
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic incorporation and utilization of an emissive 6-azauridine†
Patrycja A.
Hopkins
,
Lisa S.
McCoy
and
Yitzhak
Tor
 *
*
Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093, USA. E-mail: ytor@ucsd.edu
First published on 12th December 2016
Abstract
To display favorable fluorescent properties, the non-emissive native nucleosides need to be modified. Here we present a motif that relies on conjugating 5-membered aromatic heterocycles (e.g., thiophene) to a 6-azapyrimidine (1,2,4-triazine) core. Synthetic accessibility and desirable photophysical properties make these nucleosides attractive candidates for enzymatic incorporation and biochemical assays. While 6-azauridine triphosphate is known to be poorly tolerated by polymerases in RNA synthesis, we illustrate that conjugating a thiophene ring at position 5 overcomes such limitations, facilitating its T7 RNA polymerase-mediated in vitro transcription incorporation into RNA constructs. We further show that the modified transcripts can be ligated to longer oligonucleotides to form singly modified RNAs, as illustrated for an A-site hairpin model RNA construct, which was employed to visualize aminoglycoside antibiotics binding.
Introduction
The sensitivity and simplicity of fluorescence spectroscopy has made it one of the most invaluable techniques for exploring biomolecules, their structure and dynamics as well as their interactions with diverse ligands.1,2 Unlike many proteins, nucleic acids are not intrinsically emissive. The non-emissive pyrimidine and purine nucleobases need to be modified to display usable fluorescent properties.1 Diverse approaches for emissive nucleosides have indeed been devised over the years,1,3–8 but only a small fraction of the analogues made satisfies the criterion of isomorphicity.9–13 Isomorphic nucleobases need to closely resemble their native counterparts with respect to their size, shape and hydrogen bonding pairing abilities. While only few useful purine analogues have been reported,1,10,13–18 the largest number of chemical variations reported are for the pyrimidines, particularly 5-modified uridine analogues.19 Several groups, including ours, have investigated a variety of isomorphic nucleosides whose design relied on conjugating five-membered rings such as thiophene,11,20,21 furan,9,21–27 thiazole21,27 and oxazole9,24 to the pyrimidine core. The 5-substituted uridine cores extend the chromophore, frequently augmenting the charge separation in the excited state, yielding useful fluorophores (Fig. 1a). Interestingly, markedly fewer 6-modified uridines have been utilized in a similar manner.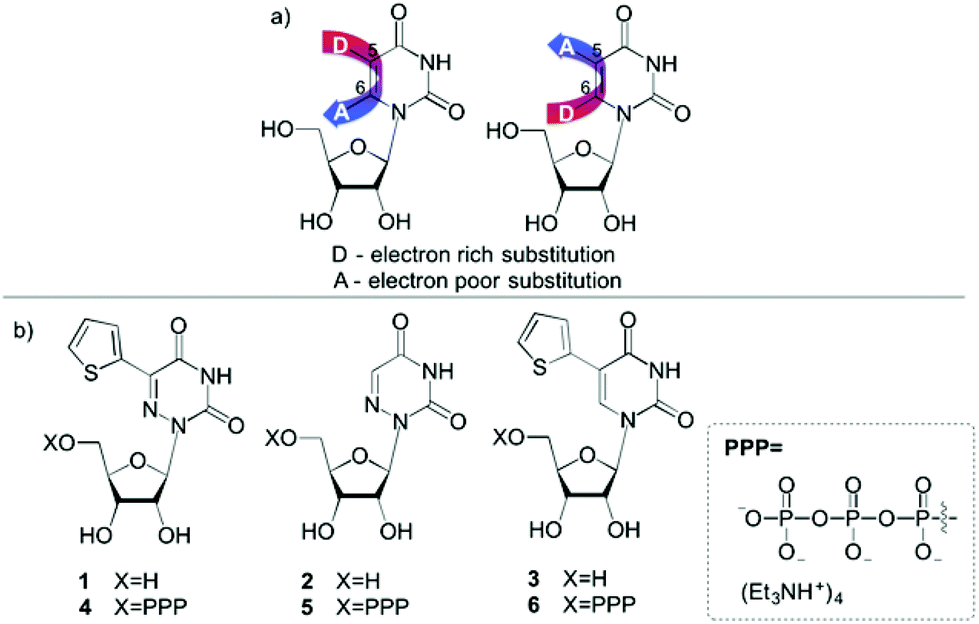 | ||
| Fig. 1 (a) Nucleoside design: enhancing the polarization of the conjugated electron poor/electron rich biaryl system. (b) Modified uridines: 5-(thiophen-2-yl)-6-azauridine (1), 6-azauridine (2), 5-(thiophen-2-yl)uridine (3) and their corresponding triphosphates 4, 5, 6. | ||
Modifying the pyrimidine's 5 and 6 positions by judiciously placing conjugated electron poor/electron rich substituents can further enhance the chromophore's “push–pull” interactions, allowing one to potentially tune their photophysical features (Fig. 1a). In our primary design, we have replaced the uracil core with the more electron-deficient 6-azauracil, while keeping the electron-rich thiophene ring, expecting red-shifted absorption and emission maxima (1, Fig. 1b). This indeed has been confirmed to be the case.12 To explore the utility of such a modification within oligonucleotides we have sought to explore the enzymatic incorporation of the corresponding triphosphates. However, enzymatic incorporation of 6-azauridine triphosphate (5) has been reported to be extremely inefficient28–30 and has been proposed to potentially be due to the nucleoside's favourable syn conformation.31–33
We have previously observed enhanced T7 RNA polymerase-mediated incorporation of 5-thiophenoUTP (6).11 We therefore hypothesized that conjugating a thiophene ring to 6-azaUTP at position 5 may partially overcome the detrimental effect of the N substituent at the 6 position, potentially providing “polymerase-friendly” substrate (4), which will allow the enzymatic fabrication of fluorescent RNAs. Here, we report the synthesis of this triphosphate and its enzymatic incorporation into oligonucleotides. The impact of the “6-aza modification” on in vitro transcription reactions was investigated by comparing the performance of native UTP, 6-azaUTP (5) and 5-thiophenoUTP (6). We illustrate that the modification at the 5 position indeed significantly improves the enzymatic incorporation with essentially doubled transcription efficiency for 5-thiopheno-6azaUTP (4) compared to 6-azaUTP (5). We further demonstrate that the resulting modified transcript can be ligated to unmodified oligonucleotides to provide longer, fluorescent RNA constructs. As a proof of concept, a singly modified A-site model RNA hairpin is assembled and used to monitor the binding of aminoglycoside antibiotics. The emissive 5-thiopheno-6-azauridine can probe antibiotics binding either directly or via FRET-monitored displacement titrations. We conclude that the efficient enzymatic incorporation of this 5-modified 6-azaUTP and its responsiveness to environmental changes make it an attractive reporter nucleoside for exploring RNA and its interactions.
