
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomistic dynamics of sulfur-deficient high-symmetry grain boundaries in molybdenum disulfide†
Jinhua
Hong‡
a,
Yanlei
Wang‡
b,
Anle
Wang
b,
Danhui
Lv
a,
Chuanhong
Jin
 *a,
Zhiping
Xu
*b,
Matt I. J.
Probert
c,
Jun
Yuan
*ac and
Ze
Zhang
a
*a,
Zhiping
Xu
*b,
Matt I. J.
Probert
c,
Jun
Yuan
*ac and
Ze
Zhang
a
aState Key Laboratory of Silicon Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou, Zhejiang 310027, P. R. China. E-mail: chhjin@zju.edu.cn
bApplied Mechanics Laboratory, Department of Engineering Mechanics, and Center for Nano and Micro Mechanics, Tsinghua University, Beijing 100084, China. E-mail: xuzp@tsinghua.edu.cn
cDepartment of Physics, University of York, Heslington, York, YO10 5DD, UK. E-mail: jun.yuan@york.ac.uk
First published on 22nd June 2017
Abstract
As a common type of structural defect, grain boundaries (GBs) play an important role in tailoring the physical and chemical properties of bulk crystals and their two-dimensional (2D) counterparts such as graphene and molybdenum disulfide (MoS2). In this study, we explore the atomic structures and dynamics of three kinds of high-symmetry GBs (α, β and γ) in monolayer MoS2. Atomic-resolution transmission electron microscopy (TEM) is used to characterize their formation and evolutionary dynamics, and atomistic simulation based analysis explains the size distribution of α-type GBs observed under TEM and the inter-GB interaction, revealing the stabilization mechanism of GBs by pre-existing sulfur vacancies. The results elucidate the correlation between the observed GB dynamics and the migration of sulfur atoms across GBs via a vacancy-mediated mechanism, offering a new perspective for GB engineering in monolayer MoS2, which may be generalized to other transition metal dichalcogenides.
1. Introduction
Grain boundaries (GBs) in general, including phase boundaries (PBs),1–7 are local structural imperfections in crystalline materials. They can profoundly influence the mechanical, electronic, optical, magnetic and chemical properties of materials.8–13 Precisely imaging the structures and/or the dynamics of GBs is essential to reveal the structure–property correlation in materials, including the development of two-dimensional (2D) materials such as graphene and MoS2. Aberration corrected scanning transmission electron microscopy (AC-STEM)14 enables direct atomic-resolution imaging of structural defects (including GBs) in 2D materials such as graphene, transition metal dichalcogenides (TMDs),15 and their atomistic dynamics16–18 under electron beam irradiation and/or at elevated temperatures. In monolayer MoS2, Lin et al.19 reported the nucleation and growth of GBs (identified as α-type GB in this work) around Re/Au dopant centers, as activated by the electron beam irradiation at elevated temperatures. The energetics and kinetics of the relevant 2H-1T phase transition were recently elucidated by Zhao and Ding based on first-principles calculations.20 In monolayer MoSe2, several groups have confirmed the formation of mirror twin-boundaries21–23 (also termed β-type GBs) due to the Se deficiency, which could be induced by the electron beam irradiation or grown intrinsically during the molecular beam epitaxy (MBE) process. For the effect of GBs on the electronic properties of 2D materials, Barja et al.24 characterized one-dimensional charge density waves at the β-type GB in MoSe2via low-temperature scanning tunneling microscopy. Huang et al.25 showed that GBs could cause a local modulation of band-gaps that depend on the mis-orientation between neighboring grains in the monolayer MoS2, introducing in-gap states that may benefit the catalytic function in hydrodesulfurization,21,24,26 and modifying the magnetic properties. Ly et al.27 confirmed experimentally that the electronic transport across GBs is highly sensitive to the inter-domain mis-orientation. All these previous results have unambiguously demonstrated that GBs have a remarkable impact on the physical and chemical properties of 2D TMDs.Despite these previous studies on GB dynamics, the S-vacancy induced atomic sliding-migration mechanism of GB evolution in the MoS2 system has not been reported due to the lack of statistically sufficient data on GBs and analysis of the inter-GB interaction and S-vacancy-assisted kinetic processes. Hence, an in-depth view of the formation and evolution of GBs in TMDs is still necessary. To address this point, we present direct characterization of the atomic processes during the formation and annihilation of high-symmetry GBs in monolayer 2H-MoS2. We focus on these GBs because of their frequent appearance under electron beam irradiation, as well as well-resolved atomic structures that can be used to construct models for atomistic simulations. The simulation results show that the GB structures with a three-Mo-column width and a length of 6–7 Mo-units are most energetically favorable for the α-type GB, which are in good agreement with our experimental evidence and the findings reported by Lin et al.19 Our TEM observations show that these GBs are always accompanied by the presence of S-vacancies near the GB, and the migration of S-atoms in the pristine lattice participates in the process of GB evolution. Further calculations demonstrate that an increase in the amount of S-vacancies and reduction in the vacancy-GB distance could dramatically reduce the formation energy of GBs, indicating that the S-vacancies induced by irradiation damage under the experimental conditions promote the nucleation and growth of GBs. By calculating the energy barriers involved in the mass transport of S-atoms across the GBs, we show that the S-vacancies aid the migration of S-atoms in the pristine lattice, leading to the observed GB dynamics, where the vacancy-mediated atomic sliding-migration process and the beam-atom interaction are responsible. This mechanism is different from the doping effect of Re impurities reported previously.19 Our investigation of GBs in monolayer 2H-MoS2 not only deepens our understanding of the energetics and dynamic evolution of structural defects in 2D materials, but also offers insights into the design of GB networks in TMDs for their catalysis function, for example in the hydrogen evolution reaction.
