
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visualization of the protein corona: towards a biomolecular understanding of nanoparticle-cell-interactions†
Maria
Kokkinopoulou‡
a,
Johanna
Simon‡
a,
Katharina
Landfester
a,
Volker
Mailänder
*b and
Ingo
Lieberwirth
 *a
*a
aMax Planck-Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: lieberw@mpip-mainz.mpg.de
bClinic of Dermatology, University Medicine Mainz, Langenbeckstraße 1, 55131 Mainz, Germany. E-mail: volker.mailaender@unimedizin-mainz.de
First published on 13th June 2017
Abstract
The use of nanocarriers in biology and medicine is complicated by the current need to understand how nanoparticles interact in complex biological surroundings. When nanocarriers come into contact with serum, proteins immediately adsorb onto their surface, forming a protein corona which defines their biological identity. Although the composition of the protein corona has been widely determined by proteomics, its morphology still remains unclear. In this study we show for the first time the morphology of the protein corona using transmission electron microscopy. We are able to demonstrate that the protein corona is not, as commonly supposed, a dense, layered shell coating the nanoparticle, but an undefined, loose network of proteins. Additionally, we are now able to visualize and discriminate between the soft and hard corona using centrifugation-based separation techniques together with proteomic characterization. The protein composition of the ∼15 nm hard corona strongly depends on the surface chemistry of the respective nanomaterial, thus further affecting cellular uptake and intracellular trafficking. Large diameter protein corona resulting from pre-incubation with soft corona or Apo-A1 inhibits cellular uptake, confirming the stealth-effect mechanism. In summary, the knowledge on protein corona formation, composition and morphology is essential to design therapeutic effective nanoparticle systems.
Introduction
On account of their small size, nanocarriers have distinct properties that make them excellent candidates for biomedical and biotechnological applications. Although their use is growing rapidly, crucial questions still arise about the interaction of nanocarriers with biological systems. When nanocarriers come into contact with biological fluids they adsorb proteins due to their high surface free energy.1,2 The proteins that are adsorbed on the surface of nanocarriers form the so-called ‘protein corona’. The protein corona thus formed alters the size, aggregation state and properties of the nanoparticles and provides them with a biological identity, which differs from their synthetic identity.3,4 The corona forms rapidly and the composition changes only quantitatively.5,6 It divides into the ‘hard’ and ‘soft’ corona, depending on the binding strength and exchange rate of the proteins. The hard corona is formed by the proteins with high binding affinities that are tightly bound and the soft corona by those proteins that are loosely bound and have high exchange rates. What the cell is finally able to recognize is the particle–protein complex.7 This means that the individual proteins present in each case are responsible for regulating the cellular uptake and the intracellular fate.8,9The proteomic composition,10 size and aggregation11 effects of the protein corona are well-known, but its morphology has still to be examined. The protein corona is usually shown either as a uniform layer or as multiple layers covering the nanoparticle. For the first time we have been able to visualize the morphology of the protein corona and provide a 3D model of its structure by using transmission electron microscopy. In contrast to existing theories and sketches, we found that the protein corona forms a loose network which is attached to the nanoparticle.
In this paper we focus on three different polystyrene nanoparticles (plain, carboxyl-functionalized and amino-functionalized). These particles are easily synthesized in a wide range of sizes/surface functionalization and are ideal candidates for studying bio–nano interactions.12 The protein corona morphology and composition of those nanoparticles were compared using TEM, DLS and LC-MS. Further we carefully monitored the process from initial corona formation directly after incubation in human serum and after each washing/centrifugation steps. Using a 3D model reconstruction, we were able to quantify the amount of adsorbed protein covering the nanoparticles. Finally, endocytosis and intracellular trafficking of the nanoparticle coated with or without protein corona was investigated after incubation with macrophages. These studies offer a better understanding of the biological identity of the nanoparticles and will therefore contribute to a safer and more effective application in nanomedicine.
Results and discussion
The research presented here was carried out on a defined set of polystyrene nanoparticles (PS-NP), synthesized by free-radical mini-emulsion polymerization13 and stabilized by the surfactant Lutensol AT-50 which has a polyethylene glycol (PEG) tail of 50 ethylene oxide units. All nanoparticles were purified under similar conditions. A detailed protocol is presented in the ESI.† In contrast to other studies where particles with different properties were used such as material, size or charge14–16 we focused on a set of PS-NPs with similar size (ESI Table 1†) and only varying surface modifications (PS, PS-COOH, PS-NH2). For flow cytometry and confocal laser scanning microscopy analysis, nanoparticles were fluorescently labeled using BODIPY (525/535 nm). Similar amounts of dye were covalently in-cooperated within the nanoparticles (ESI Table 1†). Surface functionalities were introduced by co-polymerization of monomers containing carboxy- and amino-groups (see ESI Methods†). The physico-chemical properties of nanoparticles such as charge, shape and size were characterized by ζ-potential measurements, transmission electron microscopy (TEM) and mulit-angle dynamic light scattering (DLS) in an aqueous solution and physiological buffer conditions (PBS) (see ESI Table 1†).Complementary analytical methods were applied to visualize the structure (TEM) of the protein corona and determine changes in size. The methods included multi-angle DLS as well as characterizing its composition by label-free, ultra-pressure liquid chromatography mass spectrometry (UPLC-MS).
The morphology of the protein corona was studied by TEM using a negative staining technique.17 Proteins were embedded in a thin, free standing layer of dried trehalose containing heavy metal salts (e.g. uranyl acetate) providing high contrast samples, suitable for conventional electron tomography.18,19 In order to assure that the corona morphology is not affected by the embedding into the trehalose film, additional cryo-TEM examinations have been performed. This showed no noticeable structural difference between cryo-TEM and trehalose embedding preparation (Fig. S1†). The protein corona of nanoparticles could be visualized for the first time using this technique and subsequently quantitatively analyzed using a 3-D reconstruction model.
