
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolylactic acid nano- and microchamber arrays for encapsulation of small hydrophilic molecules featuring drug release via high intensity focused ultrasound†
Meiyu
Gai
b,
Johannes
Frueh
 *a,
Tianyi
Tao
b,
Arseniy V.
Petrov
c,
Vladimir V.
Petrov
c,
Evgeniy V.
Shesterikov
d,
Sergei I.
Tverdokhlebov
d and
Gleb B.
Sukhorukov
*bce
*a,
Tianyi
Tao
b,
Arseniy V.
Petrov
c,
Vladimir V.
Petrov
c,
Evgeniy V.
Shesterikov
d,
Sergei I.
Tverdokhlebov
d and
Gleb B.
Sukhorukov
*bce
aMicro/Nano Technology Research Centre, Harbin Institute of Technology, Yikuang Street 2, Harbin 150080, China. E-mail: johannes.frueh@hit.edu.cn
bQueen Mary University of London, School of Engineering and Materials Science, Mile End, Eng, 215, London E1 4NS, UK. E-mail: g.sukhorukov@qmul.ac.uk
cRemote Controlled Theranostic Systems Lab, Educational Research Institute of Nanostructures and Biosystems, Saratov State University, 83 Astrakhanskaya Street, Saratov 410012, Russia
dExperimental Physics Department, Tomsk Polytechnic University, 30 Lenin Avenue, 634050 Tomsk, Russia
eRASA center, Tomsk Polytechnic University, 30 Lenin Avenue, Tomsk 634050, Russia
First published on 17th May 2017
Abstract
Long term encapsulation combined with spatiotemporal release for a precisely defined quantity of small hydrophilic molecules on demand remains a challenge in various fields ranging from medical drug delivery, controlled release of catalysts to industrial anti-corrosion systems. Free-standing individually sealed polylactic acid (PLA) nano- and microchamber arrays were produced by one-step dip-coating a PDMS stamp into PLA solution for 5 s followed by drying under ambient conditions. The wall thickness of these hydrophobic nano–microchambers is tunable from 150 nm to 7 μm by varying the PLA solution concentration. Furthermore, small hydrophilic molecules were successfully in situ precipitated within individual microchambers in the course of solvent evaporation after sonicating the PLA@PDMS stamp to remove air-bubbles and to load the active substance containing solvent. The cargo capacity of single chambers was determined to be in the range of several picograms, while it amounts to several micrograms per cm2. Two different methods for sealing chambers were compared: microcontact printing versus dip-coating whereby microcontact printing onto a flat PLA sheet allows for entrapment of micro-air-bubbles enabling microchambers with both ultrasound responsiveness and reduced permeability. Cargo release triggered by external high intensity focused ultrasound (HIFU) stimuli is demonstrated by experiment and compared with numerical simulations.
1. Introduction
Encapsulation systems are urgently needed for active chemicals’ delivery to encapsulate enzymes, cells, dyes, drugs or corrosion inhibitors for an extensive variety of advanced applications.1 Micropacking especially is applicable for a wide range of applications, including drug delivery,2,3 diagnostics,4 tissue engineering,5 lab-on-chip,6 corrosion inhibition,7 bio-sensors,8 membranes, templates for synthesis, optical systems,9 antifouling,10,11 self-healing materials1 and coatings.7 Small molecule (Mw < 1 kDa) encapsulation remains a bottleneck, due to the commonly employed encapsulation matrix (e.g. polyelectrolyte multilayers, liposomes) being nano-porous.12 Therefore, long term encapsulation remains a challenge especially in the field of small hydrophilic molecule delivery as well as in various microreactors and anti-corrosion applications.13A recent approach to fabricate polyelectrolyte multilayer (PEM) microchambers based on layer-by-layer (LbL) self-assembly empowered individual PEM chambers to load microparticles with a size of 2.2 μm and oil-based solutions for light sensitive release-on-demand of the said cargo.14 Nevertheless, in this method, a minimum amount of PEM bilayers is a prerequisite to ensure mechanical stability,15 resulting in time consuming (3–4 days) lab work.16 Furthermore the inherent high permeability of LbL microchambers as well as microcapsules for small molecules is only slowly reduced with increasing bilayer number.17 Other approaches utilizing biodegradable layered films produced chambers only able to encapsulate hydrophobic polymers like polystyrene.18 Current attempts to seal PEM thin films demonstrated significantly lower permeability for hydrophobic compared to hydrophilic PEM films, due to higher chain packing density, which was achieved by altering deposition conditions.19 For microcapsules, increased hydrophobic barrier properties of the PEM wall were achieved either by incorporation or in situ synthesis of nanoparticles as well as by photochemistry.20 Photochemical wall sealing relies on UV exposure for hours, whereby the in situ synthesis of carbon dots requires 20 hours at high pressure and temperature, preventing the utilization of biomolecules, yet even more their applications within biological systems.21