2. Experimental and simulation methods
2.1 Sample preparation
Monolayer MoS2 samples were prepared through standard micromechanical exfoliation of mineral-form bulk MoS2 (SPI supplies), followed by deposition onto a silicon substrate using scotch tape. Under an optical microscope, identified monolayers were transferred onto lacey carbon TEM grids via a wet-chemistry lift-off process.2.2 TEM characterization and image simulation
All the annular dark field (ADF)-STEM experiments were conducted with a spherical-aberration corrected TEM (FEI Titan ChemiSTEM, probe corrected) at an acceleration voltage of 80 kV. The spherical aberration Cs has been optimized down to 2 μm. A probe current of ∼60 pA was chosen for ADF imaging to minimize the beam damage. The convergence angle α of the incident electron probe was set to 21 mrad, and the detection angle β of the ADF detector was set to 50–200 mrad. Experimental ADF images were processed through the well-known Wiener filtering to enhance the signal-to-noise ratio. ADF image simulation was carried out by QSTEM27 under the same parameter settings as the experiments such as Cs, α, and β besides the probe size ∼1.2 Å.2.3 First-principles calculations
We explored the structural and electronic properties of monolayer 2H-MoS2 with GBs identified in the experiments by performing first-principles calculations using plane-wave basis sets based density functional theory (DFT) methods. Ultrasoft (US) pseudopotentials28 were used for the core-valence electron interaction and the Perdew–Burke–Ernzerhof (PBE) parameterization29,30 of the generalized gradient approximation (GGA) was used for the exchange–correlation functional. We used the Vienna Ab initio Simulation Package (VASP)31 for DFT calculations, with the atomic models illustrated in Fig. 1a–c. For all results presented, an energy cutoff of 280 eV was used for plane-wave basis sets. For the study of GBs, we considered a 10 × 1 supercell, and a vacuum layer of 20 Å was used to isolate the single layers. A 1 × 6 × 1 Monkhorst–Pack mesh grid for sampling k points was set up for the Brillouin zone integration. These settings were verified by achieving a total energy convergence of less than 1 meV per atom. Geometrical relaxation was carried out before calculating the structural properties and electronic structures,32 until the force on atom was converged below a threshold of 0.01 eV Å−1. The formation energy of a GB, Ef = EGB − E0, is calculated from the relaxed structure of monolayer 2H-MoS2 with GBs and compared to the native monolayer 2H-MoS2. Here, EGB and E0 are the total energy of the GB-containing and pristine structures, respectively. The DFT calculation for α-GB has also been independently checked using CASTEP43 using equivalent parameters, with a similar qualitative result. The potential energy barrier Eb for the migration of a S atom across the GBs was calculated using the nudged elastic band (NEB) technique.33,34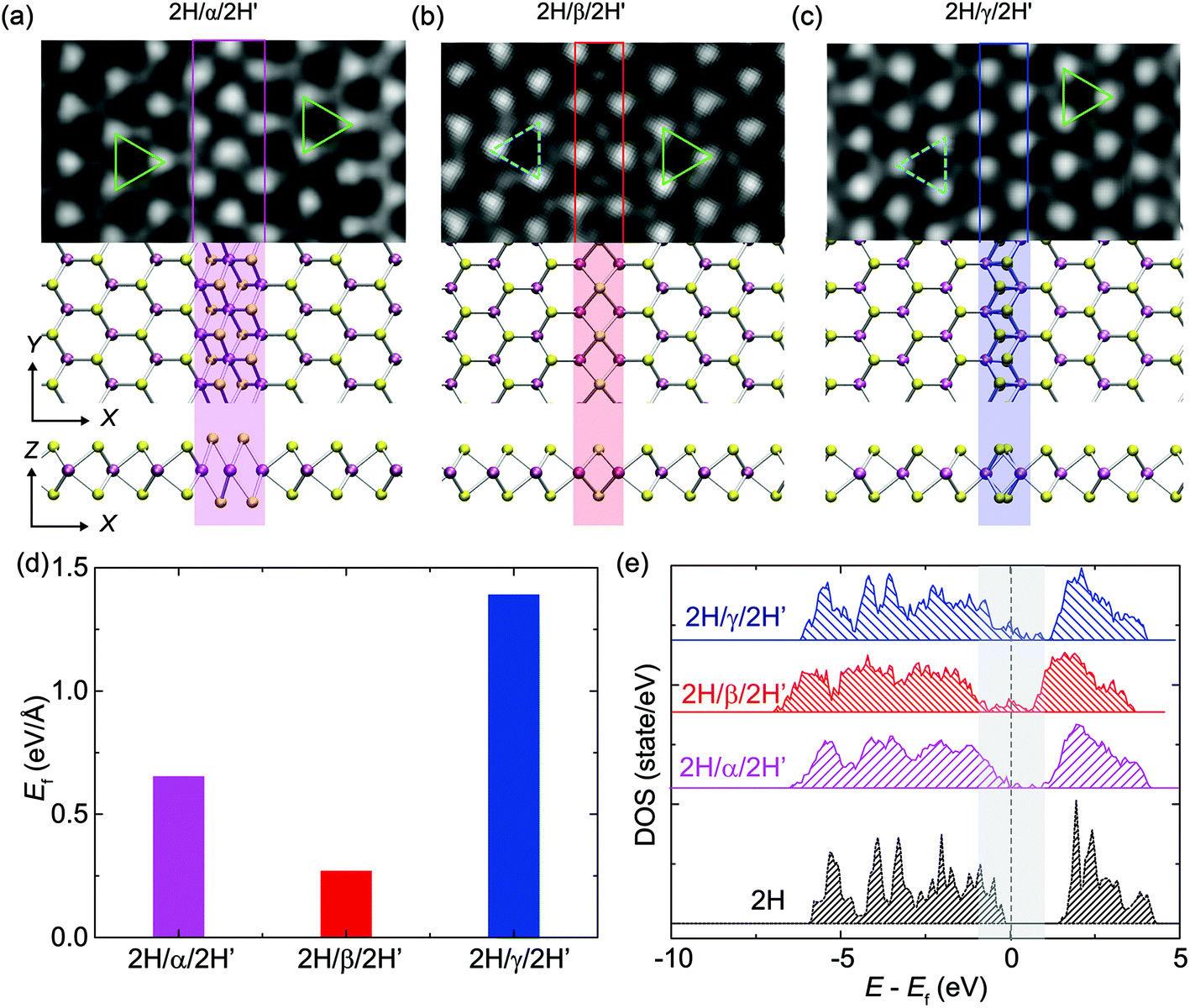 | ||
| Fig. 1 High-symmetry GBs in monolayer MoS2. (a–c) Experimental ADF-TEM images and top-/side-views of high-symmetry GBs (α, β, γ) along the zigzag direction of a hexagonal MoS2 lattice, where Mo and S atoms are colored in purple and yellow, respectively. The atomic models fit well with the ADF-STEM images. The green and cyan triangles indicate the orientation of both sides of these GBs. (d) Formation energies of the GBs obtained from DFT calculations. (e) Electronic DOS for the whole structure with GBs compared to that of crystalline 2H-MoS2. | ||
2.4 Empirical potential based calculations
DFT methods offer first-principles and an accurate description of interatomic interactions in materials, which is especially valuable for materials with complex structures and defects. However, because of their prohibitively high computational cost in the exploration of large systems with several hundreds of atoms, we used empirical potential based molecular dynamics (MD) simulations instead to study the structures for discussing the width/length distribution of GBs, and the interaction between mis-aligned GBs, where the model consists of hundreds to thousands atoms (see the ESI† for details). MD simulations are performed using the large-scale atomic/molecular massively parallel simulator (LAMMPS).35 The Stillinger–Weber (SW) potential recently developed for 2H-MoS2 was used in the calculations, which was validated by the calculations of its mechanical and thermal properties.36 Periodic boundary conditions were applied to a 2D supercell of the 2H-MoS2. In the MD simulations, the atomic structures of GBs were firstly optimized, and then equilibrated under ambient conditions (temperature T = 300 K) under a Nosé–Hoover thermostat37 for 50 ps. A time step of 0.5 fs was used to integrate the equations of motion. The atomic structures were further relaxed for 50 ps to evaluate the averaged potential energy for the formation energy Ef of 2H-MoS2 structures with GBs. The formation energies calculated from MD simulations are comparable to those obtained from DFT calculations.3. Results and discussion