Directly after incubation of nanoparticles in human serum (Fig. 1A), un-functionalized polystyrene nanoparticles (PS) were surrounded by a protein cloud, which appeared to be larger than the diameter of the nanoparticle. Given the average diameter of 140 nm for un-functionalized polystyrene nanoparticles (ESI Tables 1/2†) the average additional corona was estimated to be ∼70–100 nm thick (Fig. 1A) and referred to as the soft protein corona. Interestingly, the soft protein corona is not shown by TEM to be as uniformly distributed and well-rounded, but rather as an undefined network surrounding the nanoparticle. Generally2,20 it is described as highly dynamic layer of proteins which have high exchange rates and low binding affinities towards the nanoparticle. By adding trehalose and uranyl acetate (UA) for TEM sample preparation, the highly dynamic structure is fixed. At this stage a “snapshot” of the nanoparticles surrounded by the soft corona was taken, revealing interestingly its non-uniform structure. In addition, cryo-TEM examinations (ESI Fig. 1†) corroborated the morphology of the protein corona observed by the trehalose-UA preparation. As a result structural artifacts caused by negative staining can be excluded. While there is extensive literature dealing with the subject of hard protein corona, only limited analytic methods used to study the soft protein corona are available.21 Consequently, determining the biological relevance of the soft corona has been slowed down.22 Hence, for in vivo application of nanocarriers it is essential to concentrate on studying the interactions of nanoparticles within a given protein source (serum, plasma) and to analyze their aggregation behavior in such complex surroundings as this can highly affect the biodistribution.11 By using multi-angle dynamic light scattering (DLS) it is possible to measure the average, hydrodynamic size of nanoparticles directly incubated in the protein source.23 Multi-angle DLS was performed at 37 °C in human serum and we observed an average size increase of the hydrodynamic radius of about ∼70 nm for functionalized PS-NPs (ESI Table 2†). This size increase can be attributed to the protein corona formation and correlates well with the visualization of the protein corona in TEM images (Fig. 1A). The data evaluation procedure (ESI Material/methods†) and the auto-correlation functions respectively shown for scattering angle of 30° can be found in the ESI Fig. 5–7.†
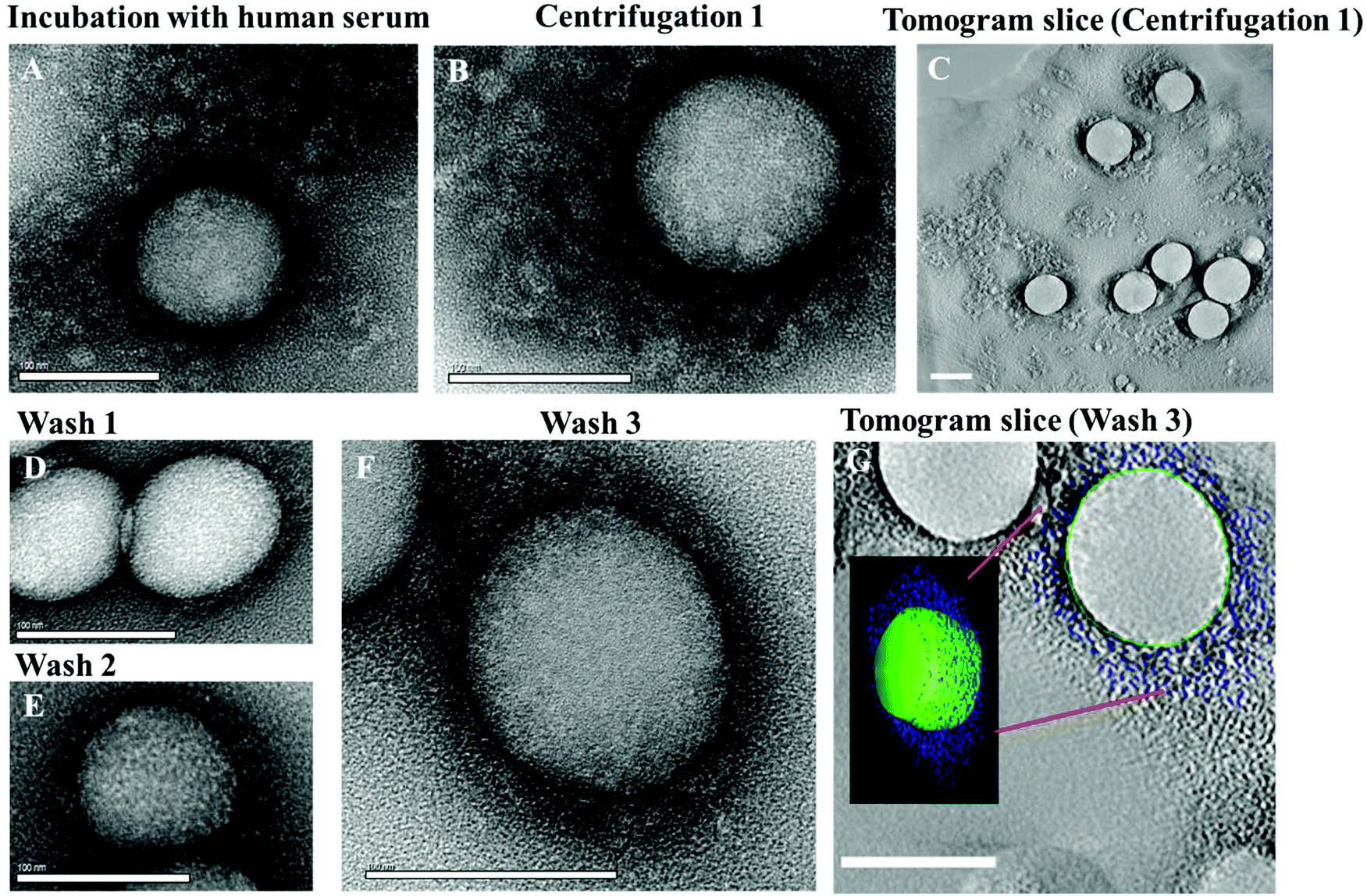 | ||
| Fig. 1 TEM micrographs of the protein corona surrounding un-functionalized polystyrene nanoparticles. A: Hard and soft corona after incubation of un-functionalized PS-NPs in human serum and B: after centrifugation and removal of the supernatant, but before washing steps. C: Tomogram slice of an area such as (B), scale bar: 100 nm. D: Hard corona after 1st wash, E: after 2nd wash, and F: after 3rd wash. G: Electron tomogram slice of the area presented in (F), superimposed by a 3D reconstruction. Scale bar: 100 nm. | ||
In order to differentiate between the loosely bound proteins forming the soft corona and those which can be assigned to the hard corona, we performed an incremental separation process. After incubation with the protein source, the PS-NPs were (i) centrifuged and the supernatant was removed and (ii) afterwards the PS-NPs were washed. The centrifugation and washing was repeated 3 times and at every step the protein corona was characterized.
As previously mentioned, in most studies dealing with the protein corona only proteins of the hard corona have been analyzed. These are tightly bound and have a high affinity towards nanoparticles.24 The hard protein corona of nanoparticles is usually isolated using multiple centrifugation and washing steps to remove loosely or unbound proteins from nanoparticles surrounded by the hard corona.25 Several other studies have compared the protein pattern obtained after centrifugation with different preparation techniques (e.g. magnetic separation, gel-filtration) and these studies have demonstrated that the general protein adsorption pattern is comparable.26,27
Our goal was to analyze the evolution of the protein corona formed directly after incubation in human serum and after each purification/washing step by isolating protein coated nanoparticles via centrifugation. We monitored the morphological development of the protein corona for un-functionalized (Fig. 1) and functionalized nanoparticles (Fig. 3A) by electron microscopy and analyzed the adsorption pattern quantitatively using LC-MS.
After the first centrifugation, un-functionalized PS nanoparticles were surrounded by a substantial protein cloud (Fig. 1B, C and ESI Fig. 2†) as observed directly after incubation (Fig. 1A). There is no significant morphological difference between the protein corona formed directly after incubation and after the first centrifugation step. Only that most of the unbound proteins that were present in the first case were washed away. The 3D reconstructions of the washed samples demonstrate the heterogeneity of the protein corona at this step (Fig. 1C). However, it is difficult to distinguish between tightly or loosely bound proteins associated to one nanoparticle and to unbound proteins. TEM images and DLS measurements reveal a high quantity of proteins adsorbing onto nanoparticles directly after incubation (Fig. 2C). This results in a marked increase in the size of the nanoparticles due to the formation of the soft corona. The soft corona was then washed off after several centrifugation steps and the nanoparticle–particle complex with tightly bound proteins (thus forming a hard corona) could then be isolated (Fig. 1D–G). This result is in agreement with the quantity of proteins quantified after each washing step which continuously decreases with washing steps (Fig. 2D). The number of loosely and unbound proteins was significantly reduced after the first wash (1st centrifugation = 20.02 ± 1.00 mg vs. 1st wash = 2.30 ± 0.10 mg m−2 un-functionalized PS-NP). After the second and third washing steps, the quantity of adsorbed proteins did not decrease significantly. After the third and last wash the protein amount in the supernatant was below the detection limit indicating that the loosely/unbound proteins were effectively washed away (ESI Table 4†). Using the 3D reconstruction presented (Fig. 1G) the number of proteins was counted every 10 nm in the z-axis (assuming that the average protein size is 10 nm). After the first centrifugation, un-functionalized nanoparticles were surrounded by ∼1200 proteins. This number drastically decreased down to ∼400 proteins after the final washing step (Fig. 2C).
