
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CaF2 nanoparticles as surface carriers of GCAP1, a calcium sensor protein involved in retinal dystrophies†
Valerio
Marino‡
 a,
Alberto
Borsatto‡
a,
Farina
Vocke
b,
Karl-Wilhelm
Koch
b and
Daniele
Dell'Orco
*a
a,
Alberto
Borsatto‡
a,
Farina
Vocke
b,
Karl-Wilhelm
Koch
b and
Daniele
Dell'Orco
*a
aDepartment of Neurosciences, Biomedicine and Movement Sciences, Section of Biological Chemistry, University of Verona, Verona, Italy. E-mail: daniele.dellorco@univr.it; Tel: +39-045-802-7637
bDepartment of Neuroscience, Biochemistry Group, University of Oldenburg, Oldenburg, Germany
First published on 8th August 2017
Abstract
CaF2-based nanoparticles (NP) are promising biocompatible tools for nanomedicine applications. The structure of the NP crystal lattice allows for specific interactions with Ca2+-binding proteins through their EF-hand cation binding motifs. Here we investigated the interaction of 23 nm citrate-coated CaF2 NP with a calcium sensor protein GCAP1 that is normally expressed in photoreceptor cells and involved in the regulation of the early steps of vision. Protein–NP interactions were thoroughly investigated for the wild type (WT) GCAP1 as well as for a variant carrying the Asp 100 to Glu mutation (D100E), which prevents the binding of Ca2+ to the highest affinity site and is linked to cone dystrophy. Circular dichroism and fluorescence spectroscopy showed that protein structure and Ca2+-sensing capability are conserved for both variants upon interaction with the NP surface, although the interaction mode depends on the specific occupation of Ca2+-binding sites. NP binding stabilizes the structure of the bound GCAP1 and occurs with nanomolar affinity, as probed by isothermal titration calorimetry. Surface plasmon resonance revealed a fully reversible binding compatible with physiologically relevant kinetics of protein release whereas biochemical assays indicated a residual capability for NP-dissociated GCAP1 to regulate the target retinal guanylate cyclase. Our study constitutes a proof of concept that CaF2 NP could be optimized to serve as biologically compatible carriers of high amounts of functional GCAP1 in photoreceptors affected by retinal dystrophies.
Introduction
Nanoparticles (NP) are increasingly used in nanomedicine for the delivery of genes, peptides and small molecules. More recently, nanodevices raised special interest for their potential use in protein therapeutics, which constitute promising candidates for the treatment of complex diseases due to the high specificity and minimal side effects.1,2 The usually high load-capacity of NP and the possibility to functionalize their surface constitute crucial aspects for applications in protein delivery and cell targeting.3,4Lanthanide-doped, citrate-coated CaF2 NP seem particularly promising as they have recently shown fully biocompatible features in in vitro experiments performed on HeLa and mesenchymal cells,5 human dendritic cells and neurons,6 and they have also shown promising antitumor activities when combined with other chemicals to form nanoscale assemblies.7,8 Interestingly, the in vivo distribution of CaF2 NP analyzed in mice showed evidence of intact blood–brain barrier crossing without functionalization,6 moreover, the luminescent features of NP upon IR two-photon excitation make them encouraging tools for fluorescence bio-imaging applications.9–11
Calcium (Ca2+) regulates many cellular and physiological processes and acts as an important second messenger in a variety of biochemical pathways,12 including the phototransduction cascade, which constitutes the framework where the early biochemical steps in vertebrate vision occur.13,14 The specific conformations adopted by Ca2+ sensor proteins in response to the changes in intracellular [Ca2+] permit the regulation of selective biological targets.15 Among Ca2+ sensor proteins operating in the phototransduction cascade, guanylate cyclase-activating proteins (GCAPs) are able to bind both Ca2+ and Mg2+ and thereby switch between activators and inhibitors of the target enzyme guanylate cyclase GC-E (from now on indicated as GC).16–20 GC uses GTP as a substrate to synthesize cyclic GMP (cGMP), another important second messenger crucial for the recovery of the photoresponse upon illumination. Indeed, cGMP synthesis is finely regulated in a Ca2+-dependent manner by GCAPs, which inhibit the GC at high [Ca2+] (∼250–800 nM), where the three functional Ca2+-binding EF hand motifs (EF2, EF3 and EF4; see Fig. 1) are fully occupied by Ca2+, and promote GC activation during the activation of the phototransduction cascade, when [Ca2+] drops down to ∼20–100 nM, and EF2 and putatively EF3 are occupied by a Mg2+ ion.13,21
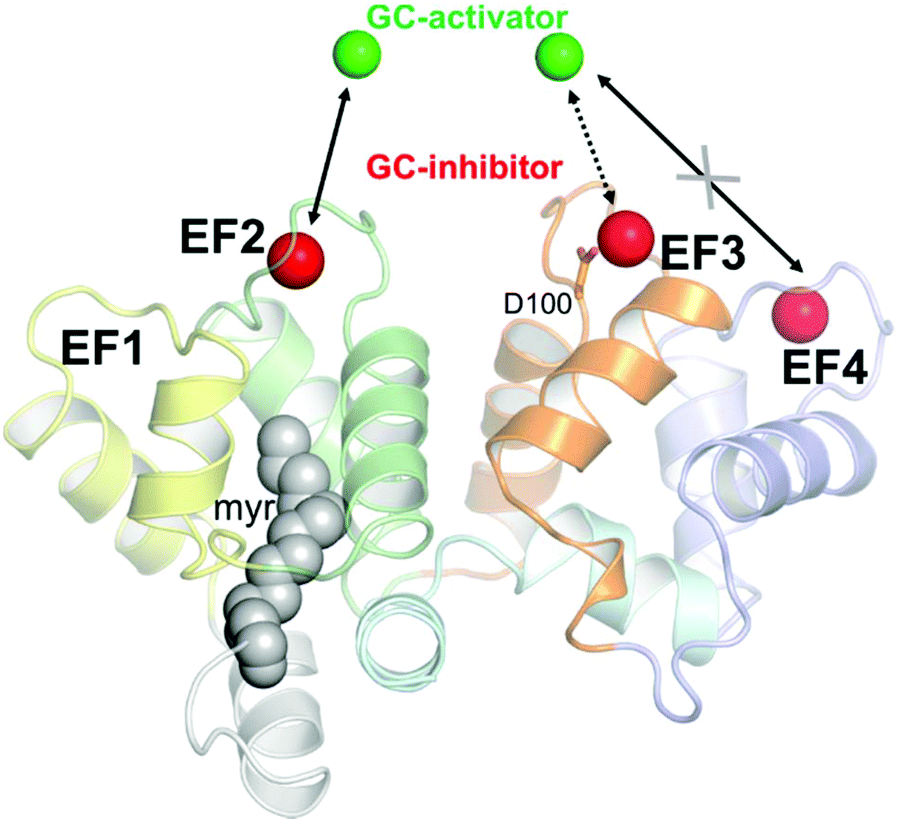 | ||
| Fig. 1 Structural model of human GCAP1 obtained by homology modeling as elucidated in ref. 24. Cartoons are colored as follows: EF1 is colored in yellow, EF2 in green, EF3 in orange and EF4 in light slate blue. N- and C-terminals are colored in pale gray and pale cyan, respectively. The myristoyl group is represented by grey spheres while calcium ions, identifying the guanylate cyclase-inhibiting state are represented by red spheres. Magnesium ions are represented by green spheres. Mg2+ ions replace Ca2+ ions in EF2 and putatively in EF3, but not in EF4, in the guanylate cyclase-activating state. The residue Asp100 coordinating Ca2+ in EF3 is represented by sticks. | ||
Very recently the possibility to exploit the high surface-to-volume ratio of CaF2 NP to develop nanocarriers for calcium sensor proteins was investigated by some of us.22,23 The effects of NP on protein structural and functional properties were found to be protein specific. Indeed, the calcium sensor recoverin (Rec), also involved in the phototransduction cascade, showed altered conformation, structural stability and Ca2+-sensing capability when incubated with CaF2 NP.23 However, when the same type of experiment was performed with the ubiquitous protein calmodulin (CaM) the interaction was shown to occur without significant alterations in protein secondary and tertiary structures, and the protein retained residual capability to function as a calcium sensor protein, thus binding to and regulating the activity of a target protein.22 Moreover, experiments performed with a CaM mutant suggested that the binding between the protein and the CaF2 NP surface was mediated by specific interactions occurring between EF-hand motifs in the protein and the Ca2+ ions within the CaF2 lattice. The relatively high affinity for the NP was hence attributed to the locally high concentration of Ca2+ ions in the crystal lattice compared to those free in solution.22
In this work, we probed the interaction between 23 nm citrate-coated CaF2 NP and two variants of GCAP1, the wild type form (WT) and a mutant, in which the aspartate residue at position 100 (Fig. 1) was replaced by a glutamic acid (D100E). The substitution is conservative with regard to the negative electric charge of the side chain, but it is located in the highest-affinity Ca2+-binding motif EF3 and reduces the affinity for Ca2+ about 270-fold.24,25 The significant reduction in Ca2+-affinity is due to the loss of the first Ca2+ coordinator, which under normal conditions provides the oxygen atom in the right orientation for optimal electrostatic interaction with the cation. The D100E mutant was also found in patients suffering from cone dystrophy and was shown to lead to constitutive GC activation at physiological Ca2+ levels.24,26,27 We performed an exhaustive biochemical and biophysical investigation and found that the high affinity for Ca2+ ions of GCAP1 reflects in a high affinity for the CaF2 NP surface. Although tight, the binding was found to be fully reversible and the dissociation kinetics was compatible with a physiologically relevant time-frame. The association between WT/D100E GCAP1 and the CaF2 NP surface occurred without major alterations in the protein structure and function, which allowed the residual regulation of the target GC, and was found to depend on the specific cation occupancy level of the interacting EF hand. Interestingly, our study shows that the interaction between CaF2 NP and GCAP1 is strongly influenced by the “protein corona” constituted by protein molecules temporarily bound to the particle surface. Since modification of NP size allows changing the number of GCAP1 binding sites on the particle surface, our results further suggest testing the site-directed transport of WT GCAP1-coated CaF2 NP into cells that express disease-associated GCAP1 mutants.