3.1 Atomic structures and electronic properties of GBs in single-layer 2H-MoS2
Three major types of high-symmetry GBs (labeled as α, β and γ-types) are frequently observed along the zigzag direction of a monolayer 2H-MoS2 matrix, as illustrated in Fig. 1, where the experimental ADF images are in good agreement with the relaxed structural models obtained from DFT calculations. The cyan and green triangles in Fig. 1a mark the orientation of both sides of these GBs. Among them, the α-type GB can be regarded as a 0° GB, or named the strained T phase in the matrix of the 2H-phase as defined by Lin et al.19 For consistency, here we have followed their terminology in naming this defect “α-type GB” where GB normally refers to the interface between grains, although no new grains are observed during the structural evolution. β-type and γ-type GBs shown in Fig. 1b and c correspond to 4|4-fold rings-4|4P and 4|4E 60° GBs, respectively.6,22 The β-type GB is parallel to the zigzag direction (named 4|4P), while the γ-type GB is characterized by the shared zigzag edges (4|4E). It should be noted that the β-type GB is also known as the inversion domain boundary (IDB)21 or mirror twin boundary (MTB)23 in the literature.DFT calculations were employed to determine the formation energies of these GBs. The results are summarized in Fig. 1d, which shows that the β-type GB has the lowest formation energy of 0.21 eV Å−1, compared to 0.32 eV Å−1 for the α-type GB and 1.22 eV Å−1 for the γ-type GB, consistent with our experimental findings that the dominant GBs are β-type rather than other types of high-symmetry GBs in the MBE-grown MoS2 and MoSe2 monolayers.21Fig. 1e shows the electronic density-of-states (DOS) of 2H-MoS2 with and without the GBs. The pristine monolayer 2H-MoS2 sheet is found to be semiconducting with a band gap of 1.62 eV, aligning with other reports.38–40 To characterize the spatial distribution of electrons near the Fermi level, the partial charge densities with energy levels ranging from EF −0.5 eV to EF +0.5 eV are plotted in Fig. S1,† where EF is the Fermi energy. This charge analysis shows that electronic states near the Fermi level are localized at the GB. These GB-induced states located within the native gap could lead to a semiconductor-to-metal transition at the GB, where one-dimensional (1D) metallic states are reported.6,21 These in-gap states induced by GBs could also, in principle, benefit the catalytic function of TMDs in hydrodesulfurization or hydrogen evolution reaction where GBs behave as the active sites for catalysis.
3.2 Size statistics of α-type GBs in 2H-MoS2
In this section, we present a quantitative analysis of the formation and structural evolution of α-type GBs in the monolayer 2H-MoS2. Fig. 2a–e show a time sequence of ADF images for the birth and subsequent growth of α-type GBs at room temperature. The α-type GB (marked by yellow polygons in Fig. 2b), or the strained-T-phase, is formed coherently within the lattice of the 2H-MoS2 matrix, along the zigzag1,10 direction. Fig. 2f shows the atomic structures of α-type GBs, where one can see significant lattice distortion near the GB. Specifically, equilateral triangles of Mo sub-lattices have turned into isosceles. To quantify the in-plane strain distribution, we employed geometric phase analysis (GPA) on the TEM images. The results are summarized in Fig. 2g, from which we conclude that a typical α-type GB leads to a shear strain εxy up to +20% (Fig. 2h–i). The nucleation process of α-type GBs at room temperature without dopants involved here is different from the thermally activated mechanism near dopant atoms as reported in the literature.19 From this experimental evidence and our ab initio energetic calculation, we conclude that the sulfur vacancies nearby, marked by red arrows in Fig. 2a–e (also see Fig. S2† for the image simulation), could serve as important stimuli to the α-type GB formation.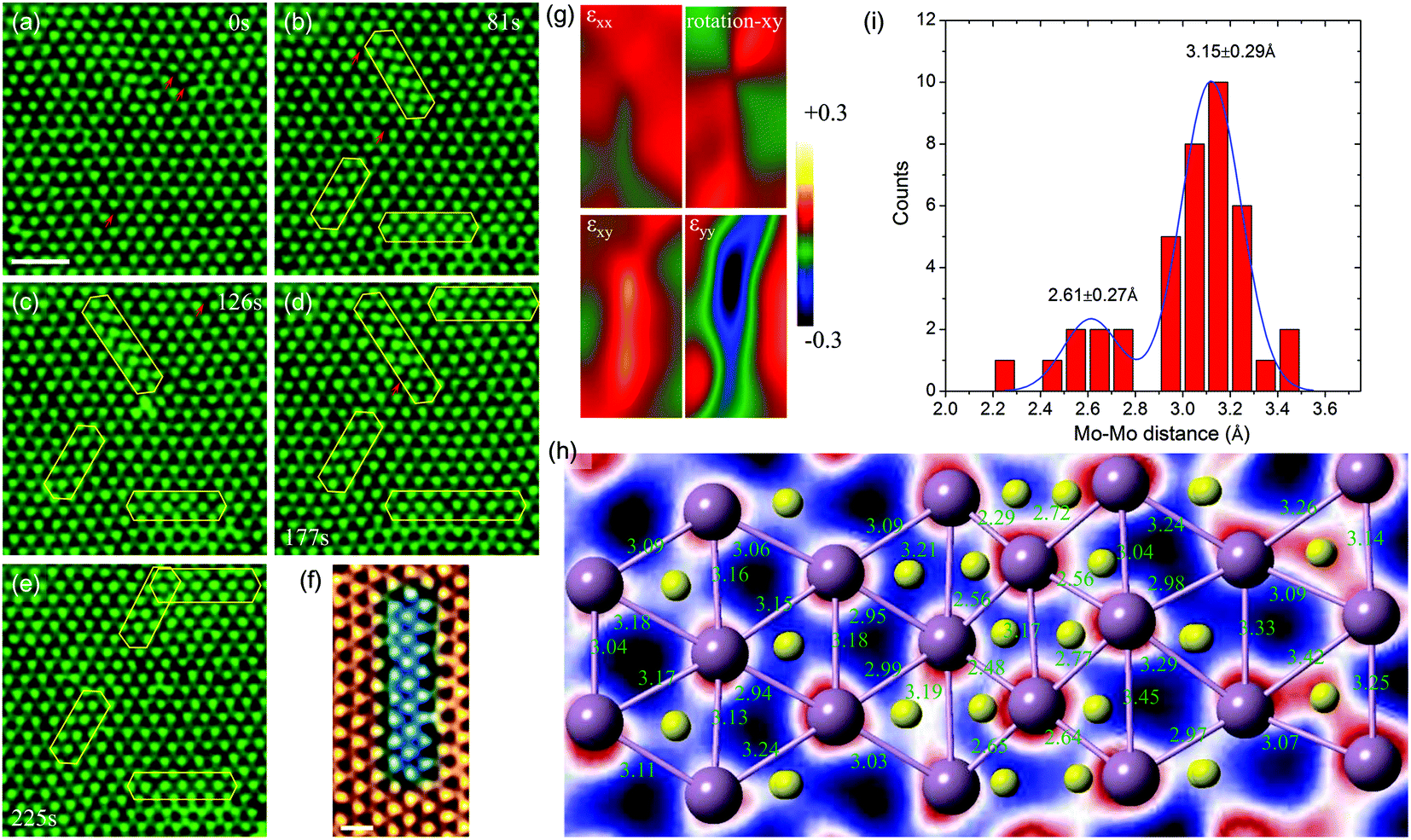 | ||
