
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA one step method for the functional and property modification of DOPA based nanocoatings†
Qinghua
Lyu
a,
Jieyu
Zhang
b,
Koon Gee
Neoh
 b and
Christina
Li Lin Chai
*a
b and
Christina
Li Lin Chai
*a
aDepartment of Pharmacy, National University of Singapore, 18 Science Drive 4, Singapore 117543. E-mail: phacllc@nus.edu.sg; Fax: +65 6779 1554; Tel: +65 6601 1061
bDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585
First published on 7th August 2017
Abstract
Biomimetic poly(catecholamine) coatings have gained much attention in recent years due to their versatility as functional materials. Despite this, only limited methods are available to modify the function and property of poly(catecholamine) coatings, primarily through post-modification methods. Our approach reported herein provides a simple approach to the fabrication of novel functionalized poly(catecholamine) coatings. The strategy employs the copolymerization of N-Ac-3,4-dihydroxyphenylalanine methyl ester (NADOPAMe) with nucleophilic additives, giving rise to nano-coatings on various surfaces including plastic, metal, glass and polymers. With the appropriate choice of nucleophilic additives, coatings with desired properties can be achieved. This is demonstrated through the fabrication of a redox responsive coating based on NADOPAMe with cysteamine as additive, which shows a concentration-dependent glutathione (GSH) responsive behavior. The ability to utilize this as a controlled release system is also demonstrated.
Introduction
Biomimetic coatings based on naturally occurring catecholamines have been the subject of extensive scientific research over the past decades.1,2 The simplicity of the dip-and-coat approach developed by Messersmith et al. enabled the deposition of poly(dopamine) (PDA) films on various types of surfaces via the self-polymerization of dopamine under alkaline conditions.3 Other related catecholamines such as L-3,4-dihydroxyphenylalanine (L-DOPA) and norepinephrine can also undergo similar oxidative self-polymerization reactions (Scheme 1a).4–6 Despite the many reported applications of such poly(catecholamines), the deposition mechanism and structures of poly(catecholamines) have yet to be unambiguously determined.7 This is true for even the most well-studied poly(catecholamine), i.e. PDA.8 PDA is believed to result from oxidative polymerization reactions, via a combination of inter- and intra-molecular reactions of intermediates derived from the dopamine quinone.1,9,10 The structure of PDA is proposed to be oligomeric in nature, with the coatings comprising of aggregates of such oligomers held through covalent and non-covalent interactions.11 The precise structure of PDA is, however, still controversial despite significant efforts to elucidate its structure. What is indisputable is that the formation of highly reactive quinone intermediates from catecholamines triggers the polymerization reaction.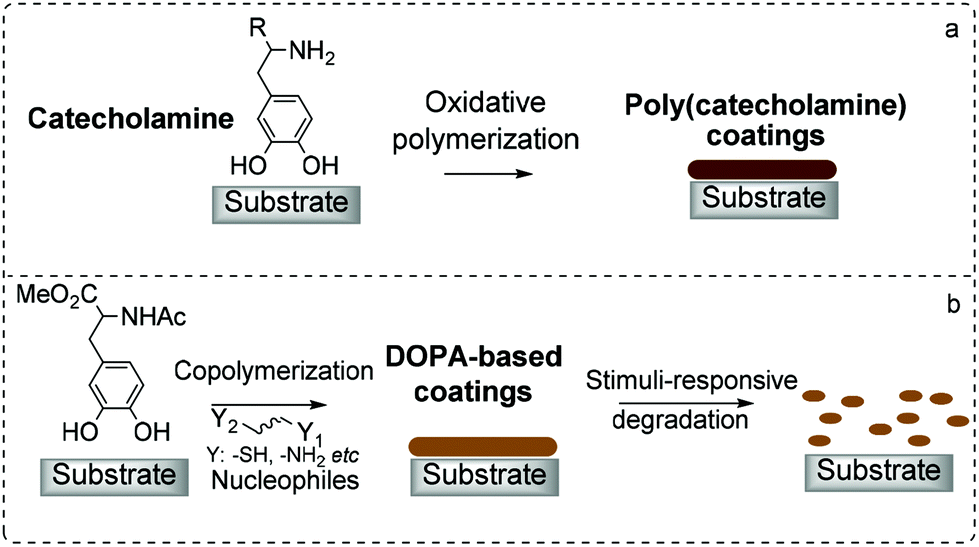 | ||
| Scheme 1 A cartoon depiction of coatings based on catecholamines formed via (a) self-polymerization and (b) copolymerization strategy. | ||
The electrophilic quinone intermediates are not only susceptible to nucleophilic attack from the free amine of catecholamine monomers, but also to external nucleophiles.12,13 Nucleophiles such as thiols and amines can undergo Michael-type addition and/or condensation reactions, respectively, with quinones. This is observed in some biomaterials, e.g. catechol–thiol conjugates in pheomelanin,14 histidinyl-dopamine conjugates in insect exoskeleton,15 and lysyl-dopa residues in Mytilus edulis foot protein-1.16 The variety of structures and functions of such biomaterials provide a significant opportunity in the development of new biomimetic functional materials. Surprisingly, there are only limited reports on the use of such a strategy in the development of new materials.9,17–21
Our interest is in the use of 3,4-dihydroxyphenylalanine (DOPA) derivatives as monomers to form coatings on substrates. In this study, N-Ac methyl ester of DOPA (NADOPAMe) was used to prevent the formation of complex eumelanin-like polymers,22 so that, the formed polymers may have more well-defined compositions compared to that of PDA. Nucleophilic additives were used for copolymerization reactions with NADOPAMe, and with the judicious choice of nucleophiles, stimuli-responsive coatings were fabricated (Scheme 1b).