Except for encapsulation, triggerable drug delivery systems are being intensively studied, which enable controlled release on-demand profiles to enhance therapeutic effectiveness and reduce systemic toxicity.22 Until now, different external stimuli for drug release, such as pH, temperature, laser and microwave radiation have been utilized to alter the drug-carrier shell permeability and facilitate controlled release.23 Of the aforementioned methods, the laser offers a low penetration depth (∼1 cm), whereby changing the pH and temperature of the human torso causes undesirable side effects. Therefore these release stimuli are expected to offer limited application for biological and medical systems.24 Microwaves offer higher penetration depths compared to the laser; however the detailed microwave–drug container interaction is not clear.25 In contrast, ultrasound is already employed as a diagnostic and therapeutic method for many diseases (i.e., prostate cancer, kidney stones). Particularly, high intensity focused ultrasound (HIFU), due to its long penetration depth and non-invasive nature has received extensive attention from biomedical scientists,26 enabling HIFU to become one of the most promising external stimuli for controlled drug release.27
Inspired by the fact that hydrophobic polymers possess efficient barrier properties for small hydrophilic molecules, a novel method to design and fabricate individually sealed polylactic acid (PLA) microchambers is reported in this work. Polylactic acid is a hydrophobic, biodegradable thermoplastic aliphatic polyester derived from naturally occurring organic acid (lactic acid).28 Due to its biocompatibility, complete biodegradability and the non-toxic nature of degradation products, PLA based materials are ideal for polymer engineering, tissue engineering and various medical implants.29 In these biomedical applications,29 implants with restricted load like rods, plates and pins in bone, surgical sutures as well as dispersed drug delivery systems are made out of PLA.30–33
We demonstrate the fabrication of sealed, biocompatible and biodegradable PLA nano- and microchamber arrays by one-step dip-coating and microcontact printing. In order to examine the feasibility of small molecule encapsulation in situ within individual microchambers and further investigate their release behaviour under HIFU treatment, the fluorescent dye Rhodamine B (RhB) (Mw = 479) and sodium chloride were chosen as model cargo substances. Furthermore, two different methods for chamber sealing were employed: dip-coating and microcontact printing,34–36 respectively, whereby microcontact printing offers cargo sealing within entrapped micro-air bubbles. Air bubbles support encapsulation of the cargo up to 14 days under submerged conditions and enhance ultrasound responsiveness, allowing for spatiotemporal release upon HIFU exposure. Experimental demonstrations are compared with numerical defined finite element (DFE) simulations.
2. Experimental section
2.1 Materials
Polylactic acid biopolymer ((PLA), 3 mm nominal granule size, weight 100 g, natural source) was purchased from Sigma-Aldrich. The poly(dimethylsiloxane) (PDMS) kit (Sylgard 184) was purchased from Dow-Corning, Midland, USA. In addition, PDMS (Elastosil, RT 602) from Wacker AG, München, Germany was used. Sodium chloride and chloroform were purchased from Sigma. Deionized (DI) water from a Milli-Q (Millipore) water purification system was utilized to make all solutions. All chemicals were used as received without further purification.2.2 Fabrication of sealed free-standing PLA microchambers
Patterned PDMS stamps with microwells were prepared by casting the PDMS pre-polymer and curing agent (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) onto silicon masters with nano- or micro-pillars, degassing it for 30 minutes in vacuum and curing it at 70 °C (Sylgard 184 needed 3 hours for curing, Elastosil RT 602 needed 1 hour). The silicon master for micropattern creation exhibits round micropillars with a diameter and a height of 5 μm and 4 μm, respectively, featuring a centre to centre distance of 15 μm (ESIFig. S1†) and it was prepared by traditional photolithography and etching processes at Shenzhen Semiconductor, Shenzhen, China. The silicon master for nanochambers (500 nm diameter, 1 μm height, 1.5 μm centre to centre distance) was prepared via electron lithography at Tomsk Polytechnic University. The PDMS was cut with a sharp surgery knife and lifted off the silicon master.
:1 ratio) onto silicon masters with nano- or micro-pillars, degassing it for 30 minutes in vacuum and curing it at 70 °C (Sylgard 184 needed 3 hours for curing, Elastosil RT 602 needed 1 hour). The silicon master for micropattern creation exhibits round micropillars with a diameter and a height of 5 μm and 4 μm, respectively, featuring a centre to centre distance of 15 μm (ESIFig. S1†) and it was prepared by traditional photolithography and etching processes at Shenzhen Semiconductor, Shenzhen, China. The silicon master for nanochambers (500 nm diameter, 1 μm height, 1.5 μm centre to centre distance) was prepared via electron lithography at Tomsk Polytechnic University. The PDMS was cut with a sharp surgery knife and lifted off the silicon master.
PLA was dissolved in chloroform at different mass concentrations (m% = 1%, 2% and 5%) to determine the influence of PLA concentration on the resulting film thickness. The PDMS stamp was dip-coated for 5 s into the PLA solution, via a dip-coating robot37 which featured an air pressure controlled robot arm performing pull-out at a speed of 8 cm s−1. By the same method, a flat PLA sheet was coated with PLA by dipping a PDMS sheet without patterns for 5 s at the same pull-out speeds. The patterned PDMS stamp@PLA microchamber was sealed by printing it onto a flat PDMS sheet@PLA under pressure (2 kg cm−2) for 10 s; subsequently the sealed PLA microchamber was collected after lifting off the PDMS stamp.