| Fig. 2 Dynamic evolution of α-type GBs and the strain in the lattice. (a) Initial state of the 2H-phase MoS2 matrix consisting of localized strained areas with sulfur vacancies (red arrows). Scale bar: 1 nm. (b–e) The evolution (birth, growth and movement) of α-type GBs. The red arrows indicate S vacancies in the lattice. (f) Detailed atomic structures of the α-type GB, which are highlighted in the blue color. Scale bar: 0.5 nm. (g) The lattice distortion induced by an α-type GB in panel (f) revealed by the geometric phase analysis (GPA), including normal strain in the x direction (εxx), the in-plane bond rotation-xy, the in-plane shear strain (εxy) and the normal strain in the y direction (εyy). The compression lattice strain is highly concentrated within the α-type GB. (h) Detailed atomic structures of the α-type GB with the nearest Mo–Mo pairs marked. (i) The histogram of Mo–Mo distances measured in panel (h). Note that the Mo–Mo distance, i.e. the lattice parameter a = 3.16 Å of the 2H phase, corresponds to the second peak, while the Mo–Mo distance within the α-type GB is reduced to 2.6 Å, leading to a large lattice strain as indicated in panel (g). | ||
TEM observation and statistics in the size distribution of over 60 α-type GBs (Fig. 3) show that these α-type GBs in the shape of nano-strips grow laterally only at the very early stage of GB evolution (Fig. 2 and 3b), while the widths are all three-Mo-column wide (Fig. 3a). This specific width preference corresponds to the width for the lowest GB formation energy ΔE in our empirical potential based calculations (Fig. 3c). According to the Boltzmann distribution factor pB = exp(−ΔE/kBT) calculated for thermal equilibrium at T = 300 K, the α-type GBs with this optimal width has ∼103 and ∼1015 higher probability to be observed than the GBs that are two- or more-than-three-Mo-columns in width (Fig. S3†). On the other hand, the length distribution of GB shown in Fig. 3b is centralized with a peak value of 6–7 Mo-sublattice units, which could be explained as follows. The formation energy of GBs increases almost linearly with the length of the embedded α-type GBs (inset of Fig. 3d), as indicated by our empirical potential based MD simulation results. However, the lateral growth/decay of GBs is a kinetic process. The nucleation of a single Mo unit with two terminations costs an energy of 2Ee = 0.021 eV per atom as the first step of α-type GB formation, where Ee is the formation energy of one Mo unit and the termination. Additional cost for increasing the GB length by one Mo unit, Eu = 0.00485 eV per atom, is however much lower. Consequently, the formation energy can be written as Ef = NuEu + 2Ee where the Nu (>0) is the length measured in the Mo unit. During the kinetic evolution process, short α-type GBs with very few (e.g. two) Mo unit lengths are very unstable due to the high formation energy of termination, and thus are not easy to be captured within the experimental time window. The longer α-type GBs, however, has a finite lifetime that could be captured in the time window of TEM observation, as thermal fluctuation can activate low-energy excitation. The abovementioned physical mechanisms can be quantified by applying a kinetic model41 and considering the Boltzmann factor of equilibrium distribution (see details in the ESI†). From the calculation results (Fig. 3d), we conclude that the probability of α-type GBs characterized in experiments is a function of the GB length, and the most probable length of GB is Nu = ∼7, which aligns with the fact that 6–7 Mo-sub-lattices are most common in the experiments.
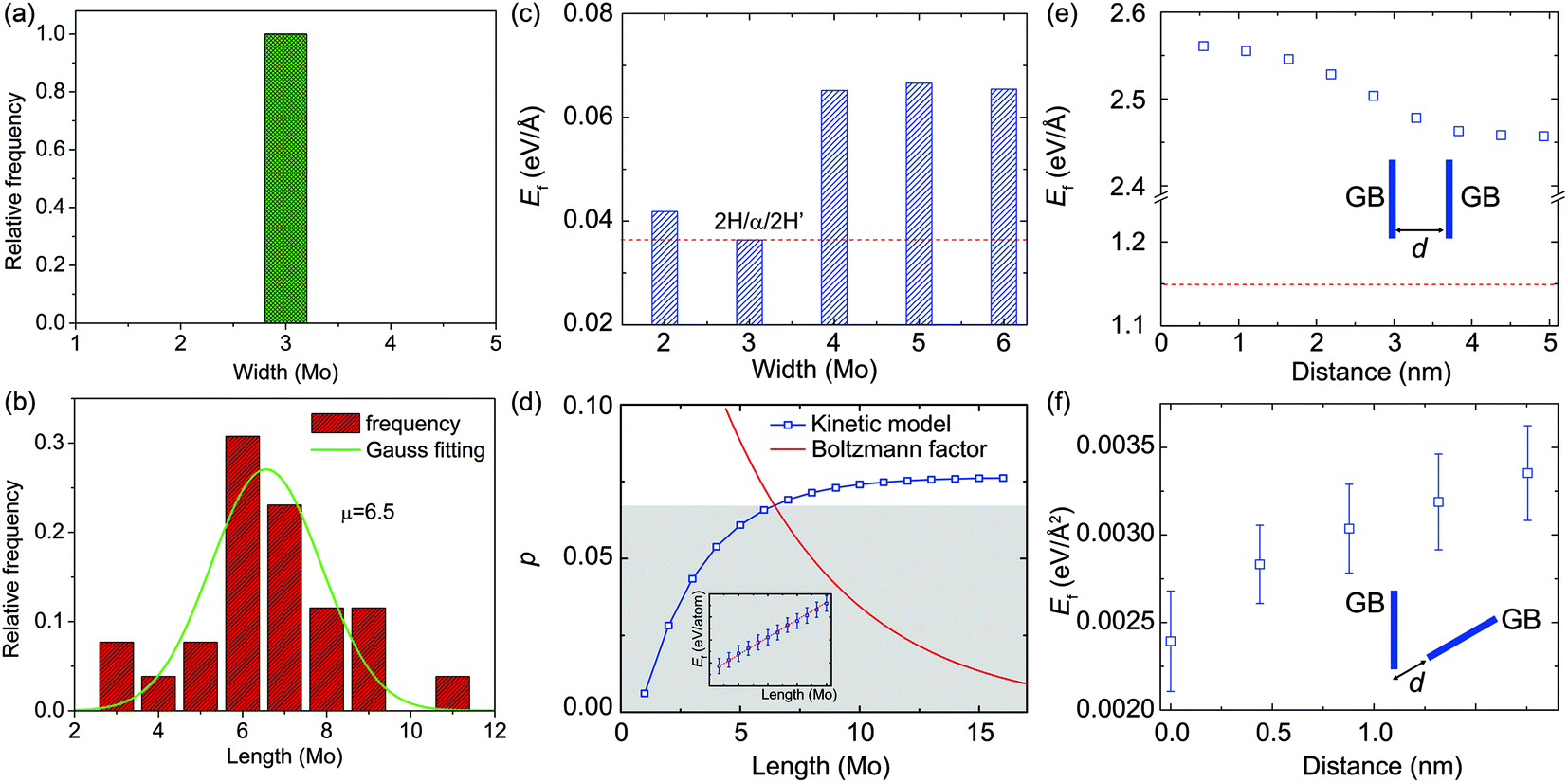 | ||
| Fig. 3 The size statistics of α-type GBs and the interactions of two α-type GBs. (a and b) Distribution of the width and length of α-type GBs characterized by TEM. The length distribution behaves as a Gaussian function with the average 6–7 Mo-units, and the width of nano-strip-like α-type GBs is always 3 Mo-units. (c) The dependence of formation energy Ef on the width of an α-type GB, calculated from empirical potential based MD simulations. (d) The probability, p, of α-type GBs plotted as a function of the length. The blue and red lines indicate the probability predicted from the kinetic model of GB dynamics and the Boltzmann factor, which suggest the most stable length of the GB to be ∼7 Mo-units. (e–f) The dependence of GB formation energy on the distance between two GBs mis-aligned by an angle of 0° and 60°, calculated from empirical potential based MD simulations. | ||