Experimental
Protein expression and purification
Plasmids containing the cDNA for bovine WT and D100E GCAP1 were prepared as per previous work.24,27 Proteins were expressed in BL21-DE3 E. coli cells co-transformed with the pBB131 plasmid encoding for S. cerevisiae N-myristoyl transferase and purified from the inclusion bodies after denaturation with 6 M guanidine-HCl, dialysis and a combination of size-exclusion and anion exchange chromatography, as elucidated in ref. 28. After purification, protein buffer was exchanged against decalcified NH4HCO3, protein samples were then concentrated, aliquoted, lyophilized and stored at −80 °C.CaF2 NP synthesis
The hydrothermal synthesis of lanthanide-doped CaF2 NP was performed by preparing a 28 ml solution of 125 mM metal chlorides (Ca/Yb/Er = 0.78/0.20/0.02 metal molar ratio), 714 mM K-citrate and 375 mM NH4F. The solution was incubated for 6 h at 190 °C in a Teflon-lined autoclave, then NP were collected by acetone precipitation followed by centrifugation for 10 minutes at 7000 rpm.5,23 A proper NP solution was finally achieved by resuspending the precipitated hydrogel in decalcified water and further decalcification was obtained through extensive dialysis using a dialysis tube containing the ion chelating resin Chelex 100 (Bio-Rad) as explained in ref. 22. The size of the NP dispersion in decalcified 5 mM Tris-HCl, pH 7.5, 150 mM KCl buffer was assessed using dynamic light scattering as described in ref. 23.Dynamic light scattering measurements
DLS experiments were carried out with a Zetasizer Nano-S (Malvern Instruments) using low volume disposable polystyrene cuvettes. Absorption and refractive index of CaF2 NP were set to 0.01 and 1.475 (http://refractiveindex.info) respectively, while viscosity and refractive index of aqueous buffer were set to default values (0.8872 cP and 1.330 respectively). All experiments were performed at 25 °C after 2 min equilibration, light was measured at a 173° backscatter angle and analyzed using a multiple narrow modes model, as in ref. 23. Protein concentration was 11.7 μM and the NP concentration was 1.1 mg ml−1. Every measurement is an average of at least 23 repetitions, each consisting of at least 12 runs.Circular dichroism spectroscopy and thermal denaturation profiles
Circular dichroism experiments were carried out with a Jasco J-710 spectropolarimeter equipped with a Peltier type thermostated cell holder. Near UV (250–320 nm) and far UV (200–250 nm) spectra were an average of 5 accumulations recorded at 25 °C with the scan rate set to 50 nm min−1, bandwidth and integration time set to 1 nm and 4 s respectively.23 Protein concentration for near UV spectra was 15.5–25.4 μM in 1 cm quartz cuvettes, while for far UV spectra it was 10–20 μM in 0.1 cm quartz cuvettes. CD spectra were recorded in 5 mM Tris-HCl, pH 7.5, 150 mM KCl buffer with a >15-fold excess EGTA with respect to the protein concentration, after sequential additions of 1 mM Mg2+ and >15-fold excess free Ca2+ with respect to the protein concentration, and the spectra with or without NP were considered as blanks and subtracted. NP concentration in CD experiments was calculated in order to have a stoichiometric ratio of GCAP1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NP = 110:1 (estimated as in ref. 23), to ensure an appropriate covering of the NP surface.
:NP = 110:1 (estimated as in ref. 23), to ensure an appropriate covering of the NP surface.
Thermal denaturation profiles were obtained by monitoring the signal at 208 nm in the 20–96 °C range, with the scan rate set to 1 °C min−1 and response time set to 4 s in a 0.1 cm quartz cuvette.23 Data fitting was performed using a function accounting for the variation of Gibbs free energy upon thermal unfolding, as explained in ref. 24.
Fluorescence titrations
The emission fluorescence spectrum (300–380 nm) of 0.7 μM WT GCAP1 in 5 mM Tris-HCl, pH 7.5, 150 mM KCl, 100 μM EGTA buffer was recorded upon excitation at 290 nm with a Jasco spectrofluorimeter FP-750 in a 1 cm quartz cuvette at a scan rate of 60 nm min−1, with excitation and emission bandwidths set to 5 nm. Mg2+ buffers used in fluorescence titration experiments in the absence and in the presence of 0.0875 mg ml−1 NP were prepared in the 90 μM–10 mM range as described in ref. 28. The signal at the wavelength of maximal fluorescence emission in the presence of 100 μM EGTA (341 nm) was monitored.Limited proteolysis
A mixture of 28.2 μM WT or D100E GCAP1 was incubated for 1 h at 25 °C with 3.4 mg ml−1 NP (GCAP1:NP = 110:1, stoichiometric ratio was estimated as in ref. 23) dissolved in decalcified 5 mM Tris/HCl, pH 7.5, 150 mM KCl, 1 mM DTT buffer and, depending on the experimental conditions, (i) 2 mM EGTA, (ii) 1 mM Mg2+ and 2 mM EGTA, (iii) 2 mM Ca2+, and (iv) 1 mM Mg2+ and 2 mM Ca2+. Protein–NP mixtures were incubated with 0.015 mg ml−1 TPCK-trypsin for 20 minutes at 25 °C (TPCK-trypsin:GCAP1 = 1:47), then the reaction was stopped by adding reducing sample buffer and boiling for 5 min. For each experimental condition a parallel sample not containing any NP was studied, another sample was prepared as the control without trypsin. Proteolytic patterns were visualized on a 15% SDS-PAGE gel with Coomassie blue staining.
Surface plasmon resonance spectroscopy
The SensiQ Pioneer surface plasmon resonance instrument was employed for the investigation of the interaction of NP with GCAP1 variants immobilized via amine coupling on a SensiQ COOH5 sensor chip coated with a carboxylated polysaccharide hydrogel spacer. Carboxyl groups were mobilized with sequential injections (5 μl min−1 flow rate) of 60 μl of 10 mM H3PO4, 60 μl HBS (10 mM Hepes, pH 7.4, 150 mM KCl, 20 mM MgCl2, 2 mM CaCl2), 60 μl 10 mM NaOH and 2 × 60 μl HBS. The sensor chip surface was then activated with a 7 min pulse of a mixture of 10 mM N-hydroxysuccinimide (NHS) and N-ethyl-N′-(dimethylaminopropyl)-carbodiimide (EDC) at a flow rate of 5 μl min−1. GCAP1 variants were dissolved in bidistilled H2O at a concentration of 1 mg ml−1, then diluted in 10 mM Na-acetate buffer (pH 3.0) to a final concentration of 7 μM. Sequential injections of proteins on the sensor chip led to the immobilization of ∼3000 RU (corresponding to ∼3 ng flow per cell, 1 RU = 1 pg mm−2, flow cell volume <40 nL), finally the surface was blocked via injection of 70 μl 1 M ethanolamine hydrochloride-NaOH pH 8.5. During the immobilization process, HBS was used as a running buffer.In all SPR experiments, one of the three flow cells was activated and then blocked without protein immobilization, and was used as a reference. All SPR interaction experiments, unless specified differently, were carried out in 5 mM Tris/HCl, pH 7.5, 150 mM KCl buffer.
Interactions between immobilized GCAP1 and NP were checked also under conditions where the particles have been previously incubated with GCAP1. Specifically, experiments were performed at a flow rate of 10 μl min−1, with sequential injections (420 s) of: (i) 0.3 mg ml−1 NP previously incubated for 5 min with 5 μM GCAP1; (ii) 0.3 mg ml−1 NP without prior protein incubation; (iii) 5 μM GCAP1 and (iv) 10 mM K-citrate during the dissociation phase. See Fig. 6 for an example of sensorgram obtained with this protocol.
NP titration experiments were performed at a flow rate of 10 μl min−1, with 10 min association time and 120 min dissociation time for NP injections, followed by 6 min injections (and 2 min equilibration/dissociation) of K-citrate to achieve complete NP-free conditions. NP concentration for each titration step was 0.1, 0.2, 0.3, 0.4 and 0.5 mg ml−1, corresponding to 8.3, 16.6, 24.9, 33.2 and 41.5 nM. Depending on the experimental setup, both running buffers and NP solutions were added with 1 mM MgCl2 or decalcified using a dialysis tube containing the ion chelating resin Chelex 100 (Bio-Rad).
Titration data for experiments without Mg2+ were fitted to a single exponential decay model:
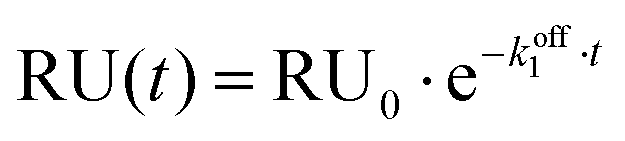
Fitting titration data for experiments in the presence of 1 mM Mg2+ to the same single exponential decay model was inaccurate, therefore a bi-exponential decay model was employed for this set of experiments:
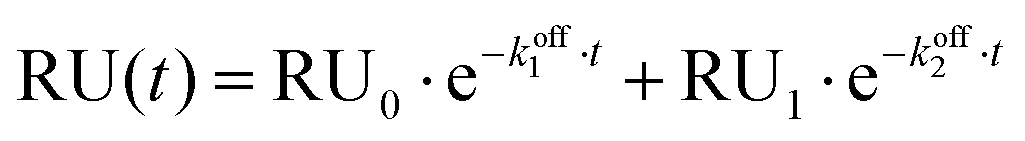
Isothermal titration calorimetry
Binding stoichiometry of GCAP1 interacting with NP was tested by isothermal titration calorimetry (ITC) as described earlier for other binding processes involving NCS proteins.18,29,30 Briefly, ITC was performed using a VP-ITC instrument from MicroCal (Northampton, MA, USA) at T = 25 °C. Purified GCAP1 or the mutant D100E was present in the recording cell in a decalcified buffer composed of 5 mM Tris-HCl pH 7.5 and 150 mM KCl. NP solution was placed in the microsyringe chamber at 4.25 μM (285.96 μl) and the titration was performed by subsequent injections of 10 μl NP solution into the recording cell keeping a time interval of 180 s between injections and an initial delay of 600 s after temperature equilibration. Control injections of NP into decalcified buffer showed constant swings from 0 to −0.01 μcal s−1. The data were fitted using a one ligand binding model employing the MicroCal software (Origin) to estimate a binding constant (KD).Guanylate cyclase expression and activation assays
Human WT photoreceptor guanylate cyclase (GC-E) was expressed in HEK293 cell membranes exactly as described before.31 Activation profile of GC was tested by two different approaches. Purified GCAP1 or D100E was added to washed HEK cell membranes at a fixed saturating concentration of 10 μM and GC activity was measured in the 20 nM–100 μM range using a Ca2+/EGTA buffer system allowing to determine the IC50 value.25–29 Secondly, we incubated GC in HEK cell membranes with increasing concentrations of WT or D100E GCAP1 (0–20 μM) under conditions, in which GCAPs were Mg2+-bound (Ca2+-free) (incubation time 5 min). Both activation series were run in the absence and presence of CaF2 NP. Western blotting was performed as described before31 using an anti-GC antibody (Santa Cruz, Heidelberg) at a dilution of 1:1000. A secondary antibody directed against IgG-POD anti rabbit was used at a dilution of 1:20000.