2.3 NaCl and RhB encapsulation
To examine the feasibility of small molecule encapsulation within individual polylactic acid (PLA) microchambers and to further investigate their release behaviour under High Intensity Focused Ultrasound (HIFU) radiation, sodium chloride (Mw = 58.44) and a fluorescent small molecular dye (Rhodamine B) RhB (Mw = 479) were chosen as model drug substances. Both NaCl (3 M) and RhB (0.2 mg mL−1) were dissolved in DI water and stored in a 25 ml polymer tube. Afterwards the PLA microchambers@PDMS stamp was dipped into the model drug solution under ultrasound sonication at 37 kHz for 30 s to remove air bubbles and to load the model drug solution into the microchambers. The sample was wiped with cotton swabs to remove the solution not filling the chambers followed by drying the chamber filling solution under ambient conditions. Such a treatment facilitates drug precipitation within the chambers. Each individual microchamber loaded 8–17 pg of RhB as well as NaCl inside.2.4 PLA microchamber drug release triggered by HIFU
The sealed PLA@RhB microchambers were inserted into a water filled quartz tube and exposed to HIFU. At specific time intervals (0 s, 5 s, 10 s, 20 s, 60 s, 100 s, 120 s, 150 s), the PLA@RhB microchambers were taken out for measurements. It is noted that PLA chambers exposed to HIFU contain no sputtered gold, as it was added later for electron microscopic observations only. The amount of released RhB was calculated by measuring the solution fluorescence intensity corresponding to the HIFU treatment duration (shown in ESI, Fig. S2†). To ensure the release was not induced by diffusion, the release of the PLA@RhB microchambers (PLA m% = 2%) kept in the dark (to prevent photobleaching of RhB) without any treatment was also monitored.2.5 Characterization
Scanning electron microscopy (SEM) (FEI, Inspect-F) was used to measure PLA microchamber morphologies after 2 min of gold sputter coating (∼10 nm of gold) to ensure conductive samples. SEM observation was carried out using an acceleration voltage of 10 kV, a spot size of 3.5, and a working distance of approximately 10 mm. Fluorescence spectrometry (LS 55, PerkinElmer) was used to analyse fluorescence from the sample within quartz cuvettes (S10C, Sigma). Confocal Laser Scanning Microscopy (CLSM) images were obtained with a Leica TS confocal scanning system (Leica, Heidelberg, Germany) equipped with a 20× objective. Chamber loading was determined via binary micrographs and analysis of the chamber content was performed via the ImageJ38 “analyse particles” plugin.For determining the effects of the PLA dipping solution's withdrawal speed, the aforementioned dipping robot with a defined withdrawal speed of 80 mm s−1 and a home build robot arm with controllable withdrawal speeds from 0 to 4.5 mm s−1 were used. The home build robot comprised of a USB-PIO digital IO card, an OR-8 relay card (both BMCM, Maisach, Germany), a ZD-6560-V3C (Shenzhen TOBY Technology Co. Ltd, Shenzhen, China) stepping motor controller, as well as a 90° angular plate on a linear optical stage which is driven by a stepping motor (TSA-150, Zolix Instruments Co., Ltd, Beijing, China). This linear optical stage was mounted via a 90° right-angle bracket to a 30 × 30 cm breadboard (PHOB-3 and OTBB33-1 from Zolix Instruments Co., Ltd, Beijing China). The control software of both robots was customized in LabView 2007 and 2011.
Release experiments were performed by a home built HIFU system, operating at 2.5 MHz with an acoustical density in the focal area of 200–300 W cm−2 displayed in ESI, Fig. S3.† The HIFU system consists of two units: the first device is a generator for radiofrequency signals that feeds the piezoelectric transducer generating the ultrasonic beam. The generator is electrically matched with the piezoelectric transducer and is obliged to generate the electrical signal with the power tunable from zero up to several hundred watts per cm2, whereby an energy density of 200–300 W cm−2 was adjusted. The generation frequency of the radio signal used corresponds to the resonance frequency of the transducer, which is equal to 2.5 MHz. The second device is the acoustical part of the set up and consists of a table supporting the piezoelectric transducer. On top of the transducer is a quartz tube which contains water for drug release studies. The said quartz tube is hermetically fixed on the plate of the table while the piezoelectric transducer represents a spherical acoustic lens made out of piezoceramics. Both surfaces of the transducer are covered by silver electrodes. Our transducer is obliged with a high acoustical quality factor Q and generates a focused ultrasonic beam with a focal distance of about 12 mm in water, used as acoustical media and the sound conductor. The diameter (D) of the sound beam in the focal plane was as small as 500 × 500 μm2.
Defined finite element simulations were performed with Comsol Multiphysics 4.7 (Comsol, Stockholm, Sweden) utilizing the acoustothermal plugin. The parameters used were obtained from Comsol's internal databases, as well as from ref. 39–41. The modelled chambers had a diameter of 5 μm and a wall thickness of 300 nm, which is in the range between 1 and 2% PLA chambers produced in this study.
3. Results
3.1. PLA microchamber fabrication
In order to produce polylactic acid (PLA) microchamber arrays, a silicone rubber (polydimethylsiloxane (PDMS)) stamp with microwells needs to be produced. The said PDMS stamp can be fabricated by curing PDMS on top of a silicon master, resulting in a negative replica of the silicon master, as displayed in ESI Fig. S1.†42 A PLA thin film was formed on top of the PDMS stamp (microwells on the surface) by dipping it into the PLA solution for 5 seconds (s), lifting the PDMS stamp@PLA out of the PLA solution, followed by air drying. Furthermore, the PDMS stamp@PLA microchamber was sonicated inside the drug solution for 30 s to remove trapped air bubbles and to load the drug solution at the same time. The PLA microchambers were sealed by microcontact printing35,43 them onto another PLA thin film by small applied pressure, after the cargo was in situ precipitated by evaporation of the solvent within the microchambers (Fig. 1A). Afterwards, the sealed microchambers were delaminated from the PDMS sheet allowing freestanding drug delivery microchamber arrays, as the top view and the backside view in Fig. 1B and C demonstrate. Sealing by microcontact printing enables the individual PLA microchambers to entrap an air bubble within its containment, which is proven by focused ion beam and scanning electron microscopy (FIB-SEM) slicing (Fig. 1D).