Once several α-type GBs are nucleated, the interaction between two α-type GBs also becomes important in generating high-density GB networks in monolayer 2H-MoS2. From our DFT calculations, we find that the average distance between neighboring Mo–Mo atoms, dMo–Mo, is 2.98 Å within the α-type GB, 3.18 Å in pristine matrix 2H-MoS2, and 3.31 Å in the GB/matrix interface. These results suggest that the GB is under compression with a strain of ∼−6.0%, while the region between the GB and pristine 2H-MoS2 is under tension with a strain of ∼4.1%. This indicates that two parallel α-type GBs are subjected to repulsion due to the tension strain in between when they are close to each other, while two non-parallel GBs could release the strain energy by approaching each other, where the compression and tension strain fields counteract. Specifically, our empirical potential based calculations show that the formation energy of two parallel α-type GBs decreases with the inter-GB distance, indicating the repulsive nature of interactions, while that of two α-type GBs mis-aligned by 60° angle increases with the inter-GB distance, which indicates an effective attraction between them (Fig. 3e and f). These results explain why (from the TEM images) two parallel GBs are all very well separated while the GBs mis-aligned by 60° angle prefer to merge by joining the ends (Fig. 2e).
3.3 Atomistic dynamics of inversion domains enclosed by β-type and γ-type GBs
It has been found that β- and γ-type GBs could be formed within the monolayer MoSe2 due to the deficiency of chalcogen atoms caused by the electron beam irradiation,22 which were also frequently observed in MoS2 under our experimental conditions (Fig. S4†). Interestingly, in addition to the GB growth, we also sometimes captured the inversed process – the annihilation of GBs, which has not been well documented for TMDs in the literature. Fig. 4 demonstrates that the inversed domain is enclosed by a triangle composed of one β-type GB and two γ-type GBs. We have resolved structural models corresponding to the experimental series (I–IV), highlighting the atomic processes involved. It should be noted that the MoS2 lattice inside the domain is still in the 2H phase, but in a 180° inversion with respect to the outer matrix. These GBs are high-symmetry 60° GBs along the zigzag direction of 2H-MoS2. From the TEM images of the domain evolution, we find that the γ-type GBs migrate towards the domain center and lengths of both the β-type and γ-type GBs decrease, through the migration of the S-atom from the γ-type GB to the domain center. As a result, the GBs are shortened, the inversed domain shrinks and eventually reconstructed to the 2H-matrix. The calculated energies along this evolution pathway, using the empirical potential method (Fig. 4), verify this mechanism and show that the total energy decreases dramatically during the annihilation of the IDB by relieving the lattice strain. As both β-type and γ-type GBs are sulfur deficient, sulfur atoms in the 2H phase outside the triangles must participate in the beam-driven conveyance of S-atoms to heal the GBs, leaving S-vacancies in the 2H-MoS2 lattices accompanied by the annihilation of GBs. This indicates that both experimentally observed beam-induced growth22 and annihilation of GB-bound inversion domains are complex dynamical processes involving different kinetic pathways. The former domain growth process is initialized at the beam-induced vacancy sites (line defects) in normal 2H lattices under beam irradiation, while the latter annihilation process starts from S-deficient GBs of the inverse domains and turns into normal 2H lattices with the S supply nearby the domains. The roles played by interaction with either the chalcogen vacancy and/or mobile chalcogen adatoms under beam irradiation will be further investigated.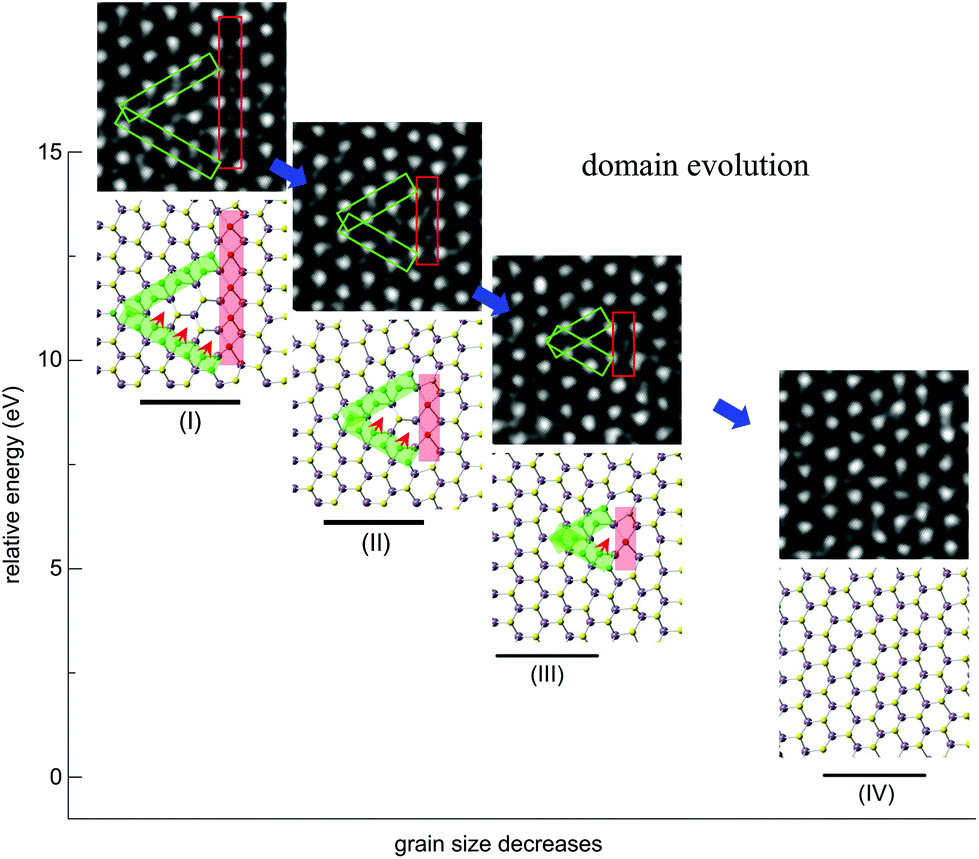 | ||
| Fig. 4 Atomic processes of the GB evolution. Experimentally observed annihilation processes (I–IV) of the inversion domain consisting of one β-type GB (the red rectangle) and two γ-type GBs (green rectangles). Scale bar: 0.5 nm. In the resolved atomic models corresponding to the experimental image series, green rectangles indicate the γ-type GBs and the red ones correspond to β-type GBs. Red arrows mark the migration path of S-atoms in the lattice, from within the γ-type boundary to the central inversion domain in the 2H phase. S-atoms in the green γ-type GBs migrate onto the hollow sites inside as indicated by the red arrows, leading to the shift of γ-type GBs. Empirical potential based MD simulation results show that the total energy decreases dramatically with the evolution of GBs. | ||
A common feature between α-, β- and γ-type GB nanostructures is the remarkable lattice distortion (Fig. 2 and 4). Compared to the α-type GBs, strain distributions at the β-type and γ-type GBs exhibit local dipoles, indicating the presence of a dislocation pair in the atomic structures (Fig. S5†). The lattice discontinuity and deformation revealed by our GPA analysis exactly reflect the intrinsic dislocation characteristics of β-type and γ-type GBs, aligning with the fact that GB is often regarded as assembled dislocations in the literature.42
3.4 The effect of S-vacancies on the GB dynamics
For the structural evolution of GBs characterized in Fig. 2 and 4, we always observed that there were sulfur vacancies nearby the GBs, which may account for the observed GB dynamics. To clarify this point, DFT calculations were carried out to reveal the dependence of the S-vacancy presence on the relative formation energy, Ef, of the GBs and the results are summarized in Fig. 5. For the sake of simplicity, we plot the dependence of Ef on the vacancy-GB distance d with the presence of only one vacancy nearby (Fig. 5a). We find that Ef is reduced as d decreases, indicating that the incorporation of S vacancy nearby stabilizes the α-type and γ-type GBs by releasing strain in the distorted lattice containing GBs. We then consider multiple S-vacancies in the 2H phase with the same distance of three Mo-units to the center of GB and the calculated values of Ef behave as a function of the number of vacancies (Fig. 5b). The formation energy of GBs decreases with the number of S-vacancies, indicating that a high concentration of S-vacancies can promote the formation of GBs.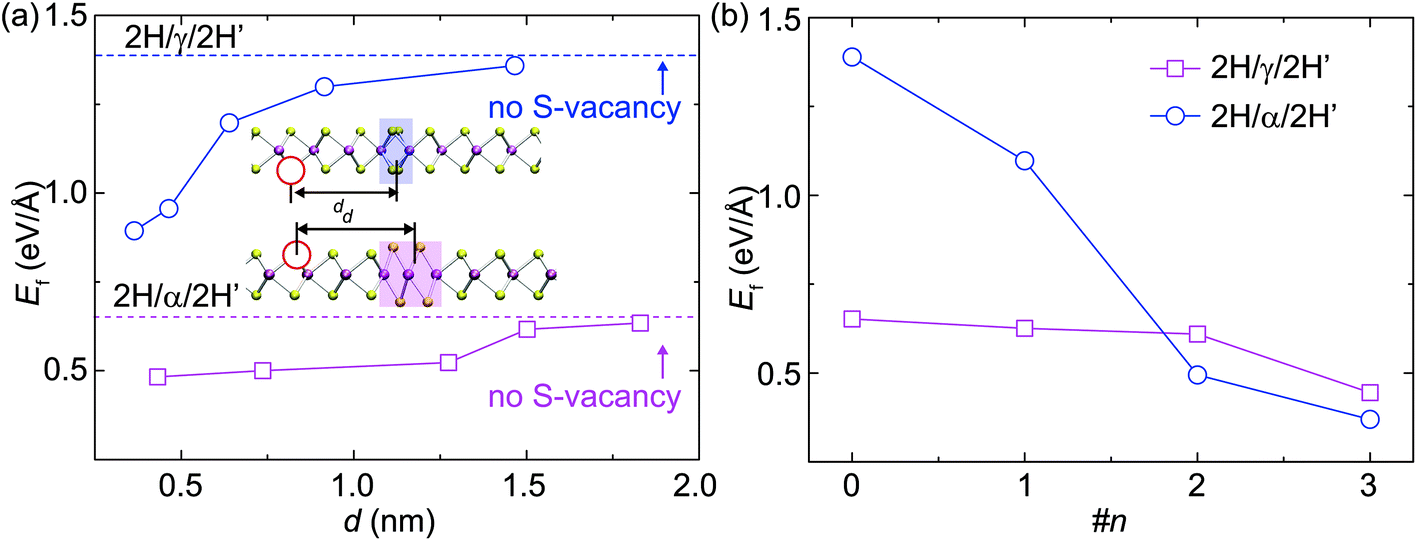 | ||
| Fig. 5 Effects of S-vacancies on the DFT-calculated formation energies of GBs. (a) The formation energies of α-type and γ-type GBs plotted as a function of the distance d between the S-vacancy and the geometrical center of GB. (b) The formation energies of α-type and γ-type GBs plotted as a function of the number of vacancies nearby. | ||
As highlighted by the red arrows in Fig. 2 and 4, the S-atom migration via a vacancy-mediated mechanism occurs during the formation of α-type GBs and in the dynamics of γ-type GBs, for example. Migration of S atoms mediated via the vacancy mechanism actually dominates the atomic processes (Fig. S6†) of α-type (Fig. 2) and γ-type (Fig. 4) GBs. To visualize this process, we highlight the slipped S atoms in green color (Fig. 4 and S6†), underneath which are other S-atoms (yellow) in their sub-lattices. Atomic migration and lattice collapse induced by these vacancies will then lead to α-type GB formation. Similarly, S atoms within the γ-type GBs migrate to the neighboring hollow sites along the red arrows in Fig. 4, leading to a collective shift of the γ-type GB. As the inversed domain shrinks, the migration of S-atoms compensates the S-deficiency in β-type and γ-type GBs and gradually turns the shortening β-type and γ-type GBs into the normal 2H phase.
To explore the energetics of S-vacancy migrating from α-type and γ-type GBs to the neighboring 2H phase of MoS2, we perform DFT calculations for the potential barriers of such processes across different types of GBs using the NEB technique. From the values of energy barriers, Eb, summarized in Fig. 6, we conclude that Eb is 0.86 and 1.97 eV per atom for S-atom migration across α-type and γ-type GBs, respectively. In our experiments, the GB dynamics observed at room temperature only occurs in the presence of an electron beam and thus the beam-specimen interaction must be a part of the driving force as well. The energy barriers for the S-atoms to migration across GBs are much higher than that available by thermal activation. Although the overall process is exothermal, beam irradiation is still necessary to accelerate the dynamics by transferring energies to GB atoms to overcome the barriers. The energy transfer due to elastic electron-nuclei collision may cause the slippage of S-layers during the GB evolution dynamics. As shown in Fig. S7,† the energy transfer onto the lattice S atom is about 5.87 eV, which is much higher than the energy barriers calculated (<2 eV) and sufficient to drive the S-atom migration via the vacancy mechanism. The beam irradiation thus assists sulfur vacancy to act as an active conveyance of S-atoms that dominates the GB evolution.