Results and discussion
Hydrodynamic properties of the GCAP1–NP complex
Dynamic Light Scattering (DLS) was used to monitor the hydrodynamic diameter of CaF2 NP in the absence and in the presence of GCAP1. Results are shown in Fig. 2.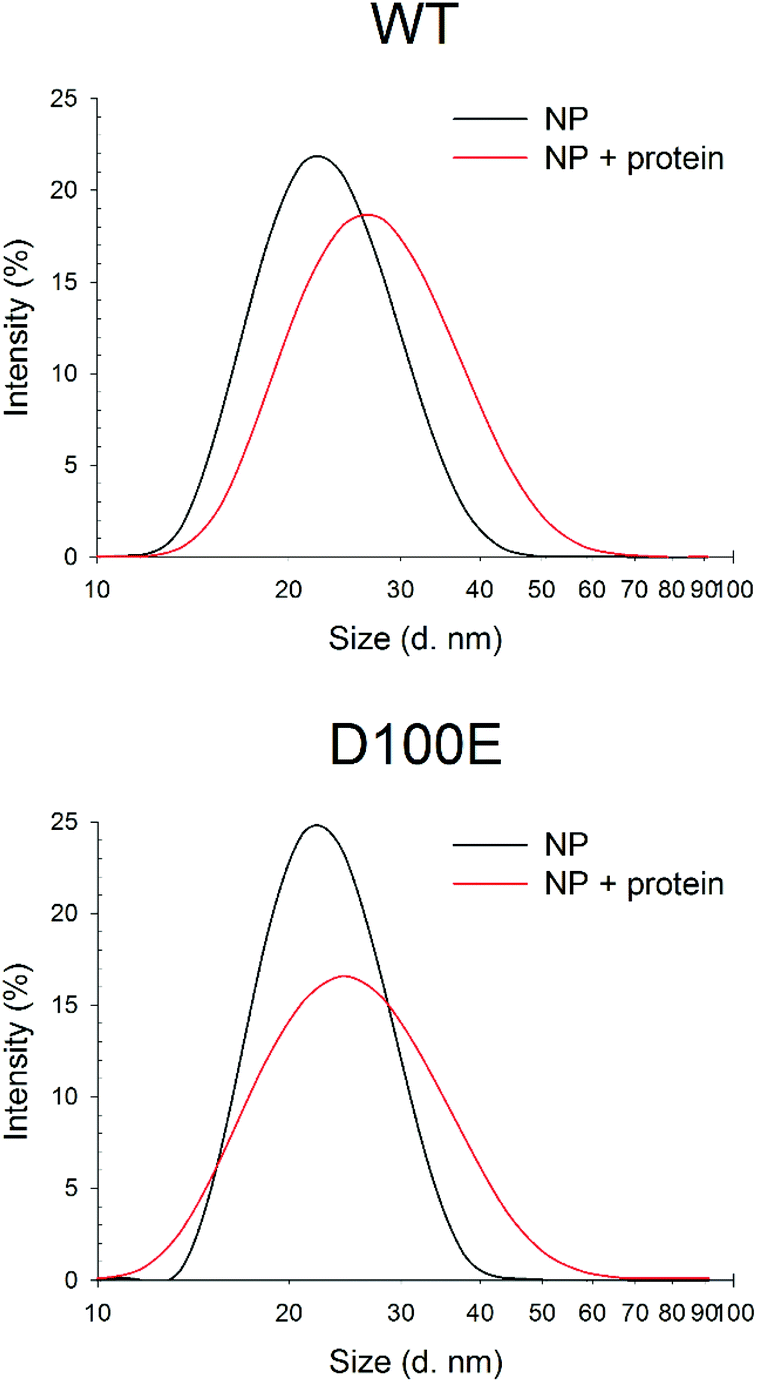 | ||
| Fig. 2 DLS measurements of CaF2 NP size upon interaction with WT and D100E GCAP1. Top: Hydrodynamic diameter of 1.1 mg ml−1 CaF2 NP (black, 30 repetitions, PdI = 0.170) and after incubation with 11.7 μM WT GCAP1 (red, 23 repetitions, PdI = 0.183). Bottom: Hydrodynamic diameter of 1.1 mg ml−1 CaF2 NP (black, 15 repetitions, PdI = 0.126) and after incubation with 11.7 μM D100E GCAP1 (red, 40 repetitions, PdI = 0.200). | ||
The NP dispersion appeared monodisperse and the measured hydrodynamic diameter was 23.1 ± 0.4 nm, with a low polydispersity index. Incubation with GCAP1 caused a 4.5 nm increase in the diameter, which was statistically significant according to a Student's t test (p < 0.001) and indicative of the binding of GCAP1 to the surface of CaF2 NP. Addition of 900 μM EGTA did not significantly change the diameter of the complex, suggesting that the chelator was unable to cause the dissociation of GCAP1 from the NP under these conditions (results not shown).
When the same experiment was performed with the D100E variant of GCAP1, in which Ca2+ binding to EF3 is prevented at physiological [Ca2+] (Fig. 1), the increase in the hydrodynamic complex, still statistically significant (p < 0.001), was approximately 3 nm (Fig. 2). The smaller increase of hydrodynamic size of the CaF2 NP–protein complex observed for D100E compared to WT GCAP1 is unlikely due to a different size or structure of the protein variants, which share very similar hydrodynamic properties32 and tertiary structures,25 but could be due to a different affinity for the NP surface, thus implying a smaller amount of mutant proteins bound to the surface at equilibrium.
Structure and thermal stability of GCAP1 in the presence of CaF2 NPs
The secondary structure of GCAP1 presented clear features of an α-helical folded protein, with typical minima at 208 and 222 nm, and it was shown to be substantially conserved in the presence of NPs (Fig. S1†). Table 1 shows the θ222/θ208 ratios used as descriptors of the spectral shape.Indeed, when incubated with decalcified CaF2 NP, the secondary structure acquired by WT GCAP1 was very similar to that of the Ca2+-saturated isolated protein (Fig. S1;†Table 1), with the same θ222/θ208 = 0.94 ratio. Sequential additions of saturating EGTA and Ca2+ showed that the protein was to some extent still able to respond to variations in Ca2+ concentration (θ222/θ208 0.93 and 0.94, respectively, Table 1), although the signal intensity slightly decreased, probably due to a destabilization of the colloidal dispersion upon further increase in free Ca2+.
Different results were obtained for the D100E mutant. Far UV CD spectra in the presence of decalcified NP show that the secondary structure was in this case different from that of the Ca2+-bound isolated protein (θ222/θ208 equal to 0.92 vs. 0.94, respectively; Table 1; Fig. S1†). While residual capability to respond to variations in [Ca2+] was observed after sequential additions of EGTA and free Ca2+ (θ222/θ208 equal to 0.94 and 0.96 respectively, Table 1), D100E seemed to acquire a slightly different conformation upon Ca2+-binding when bound to the NP (θ222/θ208 equal to 0.92 vs. 0.96, Table 1). This may suggest either a non-complete dissociation of D100E from the NP surface upon saturation with free [Ca2+], where at least an EF-hand remains occupied with the CaF2 NP lattice cations in spite of the high concentration of free Ca2+, or a complete dissociation from the NP surface, though with an altered secondary structure, at odds with the observed behavior of the WT GCAP1.
Far UV CD spectroscopy was also used to monitor alterations in protein thermal stability due to the interaction with CaF2 NP. A typical two-state transition thermal denaturation profile was observed for both protein variants in the presence of CaF2 NP and Ca2+/EGTA (Fig. 3). Interestingly, NP exerted a stabilizing effect on WT GCAP1 in the presence of EGTA (84.2 °C vs. 50.1 °C, Table 1), suggesting that the CaF2 NP crystal lattice exerts a stabilizing effect on the protein structure similar to that of isolated free Ca2+. However, the apparently strong interaction with NP eventually led to a rather abrupt transition toward a fully denatured protein, at odds with the isolated protein, which conserves the residual structure at 96 °C.24 The thermal stability of WT GCAP1 incubated with NP was instead slightly decreased in the presence of Ca2+ with respect to the isolated protein (Table 1), as shown by the thermal unfolding profiles (Fig. 3), where the transition appears to be completed in the 80–96 °C range. This could be partly due to the previously observed destabilization of the colloidal properties of the CaF2 NP dispersion at high [Ca2+]. As for the D100E GCAP1 variant, the presence of NP increased the measured protein thermal stability both in the absence (81.1 °C vs. 37.5 °C, Table 1) and in the presence (84.7 °C vs. 66.1 °C, Table 1) of free Ca2+. The measured effect of CaF2 NP on GCAP1 thermal stability points to a strong interaction, however in the case of the homolog Ca2+-sensor Rec, the same NP significantly destabilized both apo and Ca2+-bound protein forms and led to identical denaturation profiles independent of the presence of Ca2+ or EGTA.23 Altogether, these data suggest that the interaction with CaF2 NP is highly protein-specific.