Results
Triphosphate synthesis
The parent nucleoside 5-(thiophen-2-yl)-6-azauridine 1 was synthesized as reported.12 It was then converted to the 5′-triphosphate 4 using POCl3 and tributylammonium pyrophosphate (Scheme 1), following standard procedures.11,34,35 The analytically pure triphosphate 4 was obtained by ion-exchange chromatography and reverse-phase HPLC. After precipitation with 3% sodium perchlorate in acetone and treatment with Chelex 100, nucleotide 4 was characterized by mass spectrometry and 1H, 13C and 31P NMR spectroscopy (see ESI†).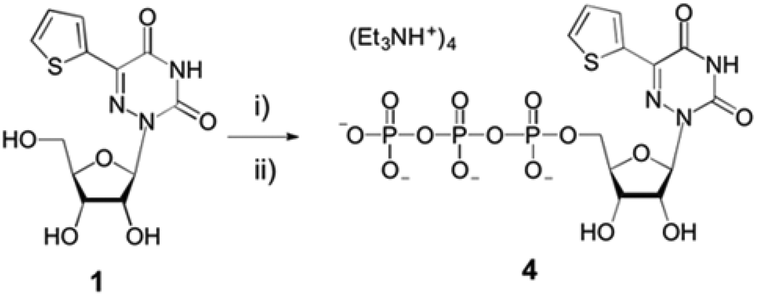 | ||
| Scheme 1 Synthesis of modified nucleoside triphosphate 4. Reagents and conditions: (i) POCl3, (MeO)3PO; (ii) tributylammonium pyrophosphate, proton sponge, 0–4 °C. See ESI† for details. | ||
T7 RNA polymerase-mediated in vitro transcription reactions
To evaluate the enzymatic incorporation of the 6-aza-modified nucleosides into short RNA oligonucleotides, transcription reactions with the analytically pure triphosphates 4–6 and T7 RNA polymerase were performed (Fig. 2a). A short DNA promoter-template duplex was assembled by annealing the 18-mer consensus promoter with commercial DNA template A. All full-length RNA transcripts 7–10 were obtained under standard in vitro transcription conditions and purified by denaturing polyacrylamide gel electrophoresis.11,14,23,36,37 UV shadowing (254 nm) was used to visualize the transcription reactions. Notably, under long UV illumination (365 nm) transcripts 8 and 10, which contain fluorescent modifications (lanes 3, 4, 7 and 8), are highly emissive (Fig. 3b).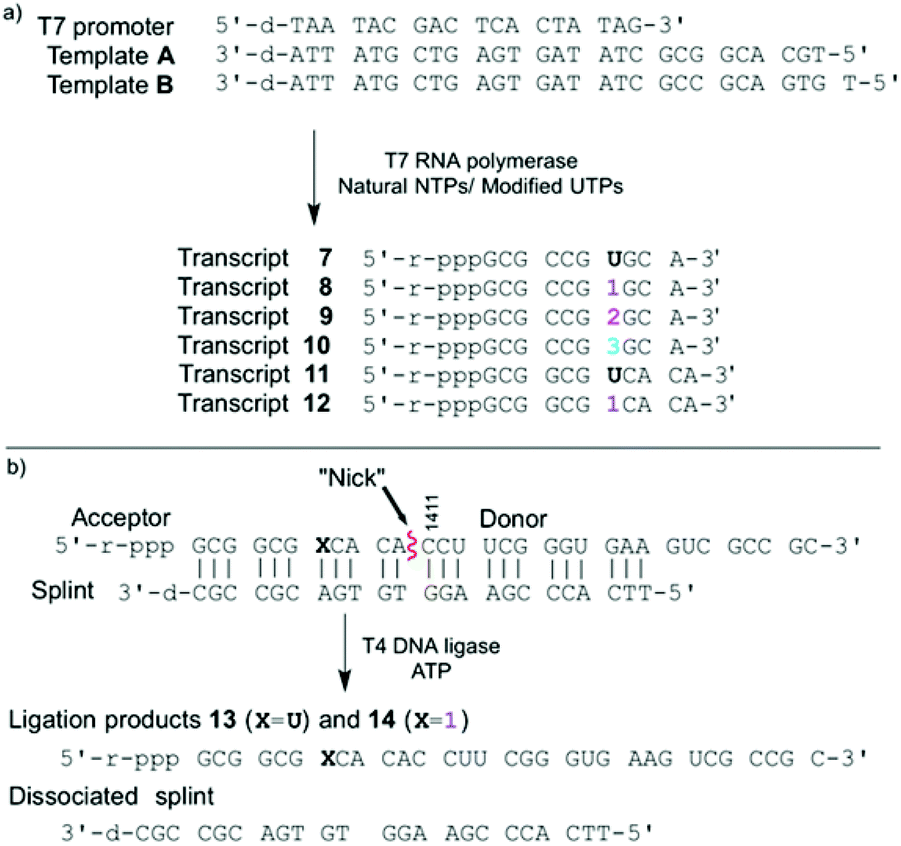 | ||
| Fig. 2 (a) Transcription reaction with T7 promoter and template A in the presence of natural NTPs and UTP or modified UTPs 4–6; resulting in transcripts 7–10 respectively. Transcription reaction with T7 promoter and template B in the presence of natural NTPs and UTP or modified 4, resulting in transcripts 11, 12 respectively. (b) Ligation reaction with T4 DNA ligase, modified acceptor, donor and splint. | ||
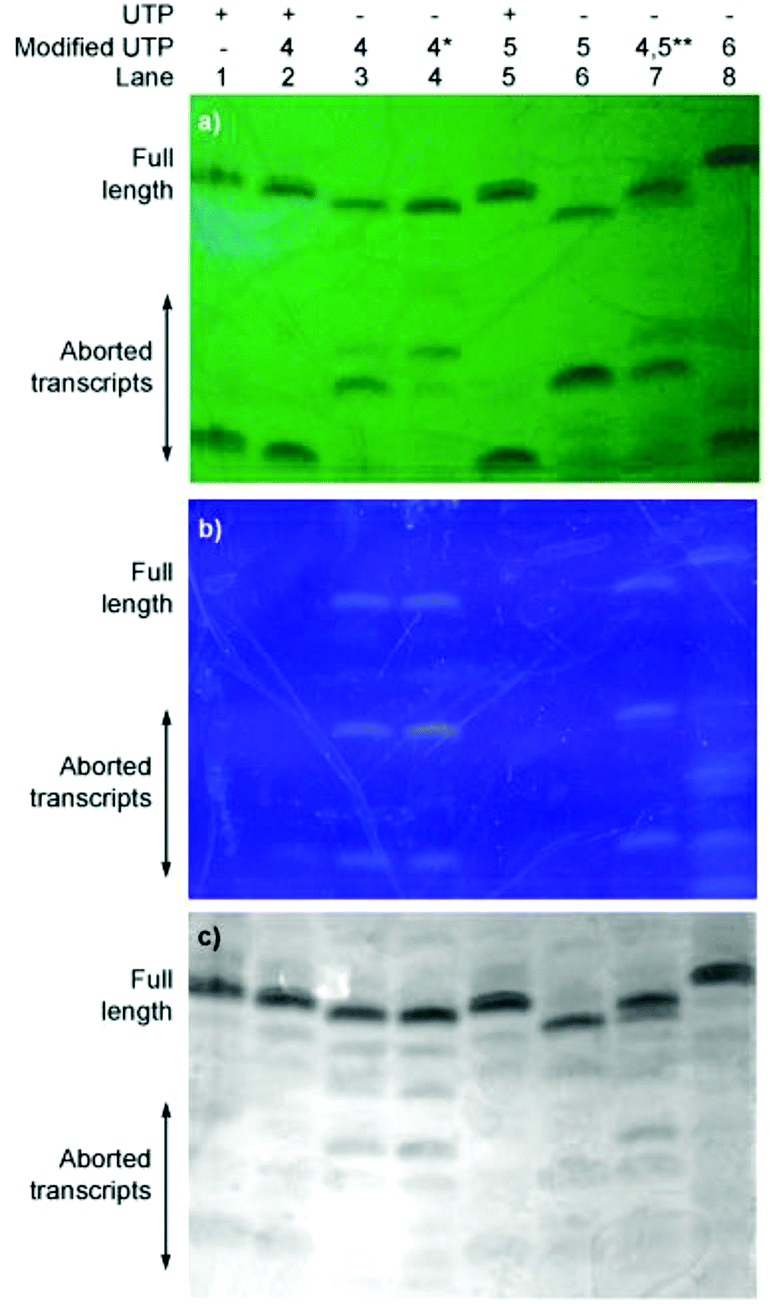 | ||
| Fig. 3 Transcription reactions were carried out with T7 promoter and template A in the presence of natural NTPs and UTP or modified UTPs 4, 5, 6. Lane 1: control reaction with all natural NTPs; lane 2: competition reaction in the presence of equimolar concentration of UTP and 4, lane 3: reaction in the presence of 4, lane 4: reaction in presence of 4 (*5 mM instead of 1 mM, as for all other reactions), lane 5: reaction in the presence of equimolar concentration of UTP and 5, lane 6: reaction in the presence of 5, lane 7**: reaction in the presence of equimolar concentration of 4 and 5, lane 8: reaction in the presence of 6. The reaction was resolved by gel electrophoresis on a denaturing 20% polyacrylamide gel; under UV light at (a) 254 nm (on TLC plate); (b) 365 nm; (c) black and white picture after Stains-all was used to visualize the oligonucleotides. | ||