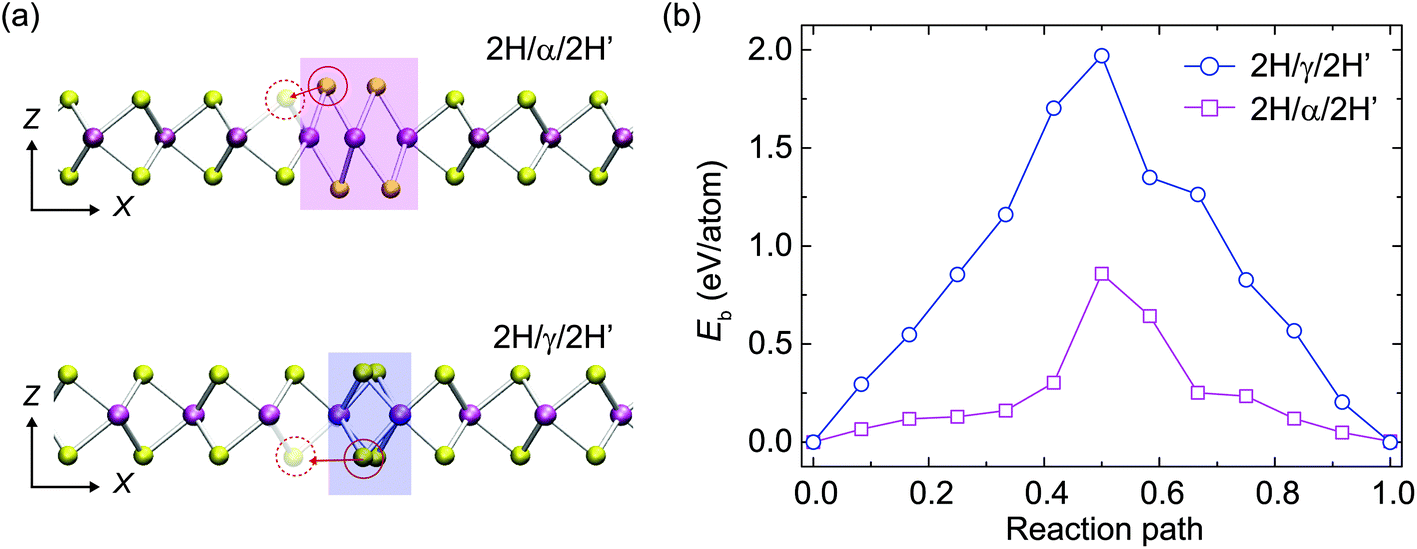 | ||
| Fig. 6 Migration of S-atoms across a GB via a vacancy-mediated mechanism, which constitutes the basic step of GB dynamics observed in the experiments. (a) S-migration from a GB to the S-vacancy nearby for α-type and γ-type GBs. (b) The energy profiles calculated from DFT for a S-atom to migrate across a GB to the S-vacancy nearby for α-type and γ-type GBs. | ||
With these results, one may wonder why the experimentally observed α-type GBs are the most common GBs characterized, as our DFT calculation shows that the β-type GBs have the lowest formation energy among all the three types. During the normal growth process of TMDs in thermal equilibrium, the β-type GBs were also observed experimentally to be the most abundant in STM and TEM for MBE-grown TMDs, which supports the conclusion from our DFT calculations. However, under the electron beam irradiation, α-GBs appear extrinsically and more frequently within the normal 2H domain. Compared to the simple S-atom migration involved in the α-type GB formation, both Mo and S atoms will have to slide (driven by beam radiation) to form the β-type and γ-type GBs. Consequently, among these extrinsic GBs induced by beam radiation, the formation of α-type GBs is more probable than the other types in in situ TEM observation.
4. Conclusions
In summary, we present a joint experiment-theory investigation of high-symmetry GBs (α-, β- and γ-types) in monolayer 2H-MoS2. The study shows that α-type GBs are always three-Mo-wide and six- or seven-Mo-long, consistent with the calculated formation energies and evolutionary kinetics of the GBs. Moreover, the interaction between parallel α-type GBs is repulsive, but attractive for those mis-aligned by 60° angle. The role of S-vacancy in GB formation is studied and we find that the formation energies of α- and γ-type GBs decrease as the density of S-vacancy increases and the vacancy-GB distance decreases. The S-vacancy helps to reduce the formation energy of GBs and thereby promotes the GB formation. The vacancy-induced sliding-migration processes of S-atoms are analyzed to understand the dynamic evolution of GBs. It is believed that electron irradiation induced S-vacancy serves as an active conveyance of S-atoms in promoting the GB evolution. In principle, our investigation on high-symmetry GBs can be extended to GBs with ordinary inter-domain mis-orientation angles in other chemical vapor deposited TMDs. The findings will also advise the control of GBs in 2D materials toward a specific GB network for catalytic applications such as the hydrogen evolution reaction.Acknowledgements
We acknowledge the financial support by the National Basic Research Program of China (Grant No. 2014CB932500 and No. 2015CB921004), the National Science Foundation of China (Grant No. 51472215 and No. 51222202) and the 111 project (Grant No. B16042). Y. W. and Z. X. acknowledge the State Key Laboratory of Mechanics and Control of Mechanical Structures, Nanjing University of Aeronautics and Astronautics (Grant No. MCMS-0414G01). J. Y. acknowledges the EPSRC (UK) funding EP/G070326 and support from Pao Yu-Kong International Foundation for a Chair Professorship in ZJU. M. P. acknowledges the EPSRC funding EP/K013564/1. The computation was performed on the Explorer 100 cluster system at Tsinghua National Laboratory for Information Science and Technology. The experimental work was carried out in the Center of Electron Microscopy of Zhejiang University.References
- C. R. M. Grovenor, J. Phys. C: Solid State Phys., 1985, 18, 4079 CrossRef CAS.
- D. MacLean, Grain Boundaries in Metals, Clarendon Press, Oxford, 1957 Search PubMed.
- W. Read and W. Shockley, Phys. Rev., 1950, 78, 275 CrossRef CAS.
- S. Najmaei, Z. Liu, W. Zhou, X. L. Zou, G. Shi, S. D. Lei, B. I. Yakobson, J. C. Idrobo, P. M. Ajayan and J. Lou, Nat. Mater., 2013, 12, 754–759 CrossRef CAS PubMed.
- A. M. van der Zande, P. Y. Huang, D. A. Chenet, T. C. Berkelbach, Y. M. You, G. H. Lee, T. F. Heinz, D. R. Reichman, D. A. Muller and J. C. Hone, Nat. Mater., 2013, 12, 554–561 CrossRef CAS PubMed.
- W. Zhou, X. L. Zou, S. Najmaei, Z. Liu, Y. M. Shi, J. Kong, J. Lou, P. M. Ajayan, B. I. Yakobson and J. C. Idrobo, Nano Lett., 2013, 13, 2615–2622 CrossRef CAS PubMed.