 | ||
| Fig. 1 Preparation and resulting PLA (m% = 2%) thin film chambers. (A) Schematic illustration of the fabrication of drug loaded polylactic acid (PLA) microchambers and drug release upon HIFU sonication. Scanning electron microscopy (SEM) image with the top view (B) and section view (C) of fabricated free-standing hollow PLA microchambers sealed by imprinting onto a flat PDMS sheet pre-coated with a PLA thin film. (D) SEM image of a PLA microchamber sealed on PLA and cut by a focused ion beam (FIB) proving that the microchamber retains its hollow structure after sealing by imprinting. Uneven wall thicknesses in (D) are caused by PLA melting upon FIB slicing. | ||
Please note that the uneven wall thicknesses in Fig. 1D are caused by PLA melting upon FIB slicing. It is worth noting that a successful PLA thin film and therefore chamber production requires a proper adjustment of the deposition conditions whereby three main parameters define the PLA thin film thickness. The first parameter is the PDMS stamp exposure time within the PLA solution: despite PDMS being crosslinked and insoluble in chloroform it can swell and deform the chamber structure44 at exposure times exceeding 10 s. Therefore, exposure times around 5 s were chosen. The second parameter is the PDMS stamp's withdrawal speed. As demonstrated in Fig. 2, PLA withdrawal speeds above 3 mm s−1 result in slightly thinner films than below, which is caused by shear force. It should be noted that the PLA thin film (m% = 2%) thickness is quite stable around 1 μm. The third and most important parameter for reproducible PLA thin film production is the PLA solution concentration, which results in PLA nanofilms (50–150 nm) for 1% up to 7 μm thick films causing filled PLA pillars instead of empty chambers at 5% (Fig. 2).
 | ||
| Fig. 2 Characterization of the PLA thin film thickness. SEM images of PLA films: (A–C) Same dip-coating pull-out speed (v = 8 cm s−1), PLA thin film thickness versus different PLA concentrations (m% = 1%, 2% and 5%); (D–F) the same PLA concentration (m% = 2%), PLA thin film thickness versus different pull-out speeds (v = 2 mm s−1, 3 mm s−1 and 4 mm s−1); (G–I) the same dip-coating pull-out speed (v = 8 cm s−1), patterned PLA microchamber thickness versus different PLA concentrations (1%, 2%, 5%). (J) Influence of pullout speed on PLA thickness with the same PLA concentration (2%), (K) PLA film thickness in relation to PLA solution concentration with the same pull-out speed (v = 8 cm s−1). | ||
Utilizing 1% PLA solution and a PDMS stamp exhibiting wells with 500 nm diameter and 1 μm length, nanochambers can be produced, as demonstrated in ESI, Fig. S4.† Such thin nanostructures were however at the stability limit as can be seen by chamber bending in ESI Fig. S4.† In addition, the films suffered from sensitivity to the electron beam, melting under zoomed in observations. For this reason we focus for the rest of this study on microchambers.
Due to films above 3% resulting in filled chambers, all the following experiments were performed with 2% PLA solution concentration (except stated otherwise), due to an optimum stability to chamber volume ratio.
3.2. Cargo loading and sealing
Cargo loading into PLA (m% = 2%) microchambers was performed via sonication within NaCl solution (3 M), and drying in air (shown in Fig. 3). The said treatment allows chamber in situ NaCl crystal growth (Fig. 3B). Utilizing 1% PLA solution, several dozens of nanometer thin PLA membranes which are vacuum stable (Fig. 3D) and mainly electron transparent (despite a 10 nm gold conduction layer) are formed.45 The electrons penetrate through the PLA sealing layer, arrive at the underlying NaCl crystal and stimulate X-ray emission, which escapes the chamber, reaching the detector.45Fig. 3E is the resulting EDX-mapping micrograph, proving successfully the presence of sealed NaCl crystals within microchambers. Subsequent FIB-cutting of loaded and sealed 2% microchambers proves the successful encapsulation of NaCl crystals (Fig. 3F).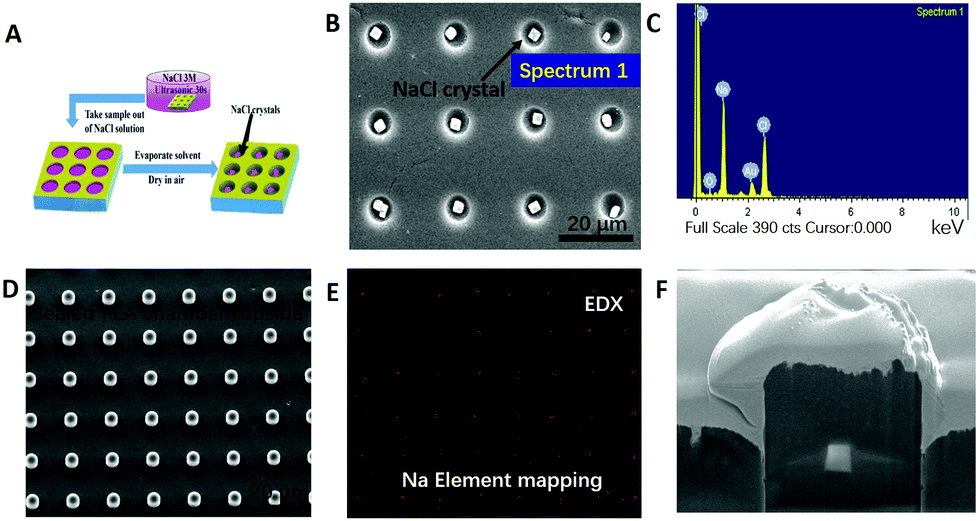 | ||
| Fig. 3 Encapsulation of small hydrophilic molecules within air bubbles in PLA thin films and proof of successful encapsulation. (A) Scheme of NaCl crystal encapsulation within hydrophobic PLA microchambers. (B) SEM image of PLA@NaCl crystals in microchambers before sealing (PLA m% = 2%), the size of NaCl crystals is 1 ± 0.2 μm. (C) EDX analysis of NaCl crystals loaded into PLA chambers. (D) SEM image of the sealed PLA@NaCl microchambers (PLA m% = 1%). (E) EDX mapping analysis of PLA@NaCl microchambers, proving that NaCl is within the chambers of (D). (F) Focused ion beam (FIB) image of a half-cut and sealed PLA@NaCl microchamber (PLA m% = 2%), uneven wall thickness from PLA melting upon FIB cutting. | ||