Following extraction and isolation, the incorporation efficiency of triphosphates 4, 5 and 6 compared to native UTP (lane 1) in the full-length product was determined to be 77 ± 2%, 37 ± 3% and 97 ± 2%, respectively (lanes 3, 6 and 8). Additionally, the aborted transcripts were extracted and characterized by MALDI-TOF MS. Nucleotide 1 produced two main truncated products, where only one is highly fluorescent (lane 3). These were assigned as the emissive 7-mer (5′-pppGCG CCG1–3′) and the shorter, non-emissive 6-mer (5′-pppGCG CCG-3′) (lane 3). The major abortive transcript for 2 was also found to correlate to a 6-mer (lane 6). Interestingly, when only native NTPs were used (lane 1) the truncated construct was characterized as a 5-mer, where the last nucleotide is A instead of C (5′-pppGCG CA-3′ instead of 5′-pppGCG CC-3′).
Additionally, competitive transcription reactions were performed in the presence of equimolar concentrations of UTP and the modified triphosphates 4 and 5 (Fig. 3, lanes 2 and 5, respectively). The 6-azauridine-containing triphosphates (4 and 5) cannot compete with UTP and only the native full length 10-mer is produced. In contrast, 5-thiophenoUTP (6), can favourably compete with UTP.11 T7 polymerase exhibits approximately 1.5× fold preference for the modified triphosphate (4), in which the thiophene ring is conjugated at position 5, over 6-azaUTP (5) (lane 7).‡
T4 DNA ligase-mediated in vitro ligation reaction
A singly modified A-site hairpin was designed for monitoring small molecule binding based on previously designed short RNA constructs (Fig. 2b and 5).23,38 A singly modified 11-nt RNA construct with 1 at position 7 was transcribed to give transcript 12 using previously described conditions (Fig. 2a and S2.2†). The modified transcript 12, serving as an acceptor, was ligated to a 5′-phosphorylated 20-nt donor oligonucleotide containing only native nucleotides (Fig. 2b) under standard in vitro ligation condition, including T4 DNA ligase, ATP and a bridging DNA splint, and resolved by 20% PAGE (Fig. S2.3†).39 The ligation product 14 was extracted and analyzed for its composition by enzymatic digestion in the presence of S1 nuclease and alkaline phosphatase. Integrated reversed-phase HPLC gave the following nucleoside ratio: C/U/A/G/1 = 10.7/4.0/4.0/12.6/1.1 (Fig. 4). Fractions corresponding to each peak were collected and subjected to mass spectrometric analysis, which confirmed the authenticity of the native and modified nucleoside 1. Thermal melting studies showed that the incorporation of 1 had a small effect on the overall stability of the folded RNA. The unmodified control RNA ligation product 13 displayed a Tm of 82.8 (±0.1) °C, while the Tm of modified RNA ligation product 14 was 79.3 (±0.3) °C (Fig. S5†).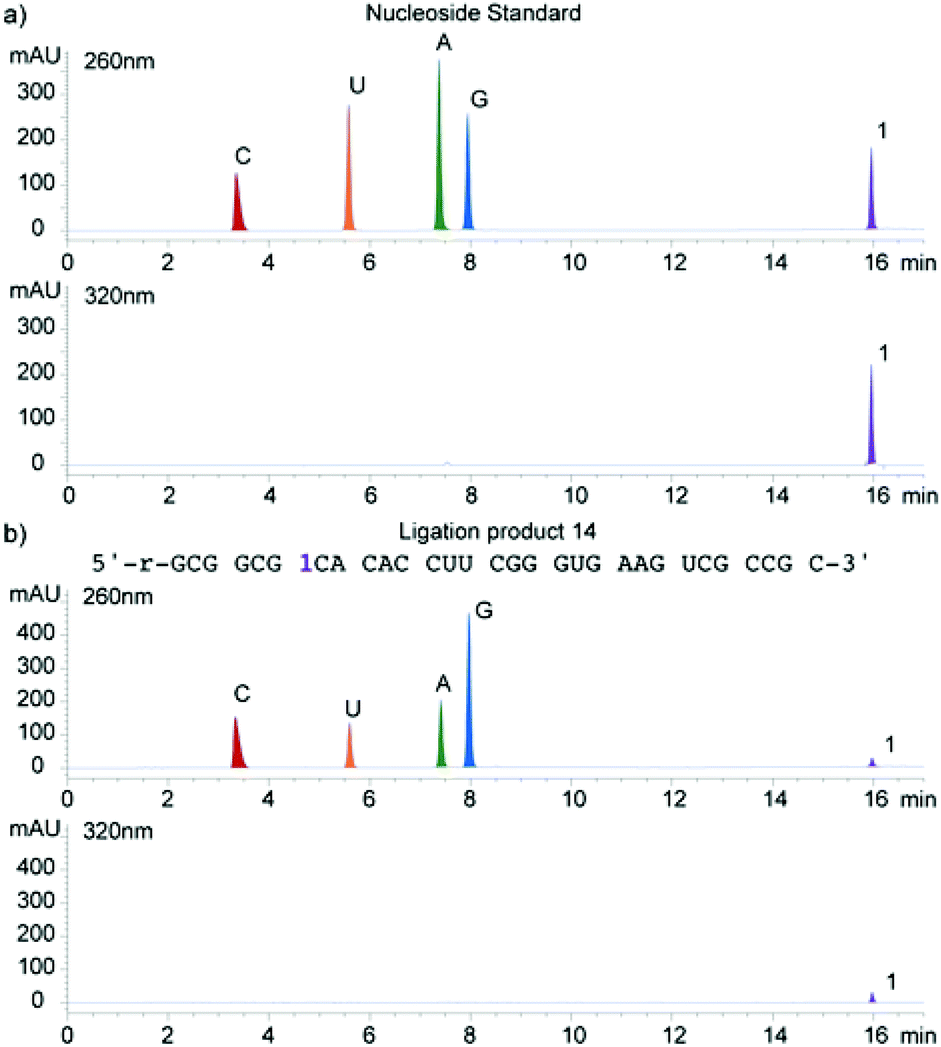 | ||
| Fig. 4 HPLC profile of enzymatic digestion reactions. (a) A mixture of nucleosides used as a standard and (b) digestion results of ligation product 14. Digestion of 1–2 nmol of transcript was carried out using S1 nuclease for 2 h at 37 °C and followed by dephosphorylation with alkaline phosphatase for 2 h at 37 °C. The ribonucleoside mixture obtained was analyzed by reverse-phase analytical HPLC with Agilent column eclipse XDB-C18 (5 μm, 4.6 × 150 mm). Mobile phase: 0–5% acetonitrile (0.1% formic acid) in water (0.1% formic acid) over 10 min and then increase 5–35% acetonitrile (0.1% formic acid) in water (0.1% formic acid) 10–17 min; flow rate 1 mL min−1. | ||
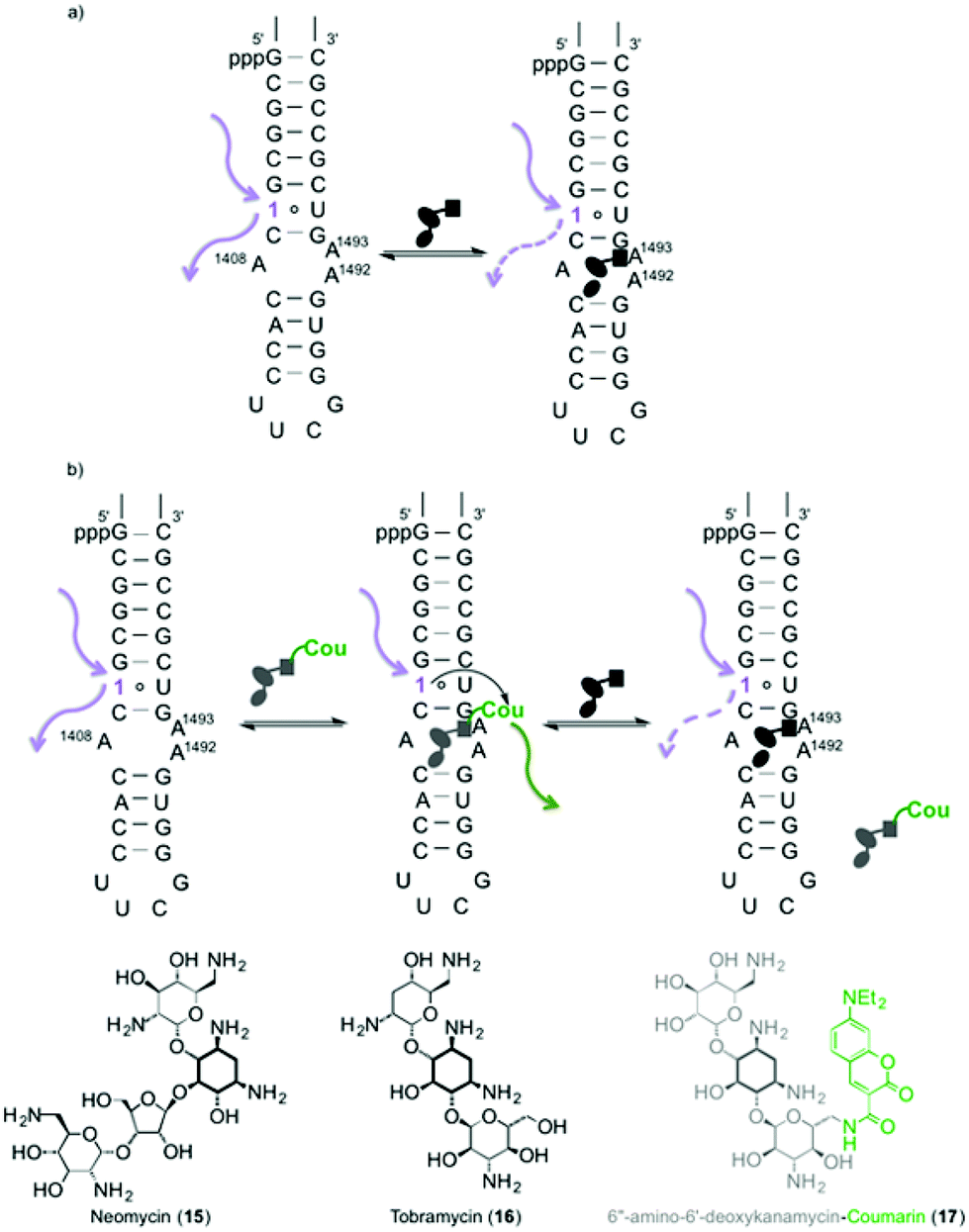 | ||
| Fig. 5 (a) Binding of aminoglycosides to a fluorescently modified A-site hairpin 14. (b) Binding and displacement events of A-site bound coumarin-aminoglycoside with unlabelled RNA binders (where 6-aza modified uridine 1 is a FRET donor and coumarin is a FRET acceptor). | ||