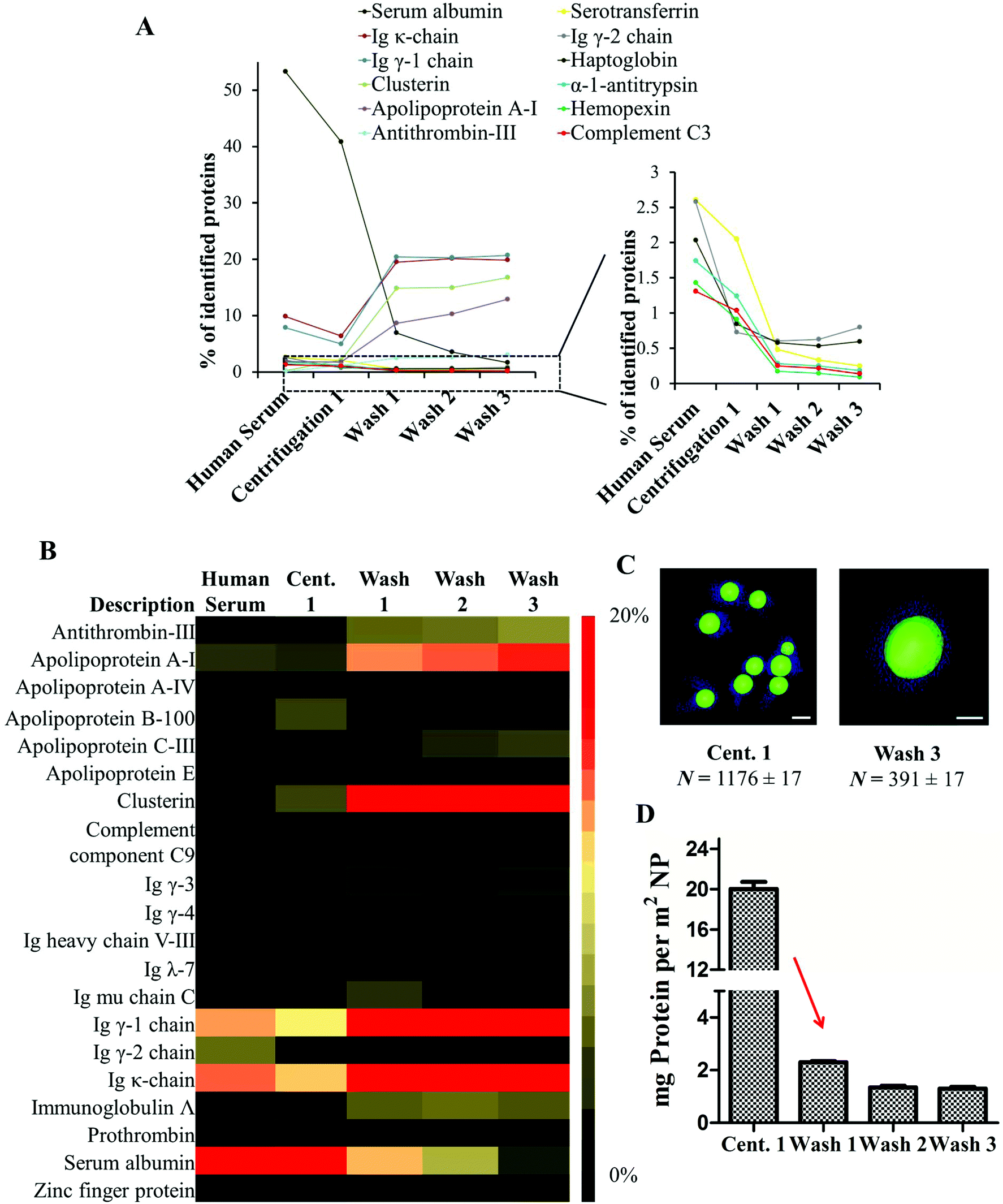 | ||
| Fig. 2 Evolution of the hard protein pattern during sample preparation. A. The number of highly abundance serum proteins decreases and the abundance of the hard corona proteins increase after each washing step. B. The heat map illustrates the relative quantity of the most abundant proteins found in the hard corona (relative quantity of proteins identified amounting to >1%, n = 4). C. 3D reconstruction of tomogram slices presented the nanoparticles surrounded by the protein corona. For TEM an average protein size of 10 nm in z-axis is assumed. D. Proteins were desorbed from un-functionalized PS nanoparticles with 2% SDS and the quantity of protein in mg per m2 NP (±SD, n = 4) was measured by Pierce Assay. | ||
There were no obvious structural differences observed in the nanoparticles–protein complexes when washed just once or three times (Fig. 1D–F). In addition, the ζ-potential of nanoparticles (−3.7 mV, measured in KCl) dramatically decreased to −21 mV after incubation in human serum, indicating protein adsorption and corona formation. Again, there were no major changes measured after the first, second and third washing steps (ESI Fig. 9†). After the third and last wash, the corona thickness measured approximately 15 nm (Fig. 1F and G) by TEM. The proteins were tightly bound to the PS nanoparticles and covered most of each periphery. In order to determine statistical robustness, the radius of more than 20 particles from the TEM micrographs was measured (Fig. 1F and ESI Fig. 3†). The hard corona radius measured by DLS was comparable (19 ± 2 nm, ESI Fig. 8†).
In order to monitor the evolution of the hard protein corona, we analyzed the protein composition of un-functionalized PS-NPs after each purification step. A detailed list of all identified proteins is found in the ESI Tables 7 and 8.† The protein composition of the protein corona is significantly altered when compared to the composition of proteins in serum.14,15 The relative amount of abundant serum proteins (>1%) was found to be drastically decreased in the case of human serum albumin, serotransferrin, haptoglobulin, hemopexin and complement C3 after the first centrifugation stage. All these proteins were detected in small quantities in the hard corona (Fig. 2B). In contrast, there was an enrichment of low abundance proteins (clusterin, apolipoprotein A1 and antithrombin-III) which are referred to as the hard corona proteins (Fig. 2B). Significantly, we were able to show that the relative abundance of the identified proteins remained stable after the first washing (Fig. 2A/B). Only slight changes in the protein pattern could be observed after the second and third washing steps. By doing this the equilibrium between proteins in solution and nanoparticle bound proteins changed rapidly and nanoparticles with tightly-bound hard corona proteins could be isolated. These results highlight the crucial purification step (after centrifugation 1 → wash 1) as here the equilibrium between unbound and nanoparticle bound proteins is critically shifted. It should be mentioned that the hard corona is stable and an exposure to new environments could only lead to partial replacement of the proteins. Since these molecules stay for longer times than the characteristic timescale of a given biological process, their presence is relevant for the biological identity of the nanoparticles.28,29 The soft corona on the other hand could be exchanged within seconds. These interactions are crucial and it should not be forgotten though that it is challenging to separate the two in a biological context.30 But in most cases the time scales for biology match the washing concepts, as in the case of particle internalization that could take place in minutes. The concept of these two time scale (hard and soft corona) in protein kinetics is described in more details elsewhere.31
In addition, it is known that the protein adsorption pattern is highly influenced by surface functionalization of nanoparticles.24,32,33 We found that the absolute number of bound proteins per defined surface of nanoparticles is significantly higher (Fig. 3C) for negatively charged nanoparticles (PS-COOH) in comparison to un-functionalized (PS), or positively charged nanoparticles (PS-NH2). In addition, vitronectin was highly enriched in the protein corona of PS-COOH (Fig. 3A) and PS-NH2 particles specifically adsorbed clusterin (ESI† detailed protein list). Clusterin was also found to be the major hard corona protein of poly(phosphoester)- and PEGylated modified PS-NPs.33 The set of PS-NPs in this experiment was stabilized with Lutensol AT-50 which is a PEG-analog surfactant. In addition, there are specific proteins (immunoglobulin k, immunoglobulin γ or apolipoprotein AI) which were identified on all polystyrene nanoparticles under investigation (protein classification ESI Fig. 11†). Previous studies14,15,34,35 support our findings as they recorded an enrichment of apolipoproteins in the hard corona of polystyrene nanoparticles.
 | ||
| Fig. 3 Analyzing the protein corona pattern around functionalized nanoparticles (PS-COOH, PS-NH2) by various analytic methods. A. NPs (total surface area of the sample: 0.05 m2) were incubated in human serum (1 mL) for 1 h, 37 °C. The protein corona was visualized by TEM after the first centrifugation and third washing step. Hard corona proteins were identified, demonstrating that the protein pattern is highly dependent on surface functionalization. Proteins very analyzed by LC-MS (average of n = 4) and visualized by SDS-PAGE. B. At a first glance the protein pattern of all nanoparticles incubated in human serum was very similar after the first centrifugation, but distinct protein bands can be seen even at this step (starred lanes “centrifugation 1”). These proteins are further identified as hard corona proteins (“wash 3”). C. The amount of bound proteins (in mg ± SD, n = 4) is quantified after desorption of proteins from nanoparticles using 2% SDS by Pierce Assay. | ||
After the first centrifugation, at a first glance, there were no major differences (SDS-PAGE, Fig. 3B) between the identified proteins with regard to the surface functionalization of the nanoparticle (PS, PS-COOH, PS-NH2). Additionally, the protein pattern is comparable to the proteins identified in human serum. The major protein band (∼62 kDa) is referred to as human serum albumin as it is the most abundant serum protein. At this stage it was not possible to clearly separate nanoparticles with tightly surrounded proteins from the remaining proteins in solution because their protein concentration was much higher.