Results and discussion
Coatings from copolymerization of NADOPAMe and various different nucleophiles
NADOPAMe was synthesized following the reported method (Scheme S1†),22 and characterized by 1H and 13C nuclear magnetic resonance spectroscopy as shown in Fig. S1.† The original dip-and-coat method pioneered by Messersmith et al.3,4 was employed in coating studies with NADOPAMe as monomer using silicon wafer as substrate. In contrast to studies with L-DOPA as monomer,4 no coating was formed on the silicon wafer when NADOPAMe was used (Fig. S2a†). This is consistent with reported studies that the self-polymerization of catechol cannot form good coatings in the absence of nucleophiles.5,9,23,24 The addition of nucleophilic additives may overcome the limitations in the use of NADOPAMe and other catechols as monomers. Thus, an amended protocol was developed (Fig. 1a) and a range of commercially available nucleophiles (Fig. 1b) was tested for their ability to form coatings with NADOPAMe on silicon wafer. Depending on the nature of the nucleophiles, cross-linking with the o-quinone of NADOPAMe may occur via Michael addition and/or condensation reactions so as to bridge the catechol moieties in the construction of the coating materials (Fig. 1a and c). In these studies, NADOPAMe was first dissolved in tris buffer (10 mM, pH 8.5) for 20 minutes. The silicon wafer was then immersed in the solution, followed by the addition of nucleophiles. The stoichiometry (1, 3, 5, 10 and 20 equivalents) of nucleophiles and the reaction times (24–72 h) were varied in order to assess their effects on coating formation. The ability to form coatings was examined visually, followed by AFM and XPS analyses wherever needed. | ||
| Fig. 1 (a) Schematic illustration of the protocol for the coating studies via copolymerization; (b) structures of nucleophiles selected for coating studies; (c) some structural units that may be present in NADOPAMe-nucleophile coatings. | ||
The choice of nucleophiles can be broadly categorized as sulfur (thiols 1–5), nitrogen (amines 6–10), bifunctional nucleophiles (11–14) and oxygen nucleophiles (alcohol 15 and carboxylic acid 16) (Fig. 1b). The copolymerization of monothiol 1 and monoamine 6 with NADOPAMe did not form any significant coating, while among the binucleophiles, the dithiols 2 and 5, the diamines 9 and 10, and cysteamine 11 formed coatings upon reaction with NADOPAMe. This suggests that a bifunctional nucleophile of appropriate length is minimally needed for cross-linking in the fabrication of coatings. The coatings that resulted from the use of the dithiols 2 and 5 (3 equivalents, tris buffer/methanol (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture, 8–10 hours reaction time) were characterized using XPS, which showed the presence of sulfur (164 eV) and nitrogen (400 eV) on the surfaces (Fig. 2d and Fig. S3a†). A minor sulfur peak (168 eV) was also observed and this may be attributed to the presence of oxidized sulfur. From AFM analyses of the samples NADOPAMe/2 (Fig. S2b†) and NADOPAMe/5 (Fig. 2a), the surfaces were not uniformly coated, possibly due to the precipitation of disulfides arising from the competing oxidation of the thiols. Water-soluble thiols DTT 3 and DMSA 4 were used as nucleophiles to circumvent this problem, but no coating was observed.