- Q. C. Li, X. L. Zou, M. X. Liu, J. Y. Sun, Y. B. Gao, Y. Qi, X. B. Zhou, B. I. Yakobson, Y. F. Zhang and Z. F. Liu, Nano Lett., 2015, 15, 5804–5810 CrossRef CAS PubMed.
- M. S. Kim, S. J. Yun, Y. Lee, C. Seo, G. H. Han, K. K. Kim, Y. H. Lee and J. Kim, ACS Nano, 2016, 10, 2399–2405 CrossRef CAS PubMed.
- K. Q. Dang and D. E. Spearot, J. Appl. Phys., 2014, 116, 013508 CrossRef.
- Z. Fei, A. S. Rodin, W. Gannett, S. Dai, W. Regan, M. Wagner, M. K. Liu, A. S. McLeod, G. Dominguez, M. Thiemens, A. H. C. Neto, F. Keilmann, A. Zettl, R. Hillenbrand, M. M. Fogler and D. N. Basov, Nat. Nanotechnol., 2013, 8, 821–825 CrossRef CAS PubMed.
- V. K. Sangwan, D. Jariwala, I. S. Kim, K. S. Chen, T. J. Marks, L. J. Lauhon and M. C. Hersam, Nat. Nanotechnol., 2015, 10, 403–406 CrossRef CAS PubMed.
- A. W. Tsen, L. Brown, M. P. Levendorf, F. Ghahari, P. Y. Huang, R. W. Havener, C. S. Ruiz-Vargas, D. A. Muller, P. Kim and J. Park, Science, 2012, 336, 1143–1146 CrossRef CAS PubMed.
- W. Bao, N. J. Borys, C. Ko, J. Suh, W. Fan, A. Thron, Y. J. Zhang, A. Buyanin, J. Zhang, S. Cabrini, P. D. Ashby, A. Weber-Bargioni, S. Tongay, S. Aloni, D. F. Ogletree, J. Q. Wu, M. B. Salmeron and P. J. Schuck, Nat. Commun., 2015, 6, 7993 CrossRef CAS PubMed.
- D. A. Muller, L. F. Kourkoutis, M. Murfitt, J. H. Song, H. Y. Hwang, J. Silcox, N. Dellby and O. L. Krivanek, Science, 2008, 319, 1073–1076 CrossRef CAS PubMed.
- H. I. Rasool, C. Ophus, Z. Zhang, M. F. Crommie, B. I. Yakobson and A. Zettl, Nano Lett., 2014, 14, 7057–7063 CrossRef CAS PubMed.
- A. Azizi, X. Zou, P. Ercius, Z. Zhang, A. L. Elías, N. Perea-López, G. Stone, M. Terrones, B. I. Yakobson and N. Alem, Nat. Commun., 2014, 5, 4867 CrossRef CAS PubMed.
- S. Kurasch, J. Kotakoski, O. Lehtinen, V. Skákalová, J. Smet, C. E. Krill III, A. V. Krasheninnikov and U. Kaiser, Nano Lett., 2012, 12, 3168–3173 CrossRef CAS PubMed.
- O. Lehtinen, S. Kurasch, A. Krasheninnikov and U. Kaiser, Nat. Commun., 2013, 4, 2098 CAS.
- Y. C. Lin, D. O. Dumcencon, Y. S. Huang and K. Suenaga, Nat. Nanotechnol., 2014, 9, 391–396 CrossRef CAS PubMed.
- W. Zhao and F. Ding, Nanoscale, 2017, 9, 2301–2309 RSC.
- H. J. Liu, L. Jiao, F. Yang, Y. Cai, X. X. Wu, W. K. Ho, C. L. Gao, J. F. Jia, N. Wang, H. Fan, W. Yao and M. H. Xie, Phys. Rev. Lett., 2014, 113, 066105 CrossRef PubMed.
- J. H. Lin, S. T. Pantelides and W. Zhou, ACS Nano, 2015, 9, 5189–5197 CrossRef CAS PubMed.
- O. Lehtinen, H. P. Komsa, A. Pulkin, M. B. Whitwick, M. W. Chen, T. Lehnert, M. J. Mohn, O. V. Yazyev, A. Kis, U. Kaiser and A. V. Krasheninnikov, ACS Nano, 2015, 9, 3274–3283 CrossRef CAS PubMed.
- S. Barja, S. Wickenburg, Z. F. Liu, Y. Zhang, H. J. Ryu, M. M. Ugeda, Z. Hussain, Z. X. Shen, S. K. Mo, E. Wong, M. B. Salmeron, F. Wang, M. F. Crommie, D. F. Ogletree, J. B. Neaton and A. Weber-Bargioni, Nat. Phys., 2016, 12, 751–756 CrossRef CAS.
- Y. L. Huang, Y. F. Chen, W. J. Zhang, S. Y. Quek, C. H. Chen, L. J. Li, W. T. Hsu, W. H. Chang, Y. J. Zheng, W. Chen and A. T. S. Wee, Nat. Commun., 2015, 6, 6298 CrossRef PubMed.
- Y. L. Huang, Z. J. Ding, W. J. Zhang, Y. H. Chang, Y. M. Shi, L. J. Li, Z. B. Song, Y. J. Zheng, D. Z. Chi, S. Y. Quek and A. T. S. Wee, Nano Lett., 2016, 16, 3682–3688 CrossRef CAS PubMed.
- T. H. Ly, D. J. Perello, J. Zhao, Q. Deng, H. Kim, G. H. Han, S. H. Chae, H. Y. Jeong and Y. H. Lee, Nat. Commun., 2016, 7, 10426 CrossRef CAS PubMed; C. T. Koch, QSTEM (2010), Determination of core structure periodicity and point defect density along dislocations, Ph.D thesis, Arizona State University, Tempe, AZ, 2002 Search PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter, 1990, 41, 7892–7895 CrossRef.
- J. P. Perdew and A. Zunger, Phys. Rev. B: Condens. Matter, 1981, 23, 5048–5079 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169–11186 CrossRef CAS.
- Y. Wang and Z. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 1970–1976 CAS.
- D. Sheppard, P. Xiao, W. Chemelewski, D. D. Johnson and G. Henkelman, J. Chem. Phys., 2012, 136, 074103 CrossRef PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- J.-W. Jiang, H. S. Park and T. Rabczuk, J. Appl. Phys., 2013, 114, 064307 CrossRef.
- D. J. Evans and B. L. Holian, J. Chem. Phys., 1985, 83, 4069–4074 CrossRef CAS.
- Q. Tang and D.-E. Jiang, Chem. Mater., 2015, 27, 3743–3748 CrossRef CAS.
- T. S. Li and G. L. Galli, J. Phys. Chem. C, 2007, 111, 16192–16196 CAS.
- S. Lebegue and O. Eriksson, Phys. Rev. B: Condens. Matter, 2009, 79, 115409 CrossRef.
- P. L. Krapivsky, S. Redner and E. Ben-Naim, A Kinetic View of Statistical Physics, Cambridge University Press, United Kingdom, 2010 Search PubMed.
- O. V. Yazyev and S. G. Louie, Phys. Rev. B: Condens. Matter, 2010, 81, 195420 CrossRef.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr02941a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |