
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A self-assembly toolbox for thiophene-based conjugated polyelectrolytes: surfactants, solvent and copolymerisation†
Judith E.
Houston
 *a,
Michèle
Chevrier
b,
Marie-Sousai
Appavou
a,
Stephen M.
King
c,
Sébastien
Clément
*b and
Rachel C.
Evans
*d
*a,
Michèle
Chevrier
b,
Marie-Sousai
Appavou
a,
Stephen M.
King
c,
Sébastien
Clément
*b and
Rachel C.
Evans
*d
aJülich Centre for Neutron Science (JCNS) at Heinz Maier-Leibnitz Zentrum (MLZ), Forschungszentrum Jülich GmbH, Lichtenbergstr. 1, 85748 Garching, Germany. E-mail: j.houston@fz-juelich.de
bInstitut Charles Gerhardt – UMR 5253, Université de Montpellier, CNRS, ENSCM, CC1701, Place Eugène Bataillon, F-34095 Montpellier Cedex 05, France. E-mail: sebastien.clement1@umontpellier.fr
cISIS Facility, STFC Rutherford Appleton Laboratory, Harwell Campus, Didcot, Oxon, OX11 0QX, UK
dDepartment of Materials Science & Metallurgy, University of Cambridge, 27 Charles Babbage Road, Cambridge, CB3 0FS, UK. E-mail: rce26@cam.ac.uk
First published on 6th November 2017
Abstract
Targeted control of the aggregation, morphology and optical properties of conjugated polymers is critical for the development of high performance optoelectronic devices. Here, self-assembly approaches are used to strategically manipulate the order, conformation and spatial distribution of conjugated polymers in solution and subsequently prepared thin films. The supramolecular complex organisation of phosphonium-functionalised homo- (P3HTPMe3) and diblock (P3HT-b-P3HTPMe3) ionic conjugated polythiophenes upon solvent-mediation and co-assembly with oppositely charged surfactants is investigated. UV/Vis absorption and photoluminescence spectroscopies, small-angle neutron scattering (SANS), cryo-transmission electron microscopy (cryo-TEM) and atomic force microscopy (AFM) are used to probe the organisation and photophysical response of the aggregates formed. Subtle differences in the surfactant mole fraction and structure, as well as the solvent polarity, yield differences in the nature of the resultant homopolyelectrolyte-surfactant complexes. In contrast, only moderate structural transformations are observed for the amphiphilic diblock copolyelectrolyte, emphasising the structure “anchoring” effect of a neutral polymer block when amphiphilic copolymers are dissolved in polar solvents. These results highlight the versatility of self-assembly to access a range of nanomorphologies, which could be crucial for the design of the next generation of organic optoelectronic devices.
Introduction
Conjugated polyelectrolytes (CPEs) are polymers with extended π-conjugated backbones and ionic pendant groups, which combine organic semiconductor properties and the charge-mediated behaviour of polyelectrolytes in a single functional macromolecule.1 They have remarkable promise as components of the active and charge transport layers of flexible optoelectronic devices, including polymer light-emitting diodes,2–4 organic field-effect transistors5–7 and organic photovoltaic devices (OPVs).8–11 Inkjet or screen-printing provide a facile route to low cost fabrication of these devices.12–14 However, the nanostructure of CPE films deposited in this manner is strongly influenced by the conformation of the polymer species in the ink. Since device performance depends on the intrinsically linked optoelectronic properties and nanoscale morphology of the polymer,15 the development of facile and reproducible processing methods that enable sophisticated control of the organisation of individual and clustered polymer chains in solution is paramount for the design of high-performance organic electronic devices.Recently, self-assembly strategies have emerged as an elegant approach for the fabrication of reproducible nanoscale architectures from CPEs.16 Due to their inherently amphiphilic structures (hydrophobic backbones and hydrophilic side groups), CPEs have a tendency to aggregate in aqueous solution or polar organic solvents, and as a result their aggregated morphology can be strongly dependent on the polarity of the medium.17,18 The addition of ionic or neutral surfactants to aqueous solutions of CPEs is known to break-up these aggregates, leading to the formation of hybrid structures with well-defined organisation.19–23 Such structural transitions result in concomitant changes in the photophysical properties, such as an increase in the emission intensity,24,25 or a shift of the emission maximum.26
Differences in the solubility and/or crystallinity of the polymer structure may also be exploited to yield more exotic structures. Block copolymers are macromolecules formed from two (or more) immiscible homopolymer chains (blocks) that are covalently linked. Due to the thermodynamic incompatibility between the adjacent blocks, these materials exhibit a natural tendency to self-assemble into nanodomains.27 The shape and size of the nanostructured morphologies are determined by the chain stiffness, molecular weight, solubility of the individual blocks and block ratio.28 Recently, particular attention has been paid to all-conjugated amphiphilic diblock copolymers, which combine ionic and neutral blocks.29,30 This introduces a further solubility gradient across the copolymer structure, leading to exotic self-assembled structures that can be conveniently modulated via solvent-mediation. Scherf et al. reported all-conjugated cationic “rod-rod” block copolyelectrolytes containing a hydrophobic polyfluorene and hydrophilic polythiophene blocks, that exhibit solvent-mediated self-assembly in mixtures of selective and non-selective solvents, such as water–methanol30,31 and water–THF.31,32 Careful selection of the solvent mixture was shown to be a convenient method to simultaneously control the nanomorphology of the self-assembled aggregates formed (e.g. vesicles, rods, etc.) and the photoluminescence properties.30–32
All-conjugated polythiophenes have attracted particular attention for applications as electron donors or interfacial layer materials in OPV devices.33–39 The analogous amphiphilic diblock polythiophene copolymers should be extremely attractive for this purpose; however, to date, there have only been a few studies dedicated to controlling the aggregate morphology and thus optoelectronic properties of amphiphilic diblock polythiophene copolymers.29,35,40,41 We recently reported the solvent-driven assembly of a family of rod–rod diblock copolymers containing a hydrophobic poly(3-hexylthiophene) (P3HT) block and a hydrophilic cationic P3HT block bearing different side-chains.29 The rigid rod-structure led to the formation of core–shell cylindrical or disc-like aggregates in different solvent mixtures.29 This investigation was followed by preliminary study of the interaction between the phosphonium-functionalised diblock copolymer poly[(3-hexylthiophene-2,5-diyl)]-block-poly[(3-(6′-(trimethylphosphoniumhexyl)thiophene-2,5-diyl))]bromide (P3HT-b-P3HTPMe3), its analogous homopolyelectrolyte poly[3-(6′-(trimethylphosphonium)hexylthiophene-2,5-diyl)]bromide (P3HTPMe3) (Scheme 1) and the anionic surfactant sodium dodecyl sulfate (SDS) in methanol–water mixtures. Small-angle neutron scattering (SANS) studies revealed that the solution structures, solvent content, and therefore hydrophobicity, were extremely dependent on both the CPE structure and counterion.35 Furthermore, a 20% increase in power conversion efficiency of an OPV device was observed after the incorporation of the CPE–surfactant complexes as cathodic interfacial layers.35
 | ||
| Scheme 1 Structures of the polythiophenes, P3HTPMe3 and P3HT-b-P3HTPMe3, and surfactants, SDS, PFOS and d25-SDS, used in this study. | ||
Here, we aim to comprehensively demonstrate the versatility of self-assembly to control the nanoscale morphology of all-conjugated homo- and diblock-polythiophene CPEs by harnessing solvent-mediation and co-assembly with ionic surfactants. The electrostatic co-assembly of the diblock copolymer P3HT-b-P3HTPMe3 and the analogous homopolymer P3HTPMe3 with the anionic surfactants, SDS, potassium heptadecafluoro-1-octanesulfonate (PFOS) and deuterated-sodium dodecyl sulfate (d25-SDS) (Scheme 1) in both water and methanol is investigated, and correlated with the nanoscale morphology of subsequently prepared thin films. Using a combination of optical, scattering and microscopic techniques, the nature of the homopolyelectrolyte-surfactant complexes is shown to be dependent on subtle differences in the surfactant mole fraction and structure, as well as the solvent polarity. In contrast, only moderate structural transformations are observed for the diblock P3HT-b-P3HTPMe3, highlighting the “structure anchoring” effect of the neutral polymer block when amphiphilic copolymers are dissolved in polar solvents.
Experimental
Materials and characterisation methods
Poly[3-(6′-(trimethylphosphonium)hexylthiophene-2,5-diyl)] bromide (P3HTPMe3),42 and poly[(3-hexylthiophene-2,5-diyl)]-block-poly[(3-(6′-(trimethylphosphoniumhexyl)thiophene-2,5-diyl))]bromide (P3HT-b-P3HTPMe3)29 were synthesised as previously reported and have number-averaged molecular weights (Mn) of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 and 13600 g mol−1, respectively, with PDIs of 1.12 and 1.36, as measured for the parent bromohexyl precursors.