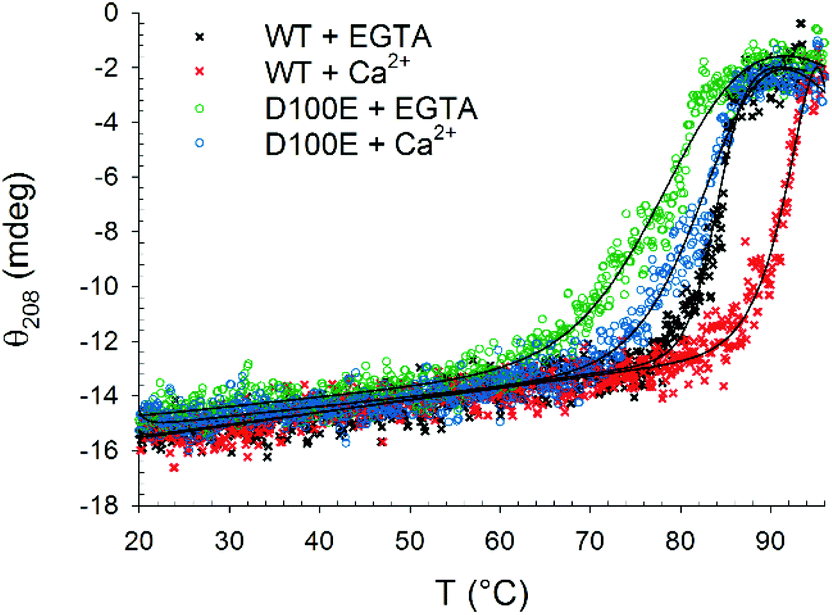 | ||
| Fig. 3 Thermal denaturation profiles of 10 μM WT and D100E GCAP1 after incubation with 1.5 mg ml−1 CaF2 NP. In detail: WT GCAP1 in the presence of 300 μM EGTA (black) or Ca2+ (red) and D100E GCAP1 in the presence of 300 μM EGTA (green) or Ca2+ (blue). The thermodynamic function (black lines) accounting for the unfolding free energy described in ref. 24 was used for data fitting. | ||
The slight but significant alterations observed in the far UV CD spectra of WT and D100E GCAP1 at 25 °C suggested a different NP interaction mode for the two protein variants. In order to further investigate the structural basis of GCAP1–NP interactions, near UV CD spectra were recorded (Fig. 4), which are sensitive to the micro-environment of aromatic residues and thus indirectly to the protein tertiary structure. The spectra show that the tertiary structure of WT GCAP1 in the presence of decalcified NP is very similar to that of the Ca2+-bound isolated protein (Fig. 4A, red curve and 4B, green curve). Additions of saturating EGTA and Ca2+ (Fig. 4B) led to the same spectroscopic changes observed for the isolated protein, thus suggesting that WT GCAP1 is still capable of acting as a Ca2+ sensor in the presence of NP. When physiological 1 mM Mg2+ was added to the NP–GCAP1 assembly, WT GCAP1 showed responsiveness (Fig. 4C, blue line vs. black line) although the spectral changes were more limited in magnitude and substantially different from those observed for the isolated protein in the presence of Mg2+,18 indicative of a slightly different tertiary structure. Interestingly, after Mg2+ addition, further saturation by Ca2+ led to a tertiary structure that differed substantially from that relative to the NP-bound state (red line, Fig. 4C). This may suggest that at least an EF-hand motif, which is not involved in the interaction with the NP, might still be occupied by Mg2+ under these conditions. Mg2+ sensing capability of WT GCAP1 in the presence of NP was also tested by intrinsic fluorescence spectroscopy (Fig. S2†). Titration experiments performed in the absence of Ca2+ showed that, within the rather narrow physiological range of [Mg2+] (500–2000 μM), WT GCAP1 was still able to respond to increasing [Mg2+] in a similar way compared to the free protein, in spite of the overall noticeable differences observed in the response pattern (red line vs. black line, Fig. S2†). Such differences are likely ascribable to the different conformations that GCAP1 acquires upon binding to CaF2 NP, which was shown to be more similar to the Ca2+-loaded form (Fig. 4).
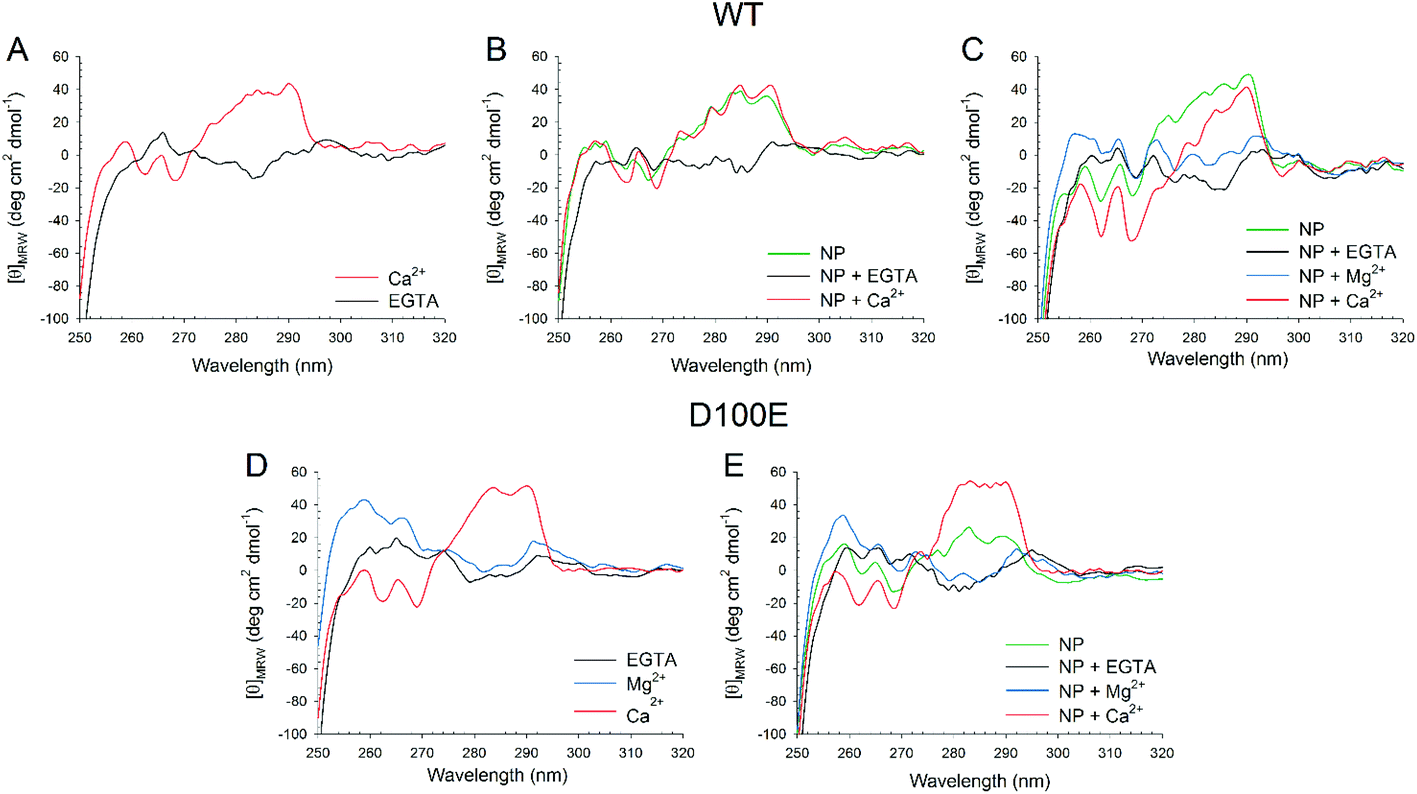 | ||
| Fig. 4 Near UV CD spectra of WT (top panels) and D100E (bottom panels) GCAP1 isolated and in complex with CaF2 NP. (A) Near UV CD spectra of 15.5 μM WT GCAP1 in the presence of 360 μM EGTA (black) and after addition of 720 μM Ca2+ (red). (B) Near UV CD spectra of 15.5 μM WT GCAP1 in the presence of 3.4 mg ml−1 NP (green) and after sequential additions of 720 μM EGTA (black) and 1.44 mM Ca2+ (red). (C) Near UV CD spectra of 15.5 μM WT GCAP1 in the presence of 3.4 mg ml−1 NP (green) and after sequential additions of 900 μM EGTA (black), 1 mM Mg2+ (blue) and 1.62 mM Ca2+ (red). (D) Near UV CD spectra of 25.4 μM D100E GCAP1 in the presence of 900 μM EGTA (black) and after sequential additions of 1 mM Mg2+ (blue) and 1.62 mM Ca2+ (red). (E) Near UV CD spectra of 21.6 μM D100E GCAP1 in the presence of 3.4 mg ml−1 NP (green) and after sequential additions of 900 μM EGTA (black), 1 mM Mg2+ (blue) and 1.62 mM Ca2+ (red). | ||
Near UV CD recorded for D100E GCAP1 highlighted a significant effect of Mg2+ on the protein tertiary structure (Fig. 4D, blue line), especially in the Phe band, which is characterized by the strongest dichroic signal so far observed for GCAP1 variants.18,28 Addition of saturating Ca2+ however restored the typical spectral shape observed for the Ca2+-bound form observed previously (Fig. 4D, red line).25 When D100E GCAP1 was incubated with decalcified NP the acquired tertiary structure significantly differed from that of the WT (compare Fig. 4E with 4C, green lines), in that the Tyr and Trp bands were significantly less intense, indicative of a different interaction mode. This could be due, at least partly, to the significantly lower affinity for Ca2+ exhibited by D100E, which might render the EF3 motif less prone to interact with the CaF2 lattice cations. NP-bound D100E GCAP1 was still able to act as a Mg2+ sensor (Fig. 4E, blue line), although to a lesser extent than the WT, while further saturation with Ca2+ resulted in CD spectra substantially overlapped with those of the isolated protein (Fig. 4E, red line), which might suggest either a complete dissociation from the NP surface, or that the EF-hands responsible for the spectral patterns observed in the near UV range are not the same that are involved in the interaction with the NP surface.