The long term encapsulation of small hydrophilic molecules was also studied in this work. Compared to single NaCl crystal encapsulation, the small molecular dye RhB (MW = 479 g mol−1) was also successfully loaded into PLA microchambers in the form of several nano-sized precipitates, as demonstrated in Fig. 4A–D. The fact that these dyes remain in the precipitated state under submerged conditions for weeks proves the entrapment of air bubbles within the microchambers. Fig. 4A–C display micrographs of the sealed PLA microchamber@RhB precipitates submerged under aqueous conditions after 2 hours, indicating that the RhB crystals are not in contact with water, otherwise the RhB crystals would be immediately dissolved. The amount of encapsulated RhB that precipitates in each microchamber is 8 × 10−12 g–1.7 × 10−11 g (or 8–17 pg), which amounts to 1.28 μg–2.72 μg cm−2 as also shown in ESI, Fig. S5.† This rationale is supported by contrast experiments, proving exclusion of aqueous RhB solution from sealed PLA chambers as CLSM micrographs in Fig. 4E and F demonstrate.
 | ||
| Fig. 4 Determination of the sealing properties of submerged PLA chambers: (A) optical micrograph of the sealed PLA@RhB precipitate within microchambers under submerged conditions after 2 hours. Confocal laser scanning microscopy (CLSM) image of the sealed PLA@RhB precipitate within microchambers in an RITC channel (B) and an overlay micrograph (C). (D) SEM image of the unsealed PLA@RhB precipitate within microchambers. (E), (F) CLSM micrographs of sealed, empty PLA microchambers in Rh-B solution in an RITC channel and an overlay image proving efficient chamber sealing. The line scan insets show the relative fluorescence intensity of the corresponding microchambers. | ||
3.3. Drug release
High intensity focused ultrasound (HIFU) is a highly efficient and non-invasive technique, which is already employed as a diagnostic and therapeutic method for many diseases.46 Here, we demonstrate that apart from long term encapsulation, one-step fabricated PLA microchambers support rapid drug release triggered by HIFU. The SEM images of sealed biodegradable PLA microchambers after exposure to different HIFU sonication times are displayed in Fig. 5, demonstrating that sealed PLA microchambers with entrapped air bubbles are responsive to ultrasound. HIFU exposure times below 5 s cause no adverse effects to the PLA chambers (Fig. 5A–F), but 10 s already open small wells in the microchamber “caps”, as indicated in Fig. 5G–I. 20 s of HIFU irradiation causes the wells in the PLA microchambers to grow slightly bigger (Fig. 5J–L). After 60 s the PLA microchamber “caps” are completely eradicated, leaving craters on the irradiated areas (D ∼500 μm) behind, as shown in Fig. 5M–O. The corresponding fluorescence images of the PLA microchamber under HIFU radiation for RhB released into water are in good agreement with the SEM data (ESI, Fig. S6†). | ||
| Fig. 5 High intensity focused ultrasound (HIFU) triggered chamber opening versus exposure time. SEM images of sealed PLA@NaCl microchambers before (0 s first row) and after HIFU exposure for 5 s (second row), the microchambers look comparable to non-ultrasound exposed chambers; after 10 s (third row) and 20 s (forth row) HIFU treatment, wells appear in chambers, whereby the absence of crystals proves cargo release; within 60 s (last row), the PLA microchamber “caps” are blown away by HIFU. The red circle in M shows the HIFU irradiation area (D ∼500 μm). The images in the same row are at different magnifications. | ||
Comparison experiments employing PLA microchambers sealed by one more dip-coating-step without air-bubble entrapment were also performed (ESI, Fig. S7†). Sealed PLA microchambers without air-bubble entrapment cannot be opened by HIFU treatment, even after increasing the exposure time up to 30 minutes. The explanation for this >300 times enhanced ultrasound sensitivity possessed by sealed PLA microchambers with air-bubble entrapment is the high refractive index differences between air and water.
The quantitative measurement of released RhB triggered by HIFU was studied (Fig. 6). To ensure the release was not induced by diffusion, the release behaviour of the PLA@RhB microchambers (PLA m% = 2%) kept in the dark (to prevent photobleaching of RhB) without any treatment was also monitored.