100 and 13600 g mol−1, respectively, with PDIs of 1.12 and 1.36, as measured for the parent bromohexyl precursors.
SDS (≥99.0%), PFOS (≥98.0%) and deuterated-methanol (d4-MeOD, 98.0%) were purchased from Sigma-Aldrich. Deuterated-sodium dodecyl sulfate (d25-SDS) was purchased from Santa Cruz Biotechnology and deuterium oxide (D2O, 99.9%) was purchased from Apollo Scientific Limited. All chemicals were used as received.
The UV/Vis absorption and fluorescence spectra were recorded at room temperature on a Shimadzu UV2401 PC UV/Vis scanning spectrometer and a Fluorolog-3 (Horiba Jobin Yvon) spectrophotometer, respectively. The emission spectra were corrected for the wavelength response of the system using correction factors supplied by the manufacturer. Samples were measured in quartz cells with an extremely short path length (0.1 mm) to prevent saturation of the detector signal.
SANS was carried out on the LOQ small-angle diffractometer at the ISIS Pulsed Neutron Source (STFC Rutherford Appleton Laboratory, Didcot, UK).43 A simultaneous q-range of ∼0.09–2.4 nm−1 was achieved utilising an incident wavelength range of 0.22–1.0 nm separated by time-of-flight and employing a fixed sample-detector distance of 4.1 m. q = (4π/λ)sin(θ/2) where λ is the wavelength and θ the scattering angle. Samples were prepared in deuterated solvents to provide good neutron scattering contrast. The samples were placed in quartz cuvettes (Hellma) of 1 mm path length and maintained at 25.0 ± 0.5 °C. Each raw scattering data set was corrected for the detector efficiencies, sample transmission and background scattering and converted to scattering cross-section data (∂Σ/∂Ω vs. q) using the instrument-specific software.44 These data were placed on an absolute scale (cm−1) using the scattering from a standard sample (a solid blend of hydrogenated and perdeuterated polystyrene) in accordance with established procedures.45 The scattering functions were fit using non-linear least-squares analysis to a Rigid Cylinder model,46,47 Lamellar Sheet model48,49 or a Core–Shell Cylinder model50 using the SasView program (version 3.1.2).51 Full details of the models and the fitting procedure can be found in the ESI.†
Cryogenic-transmission electron microscopy (cryo-TEM) was performed at the Heinz Maier Leibnitz Zentrum, Garching, Germany. Cryo-TEM measurements were carried out on concentrated samples (10 mg mL−1 in D2O). A copper grid coated with holey carbon film (Multi A, Quantifoil Micro Tools GmbH) was dipped in solution and then placed in the chamber of a cryo-plunge (EMGP Leica GmbH) maintained at 20 °C and 80% relative humidity (r.h.) The excess liquid was removed with filter paper. The samples were then cryo-fixed by rapid immersion into liquid ethane at −180 °C in the cryo-plunge. The specimens were inserted into a cryo-transfer holder (G910, Gatan, Munich, Germany) and transferred to a JEM 2200 FS EFTEM instrument (JEOL, Tokyo, Japan). Examinations were carried out at approximately −179 °C. The TEM was operated at an accelerating voltage of 200 kV. Zero-loss filtered images were taken under reduced dose conditions (<10000e− per nm2). All images were recorded digitally by a bottom-mounted 16 bit CCD camera system (TemCam-F216, TVIPS, Munich, Germany). The observable length scale range was between 5 and 500 nm.
Atomic force microscopy (AFM) measurements were performed using an Asylum Research MFP-3D™ instrument mounted on an anti-vibration plinth, in the tapping mode at room temperature under ambient conditions. Higher resolution AFM measurements were performed using diamond tips on silicon cantilevers, which were a kind gift from Adama Innovations. The silicon cantilevers had a spring constant of ∼110 nN nm−1 and resonance frequency of ∼240 kHz. All raw AFM images were analysed using the Gwyddion 2.31 software.
Results and discussion
CPE–surfactant mixtures were prepared over a range of charge ratios, x, which represents the ratio of surfactant molecules over the number of charged CPE monomers. The samples were prepared by mixing 10 mg mL−1 solutions of CPE with 10 mg mL−1 solutions of surfactant to obtain the desired charge ratio, with a total concentration of 10 mg mL−1. The value x = 1 corresponds to stoichiometric charge balance. As a representative example, the compositions of polymer–SDS mixtures are given in the ESI, Tables S1 and S2.†Optical characterisation
The optical properties of polythiophenes are well-known to be responsive to intrachain conformational changes and interchain aggregation.52,53 The addition of the anionic surfactant SDS to a solution of P3HTPMe3 in D2O at different charge ratios, x, resulted in a series of colorimetric transitions, as shown in Fig. 1. P3HTPMe3(SDS)x exhibits a series of well-defined colour transitions, from red (x = 0), to wine (x = 1/5–1), to orange (x = 5), and finally, yellow (x = 20). A similar series of colorimetric transitions were observed for the related poly[3-(6′-(N,N,N-trimethylammoniumhexyl)thiophene-2,5-diyl)]bromide (P3TMAHT) with SDS at different charge ratios.21 In contrast, for P3HT-b-P3HTPMe3(SDS)x, a colour transition from purple to red is only observed for x = 5 to x = 20.21 | ||
| Fig. 1 Photographs of (a) P3HTPMe3(SDS)x and (b) P3HT-b-P3HTPMe3(SDS)x as a function of charge ratio, x, in D2O at room temperature (total conc. = 10 mg mL−1). | ||
 | ||
| Fig. 2 Effect of the surfactant charge fraction, x, on the optical properties of CPE-SDSx complexes. (a, b) Normalised UV/Vis absorption spectra and (c, d) steady-state emission spectra for P3HTPMe3(SDS)x and P3HT-b-P3HTPMe3(SDS)x at room temperature. Total sample concentrations were 10 mg mL−1 in D2O. λex = 450 nm. | ||
The photoluminescence (PL) properties of the homopolymer and diblock copolymers are also highly sensitive to the surfactant charge ratio. Addition of SDS (x = 0.2–2) to P3HTPMe3 triggers both a narrowing of the emission band and the emergence of more resolved vibronic structure (Fig. 2c), which is assigned to the vibronic progression of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching mode (ΔE ≈ 0.15 eV).55 This is accompanied by a significant red-shift in the emission maximum (Δλem = 59 nm) by x = 1. These observations suggest that P3HTPMe3 adopts a more planar, ordered conformation in this concentration regime, which prevents free rotation around the polymer backbone.21,22 By x = 5, the emission band loses its vibronic structure, broadens and undergoes a blue-shift, which indicate the return to a more twisted conformation along the polymer backbone.21,22 In fact, the emission maximum for x = 5 is even blue-shifted when compared to that of pure P3HTPMe3. This can be attributed to a reduction of interchain interactions due to increased screening of the polymer–polymer interactions by the SDS which is present in charge excess.21,22
C stretching mode (ΔE ≈ 0.15 eV).55 This is accompanied by a significant red-shift in the emission maximum (Δλem = 59 nm) by x = 1. These observations suggest that P3HTPMe3 adopts a more planar, ordered conformation in this concentration regime, which prevents free rotation around the polymer backbone.21,22 By x = 5, the emission band loses its vibronic structure, broadens and undergoes a blue-shift, which indicate the return to a more twisted conformation along the polymer backbone.21,22 In fact, the emission maximum for x = 5 is even blue-shifted when compared to that of pure P3HTPMe3. This can be attributed to a reduction of interchain interactions due to increased screening of the polymer–polymer interactions by the SDS which is present in charge excess.21,22
The PL spectrum of P3HT-b-P3HTPMe3 has an emission maximum at 734 nm and some vibronic structure (Fig. 2d). Upon initial addition of SDS (x = 0.2–0.5), no significant spectral changes are observed, suggesting that no substantial structural reorganisation takes place in this concentration regime. However, for x = 1, a large blue-shift in the emission maximum to 672 nm results, which is accompanied by a decrease in the vibronic band at 734 nm and the emergence of a new band at 637 nm. Increasing the charge ratio further (x = 2–20) sees the complete loss of vibronic structure. The addition of the surfactant appears to reduce polymer–polymer interactions.23 The anionic surfactant is expected to associate predominantly through electrostatic interactions with the cationic P3HTPMe3 block, while polymer–polymer interactions are expected to persist in the neutral P3HT block domains of the copolymer aggregates. This may explain why some vibronic structure remains until x = 5, i.e. even when the surfactant is present in large excess.