Binding of aminoglycosides to a fluorescently modified A-site
To assess the potential of the 6-aza modified uridine 1 as a reporter nucleoside for small molecule binding assays, we chose the A-site model hairpin 14 as a test system. Note that the modified residue 1 is in close proximity to the binding site but not part of it (Fig. 5). A-site hairpin 14 was therefore excited at 320 nm and changes in emission at 420 nm were monitored upon addition of aminoglycosides. Titration curves were generated by plotting fractional fluorescence saturation versus aminoglycoside concentration. Titration of neomycin (15) resulted first in slight decrease and then more significant increase of the fluorescence intensity (Fig. 6a). The first binding event, corresponding to EC50 value of 0.9 ± 0.2 × 10−3 mM, in close agreement with literature values, reflects the A-site binding of neomycin.38 The second binding event, manifested by an increase of emission intensity, corresponding to EC50 value of 2 ± 1 × 10−2 mM, likely reflects remote neomycin–RNA association. Similarly, when tobramycin (16) was titrated into the labelled construct 14, the fluorescent intensity first slightly increased (EC50 4 ± 1 × 10−3 mM) and was then followed by more significant emission increase (EC50 0.24 ± 0.03 mM) (Fig. S6.2†).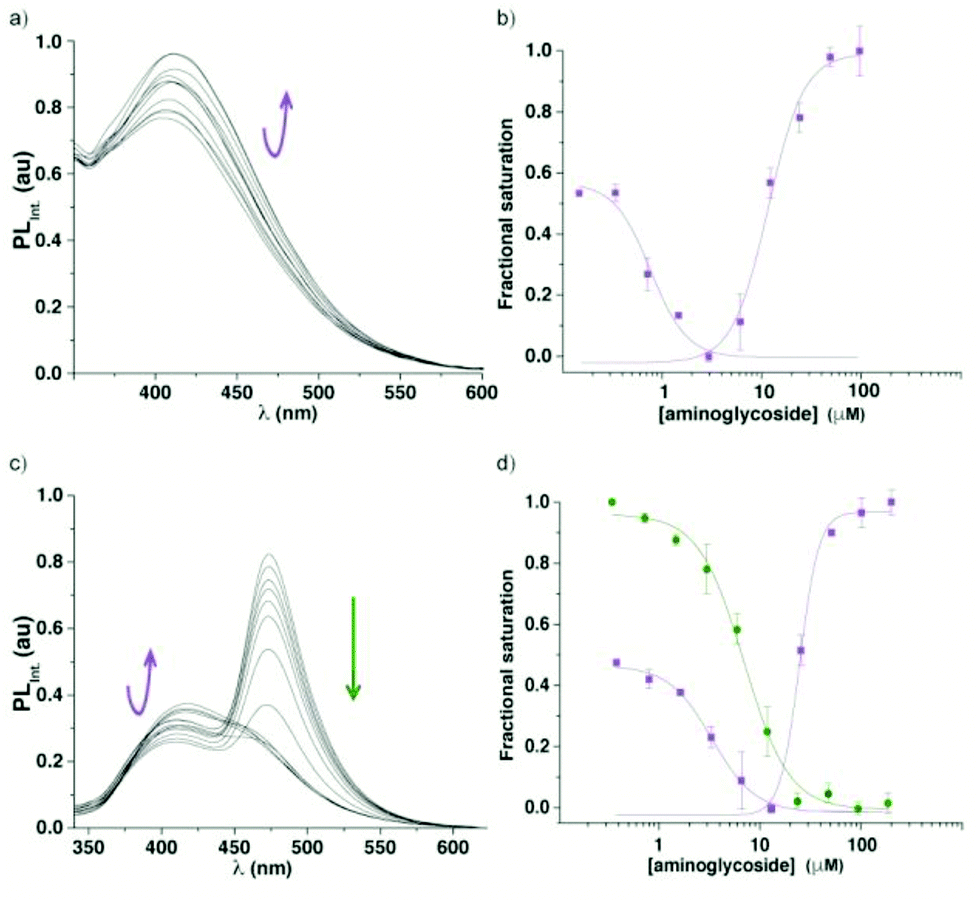 | ||
| Fig. 6 Fluorescence titrations of the A-site construct 14 with neomycin (15). (a) Representative emission spectra for titrations of A-site hairpin 14 with neomycin. (b) Curve fit for the titrations of A-site hairpin 14 with neomycin (15) with respective values EC50 0.9 ± 0.2 × 10−3 mM and 2 ± 1 × 10−2 mM (fractional saturation vs. antibiotic concentration). (c) Representative emission spectra for displacement studies: titrations of A-site hairpin 14 with neomycin (15). (d) Curve fit for the displacement studies: titrations of A-site hairpin 14 with neomycin with respective values EC50 3.0 ± 0.1 × 10−3 mM and 3 ± 1 × 10−2 mM [fractional saturation vs. antibiotic concentration; 1, FRET donor (purple), 6″-amino-6″-deoxykanamycin-coumarin conjugate, FRET acceptor (green)]. Each event was fitted separately (see ESI† for details). | ||
Displacement of A-site bound coumarin-aminoglycoside with unlabelled RNA binders
In our displacement assay format, detection of binding events is dependent on the interactions between two fluorophores, where a labelled RNA serves as a FRET donor and an appropriately labelled aminoglycoside serves as a FRET acceptor.38 The emissive 5-thiopheno-6-azauridine was found to be an excellent FRET donor for 7-diethylaminocoumarin-3-carboxylic acid (Fig. 7).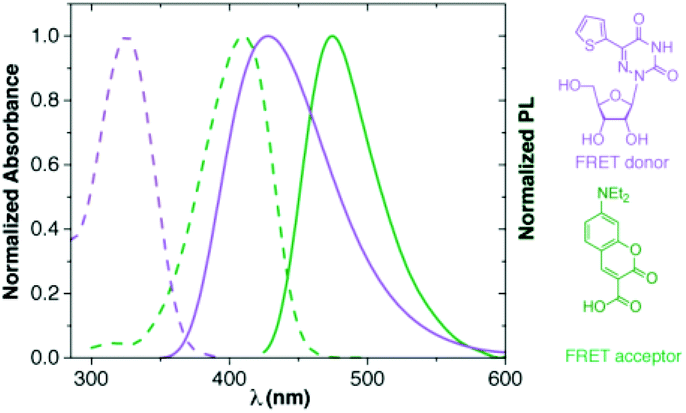 | ||
| Fig. 7 Normalized absorption (---) and emission (—) spectra of 5-thiopheno-6-azauridine in purple (1; FRET donor) and coumarin in green (FRET acceptor). | ||
Assembly of the FRET complex was accomplished by adding the coumarin conjugate of 6″-amino-6″-deoxykanamycin 17 (0.53 μM) to the modified A-site hairpin 14 (1 μM) prior to the addition of an unlabelled aminoglycoside. Titration of unlabelled neomycin (15) into a pre-formed FRET complex excited at 320 nm resulted in a slight decrease followed by an increase of emission intensity of the FRET donor at 420 nm accompanied by a concurrent decrease of the acceptor emission at 470 nm (Fig. 6). The corresponding EC50 values from the displacement assay for each event (3.0 ± 0.1 × 10−3 mM and 3 ± 1 × 10−2 mM), respectively are similar to the data obtained by the direct binding assay. Note that the first binding event is reflected by changes in the donor's and sensitized acceptor's emission, while the second event is manifested by changes in the donor's emission intensity only, as expected (Fig. 6).