Interestingly, some distinct bands were detected in the SDS-PAGE (marked by a red star, Fig. 3B) even without performing any washing steps. These bands were identified as low abundance proteins in a detailed proteomic analysis by LC-MS (e.g. vitronectin, clusterin, apolipoprotein A1). The concentration of low abundance proteins is much higher on the NP than in the pristine human serum. These proteins were further enriched in subsequent washing steps and identified as the proteins of the hard corona (Fig. 3A). There were no significant structural differences (Fig. 3A) visualized in the soft and hard protein corona of functionalized nanoparticles. Additionally the hard corona radius (ESI Table 3†) measured by TEM and DLS was comparable to un-functionalized nanoparticles (10–20 nm).
To visualize the binding of individual hard corona proteins, un-functionalized nanoparticles were incubated in the respective isolated proteins (clusterin, apolipoprotein A1 or IgG) for 1 h at 37 °C and the protein adsorption was analyzed by TEM (Fig. 5 and ESI Fig. 4†). Micrographs show that in most cases the protein was present around the entire periphery of the nanoparticles. In addition, the corona diameter was quite large (∼100 nm), as seen in the case of apolipoprotein A1 (Fig. 5A). However, the secondary structure of the proteins can be altered due to the adsorption on a surface36,37 further determining the cellular uptake.38,39
Therefore, we further studied the interactions of protein corona coated nanoparticles (Fig. 4/5) and a macrophages cell line, RAW264.7. Flow cytometry analysis, confocal laser scanning microscopy (CLSM) and TEM revealed the intracellular uptake of BODIPY-labeled PS-NPs and no adherence to the plasma membrane (Fig. 4B, ESI Fig. 14†). Further, we investigated the difference between nanoparticles surrounded by the hard corona vs. soft corona. Hard corona coated nanoparticles incubated in serum were isolated via repetitive centrifugation and washing (3 times, as prepared for protein corona analysis) and added to serum free cell culture medium (+ hard corona). Additionally, nanoparticles were directly added to serum free culture medium (− corona) or cells cultured in 100% serum (++ soft corona).
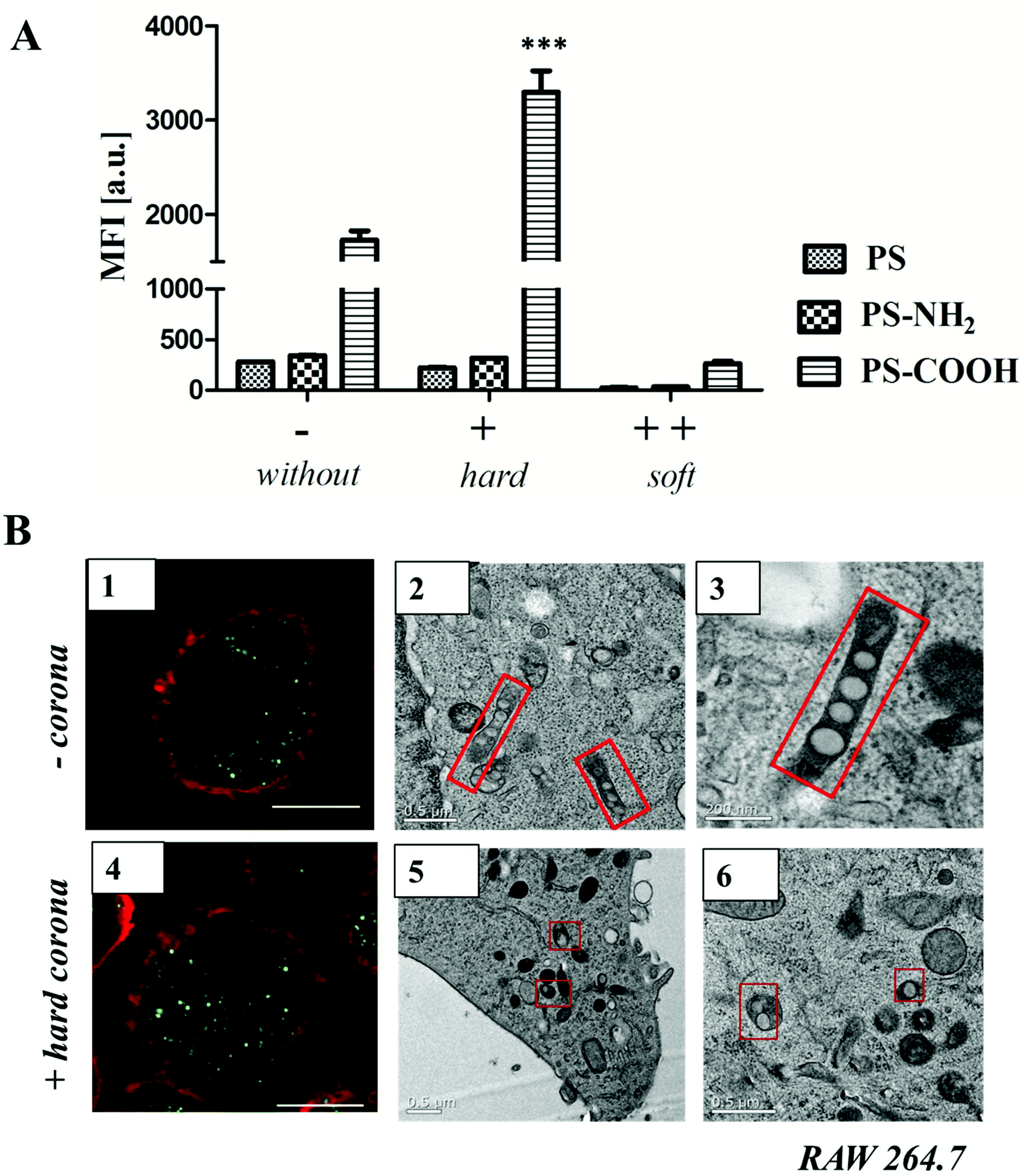 | ||
| Fig. 4 A. Flow cytometry analysis: RAW264.7 cells were incubated with un-functionalized or functionalized polystyrene nanoparticles (300 μg mL−1) for 1 h. The average of the median fluorescence intensity (MFI) of three independent experiments is shown (n = 3). Prior to cellular uptake studies, nanoparticles were incubated with human serum for 1 h at 37 °C, centrifuged and washed to remove unbound proteins. Isolated hard corona coated nanoparticles (+) or uncoated nanoparticles (−) were added to serum free cell culture medium. Additionally, uncoated nanoparticles were added to cells cultured in 100% serum (++ soft corona). B. Confocal laser scanning microscopy images (1, 4) and TEM micrographs (2, 3, 5, 6) of high-pressure frozen macrophages treated with 300 μg mL−1 of un-functionalized nanoparticles without (−) or with hard protein corona (+). Scale bar: B1, B4 = 10 μm; B3 = 200 nm; B2, B5, B6 = 0.5 μm. GraphPad Prism 5 Software was used for statistical analysis using a one-way ANOVA followed by Tukey's post-hoc multiple comparisons test. A p-value of <0.001 was considered as highly significant ***. | ||
Cellular uptake of soft corona coated nanoparticles (++) was strongly reduced in comparison to hard corona coated (+) or uncoated nanoparticles (−). Here, it was shown that the surface functionalization highly influenced uptake behavior (Fig. 4A). Un-functionalized and amino-functionalized nanoparticles were taken up to a significantly lower extent compared to carboxy-functionalized nanoparticles. Interestingly, this trend was observed with or without protein corona (Fig. 4A, ESI Fig. 15†). Additionally, it was found that cellular uptake of PS-COOH nanoparticles was significantly enhanced for nanoparticles surrounded by hard corona proteins compared to uncoated PS-COOH (p < 0.001 ***; ESI Fig. 14†). This is remarkable as we demonstrate that immunoglobulins are the major protein corona component (about 50%) for all investigated nanoparticles. As the morphology of the protein corona was visualized as an undefined loose network of proteins, this indicates that next to the presence of the protein corona the underlying surface functionalization affects cellular uptake.
Transmission electron microscopy (Fig. 4B) was used to compare the internalization mechanism and intracellular trafficking of un-functionalized nanoparticles without (−) or with hard protein corona (+). Numerous un-functionalized polystyrene nanoparticles were packed in long membrane structures (Fig. 4B 2, 3) that resemble the uncoated carriers or early endosomes present in the CLIC/GEEC endocytosis pathway.40 In the presence of protein corona, these structures were not observed. Instead, un-functionalized PS-NPs (either individually or in a group of 2–3) were packed in small vesicles, and were not found inside endosomes (Fig. 4B 5, 6). These results indicate that in the presence of protein corona, the cells choose an alternative endocytosis pathway. Additional TEM micrographs are summarized in the ESI Fig. 16/17.† The exact mechanism will be further studied with the use of antibodies and it is not further discussed here as it is beyond the scope of this paper.
Several studies have shown that the secondary structure of the proteins can be altered by adsorption on a surface36,37 and thus further determine cellular uptake.38,39 As seen in the case of apolipoprotein AI (ApoAI), we found that the corona diameter after incubation with un-functionalized PS-NP was quite large (∼100 nm, Fig. 5A). Therefore, we studied whether this affects the cellular uptake behavior. Un-functionalized nanoparticles were incubated with ApoAI and cellular uptake was analyzed by flow cytometry (Fig. 5B), cLSM (Fig. 5C), and TEM (Fig. 5D). It was found that due to pre-coating with ApoAI cellular uptake was strongly inhibited compared to uncoated (−) or hard corona coated nanoparticles (+), hence it was comparable to soft corona coated nanoparticle (++). Interestingly, the structural properties of the ApoAI and soft protein corona indicated strikingly similar properties (Fig. 5Avs.2C average corona diameter and number of corona proteins).
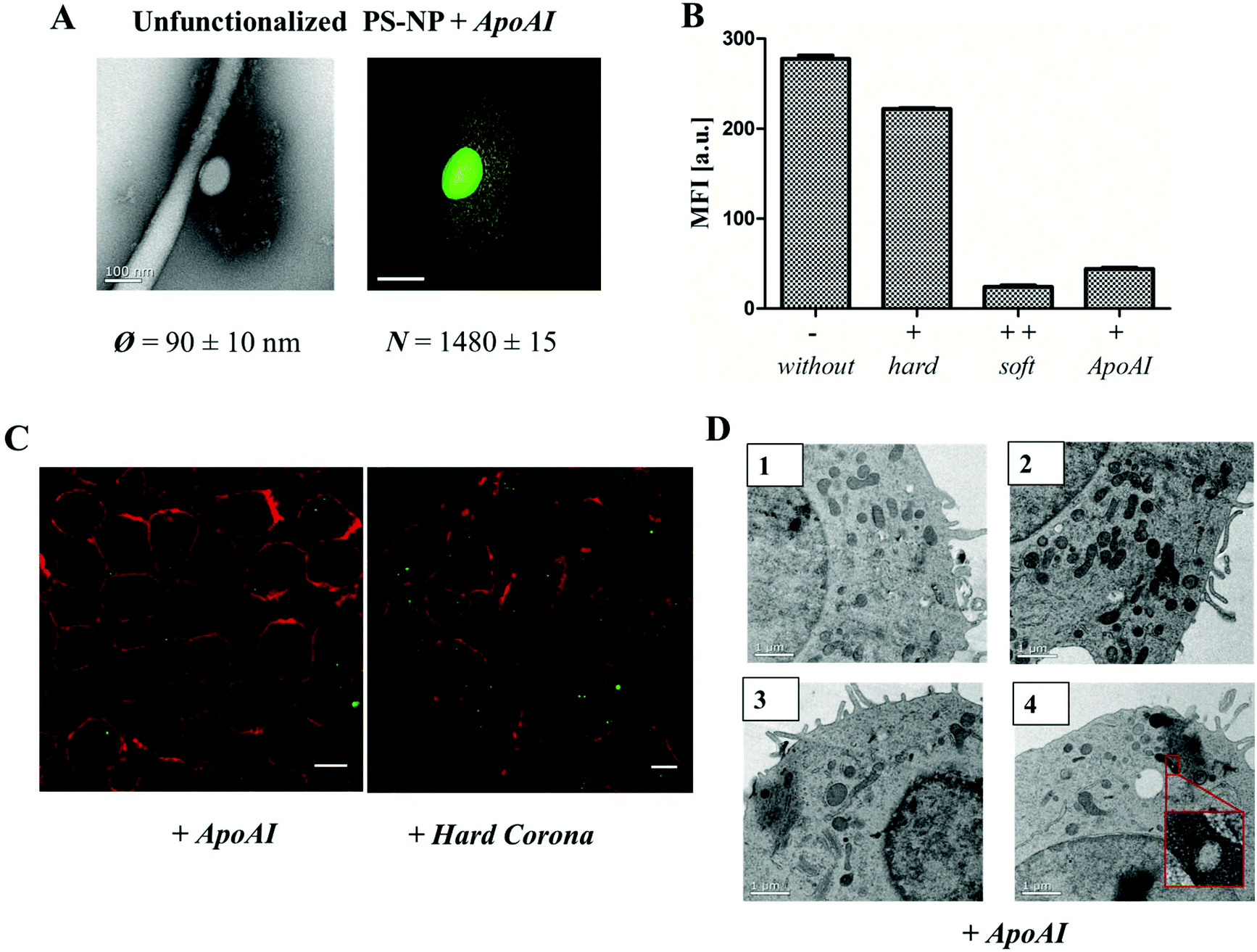 | ||
| Fig. 5 A. TEM micrographs and 3D reconstruction images of un-functionalized PS-NP (0.05 m2) incubated with ApoAI1 (100 μg) for 1 h, 37 °C. The additional corona diameter (∼100 nm) and number of ApoAI proteins (>1400 proteins) surrounding un-functionalized PS-NP is comparable to the soft corona images (Fig. 1 and 2). Scale bar: 100 nm. B. Flow cytometry analysis: RAW264.7 cells were incubated with 300 μg mL−1 of un-functionalized (−), protein corona coated (+/++) or ApoAI coated un-functionalized PS-NP for 1 h. C. Confocal laser scanning microscopy images of ApoAI or hard corona coated un-functionalized nanoparticles. The cell membrane is stained using CellmaskOrange (pseudo-coloured red) and BODIPY labeled nanoparticles are pseudo-coloured green. Scale bar: 10 μm. D. TEM micrographs of high-pressure frozen macrophages treated with 300 μg mL−1 of un-functionalized nanoparticles pre-coated with ApoAI indicating a strongly reduced uptake of nanoparticles. Scale bar: 1 μm. | ||
Experimental
Transmission electron microscopy (TEM)
In order to observe the protein corona that was formed around the PS-NPs, the samples were first diluted with 1 ml water and then 2 μl were placed onto a lacey grid and let to dry. In the case of single protein binding studies, PS-NP (0.05 m2) were incubated with individual proteins (100 μg) for 1 h, 37 °C and 2 μL of each sample was placed onto a lacey grid. The droplet method was applied with 4% uranyl acetate.19Electron micrographs were taken on an Ultrascan 1000 (Gatan) charge-coupled device (CCD) camera. The TEM was operated at 200 kV. The Digital Micrograph software (Gatan) was used to collect the images.
In order to view the protein corona in 3D, tilt series over a tilt range of −65° to +65° were recorded at a magnification of 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×. The SerialEM software (Mastronarde, 2005) was used to collect the tilt series.
000×. The SerialEM software (Mastronarde, 2005) was used to collect the tilt series.
