2014, 17, 1169–1175 CrossRef.
- S.-S. Lee, S. Park, J.-Y. Kim, H.-R. Kim, S. Lee and H. B. Oh, Phys. Chem. Chem. Phys., 2014, 16, 8376–8383 RSC.
- K. Sahra, K. Dinar, A. Seridi and M. Kadri, Struct. Chem., 2015, 26, 61–69 CrossRef CAS.
- F. O. Suliman and A. A. Elbashir, J. Mol. Struct., 2012, 1019, 43–49 CrossRef CAS.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision D.01), Gaussian Inc., Wallingford, CT, 2013 Search PubMed.
- M. Mammen, E. I. Shakhnovich, J. M. Deutch and G. M. Whitesides, J. Org. Chem., 1998, 63, 3821–3830 CrossRef CAS.
- J.-Y. Tao, W.-H. Mu, G. A. Chass, T.-H. Tang and D.-C. Fang, Int. J. Quantum Chem., 2013, 113, 975–984 CrossRef CAS.
- H.-X. Zhou and M. K. Gilson, Chem. Rev., 2009, 109, 4092–4107 CrossRef CAS PubMed.
- J. H. Jensen, Phys. Chem. Chem. Phys., 2015, 17, 12441–12451 RSC.
- C. P. Kelly, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2006, 110, 16066–16081 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- C. W. Funke, Recl. Trav. Chim. Pays-Bas, 1982, 101, 437–440 CrossRef CAS.
- J. Kelder, C. Funke, T. de Boer, L. Delbressine, D. Leysen and V. Nickolson, J. Pharm. Pharmacol., 1997, 49, 403–411 CrossRef CAS PubMed.
- S. G. Sagdinc and A. E. Sahinturk, Spectrochim. Acta, Part A, 2013, 104, 222–234 CrossRef CAS PubMed.
- W. Chen, C.-E. Chang and M. K. Gilson, Biophys. J., 2004, 87, 3035–3049 CrossRef CAS PubMed.
- T. Heine, H. F. Santos, S. Patchkovskii and H. A. Duarte, J. Phys. Chem. A, 2007, 111, 5648–5654 CrossRef CAS PubMed.
- G. A. Jeffrey, An introduction to hydrogen bonding, Oxford University Press, 1997 Search PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- E. Freire, Y. Kawasaki, A. Velázquez Campoy and A. Schön, Characterisation of Ligand Binding by Calorimetry, in Biophysical Approaches Determining Ligand Binding to Biomolecular Targets: Detection, Measurement and Modelling, ed. A. Podjarny, A. Dejaegere and B. Kieffer, Royal Society of Chemistry, 2011, ch. 9, pp. 275–299 Search PubMed.
- K. Bouchemal and S. Mazzaferro, Drug Discovery Today, 2012, 17, 623–629 CrossRef CAS PubMed.
- A. A. Saboury, M. S. Atri, M. H. Sanati and M. Sadeghi, J. Therm. Anal. Calorim., 2006, 83, 175–179 CrossRef CAS.
- S.-S. Qu, Y. Liu, T.-Z. Wang and W.-Y. Gao, Chemosphere, 2002, 46, 1211–1214 CrossRef CAS PubMed.
- D.-Z. Sun, S.-B. Wang, X.-L. Wei and B.-L. Yin, J. Chem. Thermodyn., 2005, 37, 431–436 CrossRef CAS.
- M. Ide, Y. Maeda and H. Kitano, J. Phys. Chem. B, 1997, 101, 7022–7026 CrossRef CAS.
- D. Hecht, F. Tadesse and L. Walters, J. Am. Chem. Soc., 1993, 115, 3336–3337 CrossRef CAS.
- F. Hirata, Bull. Chem. Soc. Jpn., 1998, 71, 1483–1499 CrossRef CAS.
- V. V. Yaminsky and E. A. Vogler, Curr. Opin. Colloid Interface Sci., 2001, 6, 342–349 CrossRef CAS.
- B. Pałecz, J. Am. Chem. Soc., 2005, 127, 17768–17771 CrossRef PubMed.
- B. Pałecz, S. Belica, H. Piekarski and A. Jóźwiak, Thermochim. Acta, 2009, 489, 1–4 CrossRef.
- C. Tanford, The Hydrophobic Effect: Formation of Micelles and Biological Membranes, Wiley, New York, 1980 Search PubMed.
- P. Liu, C. Chipot, X. Shao and W. Cai, J. Phys. Chem. C, 2012, 116, 17913–17918 CAS.
- Y. Kang, K. Guo, B.-J. Li and S. Zhang, Chem. Commun., 2014, 50, 11083–11092 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The DFT optimized geometries and total electronic energies for all molecules in vacuo and water. See DOI: 10.1039/c6ob02109c |
| This journal is © The Royal Society of Chemistry 2017 |