Discussion
Tuning the photophysical properties of emissive nucleoside analogues can be a challenging task. We envision that introducing an electron-withdrawing modification at position 6 can enhance the polarization of 5-modified conjugated biaryl uridines, ideally yielding red-shifted emission maxima and increased fluorescence intensity (Fig. 1a). While indeed realized with the 6-azauracil motif, the major challenge remaining was the sluggish recognition of such modified pyrimidines by common RNA polymerases.28–30 To explore the utility of such a modification within oligonucleotides we have sought to explore the enzymatic incorporation of the corresponding triphosphates, hypothesizing that the conjugated aromatic ring at the 5-position may enhance enzymatic incorporation when compared to 6-azaUTP, a rather poorly incorporating parent nucleotide.T7 RNA polymerase-mediated in vitro transcription reactions
Although in vitro transcription reactions can be very effective, the “pickiness” of RNA polymerase can be challenging. To investigate the “6-aza” effect on in vitro transcription we compared the incorporation efficiency of 5-thiopheno-6-azauridine (1), 6-azauridine (2) and 5-thiopheno-uridine (3).T7 RNA polymerase accepts the triphosphates 4–6 as substrates and effectively incorporates all three modified nucleosides 1–3. Triphosphate 6, which is lacking the nitrogen at position 6 in the uridine core is nearly as effective as the native UTP. 6-Substituted uridines are frequently forced into a less favourable syn orientation and perform less effectively in transcription reactions.31–33 Indeed, analyses of the abortive transcripts suggest the enzyme aborts transcription when in a close proximity to a 6-azauridine (2) modification, in addition to a lower overall transcription yield. Conjugation of a thiophene ring to the 6-azauridine core, restores “enzyme friendly” behaviour, facilitating the enzymatic generation of full length emissive transcripts, with 2-fold increase of incorporation efficiency compared to 6-azauridine (2). Additionally, competitive transcription reactions show, as hypothesized, that conjugating a thiophene ring to 6-azaUTP at position 5 partially overcomes the detrimental effect of the CH/N modification at the 6 position.
T4 DNA ligase-mediated in vitro ligation reaction
A singly modified A-site hairpin was designed for a potential aminoglycoside binding assay (Fig. 5). Considering that the construct contains multiple uridine residues and only one modification is desired, its preparation by an enzymatic transcription reaction is not plausible. A potential approach involves transcribing a smaller 11-nt construct with a single incorporation of 1 (transcript 12, Fig. 2a) and ligating it to a 20-nt oligonucleotide containing native uridines only (donor, Fig. 2b).Using T4 DNA ligase, an ATP-dependent enzyme, which catalyzes the joining of two duplexed oligonucleotides, an efficient ligation reaction between acceptor 12 and native donor yielded the modified A-site construct 14. Enzymatic digestion followed by reversed-phase HPLC profiling clearly shows that the nucleoside composition matches with the expected 31-nt ligation product 14's sequence (Fig. 4).
Binding of aminoglycosides to a fluorescently modified A-site
The bacterial decoding site within the 16S ribosomal RNA, also referred to as the A-site, possesses a small domain comprised of unpaired adenosines, targeted by aminoglycoside antibiotics.40–42 For biophysical studies, a short RNA construct representing a functional decoding site is commonly used.43,44 The dynamics and remodelling of this domain upon aminoglycoside binding can be exploited for fluorescence-based assays as structural changes are likely to translate into changes in the microenvironment of the fluorescent probe.45–54 Both 2-aminopurine (2-AP) and pyrroloC (pC) have shown promise as such,55–59 however, both suffer from significantly quenched emission upon incorporation into oligonucleotides.1,23 Additionally, the responsiveness of 2-aminopurine is antibiotic-dependent and appears to fail for certain binders, such as neomycin.57We hypothesized that upon titration of aminoglycoside antibiotics (e.g., neomycin, tobramycin) into the labelled RNA construct 14, the induced conformational and environmental changes would cause changes in emission intensity of the responsive fluorescent nucleoside 1 (Fig. 6a), which is sensitive to changes in polarity, viscosity, and pH.12,60–62 Indeed, upon titration of neomycin (15) and tobramycin (16) into the fluorescent A-site hairpin 14 the emission intensity has changed with increasing concentrations of the antibiotic. For neomycin, the fluorescent intensity slightly decreased initially, which was then followed by a significant increase likely reflecting two discrete binding events. Similar behaviour was observed when tobramycin was titrated. The EC50 values extracted for the first event closely corresponded to previously reported literature values for A-site–aminoglycoside binding.38 We speculate that the second event potentially reflects remote binding (such as to the hairpin stem or loop present in the RNA construct 14), which propagates to trigger an environmental change and a photophysical response of the responsive nucleoside.25,53,63,64
In a second approach to detecting aminoglycoside–RNA binding, we envisioned an assay where detection is dependent on the interaction between two fluorophores acting as a FRET pair, with 5-thiopheno-6-azauridine as a donor and 7-diethylaminocoumarin-3-carboxyamide as the acceptor. Following the binding of the singly labelled RNA 14 to a coumarin conjugate of 6″-amino-6″-deoxykanamycin 17 (Fig. 5b), which exhibit a high FRET signal, a competitor (e.g., unlabelled neomycin 15) is titrated in, displacing the fluorescent placeholder (17). The intensity of the FRET-acceptor's sensitized emission drops upon titrating neomycin, reflecting its displacement from the A-site. Further titration of neomycin, shows, as before, enhanced emission of the donor azaU nucleoside (1). This event, likely reflecting neomycin binding to remote RNA sites (e.g., the tetraloop) nevertheless propagates through the folded structure and alters the probe's immediate environment, yielding enhanced emission.
The A-site and its interactions with aminoglycoside antibiotics have been thoroughly studied.48–52 Models for specific as well as non-specific binding events have been discussed, including crystal structures showing specifically- and nonspecifically-bound neomycin.53 In this context, such fluorescence based assays are frequently used for inhibitor discovery and not necessarily for thorough biophysical analyses of RNA–ligand interactions,23,38 as the photophysical changes and their physical triggers are rarely fully interpretable, even for established probes.1
Conclusions
The design of emissive nucleosides and their implementation for biochemical and biophysical applications remain an empirical exercise. The relationship between molecular structure and photophysical properties (particularly when it comes to excited state dynamics) is hard to predict and can only be experimentally determined. This issue is particularly challenging for emissive nucleosides since their photophysical features are frequently impacted upon incorporation into oligonucleotides. Here we illustrate that 6-modified uridine analogues represent an intriguing family of emissive nucleosides with potentially valuable applications. Particularly rewarding is the observation that upon modification of the 5-position, such nucleosides can be incorporated into RNA constructs by T7 RNA polymerase-mediated in vitro transcription reactions. 5-Thiopheno-6-azauridine triphosphate (4) is a “polymerase-friendly” substrate, and is incorporated twice as efficiently as 6-azauridine triphosphate (5), the parent nucleotide (77 ± 2% vs. 37 ± 3%, respectively), which can then be utilized in RNA–ligand binding assays. We illustrate that this analogue can be used as the sole probe or in conjunction with a FRET acceptor for monitoring RNA binding by aminoglycoside antibiotics, either directly or via displacement assays, respectively. These strategies are not necessarily limited to the A-site and its binders, but can possibly be used for the analysis of other RNA targets with biochemical interest or therapeutic potential.Acknowledgements
We thank the National Institutes of Health for financial support (grant GM 069773), and the Chemistry and Biochemistry MS facility at UCSD.Notes and references
- R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579–2619 CrossRef CAS PubMed.
- M. Sameiro and T. Goncalves, Chem. Rev., 2009, 109, 190–212 CrossRef PubMed.
- L. M. Wilhelmsson, Q. Rev. Biophys., 2010, 43, 159–183 CrossRef CAS PubMed.
- Y. Tor, Pure Appl. Chem., 2009, 81, 263–272 CrossRef CAS.
- D. W. Dodd and R. H. E. Hudson, Mini-Rev. Org. Chem., 2009, 6, 378–391 CrossRef CAS.
- J. N. Wilson and E. T. Kool, Org. Biomol. Chem., 2006, 4, 4265–4274 CAS.
- A. Okamoto, Y. Saito and I. Saito, J. Photochem. Photobiol. C, 2005, 6, 108–122 CrossRef CAS.
- M. J. Rist and J. P. Marino, Curr. Org. Chem., 2002, 6, 775–793 CrossRef CAS.
- N. J. Greco and Y. Tor, J. Am. Chem. Soc., 2005, 127, 10784–10785 CrossRef CAS PubMed.
- D. Shin, R. W. Sinkeldam and Y. Tor, J. Am. Chem. Soc., 2011, 133, 14912–14915 CrossRef CAS PubMed.
- S. G. Srivatsan and Y. Tor, Chem. – Asian J., 2009, 4, 419–427 CrossRef CAS PubMed.
- R. W. Sinkeldam, P. A. Hopkins and Y. Tor, ChemPhysChem, 2012, 13, 3350–3356 CrossRef CAS PubMed.
- A. R. Rovira, A. Fin and Y. Tor, J. Am. Chem. Soc., 2015, 137, 14602–14605 CrossRef CAS PubMed.
- L. S. McCoy, D. Shin and Y. Tor, J. Am. Chem. Soc., 2014, 136, 15176–15184 CrossRef CAS PubMed.
- H. Liu, J. Gao, L. Maynard, Y. D. Saito and E. T. Kool, J. Am. Chem. Soc., 2004, 126, 1102–1109 CrossRef CAS PubMed.
- J. Gao, H. Liu and E. T. Kool, J. Am. Chem. Soc., 2004, 126, 11826–11831 CrossRef CAS PubMed.
- F. Seela and G. Becher, Helv. Chim. Acta, 2000, 83, 928–942 CrossRef CAS.
- D. C. Ward, E. Reich and L. Stryer, J. Biol. Chem., 1969, 244, 1228–1237 CAS.
- A. Fin, A. R. Rovira, P. A. Hopkins and Y. Tor, in Modified Nucleic Acids, ed. K. Nakatani and Y. Tor, Springer International Publishing, Cham, 2016, pp. 1–26 Search PubMed.
- R. S. K. Lane, R. Jones, R. W. Sinkeldam, Y. Tor and S. W. Magennis, ChemPhysChem, 2014, 15, 867–871 CrossRef CAS PubMed.
- P. Wigerinck, C. Pannecouque, R. Snoeck, P. Claes, E. De Clercq and P. Herdewijn, J. Med. Chem., 1991, 34, 2383–2389 CrossRef CAS PubMed.
- N. J. Greco and Y. Tor, Tetrahedron, 2007, 63, 3515–3527 CrossRef CAS PubMed.
- S. G. Srivatsan and Y. Tor, J. Am. Chem. Soc., 2007, 129, 2044–2053 CrossRef CAS PubMed.