Cryo-TEM
10 μl of the sample was placed onto a 400 mesh copper grid covered with lacey film. The excess dispersion was removed by blotting with filter paper. The grid is plunged into liquid ethane (automated plunging system, Vitrobot FEI) and transferred in liquid nitrogen to the TEM. Prior to the preparation, the grids were treated with oxygen plasma to make the film hydrophilic.TEM (3D reconstruction)
The alignments and the weighted back-projection-based reconstructions of raw tilt series were computed with eTomo (a program from the IMOD software package).41The diameter of the particles and the protein corona that was formed around them was calculated with ImageJ.
Dynamic light scattering
Dynamic light scattering experiments were performed with an ALV-CGS 8F SLS/DLS 5022F goniometer equipped with eight simultaneously working ALV 7004 correlators, eight QEAPD Avalanche photodiode detectors and a a HeNe laser (632.8 nm, 25 mW output power) as light source at 37 °C. Nanoparticle dispersions (1 μl, 10 mg mL−1) were measured in 1 mL of filtered Dulbecco`s magnesium- and calcium-free phosphate buffered saline (PBS) buffer solution (GIBCO, Invitrogen). Human serum was filtered through a Millex GS 220 nm filters (Millipore) into cylindrical quartz cuvettes (20 mm diameter, Hellma, Müllheim) and nanoparticles were directly (1 μL) to the cuvette and incubated with human serum for 1 h at 37 °C before the measurement.Data evaluation dynamic light scattering
Data was evaluated according to the method of Rausch et al.23 Briefly, the sum of the autocorrelation functions (ACF) of the individual components (human serum or nanoparticle) was used as fixed parameters.The ACF of human serum is approximated fitted by a sum of three exponential terms as given in eqn (S1):
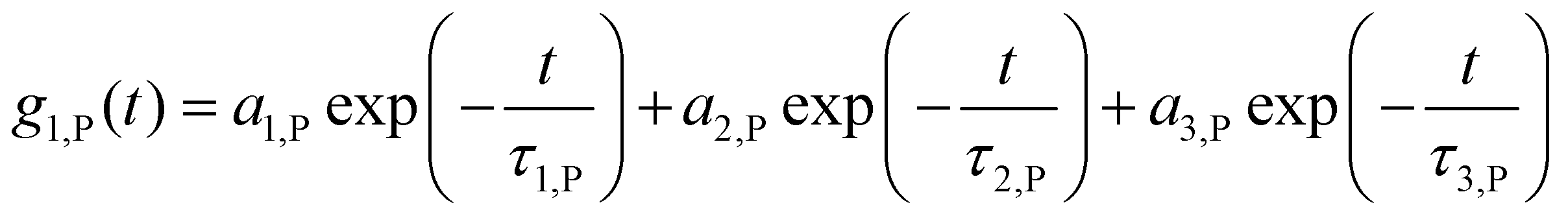 | (S1) |
 the decay times,
the decay times,  the absolute scattering angle and i, Di Brownian diffusion coefficient. The ACF of the nanoparticles is fitted by a sum of two exponential terms (S2).
the absolute scattering angle and i, Di Brownian diffusion coefficient. The ACF of the nanoparticles is fitted by a sum of two exponential terms (S2). | (S2) |
If nanoparticles are exposed to human serum the combination of the ACFs of serum and nanoparticles is analyzed. If no aggregation occurs, the resulting ACF of the mixture can be fitted by the sum of two individual ACFs g1,m(t), the so named forced fit(S3).
| g1,m(t) = fPg1,P(t) + fNPg1,NP(t) | (S3) |
If aggregation formation of nanoparticles in serum occurs, the ACF cannot be describe by sum of two components, so an additional term ACF g1,agg(t) for aggregates is needed (S4).
 | (S4) |
Therefore, the correlation function g1,m(t) consists of three terms including the aggregation terms with the intensity contribution fagg.
| g1,m(t) = fPg1,P(t) + fNPg1,NP(t) + faggg1,agg(t) | (S5) |
ζ potential and particle charge detection
The zeta (ζ) potential of the different polystyrene nanoparticle (20 μL) was measured with a Zeta Sizer Nano Series (Malvern Instrument, U.K.) in 1 mM potassium chloride solution (2 mL). A combination of a particle charge detector PCD 02 (Mütek GmbH Germany) and a Titrino Automatic Titrator 702 SM (Metrohm AG Switzerland) was used to determine the amount of surface charged groups on nanoparticles. Titration experiments were performed with nanoparticles dispersion (10 mL) with a solid content of 1 mg mL−1 (0.1 wt%) at 20 °C. Amino groups were titrated against the negatively charged polyelectrolyte standard sodium poly(ethylene sulfonate) (Pes-Na) and for carboxyl groups positively charged poly(diallyl dimethyl ammonium chloride) (PDADAMC) was used. The amount of groups was calculated as previously described.42SDS-PAGE
The protein sample (6 μg in 26 μL total volume) was mixed with 4 μL of NuPage Reducing Agent and 10 μL of NuPage LDS Sample Buffer NuPage, loaded on a 10% Bis-Tris-Protein Gels using NuPAGE MES SDS Running Buffer (all Novex, Thermo Fisher Scientific) and run for 1.5 h using SeeBlue Plus2 Pre-Stained (Invitrogen) as molecular marker. Gels were stained with SimplyBlue SafeStain (Novex, Thermo Fisher Scientific) at least for 4 h and destained in distilled water overnight.Protein quantification
The protein concentration of human serum or the desorbed hard corona proteins was quantified with the Pierce 660 nm protein Assay (Thermo Scientific; Germany) according to manufacturer's instructions and bovine serum albumin (Serva, Germany) was used a standard. Absorption was measured at 660 nm by a Tecan infinite M1000 plate reader.Human blood serum
Human blood was obtained from the Department of Transfusion Medicine Mainz from healthy donors in accordance with the Declaration of Helsinki. Blood was clotted overnight according to the standard protocol to generate human serum. A serum pool from seven volunteers was used and storaged at −80 °C. To remove any protein aggregates after thawing, human serum was centrifuged for 30 min at 20000g before usage.
Protein corona preparation
A constant ratio between particle surface area and serum concentration was chosen to ensure reproducibility. A surface area of 0.05 m2 nanoparticles (in a total volume of 300 μL) were incubated with 1 mL of human serum for 1 h, 37 °C. Previous studies have shown that the protein corona is formed stabile after 1 h.43The hard protein corona surrounding nanoparticles was isolated via centrifugation and loosely or unbound proteins were removed. Nanoparticles were centrifuged for 1 h, 20000g (4 °C). The supernatant was collected for protein quantification. The remaining nanoparticle pellet was either re-suspended in water (final nanoparticle concentration of 10 mg mL−1) and analyzed by TEM and DLS or washed with 1 mL of water. This procedure was repeated three times to ensure that all unbound proteins are removed. To elute bound proteins form the nanoparticles, the pellet was re-suspended in 2% SDS (62.5 mM Tris·HCl), heated up to 95 °C for 5 min and centrifuged for 1 h, 20000g (4 °C). The remaining supernatant was collected further analyzed by Pierce Assay, SDS-PAGE and LC-MS.
In solution digestion
Protein samples were applied to Pierce detergent removal columns (Thermo Fisher) to remove SDS prior to digestion. Proteins digestion was performed according to former instruction.44,45 Briefly, proteins were precipitated overnight using ProteoExtract protein precipitation kit (CalBioChem) according to the manufactures instructions’. Proteins were isolated via centrifugation (14000g, 10 min), washed several times and re-suspend in RapiGest SF (Waters Cooperation) dissolved in ammonium bicarbonate (50 Mm) buffer. Samples were incubated at 80 °C for 15 min. Protein disulfide bonds were reduced with dithithreitol (Sigma) at a final concentration of 5 mM. The reaction was performed at 45 min at 56 °C. Proteins were alkylated with idoacetoamide (final concentration 15 mM, Sigma) and incubated in the dark for 1 h. Digestion was carried out with a protein:trypsin ratio of 50:1 over 16 h at 37 °C and the reaction was quenched by adding 2 μL hydrochloric acid (Sigma). To remove degradation products of RapiGest SF, peptide samples were centrifuged for 14000g, 15 min (4 °C).