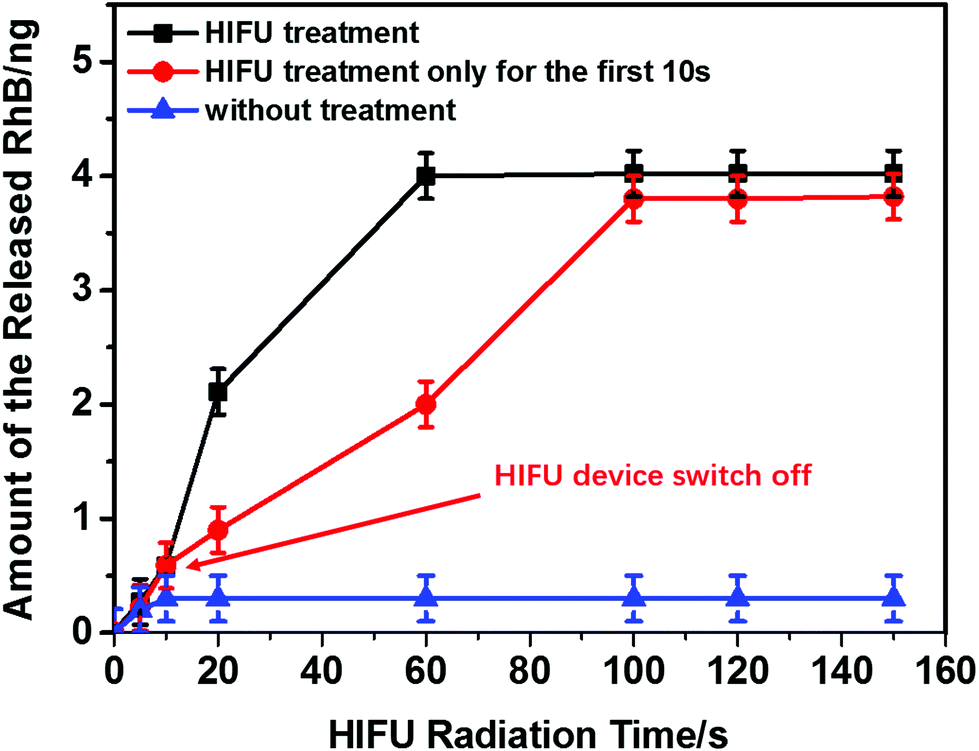 | ||
| Fig. 6 Amount of released RhB from sealed PLA@RhB microchambers triggered by HIFU. The black line shows microchambers exposed to continuous HIFU radiation and the red line shows the PLA microchambers with HIFU treatment for 10 s; then the HIFU device was switched off; the blue line visualizes a control experiment without any HIFU exposure for the PLA microchambers corresponding to the HIFU treatment duration (0 s, 5 s, 10 s, 20 s, 60 s, 100 s, 120 s, 150 s). Values from calibration and the release spectra of ESI Fig. S2.† | ||
Less than ∼5% RhB is released by diffusion within 14 days due to low PLA permeability for gases and water vapour.30 For the same reason PLA is also widely used for food packaging.47
3.4. Ultrasound harvesting mechanism
Defined finite element (DFE) simulations were used to determine the interaction between the ultrasound pressure wave and the herein utilized PLA microchambers. A significant acoustic pressure in the range of 40–160 Db (decibel) is determined within the aforementioned microchambers. This results in a maximum energy harvesting of ∼10 kW m−2 out of 1.27 MW m−2 generated by the HIFU (more device details are shown in the Experimental section and ESI, Fig. S3†), corresponding to an ∼0.8% conversion efficiency of PLA chambers with a force vector pointing outward from the chamber, even slightly deforming them (Fig. 7A). This pressure is accompanied by an 11 orders of magnitude higher air molecule speed compared to water filled chambers (Fig. 7B and C). A decreased particle speed and harvested energy for chambers not possessing air bubbles explains the failure to open them in the experiment. A comparison between Fig. 5 and 7D reveals that 3D chambers possess local pressure peaks on positions corresponding to the appearance of wells in Fig. 5; these pressure peaks facilitate a diagonal as well as a sideward chamber opening observed in the experiment. | ||
| Fig. 7 Defined finite element simulation of non-gas filled and gas filled chambers: (A) 2D simulation of acoustic pressure interaction and pressure vectors within a gas filled micro-chamber, showing slight deformations due to the pressure; (B) 2D simulation of particle velocity for an air-filled chamber proving peak maxima in the corners and slight deformations; (C) 2D simulation of the water filled microchamber proving low particle speed and no deformation; (D) air filled 3D microchamber proving pressure peaks and force vectors pointing outwards from the chamber, with a pressure peak in the upper corner. | ||
4. Conclusion
In summary, we have developed a one-step dip-coating strategy for fabricating free-standing biocompatible polylactic acid microchamber arrays (whereby the production limit is ∼500 nm sized chambers), able to encapsulate small hydrophilic molecules as well as to entrap air bubbles. To the best of our knowledge, this is the first report about the in situ crystallization of NaCl and RhB inside the individual microchambers (∼8–17 pg per chamber) with long term encapsulation in aqueous solution up to 14 days. Biodegradable PLA nano- and microchamber fabrication requires a relatively short production time (5 s, compared to several days for the present PEM chambers17 and about a day for capsules48), low permeability, and high ultrasound sensitivity due to air bubble entrapment. Numerical simulations reveal an 11 order of magnitude higher molecule speed for PLA microchambers with air bubbles compared to water filled chambers. The determined ultrasound energy harvesting efficiency is ∼0.8%. The existence of ultrasound induced local sideward directed pressure peaks was revealed to cause diagonally upwards directed chamber piercing. In addition, this highly ordered array of PLA microchambers enables the option of programmed release-on-demand for precisely defined active quantities from individual chambers in a site- and time specific manner. With these features, we expect PLA microchambers to offer considerable potential for medical applications and chemical and biochemical studies (e.g. single cell level, implant coatings, catalyst release).It is worth pointing out that the applied method might aid dispersed drug delivery systems like PEM capsules and micro motors,49 if the sealing mechanism can be transferred to disperse drug delivery systems. The approach might be used in the future also to capture desired cell types if utilized in the bloodstream, for example to capture cancer cells, or in the case of stents that capture and promote the growth of blood vessel cells. For such purposes the surface might be coated with cellular membranes and biomolecules by protocols outlined in ref. 50 and 51. Another potential application is the utilization of our system in combination with or as a replacement for electrospun PLA scaffolds which are currently used for tissue growth and as implants.52,53 In such a case electrospun fibres can be used to adjust the surface area or mechanical properties.
Acknowledgements
The authors would like to thank the following funding agencies which partly supported this work: the National Natural Science Foundation of China (grant no. 21503058) (J. F.), the State Key Laboratory Foundation (project no. 2016KM008), the Key Laboratory of Micro-/Nanotechnology Research Center, HIT, (J. F.), the Chinese Scholarship Council (CSC) Grant No. 201406120038 (M. G.), and the Queen Mary University of London (M. G.). The work was also supported by The Government of the Russian Federation (grant 14.Z50.31.0004) to support scientific research projects implemented under the supervision of leading scientists at Russian institutions and Russian institutions of higher education, A.V.P., V.V.P., G.B.S).Notes and references
- E. V. Skorb and H. Möhwald, Adv. Mater., 2013, 25, 5029–5043 CrossRef CAS PubMed.
- Z. Wu, Y. Wu, W. He, X. Lin, J. Sun and Q. He, Angew. Chem., Int. Ed., 2013, 125, 7138–7141 CrossRef.
- Y. Wu, Z. Wu, X. Lin, Q. He and J. Li, ACS Nano, 2012, 6, 10910–10916 CrossRef CAS PubMed.
- K. A. Athanasiou, G. G. Niederauer and C. M. Agrawal, Biomaterials, 1996, 17, 93–102 CrossRef CAS PubMed.
- A. H. Faraji and P. Wipf, Bioorg. Med. Chem., 2009, 17, 2950–2962 CrossRef CAS PubMed.
- G. Wei, S. Emaminejad, C. Samyuktha, C. Kevin, P. Austin, M. F. Hossain, H. Ota, H. Shiraki, D. Kiriya, D.-H. Lien, G. A. Brooks, R. W. Davis and A. Javey, Nat. Lett., 2016, 529, 509–514 CrossRef PubMed.
- D. Borisova, D. Akçakayıran, M. Schenderlein, H. Möhwald and D. G. Shchukin, Adv. Funct. Mater., 2013, 12, 3799–3812 CrossRef.
- Z. Siwy, L. Trofin, P. Kohli, L. a. Baker, C. Trautmann and C. R. Martin, J. Am. Chem. Soc., 2005, 127, 5000–5001 CrossRef CAS PubMed.
- J. Shao, M. Xuan, L. Dai, T. Si, J. Li and Q. He, Angew. Chem., Int. Ed., 2015, 54, 12782–12787 CrossRef CAS PubMed.
- J. Frueh, M. Gai, Z. Yang and Q. He, J. Nanosci. Nanotechnol., 2014, 14, 4341–4350 CrossRef CAS PubMed.
- J. A. Callow and M. E. Callow, Nat. Commun., 2011, 2, 244 CrossRef PubMed.
- S. J. Leung and M. Romanowski, ACS Nano, 2012, 6, 9383–9391 CrossRef CAS PubMed.
- M. Bédard, A. G. Skirtach and G. B. Sukhorukov, Macromol. Rapid Commun., 2007, 28, 1517–1521 CrossRef.
- M. V. Kiryukhin, S. Gorelik, S. M. Man, M. Subramanian, G. S. Antipina, H. Y. Low and G. Sukhorukov, Macromol. Rapid Commun., 2013, 34, 87–93 CrossRef CAS PubMed.
- M. V. Kiryukhin, S. M. Man, A. Tonoyan, H. Y. Low and G. B. Sukhorukov, Langmuir, 2012, 28, 5678–5686 CrossRef CAS PubMed.
- M. Gai, J. Frueh, V. L. Kudryavtseva, R. Mao, M. V. Kiryukhin and G. B. Sukhorukov, Sci. Rep., 2016, 6, 37000 CrossRef CAS PubMed.
- M. V. Kiryukhin, S. M. Man, S. R. Gorelik, G. S. Subramanian, H. Y. Low and G. B. Sukhorukov, Soft Matter, 2011, 7, 6550 RSC.
- C. Ye, D. D. Kulkarni, H. Dai and V. V. Tsukruk, Adv. Funct. Mater., 2014, 24, 4364–4373 CrossRef CAS.
- R. Klitzing and H. Moehwald, Macromolecules, 1996, 29, 6901–6906 CrossRef.
- H. Gao, A. V. Sapelkin, M. M. Titirici and G. B. Sukhorukov, ACS Nano, 2016, 10, 9608–9615 CrossRef CAS PubMed.
- Q. Yi and G. B. Sukhorukov, ACS Nano, 2013, 7, 8693–8705 CrossRef CAS PubMed.
- B. P. Timko, T. Dvir and D. S. Kohane, Adv. Mater., 2010, 22, 4925–4943 CrossRef CAS PubMed.
- M. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- H. Gao, D. Wen, N. V. Tarakina, J. Liang, A. J. Bushby and G. B. Sukhorukov, Nanoscale, 2016, 8, 5170–5180 RSC.
- D. A. Gorin, D. G. Shchukin, A. I. Mikhailov, K. Köhler, S. A. Sergeev, S. A. Portnov, I. V. Taranov, V. V. Kislov and G. B. Sukhorukov, Tech. Phys. Lett., 2006, 32, 70–72 CrossRef CAS.