:1 charge ratio of PFOS and, for comparison, SDS, are shown in Fig. 3. The normalised PL spectra of P3HTPMe3(PFOS)x and P3HT-b-P3HTPMe3(PFOS)x (x = 0.2–20) are shown in Fig. S2, ESI.†
 | ||
| Fig. 3 Effect of hydrogenated vs. perfluorinated surfactants on the optical properties of CPE-SDSx complexes. Normalised UV/Vis absorption and emission spectra for (a, c) P3HTPMe3 and (b, d) P3HT-b-P3HTPMe3 with no surfactant (black lines) and 1:1 charge ratio of SDS (red lines) or PFOS (blue lines) at room temperature. Total sample concentrations were 10 mg mL−1 in D2O. λex = 450 nm. | ||
The UV/Vis absorption spectra for P3HTPMe3(PFOS)1 and P3HT-b-P3HTPMe3(PFOS)1 are characterised by a single broad absorption band centred at 447 and 523 nm, respectively (Fig. 3a and b), similar to the spectra for the pure CPEs. The only significant difference is a reduction in the peak width for P3HT-b-P3HTPMe3(PFOS)1. This is in stark contrast to the analogous SDS systems which are significantly red-shifted upon complexation with the surfactant.
The PL spectrum of P3HTPMe3 is notably insensitive to PFOS, with only a moderate blue-shift observed (Δλem = 33 nm) by x = 2, before a red-shift back to the original emission maximum at higher x. In contrast, the PL spectrum of P3HT-b-P3HTPMe3 undergoes a large blue-shift (∼75 nm) to 657 nm by x = 1. By x = 5, λem = 614 nm, and all vibronic structure is lost. This occurs for PFOS at a lower charge ratio (x = 2) than with SDS (x = 5), which suggests that the fluorinated surfactant, with a lower head-group charge density, is more effective at reducing polymer–polymer interactions within the aggregates of P3HT-b-P3HTPMe3.23 The PL and absorption data for both systems in D2O and d4-MeOD before and after the addition of surfactant are summarised in Table 1.
:1 charge ratio). λex = 450 nm
| Solvent | λ abs (nm) | λ em (nm) | |
|---|---|---|---|
| P3HTPMe3 | D2O | 446 | 620 |
| d 4-MeOD | 445 | 592 | |
| 1:1 SDS |
D2O | 598 | 679 |
| d 4-MeOD | 446 | 582 | |
| 1:1 PFOS |
D2O | 447 | 620 |
| d 4-MeOD | 417 | 572 | |
| P3HT-b-P3HTPMe3 | D2O | 535 | 732 |
| d 4-MeOD | 518 | 630 | |
| 1:1 SDS |
D2O | 552 | 673 |
| d 4-MeOD | 516 | 636 | |
| 1:1 PFOS |
D2O | 523 | 657 |
| d 4-MeOD | 518 | 621 | |
:1 charge ratio of SDS and PFOS in d4-MeOD (10 mg mL−1) are shown in Fig. 4a. Similar to D2O, the absorption maximum of P3HT-b-P3HTPMe3 is significantly red-shifted (∼70 nm) compared to P3HTPMe3, and exhibits moderate vibronic structure. The addition of SDS results in a moderate blue-shift in λabs (∼10 nm) and a narrowing of the absorption band for the homo- and diblock polymers. However, this shift is much smaller than observed for P3HTPMe3(SDS)1 in D2O (∼150 nm), which indicates a smaller structural rearrangement in d4-MeOD. More striking distinctions are observed upon the addition of PFOS. The absorption spectrum of P3HTPMe3(PFOS)1 shows a blue-shift of ∼30 nm, while the band for P3HT-b-P3HTPMe3(PFOS)1 is significantly broader, which implies that the CPE adopts a more planar organisation.
 | ||
| Fig. 4 Effect of solvent on the optical properties of CPE-SDSx complexes. Normalised (a) UV/Vis absorption and (b) emission spectra of P3HTPMe3 (solid black lines) and P3HT-b-P3HTPMe3 (dashed black lines) and the corresponding 1:1 SDS (red lines) and PFOS (blue lines) electrostatic complexes in d4-MeOD (10 mg mL−1). | ||
P3HTPMe3 and P3HT-b-P3HTPMe3 in d4-MeOD also display distinctive PL spectra (Fig. 4b). P3HTPMe3 exhibits a broad, featureless emission band centred at 592 nm, while the emission spectrum of P3HT-b-P3HTPMe3 is broader still (520–850 nm) with well-resolved vibronic structure. The addition of SDS results in the emergence of vibronic structure for P3HTPMe3(SDS)1 and P3HT-b-P3HTPMe3(SDS)1, while the addition of PFOS results in a blue-shift in the emission maximum to 572 and 621 nm for P3HTPMe3 and P3HT-b-P3HTPMe3, respectively.
The dodecyl sulfate (DS−) and heptadecafluorooctane-sulfonate (PFOS−) counterions in complexes with P3HTPMe3 and P3HT-b-P3HTPMe3 are likely to hinder polymer–polymer interchain interactions, thereby decreasing the nominal effective conjugation length for exciton migration.25 Previously, the complexation of DS− with the related homopolymers P3TMAHT21,22 and poly[3-(6′-(N-methylimidazolium)hexyl)thiophene-2,5-diyl]bromide (P3ImiHT)23 was shown to induce significant surfactochromic transitions in aqueous solution, which could also be controlled by varying the surfactant charge ratio. However, the effect of SDS appears to be greater in D2O than d4-MeOD. There are two potential reasons for this: (1) these transitions are controlled to a large extent by the phase diagram of SDS. Micellization is known to be strongly inhibited by organic solvents, such as MeOH.58 Therefore, while the critical micelle concentration (cmc) of SDS is well-known in water (cmc = ∼8.2 mM),59 there are few reliable records of the cmc in MeOH;60 (2) the CPEs will also experience different phase transitions in methanol and water. Interestingly, PFOS has considerably more effect on the emission properties of P3HTPMe3 in d4-MeOD than in D2O. As with SDS, this is likely to be at least partially due to the difference in the cmcs of PFOS in MeOH and H2O (∼2 mM).61 Furthermore, this effect is significantly greater for the diblock copolymer vs. the homopolymer. The reduced charge density of the sulfonate head-group may enable PFOS to more effectively penetrate the neutral P3HT block core of the aggregates of P3HT-b-P3HTPMe3 than SDS. Fluorinated species, such as PFOS, may be expected to have an immiscibility gap with hydrogenated species, such as the CPES studied here, due to weaker van der Waals interactions.62 However, this may be counteracted by the high electronegativity of the fluorine atoms which will promote intermolecular interactions with P3HT through the formation of non-covalent F–S and F–H bonds.63,64 In contrast, due to its higher charge density and hydrogenated tail, SDS can form stronger ionic and van der Waals associations with the P3HTPMe3 blocks, thus, explaining why greater surfactochromic changes are observed for the P3HTPMe3(SDS)x system.
Solution-phase structures
To obtain deeper insight into the nanoscale organisation of the polymers in solution, SANS studies were performed on the P3HTPMe3-surfactant and P3HT-b-P3HTPMe3-surfactant complexes in D2O and d4-MeOD. Here, differences in the scattering length densities (SLDs) of individual components in the CPE–surfactant systems allow different zones of the complex to be probed. The SLDs of the surfactants differ significantly: SDS is closer to polythiophene, while d25-SDS and PFOS are closer to D2O and d4-MeOD (Fig. 5). Therefore, CPE-SDSx complexes will appear to neutrons as single entities, enabling the entire polymer–surfactant complex to be observed. In contrast, scattering from CPE-d25-SDS and CPE-PFOS systems originates primarily from the polymer, allowing the organisation of the polymer within the complex to be investigated. | ||
| Fig. 5 Comparison of the estimated neutron scattering length densities (SLDs) for the polymer blocks, surfactants and solvents used in the SANS studies. Materials with similar SLDs are considered to be contrast-matched. | ||
The observation window of these SANS experiments ranged from 2.6–70 nm, which covers the isolated chain lengths of the CPEs (22.2–30.4 nm) calculated from the length of the thiophene monomer (∼0.4 nm).65 The ratio of neutral:charged blocks is 59:41 which results in block lengths of 17.9 nm and 12.5 nm, respectively.35 If the CPEs were dissolved down to the single molecule level, the SANS data would level off as a Guinier plateau at an experimentally obtainable q which is not observed here for any of the CPEs in D2O or d4-MeOD. All fits to the scattering profiles are summarised in Tables S3–S10 in the ESI.† Although it was not possible to obtain unique fits to the SANS data, all the chosen fits have absolute SANS intensities consistent with the known sample concentrations (∼1% vol. dry material).