Liquid chromatography coupled to mass spectrometry (LC-MS analysis)
Samples were diluted with 0.1% formic acid and spiked with 50 fmol μL−1 Hi3 E. coli (Waters Cooperation) standard for absolute protein quantification.46 Tryptic peptides were applied to a C18 analytical reversed phase column (1.7 μm, 75 μm × 150 mm) and a C18 nanoACQUITY trap column (5 μm, 180 μm × 20 mm) in a nanoACQUITY UPLC system. Two mobile phases (A) consisting of 0.1% (v/v) formic acid in water and 0.1% (v/v) formic acid with acetonitrile and gradient of 2% to 37% of mobile phase B over 70 min were used for separation. The nanoACQUITY UPLC system was coupled with a Synapt G2- Si mass spectrometer. Electrospray ionization (ESI) was conducted in positive ion mode using a NanoLockSpray source. The sample flow rate was set to 0.3 μl min−1 and the reference component Glu-Fibrinopeptide was infused 150 fmol μl−1 at a flow rate of 0.5 μl min−1. The Synapt G2-Si was operated in resolution mode and data-independent acquisition (MSE) experiments were carried out. A mass to charge range of 50–2000 Da, scan time of 1 s, ramped trap collision energy from 20 to 40 V was set and data was acquired over 90 min. MassLynx 4.1 was used for data acquisition and processing.Protein identification
Continuum data was post lock mass corrected and further analyzed by Progenesis QI (2.0) using a reviewed human data base (Uniprot) for peptide and protein identification. Several processing parameters as noise reduction thresholds for low energy, high energy and peptide intensity were set to 120, 25, and 750 counts. The human data base was modified with the sequence information of Hi3 E. coli standard for absolute quantification. The following criteria were chosen for protein and peptide identification: one missed cleavage, maximum protein mass 600 kDa, fixed carbamidomethyl modification for cysteine, variable oxidation for methionine and protein false discovery rate of 4%. To identify a protein at least two assigned peptides and five assigned fragments are required. Peptide identification is based on three assigned fragments and identified peptides with a score parameter below 4 were discharged. Based on the TOP3/Hi3 approach the amount of each protein in fmol was provided.47A detailed overview of all identified proteins is found in separate ESI† (excel sheet).
Cell culture
The murine macrophage cell line RAW264.7 were maintained in Dulbecco's modified eagle medium (DMEM) supplemented with 10% FCS, 100 U per ml penicillin, 100 mg per ml streptomycin and 2 mM glutamine (all from Invitrogen, Germany).Cell uptake experiments: flow cytometry and confocal laser scanning microscopy
For the cell uptake experiments, cells were seeded at a density of 150000 cells per well in 24 well plates. 3 mm plasma-sterilized sapphire discs (M. Wohlwend GmbH, Sennwald, Switzerland) covered with a 20 nm carbon layer before usage were added in the well plate. After 12 h, the cells were incubated in fresh serum-free medium for 2 h, before the nanoparticle dispersions were added at a concentration of 300 μg ml−1 to the cells.
Hard protein corona coated nanoparticles were prepared as described above (Protein corona preparation). Therefore, the nanoparticles were first incubated with human serum, centrifuged and washed to remove unbound proteins. Protein coated nanoparticles were re-suspended in serum free medium (final concentration: 300 μg mL−1) and incubated with the cells for 1 h.
For flow cytometry experiments, adherent cells were washed with PBS and detached from the culture vessel with 2.5% trypsin (Gibco, Germany) and measurements were performed on a CyFlow ML cytometer (Partec, Germany) with a 488 nm laser for excitation of BODIPY and a 527 nm band pass filter for emission detection. Data analysis was performed using FCS Express V4 software (DeNovo Software, USA) selecting the cells with a FSC/SSC plot, thereby excluding cell debris. The gated events were analyzed by the fluorescent signal (FL1) expressed as median fluorescence intensity (MFI).
In order to proof intracellular localization of nanoparticles, confocal laser scanning microscopy (cLSM) experiments were performed on a LSM SP5 STED Leica Laser Scanning Confocal Microscope (Leica, Germany), consisting of an inverse fluorescence microscope DMI 6000 CS equipped with a multi-laser combination using a HCX PL APO CS 63 × 1.4 oil objective. Bodipy-labelled nanoparticles were excited with an argon laser (20 mW; λ = 514 nm), detected at 530–550 nm (pseudocolored green) and the cell membrane was stained with CellMaskOrange (2.5 μg mL−1, Invitrogen) using a laser DPSS 561 nm (≈1.3 mW), detected at 570–600 nm (pseudocolored red).
High pressure freezing and freeze substitution
For a good preservation of the structure, the specimen was frozen under high pressure (2100 bars), using the high pressure freezing machine (Engineering Office M. Wohlwend GmbH, Switzerland). The specimen (sapphire discs with cells) was enclosed and protected in a small volume between two specimen carriers and locked inside the specimen pressure chamber. Liquid nitrogen was used as cooling medium. To ensure high quality preservation, this technique was combined with freeze substitution and resin embedding. This included dehydration of the cryo-fixed samples at −90 °C by substituting the ice for an organic solvent (0.2% osmium tetroxide, 0.1% uranyl acetate and 5% water in aceton) inside the freeze substitution machine (EM, AFS 2, Leica Microsystems). After bringing the samples to room temperature, they were rinsed in pure aceton and infiltrated in EPON 812. On the next day, polymerization took place at 60 °C. Ultrathin sections were collected afterwards using a Leica ultramicrotime.Nanoparticle synthesis
Styrene (99%, Merck, Germany) and acrylid acid AA (99%, Aldrich, Germany) were freshly distilled under reduced pressure and stored at −18 °C. The following commercial products were used: 2-aminoethyl methacrylate hydrochloride (AEMH, 90%, Sigma-Aldrich, Germany), n-hexadecene (HD, Sigma-Aldrich), initiator 2,2′-azobis (2-methylbutyronitrile) (V59, Wako Chemicals, Germany) and Lutensol AT50 (BASF). The fluorescent dye Bodipy-1 was synthesized according to ref. 48, which has the maximum of absorption at 523 nm and of emission at 536 nm. A stock solution of Lutensol was prepared (1.99 g Lutensol AT50 filled to 79.65 g with steril water). The nanoparticles were synthesized by free-radical miniemulsion polymerization according to.13(i) Plain PSNPs (referred as PS): 6.0529 g of styrene, 250.72 mg of hexadecane, 103.55 mg of the initiator V-59 and 5.99 mg Bodipy-1 were added to 24 g of water containing 0.6 g of Lutensol AT50.
(ii) Carboxy-functionalized PSNPs (referred as PS-COOH):: 5.88286 g of styrene, 0.15258 g of AA, 251.28 mg of hexadecane, 99.71 mg of the initiator V-59 and 6.10 mg Bodipy-1 were added to 24 g of water containing 0.6 g of Lutensol AT50.
(iii) Amino-functionalized PSNPs (referred as PS-NH2):: 5.8887 g of styrene, 252.30 mg of hexadecane, 100.62 mg of the initiator V59 and 6.06 mg Bodipy-1 were mixed with 24 g water containing 0.12 g of AEMH and 0.6 g Lutensol AT-50.
After 1 h of stirring for pre-emulsification, the miniemulsion was prepared by ultrasonicating the mixture for 2 min by 450 W at 90% intensity (Branson sonifier W450 Digital, ½′′ tip) at 0 °C. For polymerization, the temperature was increased to 72 °C and reaction proceeded overnight.