The slight structural differences observed by CD spectroscopy between WT and D100E GCAP1 upon interaction with CaF2 NP were confirmed by limited proteolysis experiments (Fig. 5), a useful method to assess protein flexibility and folding intermediates, since the presence or absence of a bound ligand, including ions or NP, affects the accessibility of the protein structure to proteases.33–36 Proteolysis patterns of both WT and D100E GCAP1 were very similar to one another for the apo (EGTA) and Mg2+-saturated forms (lanes 1 and 2). Significant differences were noticed upon addition of Ca2+ (lanes 3 and 4, reporting on Ca2+ and Ca2+ + Mg2+, respectively), highlighting a major effect of Ca2+ on the exposure of trypsin cleavage sites for both GCAP1 variants. The comparison between WT and D100E GCAP1 in their Ca2+ and Ca2+/Mg2+-bound forms in the absence of NP highlighted a higher number of bands between 10 and 15 kDa for the mutant, and a higher molecular weight for the smallest fragment (about 12 kDa, lanes 3 and 4). In the presence of NP though, in addition to the higher number of bands in the 10–15 kDa region, D100E showed also a splitting of the band around 17 kDa in at least 2 or three different bands, and several small bands below 10 kDa (lanes 8 and 9), in line with the small but significant differences observed by CD spectroscopy.
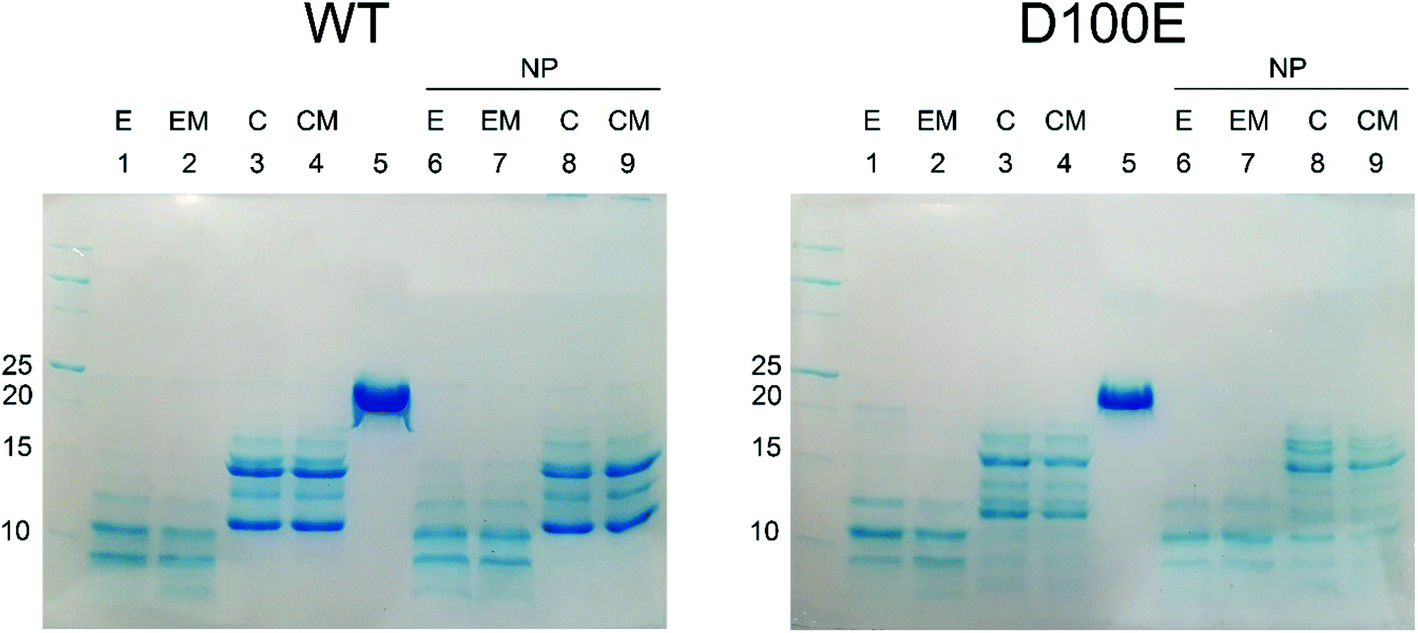 | ||
| Fig. 5 SDS-PAGE pattern following limited proteolysis of WT and D100E GCAP1 with trypsin in the presence/absence of CaF2 NP. Limited proteolysis was stopped after 20 min incubation at 25 °C with 0.015 mg ml−1 trypsin of 28.2 μM WT GCAP1 (left) and 28.2 μM D100E (right) in the absence and in the presence of 3.4 mg ml−1 CaF2 NP. Lanes in left and right panels represent the same experimental conditions: (1) 2 mM EGTA, (2) 1 mM Mg2+ and 2 mM EGTA, (3) 2 mM Ca2+, (4) 1 mM Mg2+ and 2 mM Ca2+, (5) no trypsin, (6) 3.4 mg ml−1 NP and 2 mM EGTA, (7) 3.4 mg ml−1 NP, 1 mM Mg2+ and 2 mM EGTA, (8) 3.4 mg ml−1 NP and 2 mM Ca2+, (9) 3.4 mg ml−1 NP, 1 mM Mg2+ and 2 mM Ca2+. | ||
In conclusion, limited proteolysis experiments provided independent evidence of the slightly different NP interaction mode of WT and D100E GCAP1, which corroborate the spectroscopic findings.
Stoichiometry and kinetics of GCAP1–NP interactions
In order to probe the kinetics of GCAP–NP interactions, we performed surface plasmon resonance (SPR) experiments by immobilizing both WT and D100E GCAP1 on the polysaccharide-coated surface of a gold sensor chip via amine coupling, and let CaF2 NP flow under different conditions and concentrations. An example of the obtained sensorgrams is reported in Fig. 6. Interestingly, when NP were incubated with GCAP1 for 5 min before injection no association was detected. However, if citrate-coated CaF2 NP without previous protein incubation were injected, a clear binding signal was observed, with a response of several hundred RU in amplitude, which however did not depend on the NP concentration (see Fig. 6A–C for an example of NP injections at various concentrations). The same phenomenon was observed for the D100E GCAP1 variant (results not shown).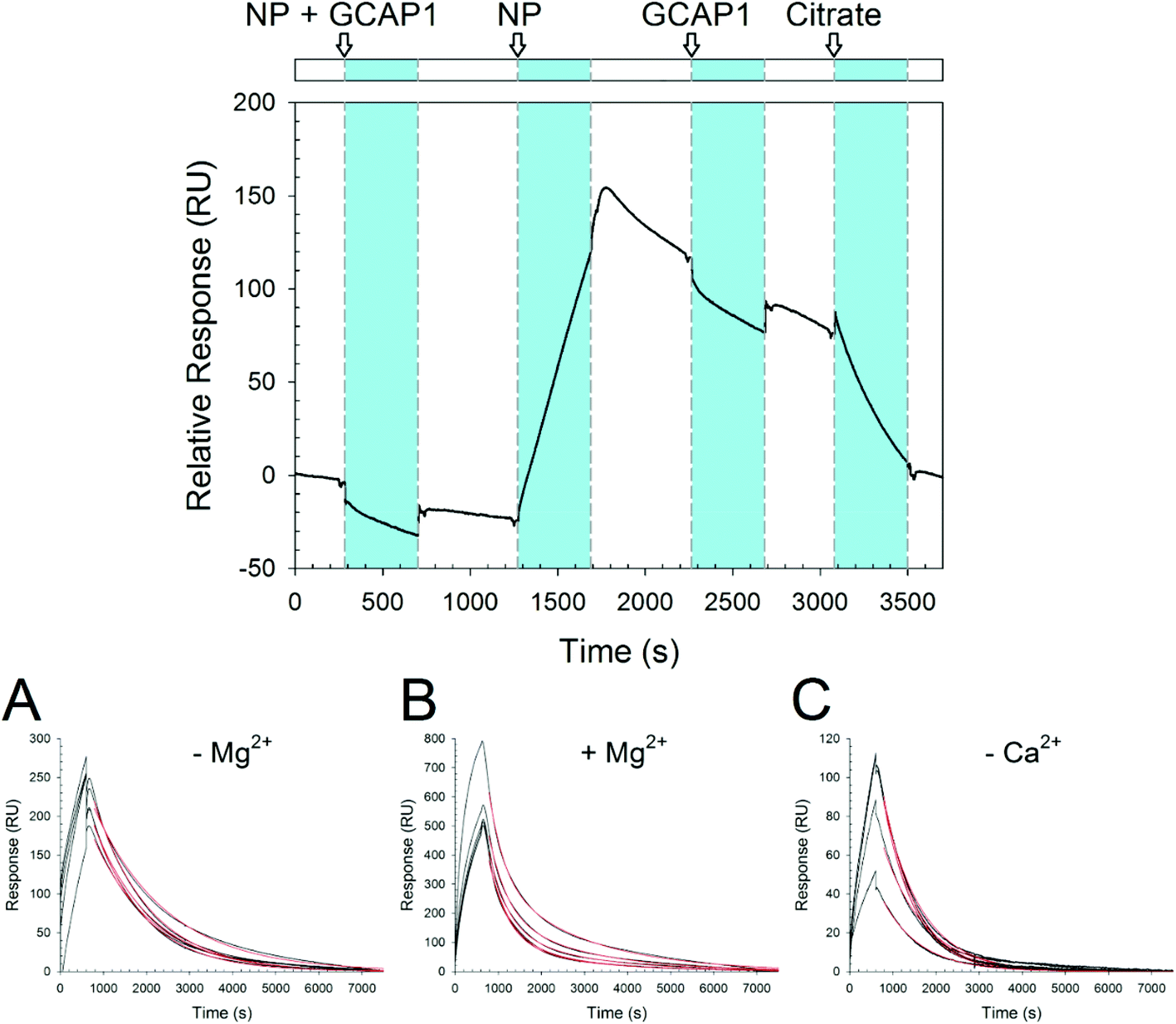 | ||
| Fig. 6 SPR experiments. Top: Sensorgram of sequential injections over immobilized WT GCAP1 of, respectively: 0.3 mg ml−1 CaF2 NP incubated with 5 μM WT GCAP1; 0.3 mg ml−1 NP; 5 μM WT GCAP1; 10 mM K-citrate. Bottom: Sensorgram of the injections of 0.1, 0.2, 0.3, 0.4, and 0.5 mg ml−1 NP over immobilized D100E GCAP1. NP were dissolved in (A) 5 mM Tris/HCl pH 7.5, 150 mM KCl, (B) 5 mM Tris/HCl pH 7.5, 150 mM KCl, 1 mM Mg2+, (C) decalcified 5 mM Tris/HCl pH 7.5, 150 mM KCl. | ||
This suggests that the binding of GCAP1 to CaF2 NP occurs only when the particle surface is available for protein binding, so that presumably the cations of the CaF2 lattice are not hindered by the presence of already bound proteins. When the injection of NP was stopped, a slow dissociation was triggered (Fig. 6), which was not significantly influenced by the subsequent injection of GCAP1, probably indicating that the NP surface available for binding was already occupied by the immobilized GCAP1 molecules, or alternatively that the injected proteins would scarcely interact with the NP due to molecular crowding on the chip surface. When 10 mM K-citrate was injected during the slow dissociation of GCAP1–NP complexes, a fast decrease in the SPR signal was observed, which resulted in the complete dissociation of the NP, as proven by the baseline returning to its initial reference. This suggests that citrate, which is used as a coating agent for CaF2 NP, competes with GCAP1 for the NP surface, hence triggering the dissociation of the complex. We point out that lower concentrations of citrate (1 mM) were not sufficient to completely dissociate the GCAP1–NP complex, and resulted in just a small decrease of the signal (data not shown).
The NP–GCAP1 interaction apparently did not follow a Langmuir adsorption model, in particular, no clear dependency of the SPR signal on NP concentration was detected, as clearly shown by the fairly similar amplitudes and shapes of the responses observed under all the tested conditions, when the particle concentration was varied in the 0.1–0.5 mg ml−1 range (Fig. 6A–C). However, it is worth noting that when the running buffer contained the physiological 1 mM Mg2+ responses were 2.5–3-fold higher in amplitude compared to the case without Mg2+ (Fig. 6Avs.6B). Although the association phase could not be possibly fit to any simple kinetic model, the dissociation phase appeared to be more regular. The lack of a plateau and the minor differences in the shape of the sensorgrams in all the association curves, even in cases where NP concentration was relatively high could be interpreted as an overall contribution of the binding and concomitant conformational changes of the NP–GCAP1 complex to the SPR signal. It is established that protein conformational changes triggered by the binding of small analytes such as Ca2+ significantly affect the SPR signal, especially under crowding conditions,37–39 and this effect could be particularly important in the case considered here due to the relatively high number of interactions involved. Moreover, CaF2 NP have a different refractive index compared to the protein/water interface, and in order for the interaction with GCAP1 to occur, they shall approach and detach from the three dimensional polysaccharide matrix on the sensor chip. All in all, the combination of protein conformational change and NP diffusion may explain why, when the injection of NP was interrupted, the SPR signal continued to increase in all cases (see Fig. 6 as an example) and reached a maximum in approximately two hundred seconds, after which the real dissociation process started.
The dissociation kinetics was found to depend on the buffer conditions, especially on the presence of free Ca2+ and Mg2+ (Fig. 6 and Table 2).
| Buffer | # Rep.a |
k
off1 × 10−4b (s−1) |
k
off2 × 10−4b (s−1) |
|---|---|---|---|
| a Number of experimental curves used to estimate koff values. b Estimated koff values obtained by fitting SPR data to a single or, if necessary, bi-exponential function. Data are presented as average ± standard deviation. | |||
| WT | |||
| Tris | 11 | 7.3 ± 0.7 | — |
| Tris + 1 mM Mg2+ | 5 | 23.1 ± 0.8 | 4.4 ± 0.3 |
| Decalcified Tris | 5 | 7.8 ± 0.8 | — |
| D100E | |||
| Tris | 6 | 7.6 ± 1.0 | — |
| Tris + 1 mM Mg2+ | 5 | 24 ± 3 | 5.2 ± 0.4 |
| Decalcified Tris | 5 | 12.0 ± 1.4 | — |
When the standard Tris buffer, without added Mg2+, was used as a running buffer the dissociation data of both GCAP1 variants could be nicely fit to a single exponential, leading to koff = 7.3 × 10−4 s−1 for the WT and koff = 7.6 × 10−4 s−1 for D100E (Table 2 and Fig. 6A). A bi-exponential function was used instead for fitting the dissociation data in the presence of 1 mM Mg2+, with satisfactory results (Table 2 and Fig. 6B). Under these conditions, the NP–protein dissociation was overall faster, without significant differences between GCAP1 variants (koff1 = 23 × 10−4 s−1 for the WT and koff1 = 24 × 10−4 s−1 for D100E, koff2 = 4.4 × 10−4 s−1 for the WT and koff2 = 5.2 × 10−4 s−1 for D100E). It is not clear why dissociation data required a fitting to a bi-exponential function. CD data indicated that the Mg2+-bound conformation of both GCAP1 variants might be slightly different in the presence CaF2 NP compared to the free protein (Fig. 3), suggesting that specific EF-hand(s) respond(s) to free Mg2+ depending on whether they are or not occupied by the Ca2+ cations of the CaF2 lattice. A speculative interpretation of the dissociation kinetics data is that there exist two different populations of GCAP1 molecules on the NP surface: some proteins have bound Mg2+, presumably via EF2 (Fig. 1) and form a less tight interaction with the NP surface that leads to faster dissociation; some other proteins may form stronger interactions with the NP surface (probably via EF2, which is not occupied by Mg2+, and EF3 and/or EF4), thus resulting in slower dissociation.
Interestingly, in the presence of decalcified Tris running buffer the dissociation kinetics followed a single exponential trend for both GCAP1 variants, although D100E GCAP1 dissociated ∼1.5 faster compared to the WT (koff1 = 12 × 10−4 s−1vs. and koff1 = 7.8 × 10−4 s−1, respectively; see Table 2 and Fig. 6C).
Overall, the dissociation kinetics assessed by surface plasmon resonance is compatible with a physiologically relevant release of GCAP1 from the surface of CaF2 NP, as under all tested conditions, in approximately two hours all the proteins coating the NP surface were completely dissociated (Fig. 6).
The interaction between GCAP1 and CaF2 NP was also investigated by isothermal titration calorimetry (ITC). Binding of NP to WT and D100E GCAP1 was in both cases exothermic (Fig. 7) and occurred with a different stoichiometry. In particular, WT GCAP1 bound to CaF2 NP with a 16:1 protein:NP stoichiometric ratio, while a higher number of D100E GCAP1 molecules could bind the NP surface (54:1 protein:NP ratio). The different stoichiometry might reflect different interaction modes of the two GCAP1 variants with the same NP, which appeared to be highly specific as shown by SPR data (Table 2). In spite of the higher stoichiometry, the affinity of D100E GCAP1 for the NP surface was approximately halved compared to that of the WT (KA = 4.2 × 107 M−1, KD = 12 nM for WT GCAP1 vs. KA = 4.3 × 107 M−1, KD = 23 nM for D100E GCAP1).
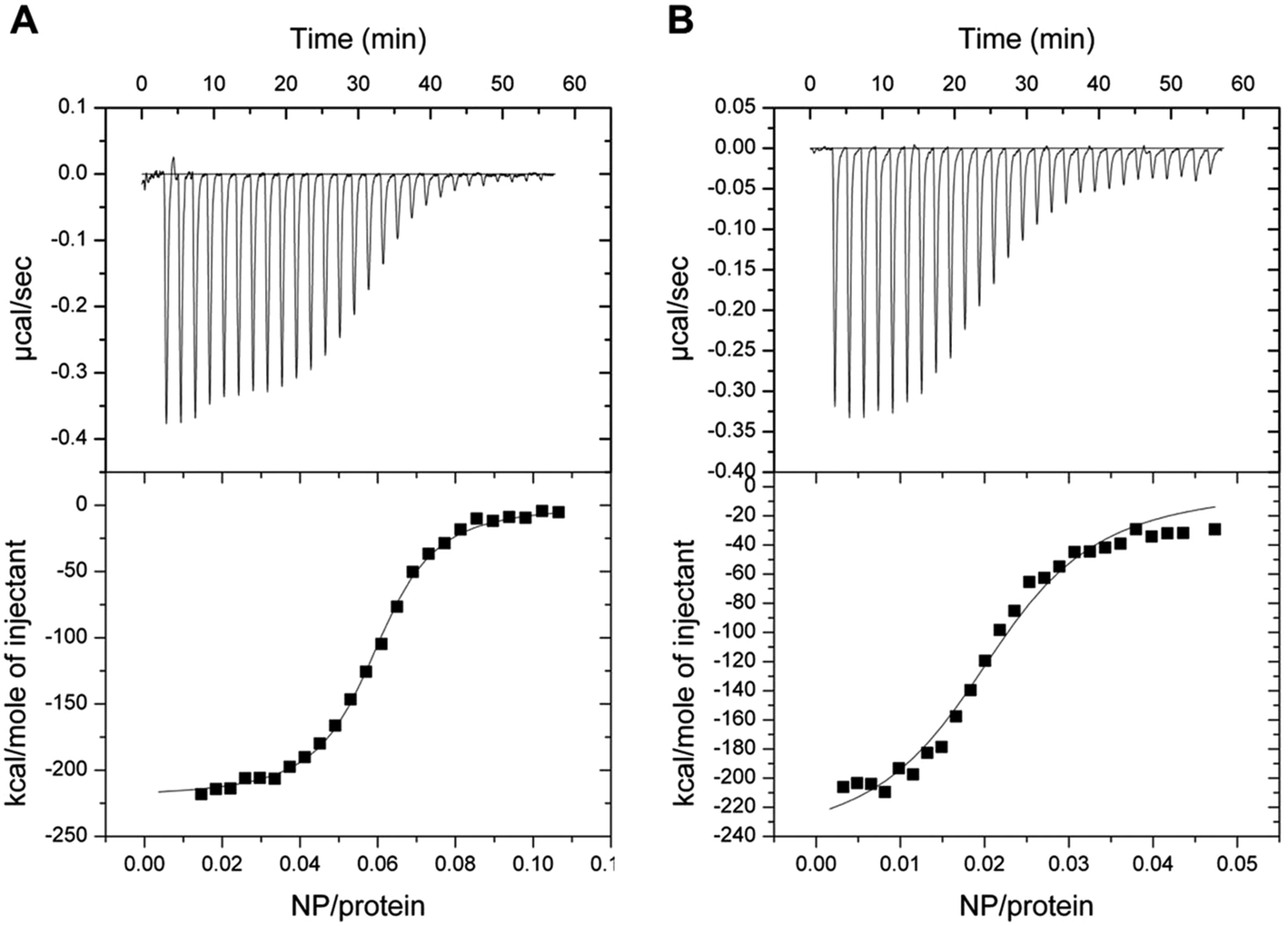 | ||
| Fig. 7 Isothermal titration calorimetry of decalcified NP in 5 mM Tris/HCl, pH 7.5, 150 mM KCl titrated into WT and D100E GCAP1. Upper panels refer to raw data, lower panels refer to the integrated peak areas. Data were fitted to a one-binding site model, both reactions were exothermic. (A) ITC of WT GCAP1 titrated with NP. (B) ITC of D100E GCAP1 titrated with NP. | ||
Overall, ITC data combined with SPR analysis support a model, in which a higher number of D100E GCAP1 molecules may bind to the CaF2 NP surface though with lower affinity, presumably due to the significantly compromised ability of EF3 to bind Ca2+ in the case of the D100E mutation.24,25 The dissociation process, at very low free [Ca2+], is significantly faster for the pathogenic D100E mutant compared to the WT.
Regulation of the target guanylate cyclase activity by GCAP1 in the presence of CaF2 NPs
The residual capability of GCAP1 to regulate the enzymatic activity of the target GC was explicitly tested in GC assays performed in the presence of CaF2 NP. Results are reported in Fig. 8 and in Table 3.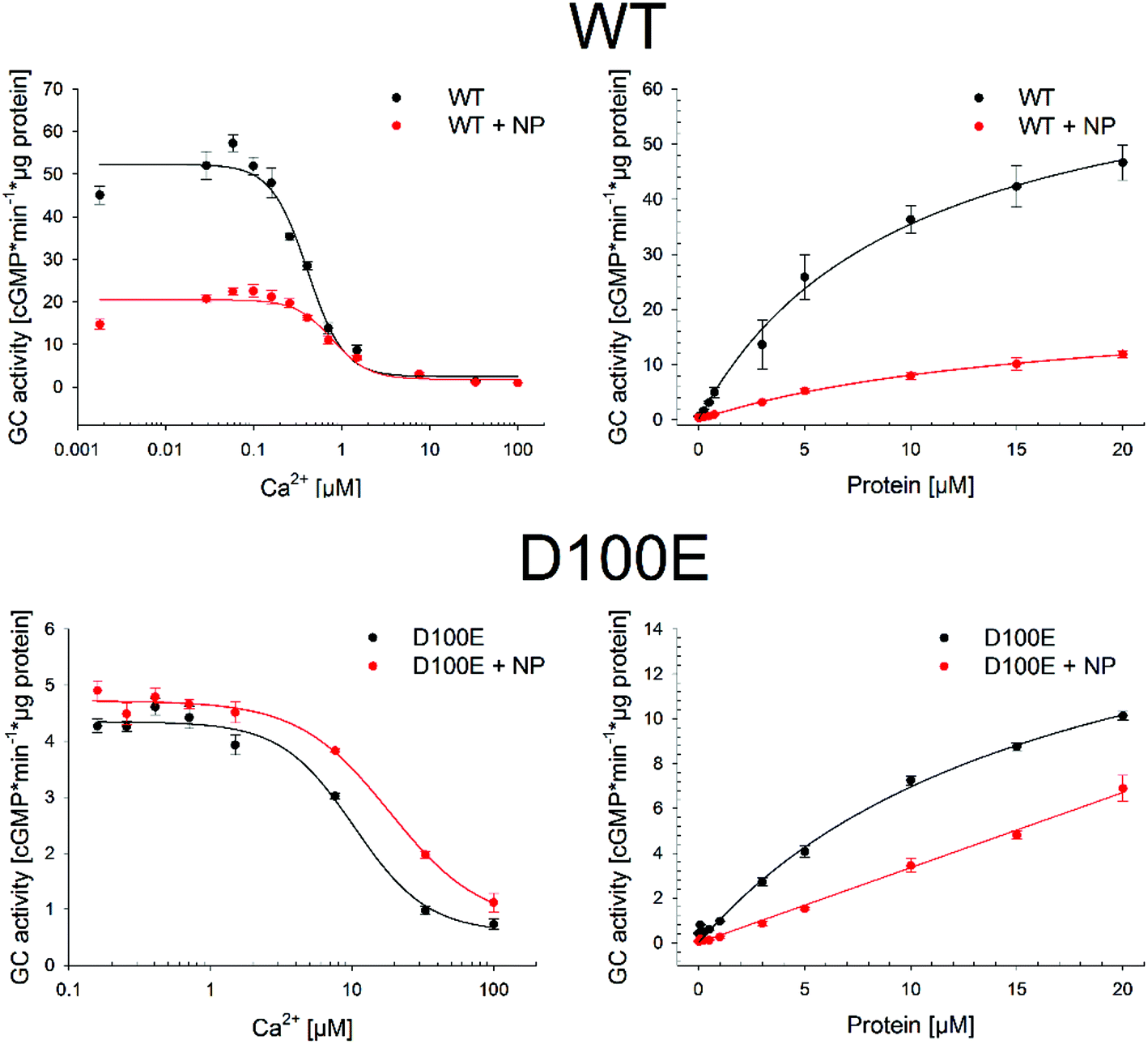 | ||
| Fig. 8 Photoreceptor guanylate cyclase GC-E (GC) regulation by WT (top panels) and D100E (bottom panels) GCAP1 in the absence and in the presence of 0.07 μM CaF2 NP. The GC activation was measured by the produced cGMP concentration within 5 min in the presence of WT or D100E GCAP1 by adding the indicated amounts of Ca2+ (left) or GCAP1 variant (right). | ||
| IC50a (μM) |
EC50b (μM) |
x-Foldc | ||||
|---|---|---|---|---|---|---|
| −NP | +NP | −NP | +NP | −NP | +NP | |
| a IC50 is the Ca2+ concentration at which the GC activity is half maximal; standard errors estimated from fitting. b EC50 is the concentration of GCAP1 at which the GC activity is half maximal; standard errors estimated from fitting. c x-Fold activation is expressed as GCmax − GCmin divided by GCmin. | ||||||
| WT | 0.37 ± 0.03 | 0.70 ± 0.07 | 9.8 ± 1.2 | 16.6 ± 1.9 | 61 | 22.9 |
| D100E | 10.4 ± 2.15 | 18.5 ± 4.1 | 17 ± 3 | N.D. | 5.3 | 3.41 |
We tested for three functional key parameters of GC activity under control of GCAP1. The IC50 value indicates the free [Ca2+], at which the GC activation by GCAP1 is half-maximal and the EC50 value is the GCAP1 concentration, at which the GC activity is half-maximal at low free [Ca2+]. The x-fold activation is a parameter reflecting the amplitude of GC activation when switching the experimental conditions from high to low free [Ca2+]. The presence of NP influenced all activation parameters for WT and for the D100E mutant, the IC50 value shifted 1.9 and 1.8 fold, respectively (Table 3 and Fig. 8) corresponding already to non-physiological high [Ca2+].24,26,27 The apparent affinity of GCAP1 interacting with the GC (expressed as EC50) shifted 1.7-fold in the presence of NP, but to an even larger degree in the case of the D100E mutant, since we observed a non-hyperbolic saturation curve over the range of tested protein concentrations (Fig. 8, red curve in lower right panel). Finally, we observed a significant reduction in x-fold activation (Table 3), which is particularly visible in Fig. 8 for WT GCAP1 (upper left panel) and to a lesser extent for D100E in Fig. 8 (lower left panel).
Altogether, these assays suggest that the presence of NP attenuates but does not prevent the regulation of the target GC by GCAP1. Indeed, the activation profile of GC largely maintained the typical regulatory features observed both in the WT and D100E case. A particular characteristic feature of GC regulation is preserved in the presence of NP, namely the apparent shift of the IC50 value towards higher free [Ca2+] observed for the D100E mutant and which might trigger the deregulation of second messenger homeostasis, eventually causing cone dystrophies.
The results obtained when monitoring the activity/regulation of GC by GCAP1 variants in the presence of CaF2 NP raised the question, whether the observed alterations could be due to the reduced availability of GCAP1, GC or both molecules as a result of the concomitant interaction with the NP. To test this hypothesis, HEK cells transfected with GC were treated with CaF2 NP for 0, 1, 2 and 4 hours, lysed and analyzed on an SDS-PAGE and western blot (Fig. S3†). The Coomassie blue stained protein band pattern after SDS-PAGE was substantially identical, indicating no protein degradation in the presence of NP (Fig. S3,† left panel). Probing the samples with an anti-GC-E antibody (western blot in Fig. S3,† right panel) revealed that the GC content (band around 100 kDa, which is framed by a red box) decreased with increasing time when the cells were treated with NP. This could be interpreted as the NP triggering GC degradation, however this hypothesis seems unlikely, since the degradation bands below the 100 kDa GC-band appeared also faint with increased incubation time. An alternative, and probably more realistic explanation is that the GC is partly interacting with the CaF2 NP, and therefore it did not run into the separating gel. The hypothesis is supported by the accumulating protein bands observed in the top slots of the SDS PAGE gel. Slots contained material that was not present in the absence of NPs. A similar phenomenon was already observed with Rec23 and calmodulin22 experiments performed with the same type of NP. We point out that, under the experimental conditions tested in GC-assays, multiple equilibria between NP, GC and GCAP1 may occur, resulting in a non-complete dissociation of GCAP1 from the NP surface, eventually leading to a decreased maximal activation of GC. It is worth noting that in these experiments, neither endogenous nor added GCAP1 was present, therefore the observed effect, i.e. a mild but significant interaction between GC and CaF2 NP, could be reduced in a cellular context in the presence of GCAP1, which would compete for the NP surface.