Homopolymer with surfactants in D2O
The related homopolymers P3TMAHT21,22 and P3ImiHT23 have previously been assigned as spherical aggregates in D2O. However, a modest upturn at q < 0.2 nm−1 of ∼q−1 implies that P3HTPMe3 may form cylindrical rather than spherical aggregates (Fig. 6a). Fitting of the SANS data (0.08 < q < 2.2 nm−1) to the Cylinder model46,47 gave an aggregate length of 8.7 nm and radius of 1.5 nm. The hump at q = 0.3 nm−1 was accounted for by including a Hayter–Penfold structure factor, which includes particle–particle repulsive interactions.66–68 Alternatively, using a Guinier plot46 (Fig. S4, ESI†) the calculated radius of gyration (Rg) of aggregates of P3HTPMe3 in D2O is 2.1 nm. | ||
| Fig. 6 Effect of surfactant type and charge ratio, x, on the solution phase structures of P3HTPMe3 and P3HT-b-P3HTPMe3. SANS data of P3HTPMe3 and P3HT-b-P3HTPMe3 in D2O with selected charge ratios of (a, b) SDS and (d, e) d25-SDS, respectively. Straight lines show q−1, q−1.5, q−2 and q−4 for comparison. (c) P3HTPMe3 and (f) P3HT-b-P3HTPMe3 (blue squares) in D2O and with 1:1 charge ratio of SDS (red circles), PFOS (pink triangle) and d25-SDS (green triangles). Solid, dashed and dot-dashed lines represent the fits described in the text. The overall concentration of each system was 10 mg mL−1. T = 25 °C. Error bars were omitted to enable the slopes to be better distinguished. Representative error bars can be seen in the ESI.† | ||
The scattering profile of P3HTPMe3 upon initial addition of hydrogenated SDS (x = 0.2) is quite different to that of the pure polymer (Fig. 6a), with an upturn at low q of ∼q−1.7. This means that only a small amount of SDS (well below its cmc) is required to weaken the interparticle electrostatic ordering within aggregates of P3HTPMe3.23 Fitting of the SANS data (0.08 < q < 2.2 nm−1) to the Cylinder model46,47 gave an aggregate length of 48.3 nm and radius of 1.8 nm. As the charge ratio is increased further from x = 0.5 to x = 2, there is a significant increase in scattering at low q (q < 0.2 nm−1). By x = 1, the scattering curve decays as q∼2.6, which is interpreted as the existence of “sheet-like” particles or larger smooth fractal aggregates, such as large or flocculated vesicles or a mixture of both.69 The power law scaling at high q (q > 1.0 nm−1) starts to decay as q−4, thus, marking the thickness of the sheet-like aggregates.23 The upturn at q = 0.8 nm−1, could be repulsion between similarly charged chains or the appearance of free SDS micelles, considering that the concentration is well above the cmc of SDS in water (8.2 mM ≈ 2.4 mg mL−1).70 Fitting with the Lamellar Sheet model48,49 gave sheet thicknesses (Tsheet) of 1.0, 2.0 and 1.4 nm for x = 0.5, 1 and 2, respectively. These thicknesses correspond to the solid state d-spacing of poly(3-hexylthiophene) (∼1.7 nm)65 or the length of individual SDS molecules (∼2.5 nm),71 implying that the polymer and surfactant must be interwoven rather than forming well-defined layers.23 At x = 5, the scattering intensity scales as q−0.5, and the data can be fitted as spherical aggregates with a radius of 2.5 nm and 65% solvent content (Fig. S7, ESI†). This is only slightly larger than pure SDS micelles, which are also spherical with radius of ∼1.9 nm (Fig. 6a, red circles). This suggests that scattering from the x = 5 sample occurs predominantly from SDS micelles, with the P3HTPMe3 associating to their surfaces, which may potentially explain the slight increase in radius.
Fig. 6d shows the SANS data and fits for P3HTPMe3(d25-SDS)x in D2O, in which the scattering profile mainly arises from P3HTPMe3 within the P3HTPMe3(d25-SDS)x associations. After addition of d25-SDS up to x = 1, the SANS data exhibit a strong upturn at low q, described empirically by a power law scaling of q−2.7, strongly resembling that of P3HTPMe3(SDS)1 (Fig. 6e). The data indicate that P3HTPMe3 forms sheet-like aggregates within the sheet-like CPE-SDSx associations and can be fitted to the Lamellar Sheet model48,49 to give a Tsheet of 3.4 nm for x = 1, surprisingly thicker than for P3HTPMe3(SDS)1 (∼2.0 nm). Interestingly, the scattering profile for P3HTPMe3(d25-SDS)5 is clearly still reminiscent of lamellar sheets (∼q−2). In contrast, the apparent spherical scattering profile in P3HTPMe3(SDS)5 is likely to be a consequence of the micelles formed from the excess SDS.
The analogous SANS profiles for P3HTPMe3(PFOS)x can be found in Fig. S7 (ESI†). For x = 1, the data show a strong upturn at low q, described by q−2.3 scaling, strongly resembling that of P3HTPMe3(SDS)1 (Fig. 6c). This implies the complexes also form sheet-like aggregates; however, fitting with the Lamellar Sheet model gave considerably thicker sheets (Tsheet = 4.8 nm).48,49 The increase is likely to be due to the increased rigidity of the surfactant tail and reduced charge density of the sulfonate head-group, making PFOS less able to penetrate the existing P3HTPMe3 aggregates compared to SDS or d25-SDS,21 and resulting in more distinct “P3HTPMe3” and “PFOS” layers.
Initially, there is practically no change in the scattering profile of P3HT-b-P3HTPMe3 upon addition of hydrogenated SDS (x = 0.2) (Fig. 6d). However, by increasing the charge ratio (x = 0.5), an increase in the scattering intensity is observed at q = 0.8 nm−1, indicative of repulsion between similarly charged chains. In addition, by x = 1 the shoulder at q = 0.2 nm−1 becomes less pronounced and the profile scales as q−2. This implies that the core–shell cylinders of the pure diblock copolymer transform into 2D sheets upon coordination with SDS. At x = 2 and x = 5 the q-scaling decreases to q−1.9 and q−1.7, respectively, indicating the progressive loss of the lamellar sheet structure upon increasing charge ratio. The SANS data (0.08 < q < 2.0 nm−1) of P3HT-b-P3HTPMe3(SDS)x were fit to the Core–Shell Cylinder,50 Lamellar Sheet48,49 or Sphere models, shown in Fig. 6b and summarised in the Table S5, ESI.† The fits for the P3HT-b-P3HTPMe3(SDS)x system proved particularly difficult to optimise. For x = 0.2 and x = 0.5, the samples were fit using the Core–Shell Cylinder model. Although the core lengths and radii are similar to those of the parent diblock copolymer, the shell appears to become thicker and drier. However, it must be noted that these fits were not perfect, and are particularly poor at q = 0.8 nm−1. At x = 1, the charge compensation point, the scattering profile was best fit to the Lamellar Sheet model,48,49 with a Tsheet of 5.9 nm. Although this fit was again not ideal, it was significantly better than the alternative fit using the Core–Shell Cylinder model.50 For x = 5 and x = 20, fits of satisfactory quality to the entire data were not possible. However, the high q data (0.27 < q < 2.4 nm−1) strongly resembled the scattering from pure SDS micelles, and a fit to the Spherical model46 resulted in radii of 2.7 and 2.3 nm for x = 5 and x = 20, respectively. This implies that by x = 5, P3HT-b-P3HTPMe3(SDS)x exists mainly as SDS micelles, potentially with associating CPE chains on the surface to account for the slight increase in radius compared to pure SDS micelles.
Interestingly, when contrast-matching with the deuterated surfactant analogue (d25-SDS) is performed, the scattering profiles of the entire P3HT-b-P3HTPMe3(d25-SDS)x series strongly resemble the core–shell cylinder profile of the parent diblock copolymer (Fig. 6e). The Core–Shell Cylinder model was fit to each of the scattering profiles and the fitting parameters are summarised in the Table S6 in the ESI.† At x = 1, the fit gave an elongated cylinder with Lcore of 47.7 nm, rcore of 4.9 nm and Tshell of 7.1 nm. Each of the fits was greatly improved by adding polydispersity to the core radius. Therefore, we propose that since lamellar sheets are observed with SDS, and yet core–shell cylinders dominate when the surfactant has been contrast-matched, the SDS may act to connect adjacent P3HT-b-P3HTPMe3 cylinders to form two-dimensional, sheet-like arrays.
Fitting with the Core–Shell Cylinder model to the P3HT-b-P3HTPMe3(PFOS)x series also gave core–shell cylinders with hydrophobic, neutral block cores and solvated, hydrophilic charged block shells (Fig. S4b, ESI†). The SLD of PFOS (2 × 10–4 nm−2) is quite well contrast-matched with D2O, and as a result the scattering is expected to mainly occur from the polymer. At x = 1, shown in Fig. 6f, the fit gave a wide, (relatively) wet core with rcore of 5.9 nm (16% wetness), a shorter Lcore of 46.6 nm, and thick, wet Tshell of 7.8 nm (85% wetness) (Fig. 8c). Notably, PFOS, unlike SDS, induced a significant increase in the “wetness” of the neutral P3HT cores.
:1 charge ratio with SDS in d4-MeOD have been discussed in depth in previous work,29 and thus will only be briefly recapped here for the sake of comparison with the analogous samples in D2O. P3HTPMe3 in d4-MeOD adopts a flexible rod-like conformation with a high solvent content (>85% solvent) and total cylinder length (90.0 nm), and a radius of 1.3 nm, while in D2O it forms charged spherical aggregates (Rg ∼2.1 nm) with interparticle interactions. P3HTPMe3 in d4-MeOD better resembles the scattering profiles obtained when this CPE or the analogous homopolymers, P3TMAHT and P3ImiHT,21,22 are combined with a small amount of SDS (CPE/surfactant charge ratio of 1:0.2) in D2O. In all three systems, the pure CPE aggregates are believed to disassemble and reorganise into CPE–surfactant cylinders.21,22 This suggests that P3HTPMe3 forms more ordered aggregates in d4-MeOD, with significant packing between CPE chains. P3HTPMe3(SDS)1 in d4-MeOD favours a lamellar-type structure (Tsheet ∼4.7 nm, ∼50% solvent) believed to be comprised of distinct P3HTPMe3 and SDS layers, rather than the interwoven sheets expected in D2O.
In contrast, both P3HT-b-P3HTPMe3 and P3HT-b-P3HTPMe3(SDS)1 in d4-MeOD form core–shell cylindrical aggregates in solution, with the P3HT block comprising the core and the P3HTPMe3 block (and associated counterions) forming the shell. Thus, unlike in D2O, counterion exchange yields only subtle changes in the scattering profile for P3HT-b-P3HTPMe3(SDS)1-d4-MeOD, without the apparent formation of lamellar sheets. These results indicate that solution structure of P3HT-b-P3HTPMe3 in d4-MeOD and D2O is primarily dictated by the hydrophobic P3HT core, which “locks-in” the cylindrical morphology of the pure diblock polymer upon surfactant addition. In comparison, the less-restricted P3HTPMe3 is able to freely transform from semi-flexible cylinders to rigid sheets upon counterion exchange in both solvents.
Cryo-transmission electron microscopy
It is important to note that when the diameter of a vesicle is well outside the q-window of a SANS experiment (3.0 to 69.8 nm here), then the scattering would appear to correspond to that of lamellar sheets (q−2). In order to investigate whether this was the case here, direct imaging by cryo-TEM has been utilised to visualise the particle size and morphology in D2O. This technique has the advantage that the micelles can be observed in their hydrated state and that the aqueous environment remains undisturbed.Cylindrical or rod-like particles were observed for P3HTPMe3-D2O, as shown in Fig. 7a, with average width of 7.5 (±2.3) nm and length of 30.6 (±14.0) nm. Such rod-like structures have previously been observed for P3HT in vitrified organic solvents such as toluene and 1,2-dichlorobenzene,73 and were attributed to π–π packing of the conjugated polythiophene backbones. Upon the addition of SDS a remarkable change in morphology is observed with the formation of two notable features: (1) large spherical aggregates, ∼73.9 (±15.3) nm in diameter, which could be vesicles, although vesicles are more typically capsule-like. More likely, these regular features are simply aggregates of CPE and/or SDS that form reasonably homogeneous nanoparticles (Fig. S8, ESI†); (2) lamellar-type striations, highlighted by the white box in Fig. 7b, with sheet thicknesses of 4.0 (±1.1) nm, which agree well the observations made by SANS. Similar thread-like micelles were previously observed in cryo-TEM images of mixtures of SDS and the organic salt 1,2-bis(2-benzylammoniumethyoxy dichloride) (BEO) in a 5:1 ratio in H2O when the concentration is high ([SDS] = 15 mM).74
 | ||
| Fig. 7 Cryo-TEM micrographs of (a) P3HTPMe3, (b) P3HTPMe3(SDS)1, (c) P3HT-b-P3HTPMe3 and (d) P3HT-b-P3HTPMe3(SDS)1. Total concentration = 10 mg mL−1. | ||
Given the difficulty in fitting unique structures to the SANS data, it is therefore not surprising that distinctive features were harder to identify in for P3HT-b-P3HTPMe3 samples. However, spherical structures, 19.8 (±3.6) nm in diameter, can definitely be identified in Fig. 7c, which are approximately the same sizes as determined from the model fitting of SANS data. The spheres become much more distinctive upon addition of SDS, with slightly larger diameters of 21.7 (±3.5) nm.
Thin film morphology
AFM was used to investigate the correlation between the nanoscale organisation of aggregates formed in solution and the morphology of subsequently cast thin films. Thin films of pure CPEs and CPE–surfactant complexes dissolved in MeOH were spin-coated onto silicon substrates. Unfortunately, it was not possible to obtain spin-coated films of samples dissolved in H2O due to the hydrophobicity of the silicon substrates.75 Therefore, all the films of the H2O samples were prepared via drop-casting.The surface morphology was found to be dependent on the type of polythiophene (homo- vs. diblock copolymer), solvent and presence and nature of surfactant. In water, P3HTPMe3 formed mainly featureless films (Fig. 8a), which is not surprising since it is expected to be well-dissolved with minimal assembly into spheres or rods. In contrast, films of P3HTPMe3(SDS)1 consisted of large aggregates with smooth flat surfaces. The inset phase image in Fig. 8b shows more clearly the formation of sheet-like domains, with a spacing of ∼50 nm. P3HTPMe3(PFOS)1 forms a collection of polydisperse spheres (Fig. 8c) that show no resemblance to the parent homopolymer or P3HTPMe3(SDS)1.
 | ||
| Fig. 8 AFM tapping-mode height images of pure CPEs and CPE–surfactant complexes at 1:1 charge ratio prepared in water (a–f) and methanol (g–l) (10 mg mL−1). (a) P3HTPMe3 and (d) P3HT-b-P3HTPMe3 with (b, e) SDS and (c, f) PFOS, respectively, drop-cast from H2O. (g) P3HTPMe3 and (j) P3HT-b-P3HTPMe3 with (h, k) SDS and (i, l) PFOS, respectively, spin-coated from MeOH. Insets: phase images at the same concentration and magnification unless otherwise stated. | ||
P3HT-b-P3HTPMe3 forms large clusters of small, polydisperse spheres, with an average diameter of 36.0 ± 7.7 nm (Fig. 8d). For P3HT-b-P3HTPMe3(SDS)1 (10 mg mL−1), it was difficult to distinguish any clear features within the extremely amorphous surface morphology (Fig. 8e). However, upon dilution (×100), we can clearly see spheres, whose core–shell structure is visible in the phase image (Fig. 8e inset). The average diameter was 55.0 ± 10.4 nm, with an average core diameter of 17.6 ± 3.9 nm. P3HT-b-P3HTPMe3(PFOS)1 forms a significantly less distinct surface, with some small spheres (∼30 nm) appearing at the edge of larger, amorphous aggregates.
Given that the concentrations of SDS and PFOS in the aqueous samples are greater than the cmcs of the pure surfactants,61,70 it is reasonable to expect the coexistence of surfactant micelles only if there is incomplete association between the CPE and surfactant. However, most of CPE-SDSx complexes formed films with distinctive morphologies when compared to the films of the pure surfactant (Fig. S9, ESI†). The only exception was P3HTPMe3(PFOS)1 which formed a collection of polydisperse spheres which resembled pure PFOS.
In MeOH, P3HTPMe3 also forms largely featureless films (Fig. 8g), with small spherical aggregates (10–20 nm) only apparent in the phase image. Complexation with SDS produces large globules (∼100 nm), which appear from the phase image potentially to be vesicles (Fig. 8h). In contrast, complexes formed with PFOS formed a generally flat plane of spheres, dispersed with significantly larger, flat structures (Fig. 8i). The high-resolution image shown in the inset clearly shows the lamellar structure within these aggregates.
P3HT-b-P3HTPMe3 in MeOH formed quite uniform films of spheres (∼30 nm in diameter), which appear to have core–shell structure in the phase image. Complexation with SDS (Fig. 8k) shows some spherical aggregates without any distinctive boundaries. There appears to be a continuous network running through this image, implying that CPE aggregates are more interdispersed with SDS. In contrast, P3HT-b-P3HTPMe3 with PFOS, which is expected to interact less effectively with the P3HTPMe3, appears to retain the spherical structure from imparted by the parent copolymer more effectively. The observed spheres of P3HT-b-P3HTPMe3-PFOS complexes are significantly larger (∼60 nm), which supports the larger core radius indicated by SANS. Pure films of pure SDS and PFOS prepared from MeOH bore no resemblance to any of the CPE–surfactant films (Fig. S10, ESI†).
Proposed self-assembly structures
Fig. 9 shows a schematic representation of the proposed aggregate structures adopted by P3HTPMe3 and P3HT-b-P3HTPMe3 upon surfactant complexation and in different solvents. The SANS fits and cryo-TEM data for P3HTPMe3 in D2O suggest it forms either short-cylinders or spherical aggregates (Fig. 9a). Upon addition of SDS, a significant colour change is observed, from the red-aggregated phase to a yellow solution of disrupted aggregates. The emergence of vibronic structure in the PL spectra for x = 0.2–2, implies the formation of rigid, compact planar aggregates. This observation agrees well with the SANS and cryo-TEM studies, which suggest the formation of sheets comprised of intimate blends of CPE and surfactant. High charge ratios led to the formation of isolated SDS micelles, although it was evident from contrast-matching SANS experiments that P3HTPMe3-SDS sheet complexes persist until x = 5.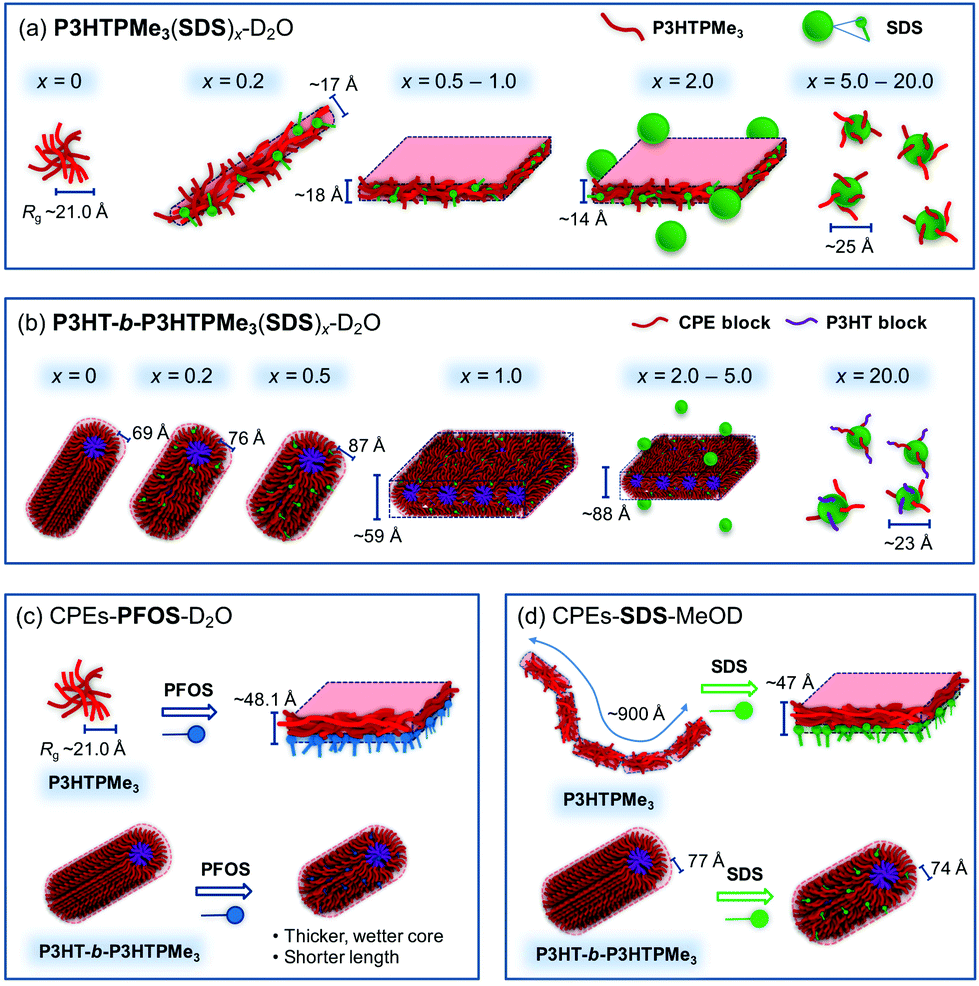 | ||
| Fig. 9 Schematic representation of proposed self-assembly structures for CPE-surfactant complexes. (a) P3HTPMe3 and (b) P3HT-b-P3HTPMe3 with increasing charge ratios, x, of SDS in D2O. P3HTPMe3 and P3HT-b-P3HTPMe3 before and after the addition of (c) 1:1 charge ratio of PFOS in D2O and (d) 1:1 charge ratio of SDS in d4-MeOD. | ||
In contrast, SDS has only a limited effect on the solution structure of P3HT-b-P3HTPMe3, with the hydrophobic P3HT core retaining the cylindrical morphology of the pure diblock copolymer (Fig. 9b). As D2O is a selective solvent for the charged CPE block, the core is expected to consist predominantly of the neutral P3HT blocks, which are surrounded by a highly solvated CPE shell. Optical and SANS studies indicated that polymer–polymer packing within the shell was affected by the presence and concentration of surfactant, which supports this assignment. With increasing SDS, an increase in the wetness and thickness of the CPE block shell was observed, which corresponds to a reorganisation of P3HTPMe3 chains. In contrast, the P3HT core was relatively unaffected, which may account for the retention of some vibronic structure in the PL spectra beyond x = 1. The SANS data suggest that the P3HT-b-P3HTPMe3(SDS)1 uniquely adopts a sheet-like structure and contrast-matching experiments with d25-SDS make it possible to see how the macromolecular structure within such aggregates may form. As such, the core–shell cylinder aggregates of P3HT-b-P3HTPMe3-SDS are proposed to bind together with the help of SDS to form lamellar sheets (Fig. 9b).
Striking differences are also observed between how the homo- and diblock CPEs interact with PFOS (Fig. 9c). Theoretical studies have shown that sulfate and sulfonate groups differ significantly in their charge density (δCD = −1.13 and −0.66, respectively).57 The reduced charge density of the sulfonate group in PFOS suggests that electrostatic interactions between P3HTPMe3 and PFOS will occur predominantly at the aggregate surface, where the interaction forms sheet-like aggregates, but does not alter the effective conjugation length of the P3HTPMe3 backbone; thus, the optical properties are essentially unchanged. This corresponds well to the sheet thickness obtained from the SANS fits which suggest that distinct PFOS/CPE layers are present. While P3HT-b-P3HTPMe3-PFOSx retains the core–shell cylinder structure, a striking increase in the wetness and radius of the P3HT core at x = 1 is observed compared to the pure CPE. Taking this into consideration with the optical observations, we propose that PFOS is able to penetrate the neutral P3HT core, causing some degree of structural rearrangement to the neutral polymer–polymer associations. The lower charge density of the sulfonate head group, coupled with the inductive effect of the fluorinated backbone, implies that weaker electrostatic interactions should be expected between the phosphonium group of the CPEs and PFOS than with SDS. Therefore, the more weakly-charged PFOS may also be expected to penetrate more deeply within the neutral P3HT blocks of the diblock copolymer than SDS.76
Methanol is a better solvent for device fabrication than water due to its higher volatility. While the optical properties of P3HTPMe3 and P3HT-b-P3HTPMe3 in MeOD showed only a minor dependence on the nature of the counterion, the aggregate structures formed strongly resemble those in D2O, although with subtle differences (Fig. 9d). P3HTPMe3 adopted a flexible rod-like conformation with high solvent content, while P3HTPMe3(SDS)1 favoured a lamellar-type structure believed to be comprised of distinct, rather than interwoven, P3HTPMe3 and SDS layers. In contrast, P3HT-b-P3HTPMe3 and P3HT-b-P3HTPMe3(SDS)1 favoured core–shell cylinder aggregates, with a P3HT core and a CPE-SDS shell.
Notably, the aggregate structures formed in solution could be effectively transferred to films deposited by both spin-coating and drop-casting. The addition of surfactants to solutions of P3HTPMe3 induced nanoscale phase separation in the resultant films, with the observation of lamellar-like regions. P3HT-b-P3HTPMe3 films formed core–shell spherical structures, somewhat in contrast to the core–shell cylinder or lamellar structures indicated by SANS. However, SANS also indicated that aggregates of P3HT-b-P3HTPMe3 possessed solvated, wet shells. It is therefore conceivable that the removal of the solvent during drying of the film may lead to partial reorganisation or shrinkage of the aggregate structure. The P3HT-b-P3HTPMe3-surfactant films exhibited less distinct spherical or sometimes amorphous structures, suggesting complete or partial collapse of the core–shell structure. It is interesting to note that the majority of core–shell structures identified in solution are retained in the films spin-coated from methanol, since one potential drawback of spin-coating is that it is known to reduce the crystallinity of P3HT thin films by preventing appropriate alignment of the polymer chains.77
Conclusions
While the optical properties of P3HTPMe3 and P3HT-b-P3HTPMe3 show a moderate dependence on the nature of the surfactant counterion, its concentration and the solvent, stark transitions in the solution structures are observed upon modifying these parameters. P3HTPMe3 has been shown to freely transform from spheres or semi-flexible cylinders to rigid sheets upon association with anionic surfactants, the size and morphology of which is dependent of the surfactant charge density, tail length and stiffness and solvent. In contrast, surfactants only have a limited effect on the solution structure of P3HT-b-P3HTPMe3, with the hydrophobic P3HT core retaining the cylindrical morphology of the pure diblock copolymer. However, contrast-matching SANS experiments with deuterated surfactants have indicated how the macromolecular organisation between P3HT-b-P3HTPMe3-SDS aggregates is formed. Furthermore, the reduced charge density of perfluorinated surfactants may offer an effective route to tune the polymer–polymer packing density within the P3HT-cores.It is perhaps unsurprising that homopolymers vs. diblock copolymers yield significantly different self-assembled structures. However, the structure “lock-in” tendency of the hydrophobic P3HT block, coupled with subtle differences in the surfactant mole fraction, chemical structure and solvent polarity (MeOH vs. H2O) give rise to remarkable variations in the range and type of complexes formed affecting both the solution phase structures and the morphology of the thin films. This has important consequences for future device preparation. For example, it has recently been shown that while charge generation occurs in the ordered domains of semi-crystalline P3HT films, the connectivity of these ordered domains through long polymer chains can strongly enhance charge transport.78 Thus, the ability to control the chain packing within CPE aggregates could be harnessed to balance the charge carrier ability with other physical properties of films currently being considered for OPV devices.77,79
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SC and MC thank the CNRS and the Université de Montpellier for financial support. Support from COST action MP1202 and the Ireland-France “Hubert Curien Ulysses” programme (grant no. 31998ZF) is gratefully acknowledged. JEH and RCE thank CRANN and the School of Chemistry, Trinity College Dublin for access to equipment for preliminary studies. We thank ISIS and STFC for the allocation of SANS beamtime (experiment RB 1510199). This work benefitted from SasView software, originally developed by the DANSE project under NSF award DMR-0520547.Notes and references
- H. Jiang, P. Taranekar, J. R. Reynolds and K. S. Schanze, Angew. Chem., Int. Ed., 2009, 48, 4300–4316 CrossRef CAS PubMed.
- Z. Ren, R. S. Nobuyasu, F. B. Dias, A. P. Monkman, S. Yan and M. R. Bryce, Macromolecules, 2016, 49, 5452–5460 CrossRef CAS.
- X. Yang, G. Zhou and W.-Y. Wong, J. Mater. Chem. C, 2014, 2, 1760–1778 RSC.
- W. Lee, J. H. Seo and H. Y. Woo, Polymer, 2013, 54, 5104–5121 CrossRef CAS.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- R. S. Ashraf, I. Meager, M. Nikolka, M. Kirkus, M. Planells, B. C. Schroeder, S. Holliday, M. Hurhangee, C. B. Nielsen, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 1314–1321 CrossRef CAS PubMed.
- C. B. Nielsen, A. Giovannitti, D.-T. Sbircea, E. Bandiello, M. R. Niazi, D. A. Hanifi, M. Sessolo, A. Amassian, G. G. Malliaras, J. Rivnay and I. McCulloch, J. Am. Chem. Soc., 2016, 138, 10252–10259 CrossRef CAS PubMed.
- J. Hwang, J. Park, Y. J. Kim, Y. H. Ha, C. E. Park, D. S. Chung, S.-K. Kwon and Y.-H. Kim, Chem. Mater., 2017, 29, 2135–2140 CrossRef CAS.
- S. Liu, P. You, J. Li, J. Li, C.-S. Lee, B. S. Ong, C. Surya and F. Yan, Energy Environ. Sci., 2015, 8, 1463–1470 CAS.
- J. Yuan, C. McDowell, C.-K. Mai, G. C. Bazan and W. Ma, Chem. Mater., 2016, 28, 7479–7486 CrossRef CAS.
- Z. Hu, K. Zhang, F. Huang and Y. Cao, Chem. Commun., 2015, 51, 5572–5585 RSC.
- J.-S. Yu, I. Kim, J.-S. Kim, J. Jo, T. T. Larsen-Olsen, R. R. Søndergaard, M. Hösel, D. Angmo, M. Jørgensen and F. C. Krebs, Nanoscale, 2012, 4, 6032–6040 RSC.
- M. Singh, H. M. Haverinen, P. Dhagat and G. E. Jabbour, Adv. Mater., 2010, 22, 673–685 CrossRef CAS PubMed.
- H. Zheng, Y. Zheng, N. Liu, N. Ai, Q. Wang, S. Wu, J. Zhou, D. Hu, S. Yu, S. Han, W. Xu, C. Luo, Y. Meng, Z. Jiang, Y. Chen, D. Li, F. Huang, J. Wang, J. Peng and Y. Cao, Nat. Commun., 2013, 4, 1971 Search PubMed.
- L. Ying, F. Huang and G. C. Bazan, Nat. Commun., 2017, 8, 14047 CrossRef CAS PubMed.
- R. C. Evans, J. Mater. Chem. C, 2013, 1, 4190–4200 RSC.
- H. D. Burrows, S. M. Fonseca, C. L. Silva, A. A. C. C. Pais, M. J. Tapia, S. Pradhan and U. Scherf, Phys. Chem. Chem. Phys., 2008, 10, 4420–4428 RSC.
- C. Tan, M. R. Pinto and K. S. Schanze, Chem. Commun., 2002, 446–447 RSC.
- H. D. Burrows, M. J. Tapia, C. L. Silva, A. A. C. C. Pais, S. M. Fonseca, J. Pina, J. Seixas de Melo, Y. Wang, E. F. Marques, M. Knaapila, A. P. Monkman, V. M. Garamus, S. Pradhan and U. Scherf, J. Phys. Chem. B, 2007, 111, 4401–4410 CrossRef CAS PubMed.
- L. Chen, S. Xu, D. McBranch and D. Whitten, J. Am. Chem. Soc., 2000, 122, 9302–9303 CrossRef CAS.
- R. C. Evans, M. Knaapila, N. Willis-Fox, M. Kraft, A. Terry, H. D. Burrows and U. Scherf, Langmuir, 2012, 28, 12348–12356 CrossRef CAS PubMed.
- M. Knaapila, R. C. Evans, V. M. Garamus, L. s. Almásy, N. m. K. Székely, A. Gutacker, U. Scherf and H. D. Burrows, Langmuir, 2010, 26, 15634–15643 CrossRef CAS PubMed.
- M. Knaapila, R. C. Evans, A. Gutacker, V. M. Garamus, N. K. Székely, U. Scherf and H. D. Burrows, Soft Matter, 2011, 7, 6863–6872 RSC.
- J. J. Lavigne, D. L. Broughton, J. N. Wilson, B. Erdogan and U. H. F. Bunz, Macromolecules, 2003, 36, 7409–7412 CrossRef CAS.
- H. D. Burrows, V. M. M. Lobo, J. Pina, M. L. Ramos, J. S. de Melo, A. J. M. Valente, M. J. Tapia, S. Pradhan and U. Scherf, Macromolecules, 2004, 37, 7425–7427 CrossRef CAS.
- M. Knaapila, L. Almásy, V. M. Garamus, C. Pearson, S. Pradhan, M. C. Petty, U. Scherf, H. D. Burrows and A. P. Monkman, J. Phys. Chem. B, 2006, 110, 10248–10257 CrossRef CAS PubMed.
- M. J. Robb, S.-Y. Ku and C. J. Hawker, Adv. Mater., 2013, 25, 5686–5700 CrossRef CAS PubMed.
- R. Verduzco, I. Botiz, D. L. Pickel, S. M. Kilbey, K. Hong, E. Dimasi and S. B. Darling, Macromolecules, 2011, 44, 530–539 CrossRef CAS.
- A. Thomas, J. E. Houston, N. Van den Brande, J. De Winter, M. Chevrier, R. K. Heenan, A. E. Terry, S. Richeter, A. Mehdi, B. Van Mele, P. Dubois, R. Lazzaroni, P. Gerbaux, R. C. Evans and S. Clément, Polym. Chem., 2014, 5, 3352–3362 RSC.
- A. Gutacker, N. Koenen, U. Scherf, S. Adamczyk, J. o. Pina, S. M. Fonseca, A. J. M. Valente, R. C. Evans, J. Seixas de Melo, H. D. Burrows and M. Knaapila, Polymer, 2010, 51, 1898–1903 CrossRef CAS.
- A. Gutacker, S. Adamczyk, A. Helfer, L. E. Garner, R. C. Evans, S. M. Fonseca, M. Knaapila, G. C. Bazan, H. D. Burrows and U. Scherf, J. Mater. Chem., 2010, 20, 1423–1430 RSC.
- G. Tu, H. Li, M. Forster, R. Heiderhoff, L. J. Balk, R. Sigel and U. Scherf, Small, 2007, 3, 1001–1006 CrossRef CAS PubMed.
- J. Kesters, S. Govaerts, G. Pirotte, J. Drijkoningen, M. Chevrier, N. Van den Brande, X. Liu, M. Fahlman, B. Van Mele, L. Lutsen, D. Vanderzande, J. Manca, S. Clément, E. Von Hauff and W. Maes, ACS Appl. Mater. Interfaces, 2016, 8, 6309–6314 CAS.
- Y. Shi, L. Tan, L. Chen and Y. Chen, J. Mater. Chem. C, 2014, 2, 8054–8064 RSC.
- M. Chevrier, J. E. Houston, J. Kesters, N. Van den Brande, A. E. Terry, S. Richeter, A. Mehdi, O. Coulembier, P. Dubois, R. Lazzaroni, B. Van Mele, W. Maes, R. C. Evans and S. Clément, J. Mater. Chem. A, 2015, 3, 23905–23916 CAS.
- K. Yao, L. Chen, Y. Chen, F. Li and P. Wang, J. Mater. Chem., 2011, 21, 13780–13784 RSC.
- X. Chen, L. Chen, K. Yao and Y. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 8321–8328 CAS.
- Y. Kim, S. Cook, S. M. Tuladhar, S. A. Choulis, J. Nelson, J. R. Durrant, D. D. C. Bradley, M. Giles, I. McCulloch, C.-S. Ha and M. Ree, Nat. Mater., 2006, 5, 197–203 CrossRef CAS.
- J. E. Houston, S. Richeter, S. Clément and R. C. Evans, Polym. Int., 2017, 66, 1333–1348 CrossRef CAS.
- E. Lee, B. Hammer, J.-K. Kim, Z. Page, T. Emrick and R. C. Hayward, J. Am. Chem. Soc., 2011, 133, 10390–10393 CrossRef CAS PubMed.
- T. Ghoos, N. Van den Brande, M. Defour, J. Brassinne, C.-A. Fustin, J.-F. Gohy, S. Hoeppener, U. S. Schubert, W. Vanormelingen, L. Lutsen, D. J. Vanderzande, B. Van Mele and W. Maes, Eur. Polym. J., 2014, 53, 206–214 CrossRef CAS.
- J. Rubio-Magnieto, A. Thomas, S. Richeter, A. Mehdi, P. Dubois, R. Lazzaroni, S. Clément and M. Surin, Chem. Commun., 2013, 49, 5483–5485 RSC.
- R. K. Heenan, J. Penfold and S. M. King, J. Appl. Crystallogr., 1997, 30, 1140–1147 CrossRef CAS.
- http://www.mantidproject.org .
- G. D. Wignall and F. S. Bates, J. Appl. Crystallogr., 1987, 20, 28–40 CrossRef CAS.
- A. Guinier and G. Fournet, Small Angle Scattering of X-rays, John Wiley and Sons, 1955 Search PubMed.
- A. A. Golosova, J. Adelsberger, A. Sepe, M. A. Niedermeier, P. Lindner, S. S. Funari, R. Jordan and C. M. Papadakis, J. Phys. Chem. C, 2012, 116, 15765–15774 CAS.
- F. Nallet, R. Laversanne and D. Roux, J. Phys. II, 1993, 3, 487–502 CrossRef CAS.
- J. Berghausen, J. Zipfel, P. Lindner and W. Richtering, J. Phys. Chem. B, 2001, 105, 11081–11088 CrossRef CAS.
- I. Livsey, J. Chem. Soc., Faraday Trans., 1987, 83, 1445–1452 RSC.
- http://www.sasview.org .
- K. K. Stokes, K. Heuzé and R. D. McCullough, Macromolecules, 2003, 36, 7114–7118 CrossRef CAS.
- K. Faied, M. Frechette, M. Ranger, L. Mazerolle, I. Levesque, M. Leclerc, T.-A. Chen and R. D. Rieke, Chem. Mater., 1995, 7, 1390–1396 CrossRef CAS.
- Y.-H. Lee, W.-C. Yen, W.-F. Su and C.-A. Dai, Soft Matter, 2011, 7, 10429–10442 RSC.
- M. Sundberg, O. Inganäs, S. Stafström, G. Gustafsson and B. Sjögren, Solid State Commun., 1989, 71, 435–439 CrossRef CAS.
- V. M. Sadtler, F. Giulieri, M. P. Krafft and J. G. Riess, Chem. – Eur. J., 1998, 4, 1952–1956 CrossRef CAS.
- P. D. T. Huibers, Langmuir, 1999, 15, 7546–7550 CrossRef CAS.
- M. Isamu, T. Akira, Y. Masakatsu and K. Hiroshi, Bull. Chem. Soc. Jpn., 1990, 63, 3502–3507 CrossRef.
- P. Mukerjee and K. J. Mysels, Critical Micelle Concentration of Aqueous Surfactant Systems, National Bureau of Standards Washington, DC, 1970 Search PubMed.
- SANS measurements performed on each of the surfactants in deuterated-methanol (conc. = [10 mg mL−1]) suggested that no micelles were formed at this concentration (see Fig. S5, ESI†).
- Q. Yuan, R. Ravikrishna and K. T. Valsaraj, Sep. Purif. Technol., 2001, 24, 309–318 CrossRef CAS.
- L. Nordstierna, I. Furó and P. Stilbs, J. Am. Chem. Soc., 2006, 128, 6704–6712 CrossRef CAS PubMed.
- H. G. Kim, B. Kang, H. Ko, J. Lee, J. Shin and K. Cho, Chem. Mater., 2015, 27, 829–838 CrossRef CAS.
- S. Dai, F. Zhao, Q. Zhang, T.-K. Lau, T. Li, K. Liu, Q. Ling, C. Wang, X. Lu, W. You and X. Zhan, J. Am. Chem. Soc., 2017, 139, 1336–1343 CrossRef CAS PubMed.
- T. J. Prosa, M. J. Winokur, J. Moulton, P. Smith and A. J. Heeger, Macromolecules, 1992, 25, 4364–4372 CrossRef CAS.
- J. B. Hayter and J. Penfold, Mol. Phys., 1981, 42, 109–118 CrossRef CAS.
- J.-P. Hansen and J. B. Hayter, Mol. Phys., 1982, 46, 651–656 CrossRef CAS.
- J. B. Hayter and J. Penfold, Colloid Polym. Sci., 1983, 261, 1022–1030 CAS.
- B. Hammouda, J. Appl. Crystallogr., 2010, 43, 716–719 CrossRef CAS.
- P. Kékicheff, C. Grabielle-Madelmont and M. Ollivon, J. Colloid Interface Sci., 1989, 131, 112–132 CrossRef.
- N. A. Mazer, G. B. Benedek and M. C. Carey, J. Phys. Chem., 1976, 80, 1075–1085 CrossRef CAS.
- P. W. Schmidt, Some Fundamental Concepts and Techniques Useful in Small-Angle Scattering Studies of Disordered Solids. In Modern Aspects of Small-Angle Scattering, Kluwer Academic Publishers: Dordrecht, The Netherlands, 1995 Search PubMed.
- M. J. M. Wirix, P. H. H. Bomans, H. Friedrich, N. A. J. M. Sommerdijk and G. de With, Nano Lett., 2014, 14, 2033–2038 CrossRef CAS PubMed.
- D. Yu, M. Tian, Y. Fan, G. Ji and Y. Wang, J. Phys. Chem. B, 2012, 116, 6425–6430 CrossRef CAS PubMed.
- Silicon wafers were cleaned before sample deposition with Piranha solution (3:1 mixture of conc. sulfuric acid and hydrogen peroxide) to remove organic residue.
- A. González-Pérez, J. M. Ruso, G. Prieto and F. Sarmiento, J. Surfactants Deterg., 2004, 7, 387–395 CrossRef.
- H. Erothu, J. Kolomanska, P. Johnston, S. Schumann, D. Deribew, D. T. W. Toolan, A. Gregori, C. Dagron-Lartigau, G. Portale, W. Bras, T. Arnold, A. Distler, R. C. Hiorns, P. Mokarian-Tabari, T. W. Collins, J. R. Howse and P. D. Topham, Macromolecules, 2015, 48, 2107–2117 CrossRef CAS.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed.
- S. Y. Son, Y. Kim, J. Lee, G.-Y. Lee, W.-T. Park, Y.-Y. Noh, C. E. Park and T. Park, J. Am. Chem. Soc., 2016, 138, 8096–8103 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV/Vis absorption and fluorescence spectra, SANS data analysis, cryo-TEM and AFM images. See DOI: 10.1039/c7nr06169b |
| This journal is © The Royal Society of Chemistry 2017 |