Conclusion
We were able to demonstrate the evolution of the protein corona during the centrifugation and washing procedures using the combined correlative approach of TEM and proteomics. By using these techniques, we could differentiate between the soft corona and the hard corona. Our findings on the morphology of the protein corona offer new insights into its real structure, which were determined to be a network-like, loosely interconnected agglomeration of proteins surrounding a nanoparticle.In order to explore the effect of surface chemistry, we presented a qualitative and quantitative analysis of the protein corona of three differently surface functionalized PS-NPs and their influence on the composition of the protein corona. Furthermore, we proved that the uptake of un-functionalized PS-NPs pre-incubated with either soft corona or Apo-A1 was prevented. Understanding how NPs interact with proteins as well as the effect of the different corona morphologies and compositions on cellular mechanisms (like uptake and trafficking) are important factors in the design of NPs. This is particularly so for those NPs with regulated biological identities and physiological effects and also for experiments based on “personalized protein corona” intended for clinical applications as suggested by Hajipour et al.49
Acknowledgements
We would like to thank Katja Klein for the synthesis of polystyrene nanoparticles, Laura K. Müller/Christine Rossenauer for the DLS measurements and Christoph Sieber for the sample preparation with HPF/FS. We gratefully acknowledge the financial support of Deutsche Forschungsgemeinschaft (DFG Sonderforschungsbereich SFB 1066, Project Q2). Open Access funding provided by the Max Planck Society.References
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS PubMed.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- P. M. Kelly, C. Åberg, E. Polo, A. O'Connell, J. Cookman, J. Fallon, Ž. Krpetić and K. A. Dawson, Nat. Nanotechnol., 2015, 10, 472–479 CrossRef CAS PubMed.
- C. D. Walkey and W. C. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- T. Cedervall, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS PubMed.
- S. Ritz, S. Schöttler, N. Kotman, G. Baier, A. Musyanovych, J. Kuharev, K. Landfester, H. Schild, O. Jahn, S. Tenzer and V. Mailaender, Biomacromolecules, 2015, 16, 1311–1321 CrossRef CAS PubMed.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Aberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS PubMed.
- D. Docter, U. Distler, W. Storck, J. Kuharev, D. Wuensch, A. Hahlbrock, S. K. Knauer, S. Tenzer and R. H. Stauber, Nat. Protoc., 2014, 9, 2030–2044 CrossRef CAS PubMed.
- K. Mohr, M. Sommer, G. Baier, S. Schöttler, P. Okwieka, S. Tenzer, K. Landfester, V. Mailänder, M. Schmidt and R. G. Meyer, J. Nanomed. Nanotechnol., 2014, 5 DOI:10.4172/2157-7439.1000193.
- C. Loos, T. Syrovets, A. Musyanovych, V. Mailander, K. Landfester, G. U. Nienhaus and T. Simmet, Beilstein J. Nanotechnol., 2014, 5, 2403–2412 CrossRef PubMed.
- K. Landfester, Annu. Rev. Mater. Res., 2006, 36, 231–279 CrossRef CAS.
- S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. Kuharev, A. Rekik, S. K. Knauer, C. Bantz, T. Nawroth, C. Bier, J. Sirirattanapan, W. Mann, L. Treuel, R. Zellner, M. Maskos, H. Schild and R. H. Stauber, ACS Nano, 2011, 5, 7155–7167 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, M. Foy, T. Berggad, S. C. Donnelly, G. Cagney, S. Linse and K. A. Dawson, Angew. Chem., Int. Ed., 2007, 46, 5754–5756 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- S. De Carlo and J. R. Harris, Micron, 2011, 42, 117–131 CrossRef CAS PubMed.
- L. Zhang, H. Tong, M. Garewal and G. Ren, Biochim. Biophys. Acta, 2013, 1830, 2150–2159 CrossRef CAS PubMed.
- P. Renz, M. Maria Kokkinopoulou, K. Landfester and I. Lieberwirth, Macromol. Chem. Phys., 2016, 217, 1879–1885 CrossRef CAS.
- S. Winzen, S. Schoettler, G. Baier, C. Rosenauer, V. Mailaender, K. Landfester and K. Mohr, Nanoscale, 2015, 7, 2992–3001 RSC.
- M. C. Lo Giudice, L. M. Herda, E. Polo and K. A. Dawson, Nat. Commun., 2016, 7 DOI:10.1038/ncomms13475.
- J. Wegener, Measuring Biological Impacts of Nanomaterials, Springer-Verlag, 2016 Search PubMed.
- K. Rausch, A. Reuter, K. Fischer and M. Schmidt, Biomacromolecules, 2010, 11 DOI:10.1021/bm100971q.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. Chan, J. Am. Chem. Soc., 2012, 134, 2139–2147 CrossRef CAS PubMed.
- M. Rahman, S. Laurent, N. Tawil, L. Yahia and M. Mahmoudi, Protein-Nanoparticle-Interactions, The Bio-Nano Interface, Springer-Verlag, 2013 Search PubMed.
- K. Thode, M. Luck, W. Semmler, R. H. Muller and M. Kresse, Pharm. Res., 1997, 14, 905–910 CrossRef CAS.
- M. P. Monopoli, S. Wan, F. B. Bombelli, E. Mahon and K. A. Dawson, Nano LIFE, 2013, 3 DOI:10.1142/S1793984413430046.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- S. Lara, F. Alnasser, E. Polo, D. Garry, M. C. Lo Giudice, D. R. Hristov, L. Rocks, A. Salvati, Y. Yan and K. A. Dawson, ACS Nano, 2017, 11, 1884–1889 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Aberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- S. Milani, F. Baldelli Bombelli, A. S. Pitek, K. A. Dawson and J. Rädler, ACS Nano, 2012, 6, 2532–2541 CrossRef CAS PubMed.
- S. Winzen, S. Schoettler, G. Baier, C. Rosenauer, V. Mailaender, K. Landfester and K. Mohr, Nanoscale, 2015, 7, 2992–3001 RSC.
- S. Schöttler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailänder and F. R. Wurm, Nat. Nanotechnol., 2016, 11, 372–377 CrossRef PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- S. Ritz, S. Schöttler, N. Kotman, G. Baier, A. Musyanovych, J. Kuharev, K. Landfester, H. Schild, O. Jahn, S. Tenzer and V. Mailaender, Biomacromolecules, 2015, 16, 1311–1321 CrossRef CAS PubMed.
- V. V. Hlady and J. Buijs, Curr. Opin. Biotechnol., 1996, 7, 72–77 CrossRef CAS PubMed.
- J. J. Gray, Curr. Opin. Struct. Biol., 2004, 14, 110–115 CrossRef CAS PubMed.
- C. C. Fleischer and C. K. Payne, J. Phys. Chem. B, 2012, 116, 8901–8907 CrossRef CAS PubMed.
- C. C. Fleischer and C. K. Payne, J. Phys. Chem., 2014, 118, 14017–14026 CrossRef CAS PubMed.
- C. G. Hansen and B. J. Nichols, J. Cell Sci., 2009, 1713–1721 CrossRef CAS PubMed.
- J. R. Kremer, D. N. Mastronarde and J. R. McIntosh, J. Struct. Biol., 1996, 116, 71–76 CrossRef CAS PubMed.
- A. Musyanovych, R. Rossmanith, C. Tontsch and K. Landfester, Langmuir, 2007, 23, 5367–5376 CrossRef CAS PubMed.
- D. Walczyk, F. Baldelli Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS PubMed.
- D. Hofmann, S. Tenzer, M. B. Bannwarth, C. Messerschmidt, S.-F. Glaser, H. Schild, K. Landfester and V. Mailänder, ACS Nano, 2014, 8, 10077–10088 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. Kuharev, A. Rekik, S. K. Knauer, C. Bantz, T. Nawroth, C. Bier, J. Sirirattanapan, W. Mann, L. Treuel, R. Zellner, M. Maskos, H. Schild and R. H. Stauber, ACS Nano, 2011, 5, 7155–7167 CrossRef CAS PubMed.
- R. A. Bradshaw, A. L. Burlingame, S. Carr and R. Aebersold, Mol. Cell. Proteomics, 2006, 5, 787–788 CAS.
- J. C. Silva, M. V. Gorenstein, G. Z. Li, J. P. C. Vissers and S. J. Geromanos, Mol. Cell. Proteomics, 2006, 5, 144–156 CAS.
- I. García-Moreno, A. Costela and L. Campo, J. Phys. Chem. A, 2004, 108, 3315–3323 CrossRef.
- M. J. Hajipour, S. Laurent, A. Aghaie, F. Rezaee and M. Mahmoudi, Biomater. Sci., 2014, 2, 1210–1221 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr02977b |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |