DOI:
10.1039/C7NR01301A
(Paper)
Nanoscale, 2017,
9, 6783-6790
Chirality as a tool for function in porous organic cages†
Received
21st February 2017
, Accepted 29th April 2017
First published on 10th May 2017
Abstract
The control of solid state assembly for porous organic cages is more challenging than for extended frameworks, such as metal–organic frameworks. Chiral recognition is one approach to achieving this control. Here we investigate chiral analogues of cages that were previously studied as racemates. We show that chiral cages can be produced directly from chiral precursors or by separating racemic cages by co-crystallisation with a second chiral cage, opening up a route to producing chiral cages from achiral precursors. These chiral cages can be cocrystallized in a modular, ‘isoreticular’ fashion, thus modifying porosity, although some chiral pairings require a specific solvent to direct the crystal into the desired packing mode. Certain cages are shown to interconvert chirality in solution, and the steric factors governing this behavior are explored both by experiment and by computational modelling.
Introduction
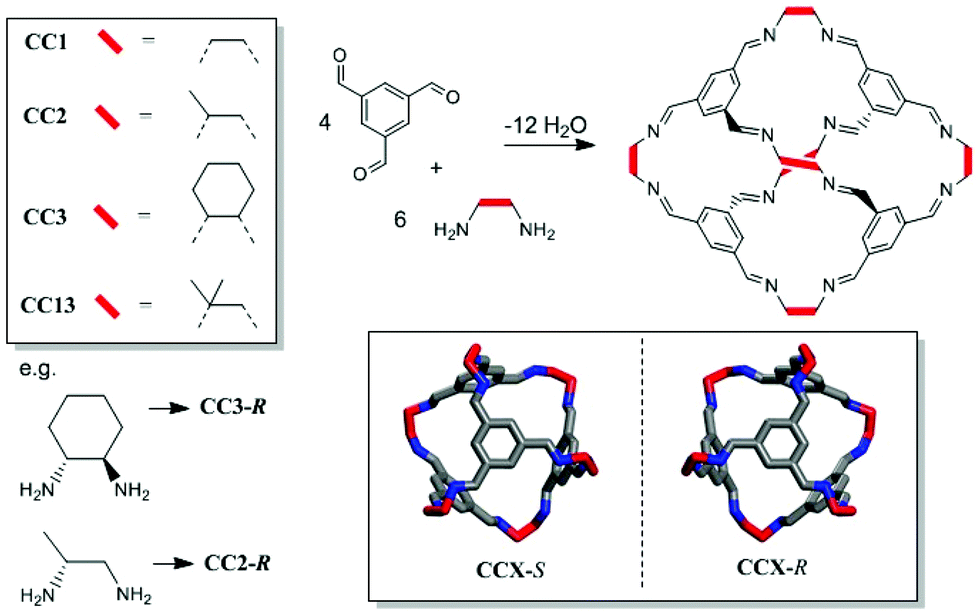
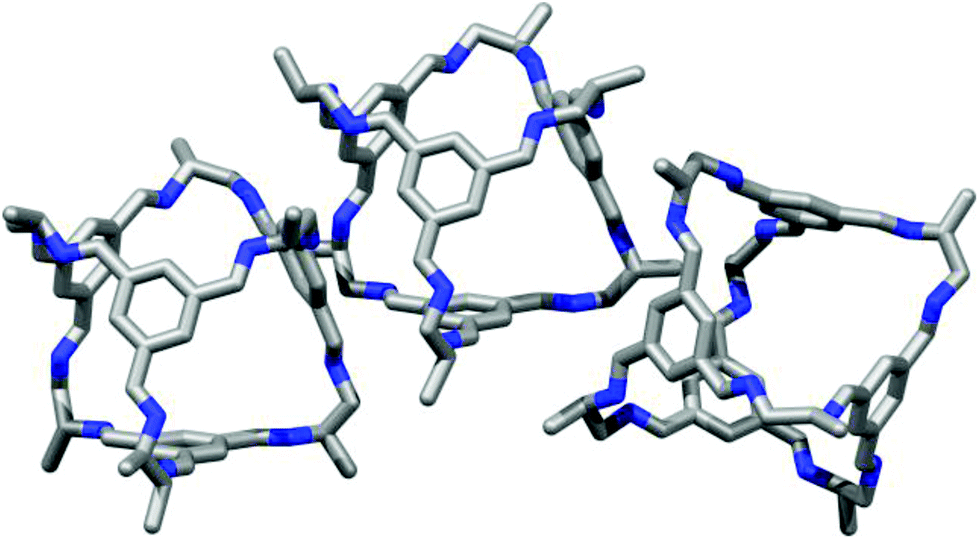
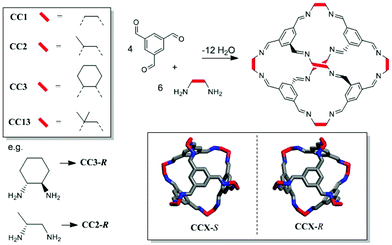
Microporous materials have potential applications in gas storage, separation, and heterogeneous catalysis.1 Most microporous solids are extended networks, such as metal–organic frameworks (MOFs),2 covalent organic frameworks (COFs),3 or organic polymer networks.4 However, there is also growing interest in porous molecular solids, such as porous organic cages.5–7 These cages comprise an internal void, open windows, and a rigid structure that prevents collapse, thus allowing accessible porosity to guest molecules. Porous organic cages have been prepared using imine condensation,8–11 boronic ester formation,12 and direct carbon–carbon bond-forming reactions.13 Molecular cage materials with apparent Brunauer–Emmett–Teller (BET) surface areas as high as 3758 m2 g−1 have been prepared.14 Since cages are discrete molecules, they are soluble in common organic solvents and can be processed into support materials or crystallized into the solid state, as required. We previously reported a class of porous [4 + 6] cycloimine cage compounds (Fig. 1).8,15 The gas sorption properties of these cages depend both on the structure of the cage itself and on its crystal packing, with multiple polymorphs being possible for most cages.15,16 Helicity, or axial chirality, is an intrinsic property of this family of cages (Fig. 1). This chirality is significant because it plays an important role in controlling the crystalline assembly of these molecules.
 |
| | Fig. 1 Chemical structure and synthesis of a range of tetrahedral 4 + 6 imine bonded covalent cages. Lower right inset illustrates helical chirality. | |
Cages of opposite chirality are often found to pack more closely, thus forming a preferred, lower energy packing. This has been observed not only for [4 + 6] imine cage structures, as shown in Fig. 1,8 but also [3 + 6] tubular imine based cages17 and [2 + 3] salicylimine cages.18 This strong chiral recognition allows different cages to be combined in a modular fashion to produce cocrystals by design. Hence, the properties of the resultant cocrystals can be tailored by the choice of the substituents on the cage building blocks. Chiral interactions can also be used to control the size and shape of the cage crystals,19 and even to produce ternary cocrystals containing three different cage molecules.20 The chirality of the cages also allows selective binding of chiral organic guest molecules such as 1-phenylethanol, thus allowing applications in enantioselective separation.21–25
Some cages have been studied as both their chiral and racemic forms, which can have different crystal packings and physical properties.19 However, cages CC2 and CC13 (Fig. 1) have only previously only been reported as racemates. We therefore set out to isolate both cages as homochiral materials, since this should allow new properties and opportunities for new cocrystal combinations.
Results and discussion
Direct synthesis of chiral CC2
We previously reported CC2 in its racemic form, and showed that it can exist as more than one racemic polymorph.8,15 Initial attempts to synthesize CC2 from its commercially-available homochiral diamine following the same protocol used for its racemic form8 failed due to the catenation (interlocking) of the cages.26 The standard synthesis for CC2 is to layer the aldehyde and amine and allow them to mix slowly, forming crystals of cage over 1–2 weeks that can then be isolated by filtration. Slow mixing of the precursors encourages the formation of discrete cages, rather than polymeric by-products.9 For racemic CC2, the formation of catenated cages, which we believe to be the thermodynamic product, requires either long equilibration times (months) or acid catalysts (trifluoroacetic acid).26 However, for chiral CC2, the products are a mixture of monomeric and catenated cages after only a few days, and after the standard reaction time for racemic CC2 (1 week), chiral CC2 forms the catenane exclusively (Fig. S1†). The chirality of the cage is important in this process. For racemic CC2, cages always ‘self sort’ to form catenanes comprised of two cages of the same chirality. This is because cages of equivalent helicity are needed for interlocking since opposite chirality would result in unfavorable close contacts.26 Hence, for the racemate to form catenated CC2 cages, each catenane must select 12 diamines of a single chirality from the racemic mixture. There is no such requirement for self-sorting in the chiral CC2 case, which rationalizes the greatly increased rate of catenation that we observe. To overcome this, the synthesis method was adapted to a high dilution dropwise addition of diamine to tri-aldehyde with stirring, with product isolation by rotary evaporation after only 24 hours. In this case, unwanted polymer formation was discouraged by the high dilution and slow reagent addition, while the mixing and short reaction time allowed monomeric cages to be produced as the kinetic product before catenanes could form. The structure and physical properties of chiral CC2 from direct synthesis were identical to those of chiral CC2 formed by separation, as described below.
Chiral CC2 separated by co-crystallisation
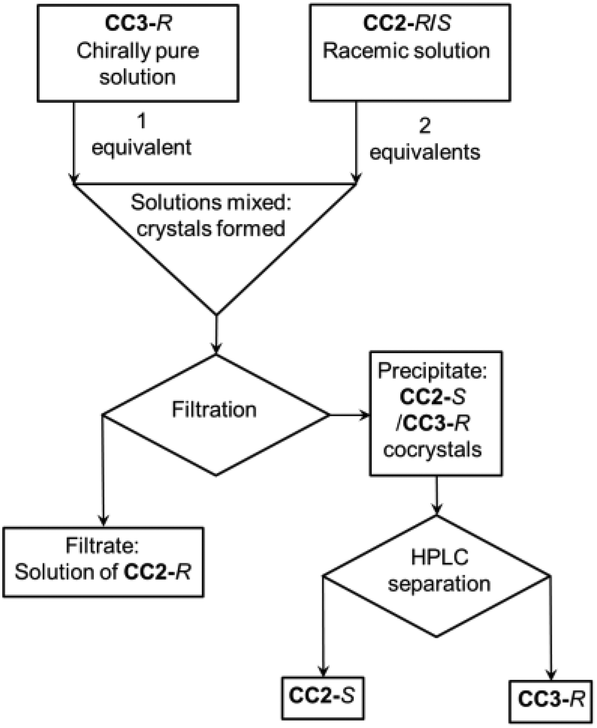
Previously, we showed that the chirality of racemic CC1 can be resolved by homochiral CC3-Rvia the formation of a (CC3-R, CC1-S) quasiracemic cocrystal.27 In that system, CC1 is flexible and its helical chirality can interconvert in solution.28 Hence, when CC3-R is crystallised in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with CC1, a cocrystal is formed in 100% yield where all the CC1 component has S chirality. By contrast, the more sterically hindered structure of CC2 (Fig. 1) cannot interconvert helical chirality in solution. We therefore hypothesized, by analogy, that a (CC3-R, CC2-S) cocrystal might be formed if racemic CC2 were mixed with homochiral CC3-R in a 2:1 ratio, leaving the CC2-R component excluded in solution (Fig. 2). This proved to be the case: when racemic CC2 and CC3-R were co-crystallised together, quasiracemic (CC3-R, CC2-S) cocrystals were isolated, which could be separated from the CC2-R, which remained in solution, by simple filtration (ESI, Fig. S2 and S3†).
:1 ratio with CC1, a cocrystal is formed in 100% yield where all the CC1 component has S chirality. By contrast, the more sterically hindered structure of CC2 (Fig. 1) cannot interconvert helical chirality in solution. We therefore hypothesized, by analogy, that a (CC3-R, CC2-S) cocrystal might be formed if racemic CC2 were mixed with homochiral CC3-R in a 2:1 ratio, leaving the CC2-R component excluded in solution (Fig. 2). This proved to be the case: when racemic CC2 and CC3-R were co-crystallised together, quasiracemic (CC3-R, CC2-S) cocrystals were isolated, which could be separated from the CC2-R, which remained in solution, by simple filtration (ESI, Fig. S2 and S3†).
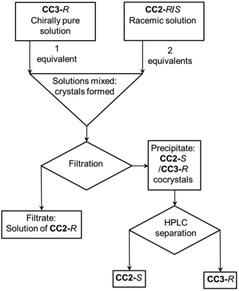 |
| | Fig. 2 Schematic representation of the process of cage separation by chiral co-crystallisation. | |
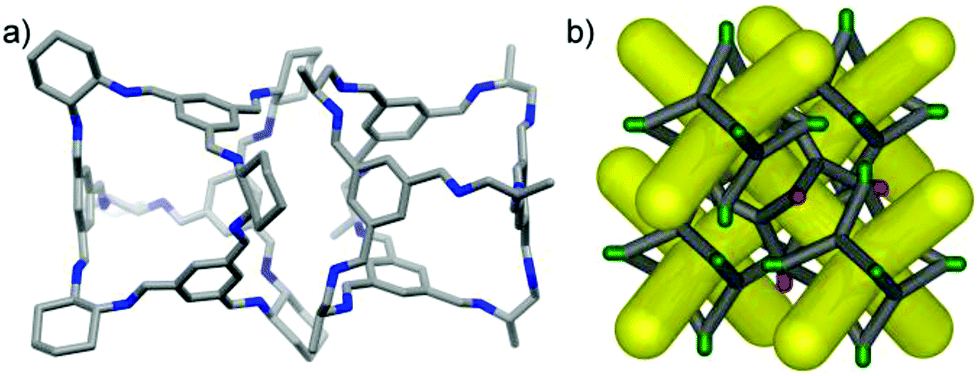
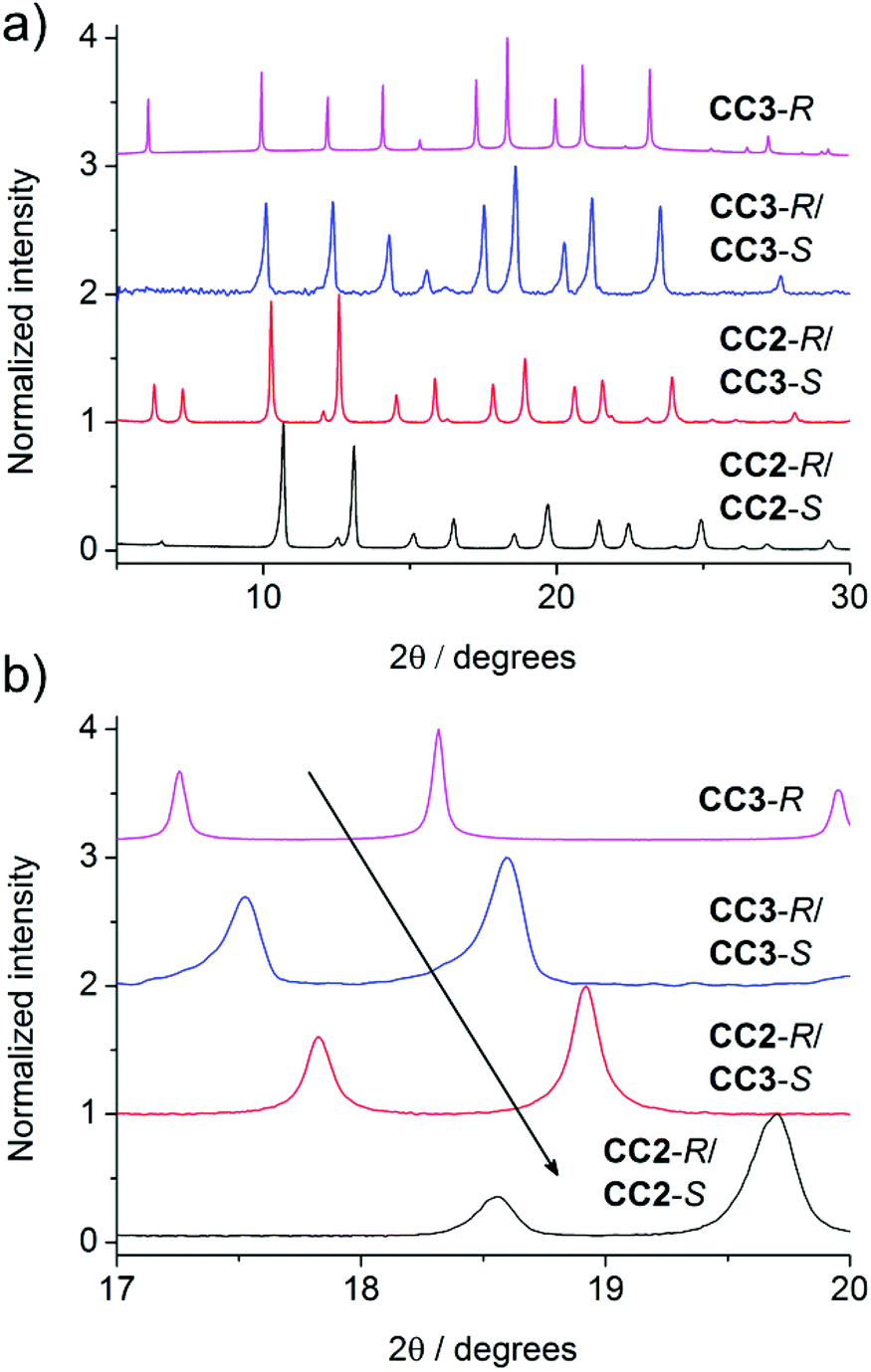
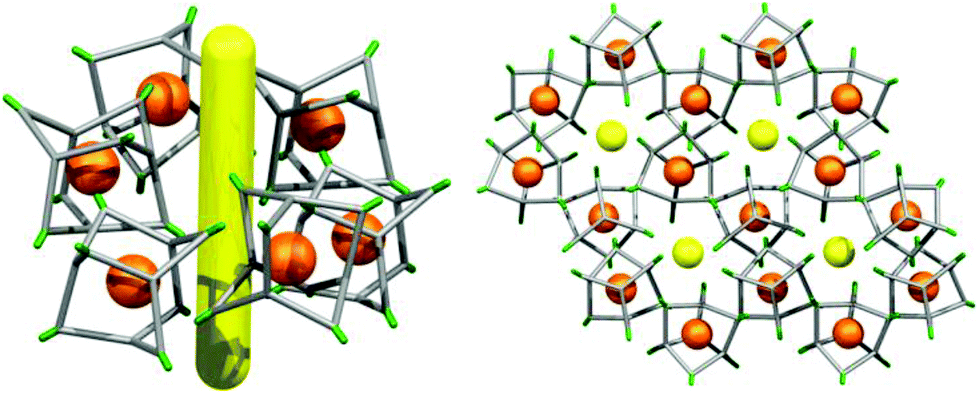
In the crystal structure of (CC3-R, CC2-S), the cage molecules pack window-to-window to generate a 3-D diamondoid pore structure running throughout the crystals (Fig. 3 and S4†). There is a clear trend in powder XRD patterns of different cages and cage combinations that adopt this isostructural, diamondoid packing mode (Fig. 4).
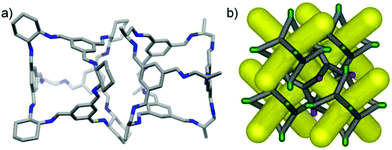 |
| | Fig. 3 (a) The heterochiral window-to-window interaction in the (CC2-R/CC3-S) crystal structure and (b) a schematic representation of the packing (CC2 and CC3 shown with green and red vertices, respectively), showing the diamondoid pore network (yellow). | |
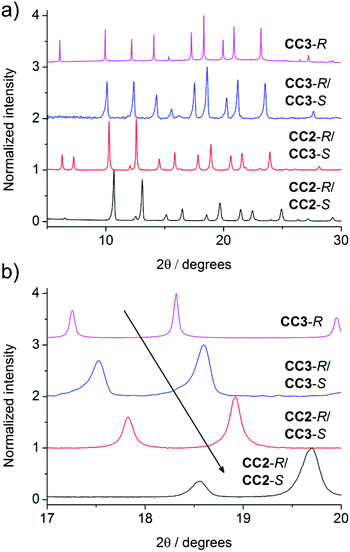 |
| | Fig. 4 PXRD patterns of cage crystals and cocrystals. (a) The similarity in powder patterns across the systems is indicative of their isoreticular structures. (b) The shift of peaks to the right (smaller unit cells) can be observed when going from a homochiral to racemic system, and as CC3 is replaced with the smaller CC2. | |
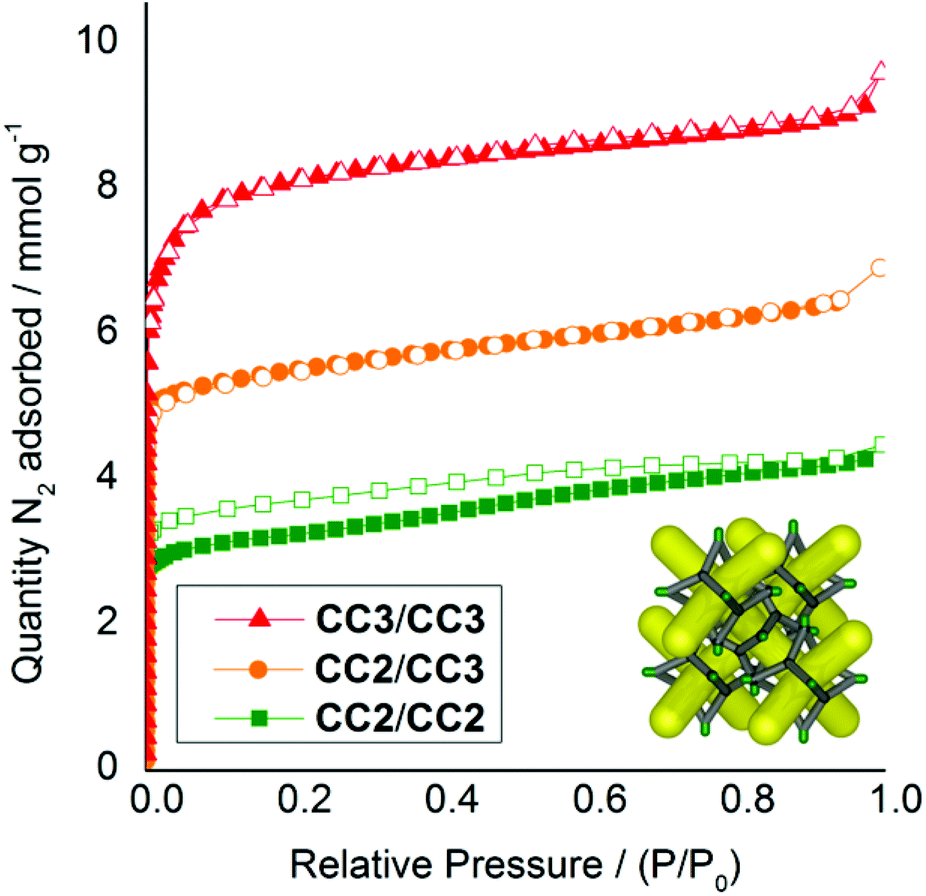
As the cages become smaller, with less sterically bulky vertices, they can pack closer together and the diffraction patterns shift to higher angles due to the smaller crystal repeat. Also, when moving from a homochiral to a heterochiral system, the cages can pack more efficiently. The cage–cage distances, and hence the unit cell dimensions, fall in the following order: homochiral CC3 > heterochiral CC3 > heterochiral CC2/CC3 > heterochiral CC2 (Fig. 4b). Gas sorption measurements for the heterochiral CC2/CC3 quasiracemic cocrystal show that its porosity also follows the trend that would be expected from an expansion in the unit cell (Fig. 5). The nitrogen uptake of the heterochiral (CC3-R, CC2-S) cocrystal falls between that of racemic CC3 and racemic CC2β (the β phase packs in the equivalent window-window packing mode).15
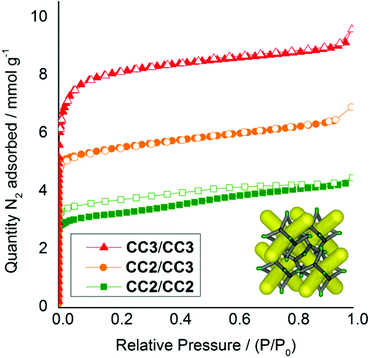 |
| | Fig. 5 Nitrogen sorption isotherms (77 K, 1 bar) for racemic and quasiracemic crystals and cocrystals of CC2 and CC3, all assembled in a diamondoid window–window packing mode (inset). CC2 racemate, green squares; (CC2/CC3) cocrystal quasiracemate orange circles, and; CC3 racemate red triangles. Adsorption isotherms shown as solid symbols; desorption curves as open symbols. | |
It is likely that defects induced by rapid crystallization also contribute to the porosity of the crystal, since we previously found that more rapid precipitation induces higher porosity.19 When opposite chiralities of CC3 are mixed, the dramatic decrease in solubility causes very rapid precipitation. By comparison, the higher solubility of CC2 leads to slower precipitation of cocrystals, and likely fewer defects.15
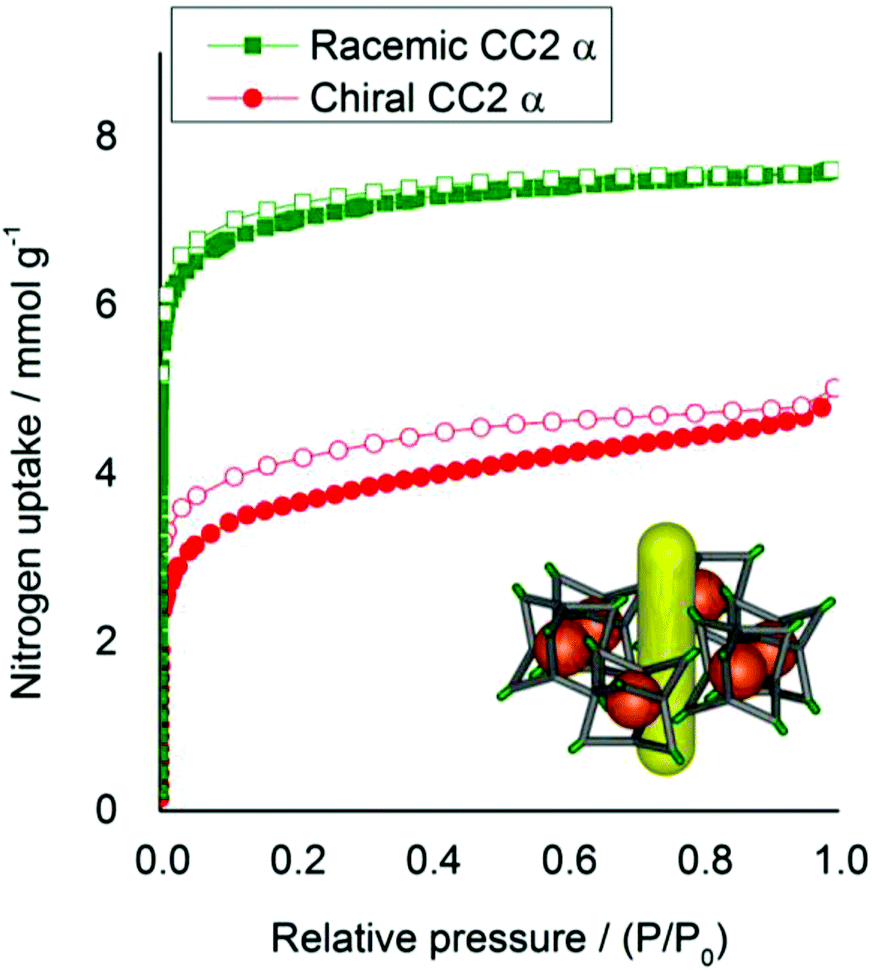
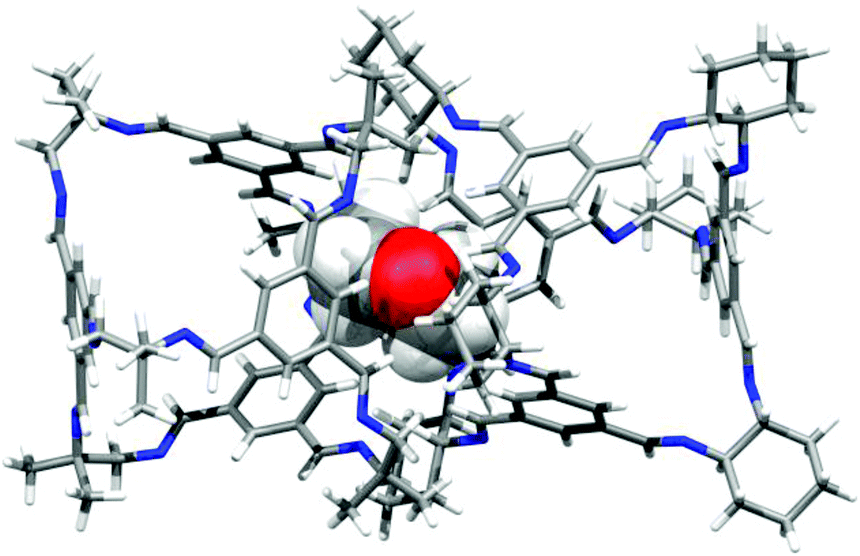
The physical properties of homochiral CC2, made by both direct synthesis and separation by co-crystallisation, were investigated with respect to racemic CC2. Homochiral CC2 is approximately 3 times more soluble than the CC2 racemate (∼600 vs. ∼200 mg mL−1 in chloroform). Given that applications of these porous materials might exploit their solution processability,5 this could provide a practical advantage. Homochiral CC2 also displays contrasting crystallization behavior to the racemate. A previous study found that racemic CC2 formed crystalline phases from all 40 solvent systems tested, most forming the α-phase that has 1-dimensional pore channels running between the cages.15 By contrast, crystallization with 1,4-dioxane afforded a window-to-window-packed β-phase with a 3-D diamondoid pore network.15 Homochiral CC2 exhibits a lower propensity to crystallize, as indicated by its higher solubility, and forms an amorphous phase from many solvent systems (Fig. S5†). However, several crystallisation solvents did generate ordered materials. Notably, 1,4-dioxane produced a homochiral solvate structure that was markedly different from the window-to-window structure observed for racemic CC2 (Fig. 6).15 In the CC2-R dioxane solvate there are no window-to-window interactions between cages; instead, the cages pack window-to-vertex and window-to-arene (Fig. 6). A mixture of ordered, and disordered 1,4-dioxane molecules occupy the cage cavities and interstitial lattice sites between inefficiently packed cages. Unsurprisingly, this structure is not stable to desolvation, but it highlights the marked difference in crystallisation behavior between chiral and racemic forms of the same cage. Some solvents tested produce PXRD patterns that suggest a packing mode that is analogous to the CC2 racemate α-phase, with 1-dimensional inter-cage channels. Crystallization with one such solvent, m-xylene, was scaled up to allow characterisation by single crystal X-ray diffraction (SCXRD) and gas sorption analysis (Fig. 7, 8 and S6†). The sorption isotherms show that chiral CC2 has a lower nitrogen uptake than racemic CC2 when they adopt the same α-packing mode (apparent BET surface areas of 307 and 532 m2 g−1, respectively). We previously investigated whether the internal cavities of the CC2 cages are accessible to gases, or if diffusion is restricted to the 1-dimensional channel running between the cages.8,29 These results suggest that the accessibility of the cage cavities to nitrogen is dependent on the chirality of the structure: that is, open in the case of the racemic CC2 crystals but closed in the homochiral form.
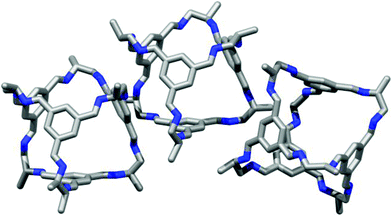 |
| | Fig. 6 Single crystal structure of the homochiral CC2-R 1,4-dioxane solvate CC2-R·4.5(1,4-dioxane)·4.25(H2O), showing window-to-vertex packing (left), and window-to-arene packing (right). | |
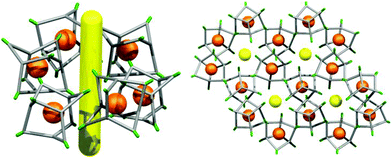 |
| | Fig. 7 Crystal packing of CC2-R molecules in the single crystal structure CC2-S·1.7(m-xylene). 1-D extrinsic voids (yellow line) are located between hexagonally arranged stacks of CC2-R molecules. | |
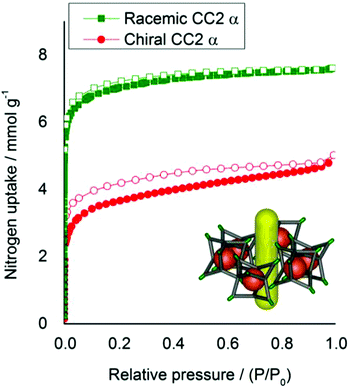 |
| | Fig. 8 Nitrogen sorption isotherms (77 K, 1 bar) for racemic and homochiral CC2, in the 1-dimensional inter-cage channel packing mode. Homochiral CC2, red circles. Racemic CC2, green squares. Adsorption isotherms shown as solid symbols, and desorption curves as open symbols. | |
Chiral CC13 separated by co-crystallisation
CC13 (Fig. 1) was previously reported as a racemate,15 which was shown to have both the highest surface area and the highest solubility of any of the [4 + 6] cages based on 1,3,5-triformylbenzene.15 By analogy with CC2, separation of CC13 into its enantiomerically-pure forms, might improve the solubility and, potentially, the porosity even further. Unlike CC2, it is not possible to synthesize homochiral CC13 directly from chiral precursors because the diamine precursors themselves are achiral.
This leaves chiral separation of the racemate into its constituent helical enantiomers as the only potential route to the chiral form of CC13. Separation of the CC13 racemate by co-crystallisation with CC3 was therefore attempted, following the same methodology validated for CC2.
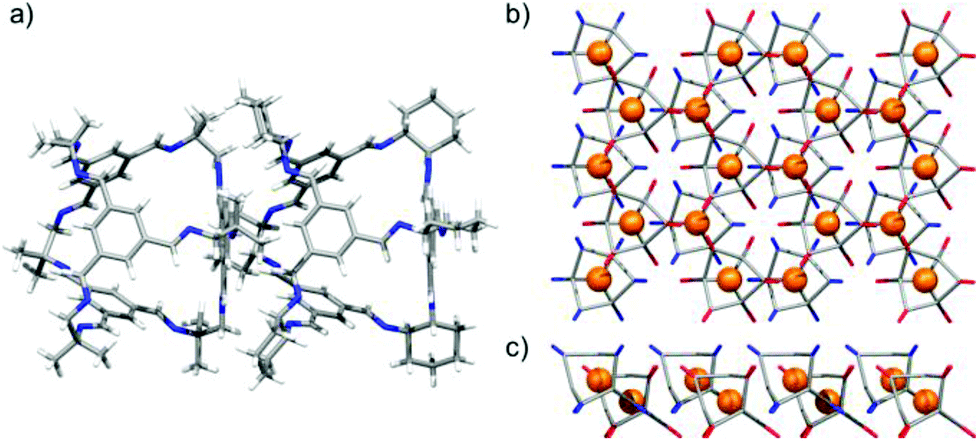
When racemic CC13 and CC3-R were mixed in a 2:1 ratio in CH2Cl2/2-propanol, a cocrystal was formed (Fig. 9 and S7†) with the two cage molecules in the desired 1:1 composition. Surprisingly, however, the expected window-to-window packing mode did not occur, but rather an arene-to-window packing was observed that was reminiscent of CC2-α (Fig. 9a). This is notable since all previous cocrystals in this cage [4 + 6] family show a strong preference for the window-to-window form.17,19,27 More surprisingly, and contrary to the CC2 case, this CC3/CC13 cocrystal selects cages of the same chirality: that is, CC3-R pairs with CC13-R. In the crystal structure, 2D networks of window-to-arene packed cages are stacked along the crystallographic c axis (Fig. 9b & c). The two novel aspects of this cage cocrystal system—the non-window packing and the selection of a single chirality—are likely correlated. The previously observed tendency of this family of cages, to form pseudoracemic cocrystals20,27 is therefore a function of the specific window-to-window interaction and is not generalizable to all analogues. This (CC3-R, CC13-R) cocrystal is less desirable in terms of porosity and it also showed poor stability to desolvation.
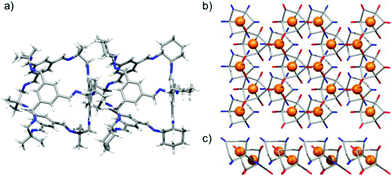 |
| | Fig. 9 Single crystal structure of CC3-R·CC13-R·6(2-propanol)·CH2Cl2·4(H2O) displaying CC3-R window to CC13-R arene packed cages (a), propagating a 2-D network structure of window-to-arene packed CC3-R (red) and CC13-R (blue) cages; perspective view [001] (b), and [010] (c). | |
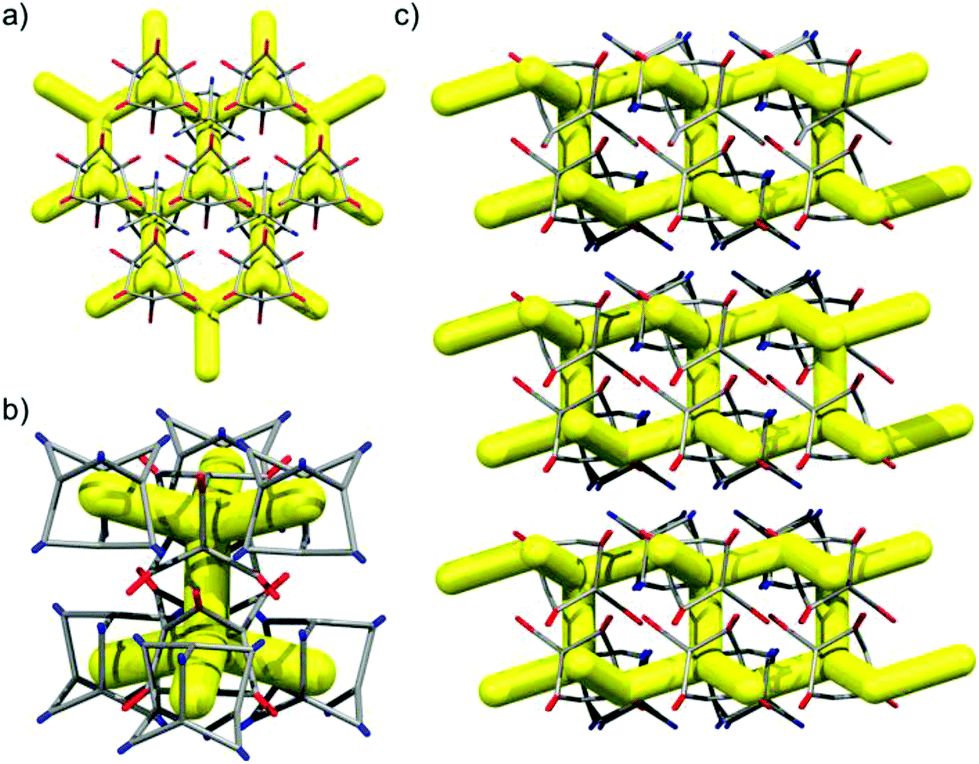
As described previously,15 1,4-dioxane can act as a directing agent to direct racemic cages to pack window-to-window, even for cages where this would not ordinarily be the lowest energy polymorph.15 This technique proved transferable to the (CC3-R, CC13-R) system, which was directed to pack window-to-window (Fig. 10). When CC3-R and CC13 were crystallized from a CH2Cl2/1,4-dioxane solution, single crystals of CC3-R·0.65(CC13-S)·0.35(CC13-R)·1.96(1,4-dioxane)·2.25(CH2Cl2)·6(H2O) were isolated. SCXRD showed that the expected 1:1 cocrystal was formed, with CC3-R and CC13 occupying separate crystallographic sites. However, the CC13 site is disordered, with occupancies of 65% and 35% for the S- and R-enantiomers, respectively. On sites occupied by CC13-S, the cages form the heterochiral window-to-window packing motif with adjacent CC3-R cages, with 1,4-dioxane located in the window cavities (Fig. 10). In contrast, when CC13-R is present, the cage is oriented to form window-to-arene interactions with its CC3 neighbors, as is observed in (CC3-R/CC13-R), rather than forming a homochiral CC3—CC13 window-to-window motif. For simplicity, if we consider CC13-S to occupy all CC13 sites in the crystal lattice, each CC3-R molecule packs in a window-to-window arrangement with three CC13-S cage molecules. This propagates 2-D hexagonal layers in the ab plane that pair with an adjacent layer via window-to-window interactions between CC3-R molecules, which bridge the two networks (Fig. 11b). These double layers stack along the c-axis (Fig. 11c), with CH2Cl2 molecules filling the space in between. For the predominant CC3—CC13 window-to-window interaction, the pairing of cages of opposing chiralities is once again observed, providing that 1,4-dioxane crystallises in the window site in 65% of the crystal structure.
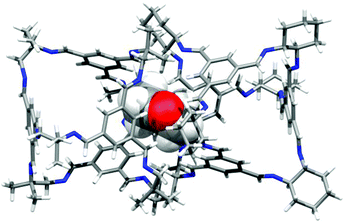 |
| | Fig. 10 1,4-Dioxane directed window-to-window pairing of CC3-R/CC13-S from the single crystal structure CC3-R·0.65(CC13-S)·0.35(CC13-R)·1.96(1,4-dioxane)·2.25(CH2Cl2)·6(H2O). | |
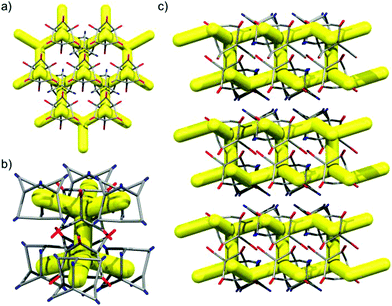 |
| | Fig. 11 Representation of the single crystal structure CC3-R·0.65(CC13-S)·0.35(CC13-R), only CC13-S is shown. Window-to-window packing between CC3-R (red) and CC13-S (blue) propagates a 2-D network structure (a). Two hexagonal 2-D networks of CC3-R–CC13-S are connected by window-to-window packed CC3-R cages (b), that are staggered along c (c). | |
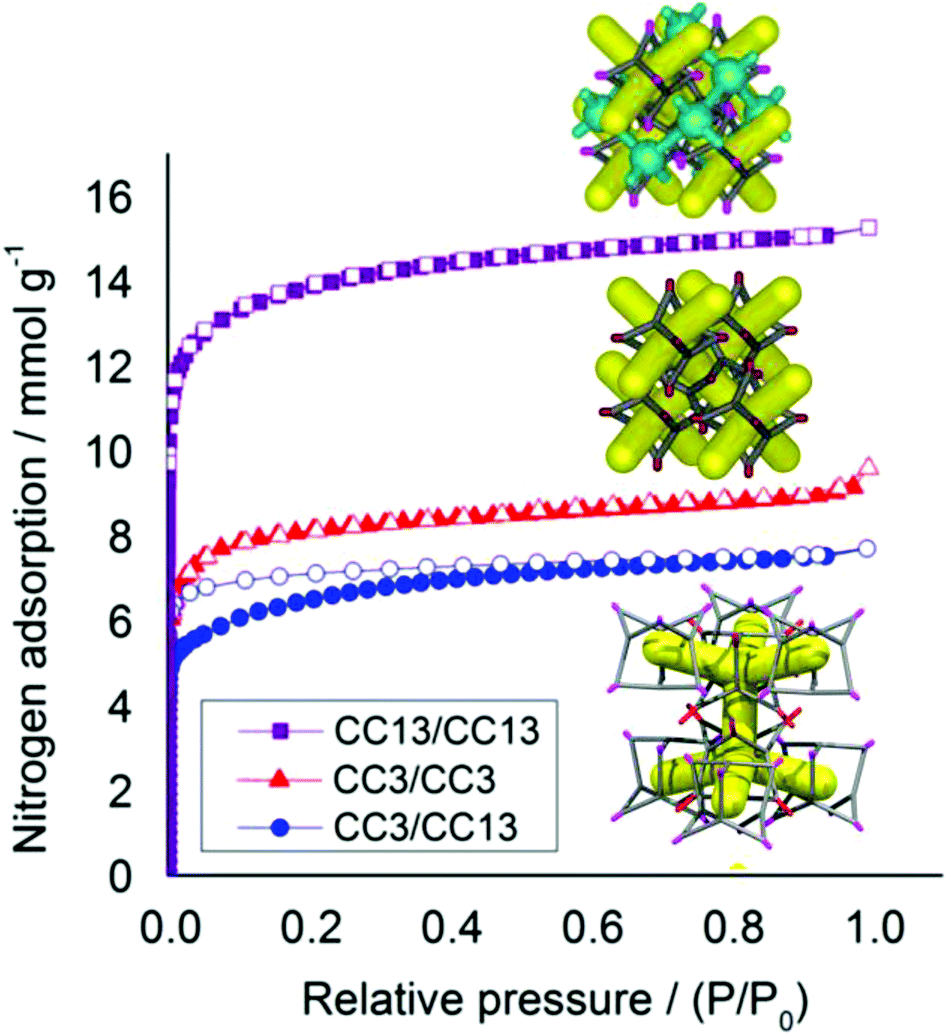
This predominantly window-to-window (CC3, CC13) cocrystal retains crystallinity on desolvation (Fig. S8 and S9†). In contrast to CC2/CC3, this structure does not exhibit sorption behavior that is intermediate between that of its respective parent cages (Fig. 11). Racemic CC13, when packed window-to-window, was shown before to possess a second 3-D pore network,15 formed from a series of connected extrinsic cavities present outside of the cages. This second network forms because the dimethyl vertices of CC13 push the cages apart and open up additional free volume.15 By contrast, when CC13 is co-crystallized with CC3, this additional pore network is blocked by the cyclohexane vertices of CC3, dramatically reducing the porosity to nitrogen in the CC3/CC13 cocrystals with respect to racemic CC13 (Fig. 12). There is also a small difference in packing between crystals of racemic CC3 and those of CC3/CC13 (Fig. 11b). The layered structure of CC3/CC13 means that although most cage windows face the window of an adjacent cage, a small proportion (approximately 1/3) face instead into three cage vertices of the opposing layer. This reduces the porosity of those window sites, slightly reducing the overall uptake in comparison to racemic CC3 (Fig. 12). Sorption of additional gases is reported in Fig. S10.†
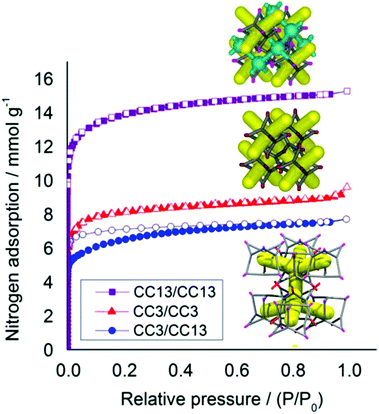 |
| | Fig. 12 Nitrogen sorption isotherms (77 K, 1 bar) for racemic and quasiracemic cocrystals of CC3 and CC13, all assembled in a window–window packing mode. CC3 racemate, red triangles; (CC3, CC13) quasiracemic cocrystal blue circles, and; CC13 racemate, green squares. Adsorption isotherms shown as solid symbols, and desorption curves as open symbols. Insets show the packing of racemic CC13, above, and both racemic CC3 and CC3/CC13, below. Only the CC13 racemate has a second interpenetrating pore channel (shown in blue), approximately doubling its porosity. | |
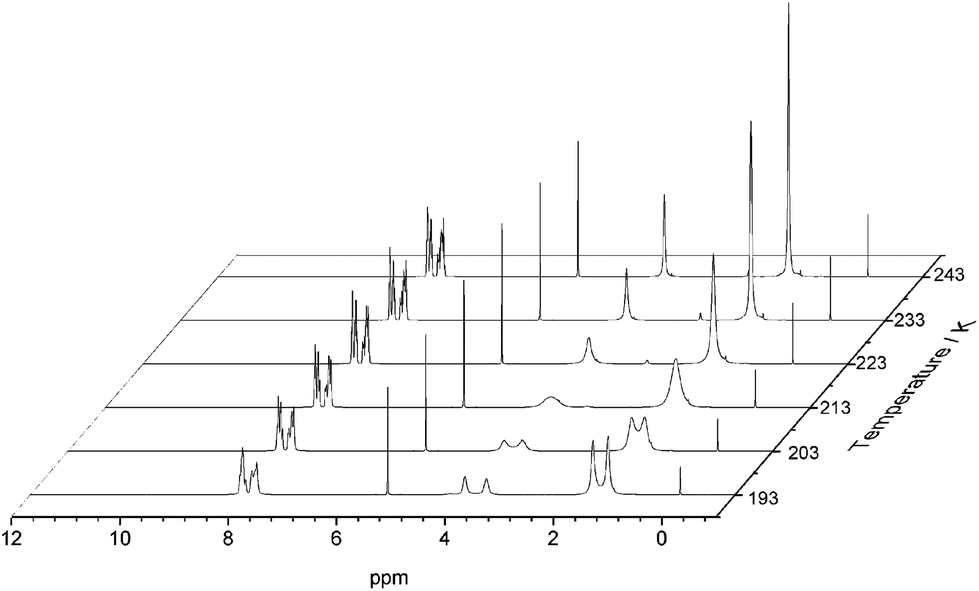
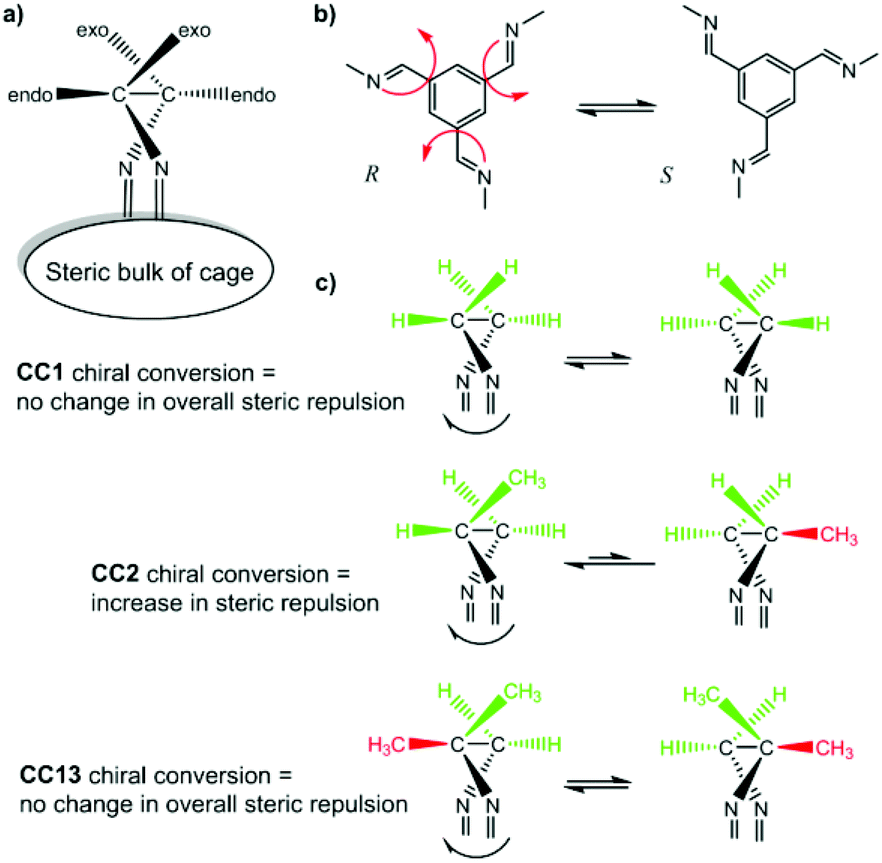
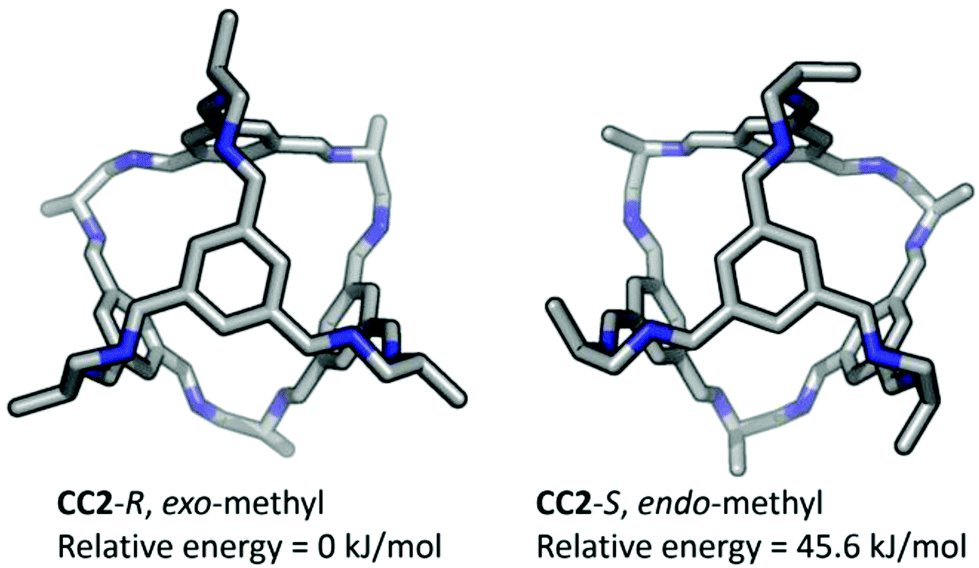
After removal of the cocrystalline CC3-R/CC13-R precipitate, the residual cage present in the filtrate was crystallized. However, SCXRD structural determination revealed the resultant material to be racemic CC13, rather than the expected homochiral form. Racemic material was also obtained after separation of CC13 from the cocrystal by HPLC. Since the CC13 in the cocrystal was shown to be homochiral, this suggests that CC13 can interchange chirality in solution, and revert to a racemic mixture. Variable temperature NMR confirmed that at <203 K the vertex protons of CC13 are resolved, but that they are merged at higher temperatures, indicating that this chiral exchange occurs rapidly at room temperature in solution (Fig. 13 and S11†). Chiral interconversion has been previously documented for CC1.28 However, interconversion does not occur for CC2, even in solution. There is a clear progression in the steric demand of the vertex groups from CC1 (non-functionalised vertices) to CC2 (methyl functionalised vertices) to CC13 (dimethyl functionalised vertices). Why, then, do the single methyl groups of CC2 act to prevent interconversion while the dimethyl groups of CC13 allow it? The methyl groups of CC2 have been shown to exclusively occupy the exo position (away from the cage) in preference to the endo position (towards the cage) (Fig. 14).26 Therefore, for CC2-R (formed with all methyl groups in the more favorable exo position) to ‘flip’ chirality to S requires six methyl groups to occupy the unfavorable endo position. This is not the case for CC1, as it has no sterically unfavorable methyl-cage contacts. Nor is it the case for CC13, because this cage must have an unfavorable methyl-cage contact for either enantiomer. This unfavorable repulsion of the methyl group from the cage may contribute to the lower activation energy for the chiral interconversion of CC13 in comparison to CC1 (24 and 35 kJ mol−1, respectively), as the energy well at either side of the barrier to conversion for CC13 would be expected to be shallower. Therefore, while CC1 and CC13 would be expected to display a symmetric energy landscape to interconversion, with a favorable energy well at either side and a surmountable energetic barrier separating them, CC2 is expected to show an asymmetric energy landscape. This could explain why chiral interconversion of CC2 does not occur at room temperature: the resultant structure is unfavorable, and would revert back to the original chirality. A computational investigation was carried out to confirm this hypothesis. CC2 structures corresponding to different steps of the chiral interconversion were analyzed using density functional theory and their relative energies were compared (see ESI,† computational methods, and Fig. S12 and S13, Table S1†). Results show that flipping the chirality of CC2-R but leaving all the methyl groups in the exo position leads to an extremely strained structure, which lies 135.5 kJ mol−1 above the initial one. Moving all the methyl groups from the exo to the endo position releases the structural strain somewhat, but still leaves the structure 45.6 kJ mol−1 higher than the exo structure of opposite chirality (Fig. 15).
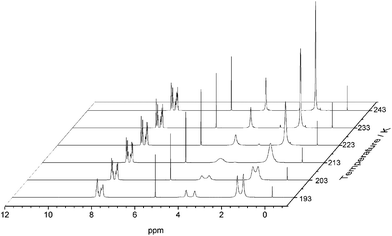 |
| | Fig. 13 Variable temperature 1H NMR of CC13 recorded in CD2Cl2. Splitting of the peaks at 3.80 ppm (s, 1 H, N–CH2–C), and 1.51 ppm (s, 3 H, –C(CH3)2) can be observed at lower temperatures. | |
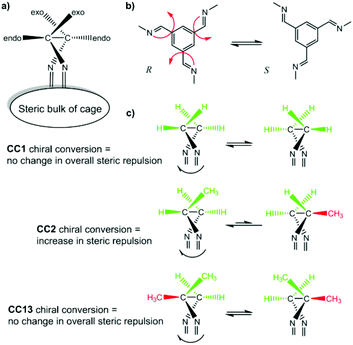 |
| | Fig. 14 Schematic of CC1, CC2, and CC13 chiral interconversion. Both CC1 and CC13 should exhibit symmetrical energy landscapes when interchanging from R to S chirality, whereas CC2 favours the as-formed chirality with all methyl groups in the favourable exo position. | |
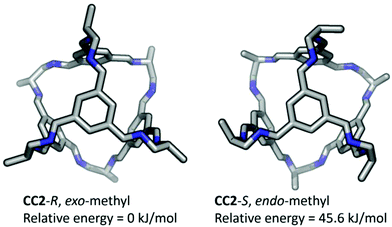 |
| | Fig. 15 Simulated structures of CC2-R with methyl groups shown in the exo position, as observed in the crystal structure, and after conversion to CC2-S, with the methyl groups moved to the endo position, which results in a higher relative energy. | |
Conclusions
A homochiral form of porous organic cage CC2 is reported for the first time. It is possible to isolate this chiral form from a racemic CC2 mixture by selective crystallization as well as by direct synthesis from the chiral diamine precursor. Homochiral CC2 was found to have improved solubility, and both its crystal packing and resultant sorption properties were modified with respect to racemic CC2. The chirality of the CC2 cages was also shown to strongly affect their tendency to catenate because only homochiral catenanes are formed. The synthesis of two additional cocrystal systems, CC2/CC3 and CC3/CC13, illustrates the transferability of this rational design approach, although in the case of CC3/CC13, a directing solvent, dioxane, was needed to achieve an isoreticular window-to-window packing mode. Chiral interconversion occurs in solution for CC1 and CC13, but not for CC2 where the single vertex methyl groups lock the chirality in place. As the field of porous organic cages becomes more developed, the understanding of subtle relationships between factors such as chirality, crystal packing, and cage flexibility will play an increasingly important role. Moreover, the strong effect of chirality on solubility is relevant to the design of next-generation porous organic liquids, where porosity depends on producing a liquid with a high concentration of cage cavities.
Acknowledgements
We acknowledge the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013)/ERC through grant agreement n. 321156 (ERC-AG-PE5-ROBOT) and EPSRC (grants EP/N004884/1) for funding. T. H. and K. E. J. thank the Royal Society for University Research Fellowships. We acknowledge Rob Clowes for assistance with sorption measurements, Dan Holden and Rebecca Greenaway for useful discussions, and Marc Schmidtmann for preliminary crystallographic investigations. We thank Diamond Light Source for access to beamline I11 (EE12336) and I19 (MT11231) that contributed to the results presented here.
Notes and references
-
Handbook of Porous Solids, ed. F. Schüth, K. S. W. Sing and J. Weitkamp, Wiley VCH, Heidelberg, 2002 Search PubMed
 .
.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed .
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortes, A. P. Cote, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed .
- Y. H. Xu, S. B. Jin, H. Xu, A. Nagai and D. L. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC .
- T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS .
- J. Tian, P. K. Thallapally and B. P. McGrail, CrystEngComm, 2012, 14, 1909–1919 RSC .
- G. Zhang and M. Mastalerz, Chem. Soc. Rev., 2014, 43, 1934–1947 RSC .
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed .
- M. E. Briggs and A. I. Cooper, Chem. Mater., 2017, 29, 149–157 CrossRef CAS PubMed .
- Y. H. Jin, B. A. Voss, R. D. Noble and W. Zhang, Angew. Chem., Int. Ed., 2010, 49, 6348–6351 CrossRef CAS PubMed .
- Y. Jin, Y. Zhu and W. Zhang, CrystEngComm, 2013, 15, 1484–1499 RSC .
- M. Mastalerz, Angew. Chem., Int. Ed., 2010, 49, 5042–5053 CrossRef CAS PubMed .
- A. Avellaneda, P. Valente, A. Burgun, J. D. Evans, A. W. Markwell-Heys, D. Rankine, D. J. Nielsen, M. R. Hill, C. J. Sumby and C. J. Doonan, Angew. Chem., Int. Ed., 2013, 52, 3746–3749 CrossRef CAS PubMed .
- G. Zhang, O. Presly, F. White, I. M. Oppel and M. Mastalerz, Angew. Chem., Int. Ed., 2014, 53, 1516–1520 CrossRef CAS PubMed .
- T. Hasell, J. L. Culshaw, S. Y. Chong, M. Schmidtmann, M. A. Little, K. E. Jelfs, E. O. Pyzer-Knapp, H. Shepherd, D. J. Adams, G. M. Day and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 1438–1448 CrossRef CAS PubMed .
- J. T. A. Jones, D. Holden, T. Mitra, T. Hasell, D. J. Adams, K. E. Jelfs, A. Trewin, D. J. Willock, G. M. Day, J. Bacsa, A. Steiner and A. I. Cooper, Angew. Chem., Int. Ed., 2011, 50, 749–753 CrossRef CAS PubMed .
- A. G. Slater, M. A. Little, A. Pulido, S. Y. Chong, D. Holden, L. Chen, C. Morgan, X. Wu, G. Cheng, R. Clowes, M. E. Briggs, T. Hasell, K. E. Jelfs, G. M. Day and A. I. Cooper, Nat. Chem., 2017, 9, 17–25 CAS .
- D. Beaudoin, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2017, 56, 1244–1248 CrossRef CAS PubMed .
- T. Hasell, S. Y. Chong, K. E. Jelfs, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2012, 134, 588–598 CrossRef CAS PubMed .
- T. Hasell, S. Y. Chong, M. Schmidtmann, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2012, 51, 7154–7157 CrossRef CAS PubMed .
- L. Chen, P. S. Reiss, S. Y. Chong, D. Holden, K. E. Jelfs, T. Hasell, M. A. Little, A. Kewley, M. E. Briggs, A. Stephenson, K. M. Thomas, J. A. Armstrong, J. Bell, J. Busto, R. Noel, J. Liu, D. M. Strachan, P. K. Thallapally and A. I. Cooper, Nat. Mater., 2014, 13, 954–960 CrossRef CAS PubMed .
- A. Kewley, A. Stephenson, L. Chen, M. E. Briggs, T. Hasell and A. I. Cooper, Chem. Mater., 2015, 27(9), 3207–3210 CrossRef CAS .
- S.-M. Xie, J.-H. Zhang, N. Fu, B.-J. Wang, L. Chen and L.-M. Yuan, Anal. Chim. Acta, 2016, 903, 156–163 CrossRef CAS PubMed .
- J.-H. Zhang, S.-M. Xie, L. Chen, B.-J. Wang, P.-G. He and L.-M. Yuan, Anal. Chem., 2015, 87, 7817–7824 CrossRef CAS PubMed .
- J.-H. Zhang, S.-M. Xie, B.-J. Wang, P.-G. He and L.-M. Yuan, J. Chromatogr. A, 2015, 1426, 174–182 CrossRef CAS PubMed .
- T. Hasell, X. F. Wu, J. T. A. Jones, J. Bacsa, A. Steiner, T. Mitra, A. Trewin, D. J. Adams and A. I. Cooper, Nat. Chem., 2010, 2, 750–755 CrossRef CAS PubMed .
- J. T. A. Jones, T. Hasell, X. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Nature, 2011, 474, 367–371 CrossRef CAS PubMed .
- K. E. Jelfs, F. Schiffmann, J. T. A. Jones, B. Slater, F. Cora and A. I. Cooper, Phys. Chem. Chem. Phys., 2011, 13, 20081–20085 RSC .
- T. Hasell, J. A. Armstrong, K. E. Jelfs, F. H. Tay, K. M. Thomas, S. G. Kazarian and A. I. Cooper, Chem. Commun., 2013, 49, 9410–9412 RSC .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr01301a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
M. A.
Little
*a,
M. A.
Little