:1) mixture, 8–10 hours reaction time) were characterized using XPS, which showed the presence of sulfur (164 eV) and nitrogen (400 eV) on the surfaces (Fig. 2d and Fig. S3a†). A minor sulfur peak (168 eV) was also observed and this may be attributed to the presence of oxidized sulfur. From AFM analyses of the samples NADOPAMe/2 (Fig. S2b†) and NADOPAMe/5 (Fig. 2a), the surfaces were not uniformly coated, possibly due to the precipitation of disulfides arising from the competing oxidation of the thiols. Water-soluble thiols DTT 3 and DMSA 4 were used as nucleophiles to circumvent this problem, but no coating was observed.
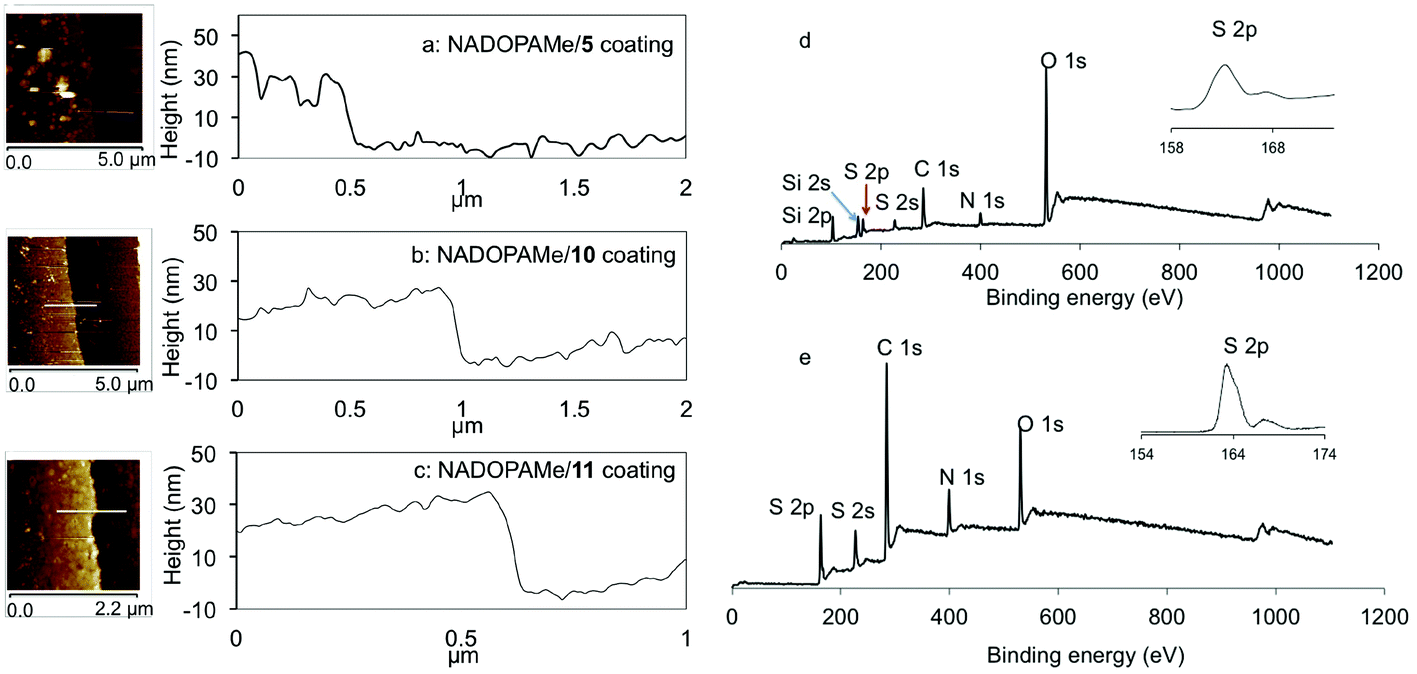 | ||
| Fig. 2 (a–c) AFM analyses of coatings from copolymerization of dithiol 5 (3 eq., 18 h), diamine 10 (10 eq., 36 h) and cysteamine 11 (3 eq., 48 h) with NADOPAMe. (d & e) XPS wide scan and S 2p core level spectra for coatings of NADOPAMe/5 and NADOPAMe/11. | ||
The use of amines 6–8 (3 eq., tris buffer pH 8.5, 72 h) as nucleophiles did not lead to the formation of coatings. When the concentrations of amines 7 and 8 were increased to 20 equivalents (72 h), large aggregates were observed on the surfaces, but these could be washed away with water during the washing step. In contrast, under similar conditions (20 eq., 56 hours), copolymerizing diamine 9 with NADOPAMe gave a thin, uneven coating of 8–10 nm (Fig. S2c†), which was analyzed by XPS (Fig. S3b†). A thick light-yellow coating (30 nm) was obtained when the diamine 10 was used as nucleophile (3 eq., 36 hours) as shown in Fig. 2b. The use of cysteamine 11 as a bifunctional nucleophile with NADOPAMe led to the formation of a 25 nm-thick yellow-brown coating after 48 h of copolymerization (Fig. 2c). XPS analysis (Fig. 2e) confirmed the presence of sulfur on the surface (binding energy of 164 eV attributed to S 2p). Due to the complex copolymerization process, the precise structure and/or components of the coating film remain unsolved. In addition to the intermolecular connections proposed in Fig. 1c, the copolymerization of NAODPAMe/11 may also undergo intramolecular cyclization to produce some benzothiazine-like intermediates (Scheme S2†), similar to the biosynthesis of pheomelanin.14,19 This successful formation of a coating with cysteamine 11 as compared to cysteine 12 and cysteine methyl ester 13 where no coating was formed suggests that the structure of the bifunctional nucleophiles is critical for coating formation. This may be related to steric and/or electronic effects of the nucleophilic entities.
To assess the versatility of the coating process on different substrates, glass, quartz, polystyrene, stainless steel and poly(lysine) were coated with the polymer of NADOPAMe/10 using the same protocol as outlined in Fig. 1. XPS characterization of these surfaces showed the presence of sulfur and nitrogen (Fig. S4†), which confirmed the successful coatings on these substrates. The water contact angles (CA) of all the coated substrates were measured. As shown in Fig. 3, the CA values of these substrates before coating varied from 20 to 85°, but after coating the CA values were consistently 49 ± 3°. Similar experiments were carried out with NADOPAMe/11, in which the measured CA was 41 ± 4° on the different surfaces. These studies show that a variety of organic or inorganic surfaces can be modified using this copolymerization approach, similar to the universality of PDA surface modification.
 | ||
| Fig. 3 Water contact angles of NADOPAMe/10 and NADOPAMe/11 coatings on different surfaces. | ||
In order to gain some understanding into the structures of the oligomers/polymers that are involved in the formation of the coatings, attenuated total reflection-infrared spectroscopy (ATR-IR) and matrix assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) analysis of samples from the copolymerization of NADOPAMe with 2, 5, 9, 10 and 11, respectively, were carried out. As shown in Fig. 4, a broad band around 3400–3200 cm−1 region was assigned to O–H and/or N–H stretching vibrations of the catechol and/or amine groups. This IR absorbance is stronger for coatings formed using amines 9–11 than that with thiols 2 and 5, presumably due to the presence of free amine functionalities. The absorption bands around 3000–2850 cm−1 were assigned to the C–H asymmetric and symmetric stretching vibration of CH2 aliphatic moiety. The dominant bands of 1760–1640 cm−1 were assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrations of carboxylic ester (1735 cm−1), amide (1625 cm−1) and o-quinone functionality (1710–1665 cm−1), and the major absorbance at 1600–1510 cm−1 was ascribed to aromatic ring vibrations. Other vibrations in the 1450–1400 cm−1 region were associated with CH2 aliphatic vibrations from both NADOPAMe and the nucleophiles.
O vibrations of carboxylic ester (1735 cm−1), amide (1625 cm−1) and o-quinone functionality (1710–1665 cm−1), and the major absorbance at 1600–1510 cm−1 was ascribed to aromatic ring vibrations. Other vibrations in the 1450–1400 cm−1 region were associated with CH2 aliphatic vibrations from both NADOPAMe and the nucleophiles.