Further control experiments were performed to ensure a normal expression of GC in the transfected cells, in the presence of CaF2 NP. The GC vector contained also a GFP, which was co-expressed and similar in all cases, thus indicating normal expression levels (results not shown).
Conclusions
Our results show that GCAP1 interacts with biocompatible CaF2 NP by forming a reversible protein–particle complex, which dissociates over physiologically compatible time frames, does not alter the protein structure and retains the GCAP1 capability to regulate the target GC. The binding was shown to depend on specific interactions, and even a single point mutation in the protein, such as in the case of D100E GCAP1, may significantly affect the binding mode as well as the affinity for the particle.Nanodevices in general offer the unique feature of having a large surface-to-volume ratio. The possibility to safely carry a functional protein on the surface of the device and release it at appropriate cell locations would constitute a promising premise for the potential treatment of diseases related to mutated proteins. Indeed, recombinant functional proteins carried by the device may be released in high amounts and compensate for the lack of native proteins and even outcompete the dysfunctional ones. The retinal delivery of functional recombinant proteins by nanovesicles has been shown to be able to modify the phototransduction cascade in mouse photoreceptors in a quantitatively controlled manner.40 Although our study mainly focused on the physicochemical details of the interaction and on assessing the functional consequence of protein–NP interaction, our results constitute a solid basis for future studies that specifically address the potential applicability of CaF2 NP as surface carriers of functional GCAP1 in photoreceptor cells, therefore opening new scenarios for the treatment of cone dystrophies, which have been largely associated with mutations in the gene encoding GCAP1. Optimization of physicochemical parameters such as NP size, coating and functionalization for selective targeting would all be necessary steps in this respect.
Acknowledgements
The financial support of Fondazione Telethon – Italy (Grant no. GGP16010 to DDO) is gratefully acknowledged. This work was also supported by a grant from the Deutsche Forschungsgemeinschaft DFG (KO 948/10-2) to KWK.References
- P. J. Carter, Exp. Cell Res., 2011, 317, 1261–1269 CrossRef CAS PubMed.
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- D. A. Christian and C. A. Hunter, Immunotherapy, 2012, 4, 425–441 CrossRef CAS PubMed.
- G. V. Deodhar, M. L. Adams and B. G. Trewyn, Biotechnol. J., 2017, 12(1) DOI:10.1002/biot.201600408.
- N. N. Dong, M. Pedroni, F. Piccinelli, G. Conti, A. Sbarbati, J. E. Ramirez-Hernandez, L. M. Maestro, M. C. Iglesias-de la Cruz, F. Sanz-Rodriguez, A. Juarranz, F. Chen, F. Vetrone, J. A. Capobianco, J. G. Sole, M. Bettinelli, D. Jaque and A. Speghini, ACS Nano, 2011, 5, 8665–8671 CrossRef CAS PubMed.
- C. Portioli, M. Pedroni, D. Benati, M. Donini, R. Bonafede, R. Mariotti, L. Perbellini, M. Cerpelloni, S. Dusi, A. Speghini and M. Bentivoglio, Nanomedicine, 2016, 11, 3039–3051 CrossRef CAS PubMed.
- X. Deng, Y. Dai, J. Liu, Y. Zhou, P. Ma, Z. Cheng, Y. Chen, K. Deng, X. Li, Z. Hou, C. Li and J. Lin, Biomaterials, 2015, 50, 154–163 CrossRef CAS PubMed.
- Y. Li, Y. Zhou, X. Li, J. Sun, Z. Ren, W. Wen, X. Yang and G. Han, RSC Adv., 2016, 6, 38365–38370 RSC.
- A. A. Ansari, A. K. Parchur, B. Kumar and S. B. Rai, J. Mater. Sci. Mater. Med., 2016, 27, 178 CrossRef PubMed.
- Z. Li, Y. Zhang, L. Huang, Y. Yang, Y. Zhao, G. El-Banna and G. Han, Theranostics, 2016, 6, 2380–2393 CrossRef CAS PubMed.
- W. Yin, G. Tian, W. Ren, L. Yan, S. Jin, Z. Gu, L. Zhou, J. Li and Y. Zhao, Dalton Trans., 2014, 43, 3861–3870 RSC.
- M. J. Berridge, M. D. Bootman and H. L. Roderick, Nat. Rev. Mol. Cell Biol., 2003, 4, 517–529 CrossRef CAS PubMed.
- K. Nakatani, C. Chen, K. W. Yau and Y. Koutalos, Adv. Exp. Med. Biol., 2002, 514, 1–20 CrossRef CAS PubMed.
- E. N. Pugh Jr. and T. D. Lamb, Phototransduction in Vertebrate Rods and Cones: Molecular Mechanisms of Amplification, Recovery and Light Adaptation, Elsevier Science B.V., 2000 Search PubMed.
- E. A. Permyakov and R. H. Kretsinger, Calcium binding proteins, John Wiley & Sons, Hobok, New Jersey, 2011 Search PubMed.
- A. M. Dizhoor, E. V. Olshevskaya and I. V. Peshenko, Mol. Cell. Biochem., 2010, 334, 117–124 CrossRef CAS PubMed.
- S. Lim, I. Peshenko, A. Dizhoor and J. B. Ames, Biochemistry, 2009, 48, 850–862 CrossRef CAS PubMed.
- V. Marino, S. Sulmann, K. W. Koch and D. Dell'Orco, Biochim. Biophys. Acta, 2015, 1853, 2055–2065 CrossRef CAS PubMed.
- I. V. Peshenko and A. M. Dizhoor, J. Biol. Chem., 2004, 279, 16903–16906 CrossRef CAS PubMed.
- I. V. Peshenko and A. M. Dizhoor, J. Biol. Chem., 2006, 281, 23830–23841 CrossRef CAS PubMed.
- K. W. Koch and D. Dell'Orco, ACS Chem. Neurosci., 2013, 4, 909–917 CrossRef CAS PubMed.
- A. Astegno, E. Maresi, V. Marino, P. Dominici, M. Pedroni, F. Piccinelli and D. Dell'Orco, Nanoscale, 2014, 6, 15037–15047 RSC.
- V. Marino, A. Astegno, M. Pedroni, F. Piccinelli and D. Dell'Orco, Nanoscale, 2014, 6, 412–423 RSC.
- D. Dell'Orco, P. Behnen, S. Linse and K. W. Koch, Cell. Mol. Life Sci., 2010, 67, 973–984 CrossRef PubMed.
- D. Dell'Orco, S. Sulmann, P. Zagel, V. Marino and K. W. Koch, Cell. Mol. Life Sci., 2014, 71, 3829–3840 CrossRef PubMed.
- P. Behnen, D. Dell'Orco and K. W. Koch, Biol. Chem., 2010, 391, 631–637 CrossRef CAS PubMed.
- V. B. Kitiratschky, P. Behnen, U. Kellner, J. R. Heckenlively, E. Zrenner, H. Jagle, S. Kohl, B. Wissinger and K. W. Koch, Hum. Mutat., 2009, 30, E782–E796 CrossRef PubMed.
- V. Marino, A. Scholten, K. W. Koch and D. Dell'Orco, Hum. Mol. Genet., 2015, 24, 6653–6666 CrossRef CAS PubMed.
- S. Sulmann, A. Kussrow, D. J. Bornhop and K. W. Koch, Sci. Rep., 2017, 7, 45515 CrossRef PubMed.
- S. Sulmann, F. Vocke, A. Scholten and K. W. Koch, Sci. Rep., 2015, 5, 11228 CrossRef CAS PubMed.
- P. Zägel, D. Dell'Orco and K. W. Koch, Biochemistry, 2013, 52, 5065–5074 CrossRef PubMed.
- S. Sulmann, D. Dell'Orco, V. Marino, P. Behnen and K. W. Koch, Chemistry, 2014, 20, 6756–6762 CrossRef CAS PubMed.
- R. Cukalevski, M. Lundqvist, C. Oslakovic, B. Dahlback, S. Linse and T. Cedervall, Langmuir, 2011, 27, 14360–14369 CrossRef CAS PubMed.
- A. Fontana, P. P. de Laureto, B. Spolaore, E. Frare, P. Picotti and M. Zambonin, Acta Biochim. Pol., 2004, 51, 299–321 CAS.
- C. Lange and K. W. Koch, Biochemistry, 1997, 36, 12019–12026 CrossRef CAS PubMed.
- S. Pedigo and M. A. Shea, Biochemistry, 1995, 34, 1179–1196 CrossRef CAS PubMed.
- D. Dell'Orco and K. W. Koch, ACS Chem. Biol., 2016, 11, 2390–2397 CrossRef PubMed.
- D. Dell'Orco, M. Muller and K. W. Koch, Chem. Commun., 2010, 46, 7316–7318 RSC.
- D. Dell'Orco, S. Sulmann, S. Linse and K. W. Koch, Anal. Chem., 2012, 84, 2982–2989 CrossRef PubMed.
- S. Asteriti, G. Dal Cortivo, V. Pontelli, L. Cangiano, M. Buffelli and D. Dell'Orco, Biochem. Biophys. Res. Commun., 2015, 461, 665–670 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supporting Fig. S1–S3. See DOI: 10.1039/c7nr03288a |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2017 |