- S. G. Srivatsan and Y. Tor, Tetrahedron, 2007, 63, 3601–3607 CrossRef CAS PubMed.
- R. W. Sinkeldam, N. J. Greco and Y. Tor, ChemBioChem, 2008, 9, 706–709 CrossRef CAS PubMed.
- N. J. Greco, R. W. Sinkeldam and Y. Tor, Org. Lett., 2009, 11, 1115–1118 CrossRef CAS PubMed.
- A. J. Gutierrez, T. J. Terhorst, M. D. Matteucci and B. C. Froehler, J. Am. Chem. Soc., 1994, 116, 5540–5544 CrossRef CAS.
- F. M. Kahan and J. Hurwitz, J. Biol. Chem., 1962, 237, 3778–3785 CAS.
- L. Shapiro and J. T. August, J. Mol. Biol., 1965, 14, 214–220 CrossRef CAS PubMed.
- S. Asano, Y. Kurashin, Y. Anraku and D. I. Mizuno, J. Biochem., 1971, 70, 9–20 CAS.
- V. Cody and T. I. Kalman, Nucleosides Nucleotides, 1985, 4, 587–594 CAS.
- K. Felczak, A. K. Drabikowska, J. A. Vilpo, T. Kulikowski and D. Shugar, J. Med. Chem., 1996, 39, 1720–1728 CrossRef CAS PubMed.
- E. Sochacka, G. Czerwinska, R. Guenther, R. Cain, P. F. Agris and A. Malkiewicz, Nucleosides, Nucleotides Nucleic Acids, 2000, 19, 515–531 CAS.
- S. G. Srivatsan, H. Weizman and Y. Tor, Org. Biomol. Chem., 2008, 6, 1334–1338 CAS.
- J. Ludwig, Acta Biochim. Biophys., 1981, 16, 131–133 CAS.
- J. F. Milligan and O. C. Uhlenbeck, Methods Enzymol., 1989, 180, 51–62 CAS.
- J. F. Milligan, D. R. Groebe, G. W. Witherell and O. C. Uhlenbeck, Nucleic Acids Res., 1987, 15, 8783–8798 CrossRef CAS PubMed.
- Y. Xie, A. V. Dix and Y. Tor, J. Am. Chem. Soc., 2009, 131, 17605–17614 CrossRef CAS PubMed.
- K. Lang and R. Micura, Nat. Protocols, 2008, 3, 1457–1466 CAS.
- D. Moazed and H. F. Noller, Nature, 1987, 327, 389–394 CrossRef CAS PubMed.
- Y. Tor, Angew. Chem., Int. Ed., 1999, 38, 1579–1582 CrossRef CAS.
- L. S. McCoy, Y. Xie and Y. Tor, WIREs RNA, 2011, 2, 209–232 CrossRef CAS PubMed.
- P. Purohit and S. Stern, Nature, 1994, 370, 659–662 CrossRef CAS PubMed.
- M. I. Recht, D. Fourmy, S. C. Blanchard, K. D. Dahlquist and J. D. Puglisi, J. Mol. Biol., 1996, 262, 421–436 CrossRef CAS PubMed.
- M. L. Zapp, S. Stern and M. R. Green, Cell, 1993, 74, 969–978 CrossRef CAS PubMed.
- H. Y. Mei, D. P. Mack, A. A. Galan, N. S. Halim, A. Heldsinger, J. A. Loo, D. W. Moreland, K. A. SannesLowery, L. Sharmeen, H. N. Truong and A. W. Czarnik, Bioorg. Med. Chem., 1997, 5, 1173–1184 CrossRef CAS PubMed.
- T. Hermann and E. Westhof, Biopolymers, 1998, 48, 155–165 CrossRef CAS PubMed.
- Y. Tor, T. Hermann and E. Westhof, Chem. Biol., 1998, 5, R277–R283 CrossRef CAS PubMed.
- T. Hermann and E. Westhof, J. Mol. Biol., 1998, 276, 903–912 CrossRef CAS PubMed.
- Q. Vicens and E. Westhof, Biopolymers, 2003, 70, 42–57 CrossRef CAS PubMed.
- Q. Vicens and E. Westhof, ChemBioChem, 2003, 4, 1018–1023 CrossRef CAS PubMed.
- Y. Tor, ChemBioChem, 2003, 4, 998–1007 CrossRef CAS PubMed.
- F. Zhao, Q. Zhao, K. F. Blount, Q. Han, Y. Tor and T. Hermann, Angew. Chem., Int. Ed., 2005, 44, 5329–5334 CrossRef CAS PubMed.
- J. R. Thomas and P. J. Hergenrother, Chem. Rev., 2008, 108, 1171–1224 CrossRef CAS PubMed.
- M. Kaul, C. M. Barbieri and D. S. Pilch, J. Am. Chem. Soc., 2004, 126, 3447–3453 CrossRef CAS PubMed.
- S. Shandrick, Q. Zhao, Q. Han, B. K. Ayida, M. Takahashi, G. C. Winters, K. B. Simonsen, D. Vourloumis and T. Hermann, Angew. Chem., Int. Ed., 2004, 43, 3177–3182 CrossRef CAS PubMed.
- M. Kaul, C. M. Barbieri and D. S. Pilch, J. Am. Chem. Soc., 2006, 128, 1261–1271 CrossRef CAS PubMed.
- C. M. Barbieri, M. Kaul and D. S. Pilch, Tetrahedron, 2007, 63, 3567–3574 CrossRef CAS PubMed.
- A. A. Marti, S. Jockusch, Z. M. Li, J. Y. Ju and N. J. Turro, Nucleic Acids Res., 2006, 34, e50 CrossRef PubMed.
- S. K. Pal and A. H. Zewail, Chem. Rev., 2004, 104, 2099–2123 CrossRef CAS PubMed.
- B. Nguyen, S. Neidle and W. D. Wilson, Acc. Chem. Res., 2009, 42, 11–21 CrossRef CAS PubMed.
- E. Duboue-Dijon, A. C. Fogarty, J. T. Hynes and D. Laage, J. Am. Chem. Soc., 2016, 138, 7610–7620 CrossRef CAS PubMed.
- J. H. Cho and R. R. Rando, Biochemistry, 1999, 38, 8548–8554 CrossRef CAS PubMed.
- K. F. Blount, F. Zhao, T. Hermann and Y. Tor, J. Am. Chem. Soc., 2005, 127, 9818–9829 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob02080a |
| ‡ For full characterization of transcripts 7–10 by MALDI-TOF MS and digestions profiles see ESI sections S3 and S4.† |
| This journal is © The Royal Society of Chemistry 2017 |