- M. Ma, H. X. Xu, H. R. Chen, X. Q. Jia, K. Zhang, Q. Wang, S. G. Zheng, R. Wu, M. H. Yao, X. J. Cai, F. Q. Li and J. L. Shi, Adv. Mater., 2014, 26, 7378–7385 CrossRef CAS PubMed.
- X. Wang, H. Chen, Y. Chen, M. Ma, K. Zhang, F. Li, Y. Zheng, D. Zeng, Q. Wang and J. Shi, Adv. Mater., 2012, 24, 785–791 CrossRef CAS PubMed.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- R. P. Pawar, S. U. Tekale, S. U. Shisodia, J. T. Totre and A. J. Domb, Recent Pat. Regener. Med., 2014, 4, 40–51 CrossRef CAS.
- M. Jamshidian, E. A. Tehrany, M. Imran, M. Jacquot and S. Desobry, Compr. Rev. Food Sci. Food Saf., 2010, 9, 552–571 CrossRef CAS.
- K. Hamad, M. Kaseem, H. W. Yang, F. Deri and Y. G. Ko, eXPRESS Polym. Lett., 2015, 9, 435–455 CrossRef CAS.
- D. Shumigin, E. Tarasova, A. Krumme and P. Meier, Mater. Sci., 2011, 17, 32–37 CrossRef.
- F. Rancan, D. Papakostas, S. Hadam, S. Hackbarth, T. Delair, C. Primard, B. Verrier, W. Sterry, U. Blume-Peytavi and A. Vogt, Pharm. Res., 2009, 26, 2027–2036 CrossRef CAS PubMed.
- M. Gai, J. Frueh, G. Sukhorukov, A. Girard-Egrot, S. Rebaud, B. Doumeche and Q. He, Colloids Surf., A, 2015, 483, 271–278 CrossRef CAS.
- M. Gai, J. Frueh, A. Girard-Egrot, S. Rebaud, B. Doumeche and Q. He, RSC Adv., 2015, 5, 51891–51899 RSC.
- P. T. Hammond, Adv. Mater., 2004, 16, 1271–1293 CrossRef CAS.
- S. A. Portnov, A. M. Yashchenok, A. S. Gubskii, D. A. Gorin, A. A. Neveshkin, B. N. Klimov, A. A. Nefedov and M. V. Lomova, Instrum. Exp. Tech., 2006, 49, 849–854 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- H. Ma, P. Tian, J. Pello, P. M. Bendix and L. B. Oddershede, ACS Nano Lett., 2014, 14, 612–619 CrossRef CAS PubMed.
- P. V. Ruijgrok, N. R. Verhart, P. Zijlstra, A. L. Tchebotareva and M. Orrit, Phys. Rev. Lett., 2011, 107, 1–4 CrossRef PubMed.
- L. Jauffred, S. M.-R. Taheri, R. Schmitt, H. Linke and L. B. Oddershede, Nano Lett., 2015, 15, 4713–4719 CrossRef CAS PubMed.
- H. A. Biebuyck, N. B. Larsen, E. Delamarche and B. Miche, IBM J. Res. Dev., 1997, 41, 159–170 CrossRef CAS.
- P. T. Hammond, Adv. Mater., 2004, 16, 1271–1293 CrossRef CAS.
- J. N. Lee, C. Park and G. M. Whitesides, Anal. Chem., 2003, 75, 6544–6554 CrossRef CAS PubMed.
- Electron scattering calculation, National Institute of Standards and Technology Webpage: http://physics.nist.gov/PhysRefData/Star/Text/ESTAR.html last accessed 13.04.2017.
- R. O. Illing, J. E. Kennedy, F. Wu, G. R. ter Haar, A. S. Protheroe, P. J. Friend, F. V. Gleeson, D. W. Cranston, R. R. Phillips and M. R. Middleton, Br. J. Cancer, 2005, 93, 890–895 CrossRef CAS PubMed.
- H. Tsuji, R. Okino, H. Daimon and K. Fujie, J. Appl. Polym. Sci., 2006, 99, 2245–2252 CrossRef CAS.
- G. Sukhorukov, A. Fery and M. Brumen, Phys. Chem., 2004, 6, 4078–4089 CAS.
- X. Lin, Z. Wu, Y. Wu, M. Xuan and Q. He, Adv. Mater., 2016, 28, 1060–1072 CrossRef CAS PubMed.
- M. Xuan, J. Shao, L. Dai, Q. He and J. Li, Adv. Healthcare Mater., 2015, 4, 1645–1652 CrossRef CAS PubMed.
- J. Shao, M. Xuan, T. Si, L. Dai and Q. He, Nanoscale, 2015, 7, 19092–19098 RSC.
- M. S. Savelyeva, A. A. Abalymov, G. P. Lyubun, I. V. Vidyasheva, A. M. Yashchenok, T. E. L. Douglas, D. A. Gorin and B. V. Parakhonskiy, J. Biomed. Mater. Res., Part A, 2017, 105, 94–103 CrossRef CAS PubMed.
- Y. Shapovalova, D. Lytkina, L. Rasskazova, A. Gudima, V. Ryabov, A. Filimoshkin, I. Kurzina and J. Kzhyshkowska, Eur. J. Cancer Suppl., 2015, 13, 49–50 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: A figure of the silicon master and PDMS stamps, PLA nanochambers, layout of the PLA chambers and content, fluorescence images of opened chambers, effects of HIFU on non-air bubble sealed chambers, HIFU device and RhB fluorescence spectra. See DOI: 10.1039/c7nr01841j |
| This journal is © The Royal Society of Chemistry 2017 |