 | ||
| Fig. 4 ATR-FTIR spectra of copolymer coatings on poly(lysine) surfaces. | ||
The mass spectra of the copolymer samples (Fig. S5 and 6†) were compared with that derived from the self-polymerization of NADOPAMe and DOPA methyl ester. Spectra of poly-NADOPAMe showed a lower intensity of oligomeric composition as compared to that from the polymerization of DOPA methyl ester (Fig. S5†). This is consistent with the rationale that protection of the amino groups of DOPA will reduce competing reactions of intramolecular cyclization to form reactive dihydroxyindole intermediates and their subsequent polymerization.25 In general, the maximum m/z values in the mass spectra of these copolymer coatings were less than 1000 (Fig. S6†). However, it should be noted that MALDI-TOF mass analysis is only able to provide information on oligomers that were dissolved in water/dimethyl formamide mixture, and not suitable for insoluble particles which may contain large oligomers. The common peaks observed in the spectra of the samples obtained from self-polymerization of NADOPAMe and its copolymers with nucleophiles suggest that the copolymerized coatings may contain some oligomers of NADOPAMe. For example, the m/z value of 503.8 Da is assigned to the dimer of NADOPAMe (Scheme S3†), which can arise from aryl–aryl coupling reaction.13,26–28
The above findings demonstrate the promise of using thiol linkers as additives in the construction of robust coatings with catecholamines. The interference of insoluble oxidized disulfides in aqueous solutions precludes the use of certain dithiol linkers but it is envisaged that these limitations can be overcome with careful design of linker structures and control of reaction conditions. Amino nucleophiles have been shown to play a crucial role in the formation of stable coatings via cation–π interaction during the self-polymerization of catecholamines or copolymerization with catechols.5,29 Our studies show that the length and the structure of the tether (e.g.7–10) can affect non-covalent/covalent interactions between the oligomers formed and thus significantly influence the formation of the coatings. Our investigations also indicate that groups adjacent to the amino functionalities have a profound influence on the copolymerization process, as illustrated by the vastly different outcomes when cysteamine 11, cysteine 12 and cysteine methyl ester 13 were used as additives.
Proof-concept study on the fabrication of redox-responsive coating
Our methodology as described above can be applied to the fabrication of stimuli-responsive coatings in a simple and versatile manner, as outlined in Scheme 2. To demonstrate this, the redox responsive behavior of NADOPAMe/10 coating was studied. Glutathione (GSH) as reductant at concentrations of 1, 5, 20 mM were chosen to provide the stimulus for studying degradation kinetics.30–32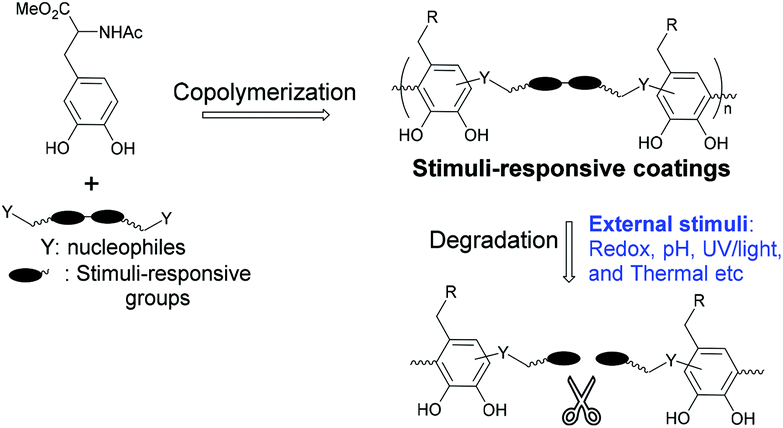 | ||
| Scheme 2 Construction of stimuli-responsive coatings via copolymerization strategy. | ||
Silicon wafer coated with NADOPAMe/10 was treated with 20 mM GSH, and the degradation of the coating was monitored by XPS (Fig. 5a). The intensity of the disulfide signal, S 2p (164 eV), decreased with time of treatment while that of the substrate Si 2p (99.5 eV) increased with time. This suggests that the integrity of the film is compromised upon treatment with GSH. The degradation is also confirmed by FTIR analysis of NADOPAMe/10 coated poly(lysine) surfaces as shown in Fig. 5b. The IR peaks of the untreated coating observed at 3261 (ν(N–H) and ν(O–H)), 2951 (ν(C–H)), 1735 (ν(CO)), and 1525 (ν(CC)ring) cm−1 were barely observable after treatment with GSH solution (20 mM, 4 h). An analogous experiment was carried out to monitor coating degradation in situ using UV analysis (Fig. S7 and 8†). In the presence of 1 mM GSH (8 h), the absorbance at 380 nm showed a slight decrease but when the concentration of GSH increased to 5 mM, the absorbance at 380 nm decreased by approximately half in 4 h. In the presence of 20 mM of GSH, almost all the coating was removed from the poly(lysine) surface by 4 h. In a control experiment, the NADOPAMe/10 coating soaked in a pH 7.4 tris buffer did not show significant changes over 8 h as indicated by XPS or UV analysis.
 | ||
| Fig. 5 (a) XPS spectra of NADOPAMe/10 on Si surface and (b) ATR-FTIR spectra of NADOPAMe/10 on poly(lysine) surface before and after treatment with 20 mM GSH in tris buffer (10 mM, pH 7.4). | ||
Loading and release of test compound in redox-responsive coating
To demonstrate that this stimuli-responsive coating film can be used as a release system, fluorescein dye was added together with the additive 10 to the tris solution of NADOPAMe following the same copolymerization protocol shown in Fig. 1. It is expected that fluorescein will be incorporated into the NADOPAMe/10 film when formed and will be released with the degradation of the coating upon GSH treatment.31 The incorporation of fluorescein into the coating was characterized by a new strong absorbance at 495 nm (Fig. 6a). The release kinetics of fluorescein upon GSH treatment was further monitored using fluorescence spectrometry. In the presence of 5 mM or 20 mM GSH solution, an initial fast release of fluorescein was observed due to the degradation of coatings and the cumulative release reached a plateau after 4 h (Fig. 6b). In contrast, the coating in pH 7.4 buffer solution was stable over 8 h, and the fluorescein was released at a much slower rate, presumably due to the swelling of the coating. These findings suggest that compounds can be successfully incorporated into these coatings via the copolymerization protocol, and their release can be triggered by appropriate stimuli, in our case, GSH as the reductant of the disulfide linkage. | ||
| Fig. 6 (a) UV-vis absorbance of NADOPAMe/10/fluorescein on poly(lysine) before and after treatment with pH 7.4 tris buffer (8 h), 5 mM GSH (4 h) or 20 mM GSH (4 h) in tris buffer; (b) fluorescence-spectrometric analyses of fluorescein released from the coatings in the presence of different concentrations of GSH. | ||
Conclusions
In summary, we have developed a new strategy using nucleophilic additives to facilitate the polymerization of NADOPAME to form coatings. As demonstrated, stimuli-responsive coatings can be fabricated using this facile dip-and-coat process, which are potentially promising in applications such as release systems and for drug delivery. In contrast to poly(catecholamine) coatings, where the choice of precursors is limited, the copolymerization of varied catechol(amine)s and functional additives can offer coatings with tailored chemical and physical properties.33,34 It is expected that this low-cost and facile bioinspired copolymerization approach will open new opportunities for the development of novel and versatile functional biomimetic materials for a range of applications.Experimental
Nucleophilic additives used for coating studies
All the additives are commercially available and used without further purification: additives 1, 3–6, 9 and 12–14 were purchased from Sigma-Aldrich Pte Ltd; 2, 7, 8, 10 and 16 were purchased from Alfa Aesar; 11 and 15 were purchased from Tokyo Chemical Industry Co., Ltd (Singapore).Substrates used for coating studies
Silicon (Si) wafers were obtained from Mitsubishi Silicon America, USA. AISI type 304 stainless steel foils (0.05 mm in thickness) were purchased from Goodfellow Ltd of Cambridge, U.K., Quartz microscope slides (fused, 76.2 mm × 25.4 mm × 1.0 mm) and glass slides (76.2 mm × 25.4 mm, 1–1.2 mm thick) were purchased from Alfa Aesar. Poly(lysine) slides (Thermo Scientific™ 25 mm × 75 mm × 1.0 mm) and polystyrene dishes (60 mm × 15 mm) were purchased from Fisher Scientific.General dip-and-coat method
Si wafers were pre-cleaned with a solution of 4.8% HF and 7% HNO3 in water, and then cleaned using ultrasound irradiation in deionized (DI) water and ethanol for 10 min each. Other substrates were first cleaned in an ultrasonic water bath for 30 min, rinsed with ethanol, and then blown dry by nitrogen gas. The substrates, Si wafer (1 cm × 1 cm), glass (2.5 cm × 2.5 cm), poly(lysine) (2.5 cm × 2.5 cm), stainless steel (1 cm × 1 cm), polystyrene (1 cm × 1 cm), quartz slides (7.6 cm × 2.5 cm) were then immersed in 20 ml of tris buffer (10 mM, pH 8.5) with 10 mM NADOPAMe for 20 min, and additives were added at specified stoichiometries at room temperature. After coating for a specified time, the substrates were rinsed with water and ethanol, and dried by a stream of nitrogen gas. These samples were subsequently characterized as described below.Characterization of coatings
Atomic force microscopy (AFM) analyses were carried out on Bruker Dimension ICON. X-ray photoelectron spectroscopy (XPS) measurements were performed on a Kratos AXIS Ultra HAS spectrometer with a monochromatized Al Kα X-ray source (1486.6 eV photons), at a constant dwell time of 100 ms and pass energy of 40 eV. All binding energies were referenced to the C 1s hydrocarbon peak at 284.6 eV. Attenuated total reflection (ATR)-Fourier transform infrared spectroscopy (FTIR) analysis was carried out on a Bruker Alpha spectrometer equipped with (RT) DLaTGS detector (Bruker, USA) and a platinum ATR accessory with a 2 mm diameter single reflection diamond crystal. A clean silicon wafer and poly(lysine) slide were used as references respectively. Spectroscopic information was collected in the 1200–3600 cm−1 range with a 4 cm−1 resolution and 680 acquisitions per sample and corrected for baseline drift. Static water contact angles were measured using the sessile drop method with a 2 μL water droplet in a telescopic goniometer (Rame-Hart Model 10000-(230)). Each static water contact angle reported was the mean value from three surfaces, with the value from each surface obtained by averaging the contact angles from at least three locations.Preparation of redox-responsive coating samples
Different concentrations of GSH (1, 5, or 20 mM) in tris solution (10 mM, pH 7.4) were prepared for the degradation study of NADOPAMe/10 film on Si wafer and poly(lysine) slides. Coated substrates, Si wafer (1 cm × 1 cm) and poly(lysine) slides (2.5 cm × 2.5 cm), were immersed in the above-mentioned GSH solutions or a control pH 7.4 buffer for a predetermined time, followed by washing with DI water and drying under nitrogen, and finally subjected to XPS, ATR-FTIR, and UV/Vis analysis. XPS and ATR-FTIR analysis of coated Si samples were carried out as described in the coating studies above. UV/Vis analysis of coated poly(lysine) samples were carried out using a Shimadzu UV-1800 UV-Visible spectrophotometer.Loading and release of fluorescein
NADOPAMe/10 coatings with loaded fluorescein were prepared as follows: the pre-cleaned substrates, Si wafer (1 cm × 1 cm) and poly(lysine) (2.5 cm × 2.5 cm), were immersed in 20 ml of tris buffer (10 mM, pH 8.5) with 10 mM NADOPAMe for 20 min, and then the nucleophile 10 (30 mM) and fluorescein sodium salt (5 mM) were added at room temperature. After 36–40 h, the substrates (Si wafer and poly(lysine)) slides were then taken out, washed with DI water and dried under nitrogen gas. The coated substrates were incubated in buffer solution (10 mM tris buffer, pH 7.4) containing different concentrations of GSH (5, or 20 mM) as well as in blank tris buffer (10 mM, pH 7.4). Time course measurements of the fluorescence intensity of these buffer solutions were carried out using a Hitachi F-7000 fluorescence spectrophotometer (λex = 494 nm; λem = 512 nm) to compare the release kinetics of fluorescein under different GSH concentrations.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank SERC-A*STAR for financial support of this project (Project no: 1321202069).Notes and references
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- M. Y. Liu, G. J. Zeng, K. Wang, Q. Wan, L. Tao, X. Y. Zhang and Y. Wei, Nanoscale, 2016, 8, 16819–16840 RSC.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- J. H. Kuang, J. L. Guo and P. B. Messersmith, Adv. Mater. Interfaces, 2014, 1, 6 Search PubMed.
- C. Lim, J. Huang, S. Kim, H. Lee, H. Zeng and D. S. Hwang, Angew. Chem., Int. Ed., 2016, 55, 3342–3346 CrossRef CAS PubMed.
- S. M. Kang, J. Rho, I. S. Choi, P. B. Messersmith and H. Lee, J. Am. Chem. Soc., 2009, 131, 13224–13225 CrossRef CAS PubMed.
- R. Mrowczynski, R. Markiewicz and J. Liebscher, Polym. Int., 2016, 65, 1288–1299 CrossRef CAS.
- J. Liebscher, R. Mrowczynski, H. A. Scheidt, C. Filip, N. D. Hadade, R. Turcu, A. Bende and S. Beck, Langmuir, 2013, 29, 10539–10548 CrossRef CAS PubMed.
- S. Chen, X. Li, Z. L. Yang, S. Zhou, R. F. Luo, M. F. Maitz, Y. C. Zhao, J. Wang, K. Q. Xiong and N. Huang, Colloids Surf., B, 2014, 113, 125–133 CrossRef CAS PubMed.
- N. F. Della Vecchia, R. Avolio, M. Alfè, M. E. Errico, A. Napolitano and M. d'Ischia, Adv. Funct. Mater., 2013, 23, 1331–1340 CrossRef CAS.
- Y. H. Ding, L. T. Weng, M. Yang, Z. L. Yang, X. Lu, N. Huang and Y. Leng, Langmuir, 2014, 30, 12258–12269 CrossRef CAS PubMed.
- S. Bittner, Amino Acids, 2006, 30, 205–224 CrossRef CAS PubMed.
- J. Yang, M. A. C. Stuart and M. Kamperman, Chem. Soc. Rev., 2014, 43, 8271–8298 RSC.
- L. Mosca, C. Foppoli, R. Coccia and M. A. Rosei, Pigm. Cell Res., 1996, 9, 117–125 CrossRef CAS.
- X. Huang, R. D. Xu, M. D. Hawley and K. J. Kramer, Bioorg. Chem., 1997, 25, 179–202 CrossRef CAS.
- S. X. Wang, N. Nakamura, M. Mure, J. P. Klinman and J. SandersLoehr, J. Biol. Chem., 1997, 272, 28841–28844 CrossRef CAS PubMed.
- X. N. Yin, J. Wang, J. J. Zhou and L. Li, Chin. J. Polym. Sci., 2015, 33, 1721–1729 CrossRef CAS.
- Y. Lee, H. J. Chung, S. Yeo, C. H. Ahn, H. Lee, P. B. Messersmith and T. G. Park, Soft Matter, 2010, 6, 977–983 RSC.
- N. F. Della Vecchia, R. Marega, M. Ambrico, M. Iacomino, R. Micillo, A. Napolitano, D. Bonifazi and M. d'Ischia, J. Mater. Chem. C, 2015, 3, 6525–6531 RSC.
- H. S. Kim, H. O. Ham, Y. J. Son, P. B. Messersmith and H. S. Yoo, J. Mater. Chem. B, 2013, 1, 3940–3949 RSC.
- W. Z. Qiu, Y. Lv, Y. Du, H. C. Yang and Z. K. Xu, RSC Adv., 2016, 6, 34096–34102 RSC.
- Y. S. Cheah, S. Santhanakrishnan, M. B. Sullivan, K. G. Neoh and C. L. L. Chai, Tetrahedron, 2016, 72, 6543–6550 CrossRef CAS.
- J. Saiz-Poseu, J. Sedo, B. Garcia, C. Benaiges, T. Parella, R. Alibes, J. Hernando, F. Busque and D. Ruiz-Molina, Adv. Mater., 2013, 25, 2066–2070 CrossRef CAS PubMed.
- Y. J. Liu, R. F. Luo, F. Y. Shen, L. L. Tang, J. Wang and N. Huang, Appl. Surf. Sci., 2015, 328, 163–169 CrossRef CAS.
- M. d'Ischia, A. Napolitano, V. Ball, C. T. Chen and M. J. Buehler, Acc. Chem. Res., 2014, 47, 3541–3550 CrossRef PubMed.
- M. Yu, J. Hwang and T. J. Deming, J. Am. Chem. Soc., 1999, 121, 5825–5826 CrossRef CAS.
- L. M. McDowell, L. A. Burzio, J. H. Waite and J. Schaefer, J. Biol. Chem., 1999, 274, 20293–20295 CrossRef CAS PubMed.
- L. A. Burzio and J. H. Waite, Biochemistry, 2000, 39, 11147–11153 CrossRef CAS PubMed.
- H. M. Hu, J. C. Dyke, B. A. Bowman, C. C. Ko and W. You, Langmuir, 2016, 32, 9873–9882 CrossRef CAS PubMed.
- A. Pompella and A. Corti, Front. Pharmacol., 2015, 6, 98 Search PubMed.
- D. Hong, H. Lee, B. J. Kim, T. Park, J. Y. Choi, M. Park, J. Lee, H. Cho, S. P. Hong, S. H. Yang, S. H. Jung, S. B. Ko and I. S. Choi, Nanoscale, 2015, 7, 20149–20154 RSC.
- A. P. R. Johnston, G. K. Such and F. Caruso, Angew. Chem., Int. Ed., 2010, 49, 2664–2666 CrossRef CAS PubMed.
- E. Faure, C. Falentin-Daudre, C. Jerome, J. Lyskawa, D. Fournier, P. Woisel and C. Detrembleur, Prog. Polym. Sci., 2013, 38, 236–270 CrossRef CAS.
- J. Sedo, J. Saiz-Poseu, F. Busque and D. Ruiz-Molina, Adv. Mater., 2013, 25, 653–701 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr05293f |
| This journal is © The Royal Society of Chemistry 2017 |